

Abstract
Conversion of mesenchymal stem cells into terminally differentiated adipocytes progresses sequentially through regulated transcriptional steps. While it is clear that the late phases of adipocyte maturation are governed by the nuclear receptor peroxisome proliferator-activated receptor gamma (PPARγ), less is known about the transcriptional control of the initial stages of differentiation. To identify early regulators, we performed a small interfering RNA (siRNA) screen of Forkhead-box genes in adipocytes and show here for the first time that the winged helix factor Foxa3 promotes adipocyte differentiation by cooperating with C/EBPβ and -δ to transcriptionally induce PPARγ expression. Furthermore, we demonstrate that mice with genetic ablation of Foxa3 have a selective decrease in epididymal fat depot and a cell-autonomous defect to induce PPARγ specifically in their visceral adipocytes. In obese subjects, FOXA3 is differentially expressed in visceral and subcutaneous adipose depots. Overall, our study implicates Foxa3 in the regulation of adipocyte differentiation and depot-selective adipose tissue expansion.
INTRODUCTION
Adipose tissue is a critical organ for maintaining energy homeostasis. Mammals have several types of fat tissues, which differ primarily in their ability to store and utilize lipids as fuel (1). Although, in general, white fat contains cells specialized in energy storage and secretion of signaling molecules (2), adipocytes from distinct anatomic depots differ significantly in their gene expression profiles and their adipokine repertoire (3). These intrinsic differences are thought to be critical to how adipose depots contribute differentially to the etiology of the metabolic syndrome and diabetes (4).
Fat cells develop through a sequential series of molecular events orchestrated in response to developmental cues or select nutritional and hormonal stimuli. The differentiation of a preadipocyte into a bona fide adipocyte is regulated at the transcriptional level by the nuclear receptor peroxisome proliferator-activated receptor gamma (PPARγ), which acts as a central regulatory node for the induction and maintenance of fat cell differentiation and function (5). Several transcription regulators have been implicated in the control of early differentiation by modulating the upstream events leading to the induction or the suppression of PPARγ expression (2, 6). However, little is known about the specific contribution of these factors to depot-specific lipid accumulation.
Forkhead-box (Fox) proteins are a large family of transcription factors shown to be critically involved in the regulation of aging, organ development, and cell and organismal survival as well as in metabolism (7). Fox family members contain a conserved Forkhead-box motif and a DNA binding domain but otherwise diverge in their remaining regions. A number of Fox factors play critical roles in early developmental specification events of organs, such as the members of the Foxa subfamily in liver and pancreas development (8–10). Although a few Fox proteins have been shown to influence adipose tissue biology by inhibiting white fat differentiation (11–13), to the best of our knowledge, a systematic analysis of the role of this family of factors in early events of fat development has not been performed. Given the critical importance of Forkhead proteins in the development and the differentiation of other tissues and organs (7, 14), we systematically investigated their function in adipocyte differentiation by performing a genetic screen to assess the specific role of each family member in this process. Our analysis identified Foxa3 as a positive regulator of adipocyte differentiation and lipid accumulation and demonstrated that Foxa3 modulates PPARγ expression in vitro and in vivo and that its ablation in mice selectively decreases epididymal adipose tissue expansion.
MATERIALS AND METHODS
siRNA reagents and plasmids.
Small interfering RNAs (siRNAs) targeting each individual Forkhead factor or PPARγ, C/EBPα, -β, or -δ and an siRNA control (siGENOMEsiRNA reagents, SMARTpool, and siCONTROL nontargeting siRNA) were purchased from Dharmacon. Foxa3 cDNA was amplified from a mouse liver cDNA library with primers containing a Kozak and a Flag sequence, namely, F (5′-AACAGAATTCGCCACCATGGACTACAAAGACGATGACGATAAACT GGGCTC AGTGAAGAT-3′) and R (5′-CCCGCTCTCTGCTTAATGCATCCTAGGATATCACAA-3′), cloned into pcDNA3.1 (Invitrogen) at the EcoRI and EcoRV sites and subcloned into pMSCV retroviral vector (Clontech) at the EcoRI site. Foxa3-DBD mutant R162P/N165I/M202R/R210P was generated by site-directed mutagenesis (Stratagene) with primers listed in Table S1 in the supplemental material. Plasmids expressing either C/EBPβ or C/EBPδ were purchased from Addgene. The C/EBPα plasmid was a gift of Kai Ge. The mouse PPARγ promoter (−2200 to +1) was amplified from genomic DNA with primers containing NheI and HindIII sites, namely, F (5′-AACAGCTAGCCCCCCACTTTCACCATAGTC-3′) and R (5′-TTGTAAGCTTAACAG CATAAAACAGAGATT-3′, and cloned into pGL3-basic vector (Promega).
Differentiation assays.
To induce adipocyte differentiation, confluent 10T1/2 cells, either transfected with 100 nM siRNA or overexpressing Foxa3, PPARγ, or vector, were cultured in high-glucose Dulbecco's modified Eagle's medium (DMEM) containing 10% fetal bovine serum (FBS) supplemented with 5 μg/ml insulin and 10 μM troglitazone, while 3T3-L1 cells were stimulated with DMEM containing 10% FBS and MDI (5 μg/ml insulin, 0.5 mM isobutylmethylxanthine, and 5 mM dexamethasone) for 48 h and subsequently cultured in DMEM containing 10% FBS supplemented with 5 μg/ml insulin (maintenance medium). To generate wild-type (WT) and Foxa3-null stromal-vascular fractions (SVF) of cells from inguinal and visceral depots, epididymal and inguinal adipose tissues were isolated from WT and Foxa3-null mice, minced, and subjected to collagenase digestion (1 mg/ml) for 45 min in a buffer containing 0.123 M NaCl, 5 mM KCl, 1.3 mM CaCl2, 5 mM glucose, 100 mM HEPES, and 4% bovine serum albumin (BSA). Digested tissues were filtered through a 100-μm-pore-size nylon screen and centrifuged for 5 min at 150 × g. Cell pellets were resuspended in DMEM containing 20% FBS, 25 mM glucose, 20 mM HEPES, and 1% penicillin and streptomycin (pen/strep), and the culture medium was changed daily. For differentiation assays, SVF obtained from visceral and subcutaneous fat depots were stimulated for 48 h with medium containing 10% FBS, MDI, and 10 μM troglitazone and subsequently cultured in maintenance medium.
Protein analysis.
Immunoprecipitations were performed according to protocols described previously (15). Proteins were separated by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membranes (Pierce). Blots were incubated with primary antibodies overnight at 4°C and at room temperature for 1 h with secondary antibodies. Immune complexes were visualized by using ECL Plus (Pierce), following the manufacturer's instructions.
EMSA.
For electromobility shift assays (EMSA), nuclear extracts obtained from U2OS cells transfected with Foxa3-WT or Foxa3-DBD mutant expression plasmids were incubated with a biotin-labeled oligonucleotide (5′-AACTATTCCTTTTTATAGAATTTGGATAGCAGTAACA-3′) corresponding to the putative Foxa binding site identified in the PPARγ promoter. Supershift was obtained by adding 0.4 μg of Foxa3 antibody, and a competition assay was performed using 200-fold unlabeled probe. After incubation, DNA-protein complexes were separated on a 4% nondenaturing polyacrylamide gel followed by transfer to a positively charged nylon membrane (Pierce). After UV cross-linking, detection was performed following the manufacturer's protocol (Pierce).
ChIP assays.
Chromatin immunoprecipitation (ChIP) assays were performed using a ChIP assay kit (Upstate), according to the manufacturer's instructions. PCR was performed using primers amplifying the C/EBP binding site (5′-GGCCAAATACGTTTATCTGGTG-3′ and 5′-TCACTGTTCTGTGAGGGGC-3′), the Foxa3 binding site (5′-TCACTTAAACATCAACCATTGGA-3′ and 5′-GGTCCAAAATGTTACTGCTATCC-3′), or the TATA box site (5′-GCCCCTCACAGAACAGTGAA-3′ and 5′-AACAGCATAAAACAGAGATTT-3′). PCR products were run on an agarose gel and visualized by ethidium bromide staining. Relative enrichment was quantified by real-time PCR.
Gene expression analysis.
RNA was extracted using TRIzol (Invitrogen), cDNA was generated according to manufacturer's instructions (ABI), and real-time PCR was carried out with SYBR green (Roche). 18S was used for normalization. Mouse Foxa3 mRNA levels were detected with the following primers: F (5′-TGAATCCTGTGCCCACCAT) and R (3′-AGCTGAGTGGGTTCAAGGTCAT). Human FOXA3 was detected with TaqMan primer sets (ABI).
Transfections and luciferase assays.
10T1/2 and 3T3-L1 cells were transfected using a Nucleofector 96-well system (Amaxa) and U2OS cells (ATCC) with FuGENE 6 (Roche), according to the manufacturer's instructions. Luciferase activity was assayed 48 h after transfection, according to the manufacturer protocol (Promega Corporation), using Victor3V (PerkinElmer).
Antibodies.
Anti-Flag M2 beads and anti-Flag antibody were purchased from Sigma, and anti-C/EBPα, anti-C/EBPβ, anti-C/EBPδ, and anti-HNF3γ (Foxa3) antibodies were purchased from Santa Cruz. Secondary antibodies were purchased from Amersham and Vector Labs.
Mice.
All animal experiments were performed according to the guidelines of the National Institute of Diabetes and Digestive and Kidney Disease Animal Care and Use Committee (ACUC). Mice were housed in 12-h light/dark cycles (light on at 6 a.m.) and allowed ad libitum access to food and water. Mice were genotyped by PCR with the following primers: Foxa3-F (forward) (5′-TCCCAAGCTTGGGCACTGGTG GCCA-3′), Foxa3-R (reverse) (5′-GTGGCAGCTGTAGTGGTGGCAG-3′), and lacZ (5′-CGCCATTCGCCATTCAGGCTGC-3′). Mice were sacrificed by ketamine (90 mg/kg of body weight) and xylazine (10 mg/kg) intraperitoneal (i.p.) injection according to NIH ACUC animal study approved procedures. Harvested tissues were snap-frozen in liquid nitrogen or fixed using 4% paraformaldehyde (EMS) and embedded in paraffin for hematoxylin and eosin staining. Adult WT and Foxa3-null mice were fed a high-fat diet (HFD) for 22 weeks (Research Diets, D12492). C57BL/6J mice fed a normal diet or a HFD were purchased from The Jackson Laboratory.
Immunohistochemistry.
Tissues were dissected and fixed in 10% formalin and embedded in paraffin according to standard procedures. Paraffin-embedded tissues were cut in sections of 5 μm thickness and stained with hematoxylin and eosin (Histoserv) for morphological analysis, following the manufacturer's instructions (Vector Labs). Stained slides were analyzed using a microscope at ×200 magnification (Olympus). Adipocyte sizes were measured using ImageJ software (version 1.45s). At least 200 cells from each animal group were measured.
Human adipose tissue samples.
Fat biopsy specimens from the subcutaneous adipose tissue (SAT) of the midanterior abdominal region and visceral adipose tissue (VAT) of the omental region were obtained from 14 obese women with a body mass index (BMI) of 45.6 kg/m2 (range, 36.1 to 55.6) recruited at the Stanford's Bariatric Surgery Program, with a mean age of 40 years (range, 23 to 59), as previously described (16). The study was approved by the Stanford University Human Subjects Committee. All subjects gave written informed consent.
ITT, GTT, and serum tests.
For insulin tolerance tests (ITT), mice received an intraperitoneal injection of insulin (1 mU/kg). For glucose tolerance tests (GTT), mice were fasted overnight and injected intraperitoneally with a glucose solution in saline solution (2 g/kg). Plasma glucose levels were measured from tail blood before or at 15, 30, 60, 90, and 120 min after insulin or glucose injections. Blood samples for measurements of serum adiponectin, insulin, and cholesterol levels were collected using a back-eye needle. Serum parameters were measured with kits obtained from Linco Research and Thermo.
Statistical analysis.
All experiments were repeated at least three times. Results are presented as means ± standard errors of the means (SEM). Student's t test and one- or two-way analysis of variance (ANOVA) followed by appropriate posttests were performed with GraphPad software (GraphPad). A P value of less than 0.05 was considered statistically significant.
RESULTS
siRNA screening of Forkhead family members identified Foxa3 as a positive regulator of adipocyte differentiation.
Forkhead-box proteins regulate development and metabolism, and several of them have been previously shown to be critically involved in lineage determination of liver and pancreas (8–10). To date, only three members of the Fox family, Foxo1, Foxa2, and Foxc2, have been studied in the context of adipocyte physiology and shown to affect adipocyte differentiation negatively (11–13). To systematically assess the impact of each member of the mammalian Fox family on adipocyte differentiation, we expressed 35 siRNAs targeting each of the mouse Fox genes in the 10T1/2 mesenchymal stem cell line and measured the induction of the adipocyte marker aP2 as a molecular readout for adipose differentiation. Figure S1 in the supplemental material shows that the mRNA levels of Fox factors were lowered by their respective siRNAs, indicating that knockdown occurred successfully. As shown in Fig. 1A, knockdown of 18 Fox proteins affected adipocyte differentiation. Among those, we also identified Foxo1, Foxa2, and Foxc2, which acted as negative regulators of differentiation, as demonstrated in previous studies. Interestingly, in sharp contrast to the positive effects observed with the majority of Fox siRNAs, knockdown of Foxa3 and Foxn2 decreased adipocyte differentiation (Fig. 1A). We then tested the effects of the eight siRNAs exerting the strongest positive and negative effects in our primary screen in the 3T3-L1 preadipocyte cell line and demonstrated that only knockdown of Foxd4, Foxi1, and Foxa3 altered differentiation (Fig. 1B). Given the strong effects of Foxa3 siRNA on two in vitro models of adipocyte differentiation and our interest in understanding how Fox proteins promote this process, we focused our investigation on the function of Foxa3.
Fig 1.

Global analysis of Forkhead-box proteins reveals a role for Foxa3 in promoting adipocyte differentiation. (A) aP2 mRNA levels in 10T1/2 cells expressing 35 distinct siRNAs targeting Forkhead genes relative to siRNA control, 72 h after induction of differentiation. (B) aP2 mRNA levels in 3T3-L1 cells after siRNA knockdown of selected Forkhead factors, 72 h after induction of differentiation. (C) Relative mRNA levels of adipocyte markers in 10T1/2 cells expressing either control siRNA (si-control) or Foxa3-siRNA (si-Foxa3), 72 h after induction of differentiation. (D) Oil Red O staining of differentiated 10T1/2 cells expressing either vector or Foxa3 and relative mRNA levels of adipocyte differentiation markers in 10T1/2 cells expressing either vector or Foxa3, at day 6 of differentiation. Data represent means ± SEM (**, P < 0.01; ***, P < 0.001).
To fully characterize the molecular details of the regulation of adipocyte differentiation by Foxa3, we analyzed the effects of silencing or ectopically expressing Foxa3 in 10T1/2 cells. As shown in Fig. 1C, Foxa3 knockdown led to a significant decrease in mRNA levels of adipogenic markers such as aP2, PPARγ, C/EBPα, -β, and -δ, perilipin, and adiponectin. Conversely, when Foxa3 was ectopically expressed in 10T1/2 cells, we observed an increase in lipid-storing cells (Fig. 1D). These morphological changes were paralleled by an increase in mRNA levels of adipocyte markers, as shown in Fig. 1D. Overall, our data demonstrated a proadipogenic role of Foxa3 in vitro.
Foxa3 is an upstream regulator of PPARγ.
To gain insights into the mechanisms of the action of Foxa3, we first examined the temporal expression pattern of Foxa3 during adipogenic differentiation of 10T1/2 cells. Foxa3 mRNA levels were induced as early as day 2 of adipocyte differentiation (Fig. 2A), paralleled the expression of the early regulators C/EBPβ and C/EBPδ, and preceded the expression of C/EBPα and PPARγ. To further determine when Foxa3 would exert its critical function in the course of 3T3-L1 differentiation, we analyzed the temporal expression pattern of PPARγ and C/EBP family members during the differentiation of si-control- and si-Foxa3 3T3-L1-expressing cells. As shown in Fig. S2 in the supplemental material, PPARγ and C/EBPα mRNA levels were decreased when Foxa3 was knocked down, while no effects were observed on the upstream PPARγ regulators, C/EBPβ and C/EBPδ. The sequence of Foxa3 expression observed in 10T1/2 and 3T3-L1 cells suggested that Foxa3 may lie upstream of C/EBPα and PPARγ. To address this experimentally, we analyzed the effects of siRNA-mediated suppression of early and late effectors of differentiation in 10T1/2 cells ectopically expressing either vector, Foxa3 or PPARγ. While siRNA-mediated suppression of either early (C/EBPβ and -δ) or late (C/EBPα and PPARγ) proadipogenic factors decreased the differentiation of 10T1/2-control or 10T1/2-Foxa3 cells, as measured by the mRNA levels of the adipocyte marker aP2, the differentiation of 10T1/2-PPARγ cells was impaired only when the expression of late regulators was reduced (Fig. 2B). These data suggested that Foxa3 lies upstream of PPARγ and that it requires C/EBPα and PPARγ for its full effects on differentiation.
Fig 2.

Foxa3 is involved in early stages of adipocyte differentiation. (A) Relative mRNA levels of Foxa3 and of adipocyte markers during 10T1/2 cell differentiation. (B) aP2 mRNA levels in 10T1/2 cells expressing vector, Foxa3, or PPARγ in the presence of either control siRNA or siRNAs targeting C/EBPs or PPARγ, 72 h after induction of differentiation. Values are expressed as relative to the control siRNA values. Data represent means ± SEM (*, P < 0.05; **, P < 0.01; ns, nonsignificant).
To determine whether Foxa3 is involved in the regulation of PPARγ levels, we analyzed the PPARγ promoter to identify putative Foxa3 binding sites. Through in silico analysis, we found a putative Foxa response element in close proximity to a C/EBP binding region, within 2 kb upstream of the PPARγ start site (Fig. 3A). To investigate whether Foxa3 could regulate PPARγ expression through this site, we cloned 2,000 bp of the PPARγ2 promoter region upstream of the ATG site in a luciferase reporter vector and assessed the ability of Foxa3 to activate transcription through the putative Foxa-responsive element. Luciferase reporter assays showed that while Foxa3 alone cannot activate transcription, it acts synergistically with C/EBPβ or C/EBPδ to activate the PPARγ2-promoter-driven reporter (Fig. 3B). To further investigate the mechanism of cooperation between Foxa3 and C/EBP factors, we assessed their direct interaction. Immunoprecipitation assays revealed that Foxa3 and C/EBPβ and C/EBPδ exist in the same protein complex (Fig. 3C), suggesting that these factors act coordinately at the PPARγ2 promoter. ChIP assays confirmed that Foxa3 binds to the PPARγ2 promoter at both the Foxa- and C/EBP-responsive elements (Fig. 3D). To determine whether the modulation of Foxa3 levels has effects on the binding of C/EBPβ and -δ to the PPARγ2 promoter, we performed Foxa3 knockdown experiments followed by ChIP assays. As shown in Fig. 3E, ChIP assays revealed that a reduction in Foxa3 levels led to decreased C/EBPβ and -δ occupancy at the C/EBP-responsive element. These data suggest a role of Foxa3 as a pioneer factor.
Fig 3.

Foxa3 regulates PPARγ expression in cooperation with C/EBPβ and C/EBPδ. (A) C/EBP and Foxa DNA binding sites at the PPARγ2 promoter. (B) Analysis of the transcriptional activity of Foxa3 and C/EBPs on a PPARγ2-promoter luciferase reporter. a.u., arbitrary units. (C) Foxa3 and C/EBPα, -β, or -δ coimmunoprecipitations (IP). WB, Western blot. (D) ChIP assay on Foxa and C/EBP sites at the PPARγ2 promoter at day 2 of adipocyte differentiation. (E) ChIP assay at the C/EBP site present at the PPARγ2 promoter in si-control- and si-Foxa3 3T3-L1-expressing cells at day 2 of adipocyte differentiation. (F) EMSA performed at the Foxa3 binding site using a nuclear extract obtained from U2OS cells expressing either the Foxa3 wild type (Foxa3-WT) or a Foxa3 DNA binding domain mutant (Foxa3-DBD). Supershift (SS) analysis was performed using an anti-Foxa3 antibody. (G) Coimmunoprecipitation of Foxa3-DBD mutant with C/EBPδ. (H) Luciferase activity of Foxa3-WT and Foxa3-DBD mutant on the PPARγ2-promoter-driven luciferase reporter. (I) Oil Red O staining of 10T1/2 cells expressing vector, Foxa3-WT, or Foxa3-DBD mutant at day 6 after induction of adipocyte differentiation. (J) Relative aP2 mRNA levels of 10T1/2 cells expressing vector, Foxa3-WT, or Foxa3-DBD mutant at day 6 of adipocyte differentiation. Data represent means ± SEM (***, P < 0.001; ###, P < 0.001; **, P < 0.01; ns, nonsignificant).
In order to determine whether binding to DNA was required for the transcriptional cooperation between Foxa3 and C/EBPs, we generated a Foxa3 mutant lacking the DNA binding domain (Foxa3-DBD) and confirmed its inability to bind to DNA by EMSA (Fig. 3F). While Foxa3-DBD retained its capacity to dock C/EBPδ (Fig. 3G), it no longer cooperated with C/EBPδ to activate transcription (Fig. 3H), suggesting that DNA binding is required for Foxa3 transcriptional cooperation with C/EBPs. To determine whether Foxa3 required DNA binding for its proadipogenic function, we ectopically expressed Foxa3-WT and Foxa3-DBD in 10T1/2 cells and induced differentiation. Figure 3I and J show that Foxa3 was no longer able to promote adipocyte differentiation in the absence of its DNA binding domain, as demonstrated by Oil red O staining and by aP2 mRNA levels. Together, these data indicate that Foxa3 promotes adipocyte differentiation through direct DNA binding and via physical and functional cooperation with selected C/EBPs.
Foxa3-null mice have a selective decrease in epididymal adipose depot.
Foxa3-null mice have been previously shown to be viable without abnormalities when fed a normal diet (17, 18). Given our results, we tested the role of Foxa3 in vivo under conditions of nutritional challenge. First, we analyzed wild-type (WT) and Foxa3-null mice maintained on a milk diet prior to weaning. Detailed analysis of fat tissues obtained from 2-week-old WT and Foxa3-null mice showed a selective decrease in the amount of epididymal fat in Foxa3-null mice compared to WT mice (Fig. 4A and B), while no differences were detected in inguinal fat tissues from either WT or Foxa3-null mice, suggesting a depot-specific defect. Further molecular analysis also revealed reduced levels of adipocyte differentiation markers in the epididymal depot of Foxa3-null mice compared to WT mice (Fig. 4C), while no differences in the levels of these markers were found in inguinal fat obtained from WT and Foxa3-null mice. Similarly, we observed a selective decrease in perigonadal fat depot and a reduction in the levels of PPARγ and of its target genes also in Foxa-null females (see Fig. S3A and B in the supplemental material). These data suggest that Foxa3 ablation in vivo selectively impacts epididymal and perigonadal adiposity and reduces the levels of PPARγ and its target genes in these visceral fat depots. To assess whether Foxa3 exerted a depot-selective control on adiposity by controlling the size of the progenitor pool in the SVF, we compared the amounts of adipocyte precursors cells (APC) present in the epididymal depot of WT and Foxa3-null mice. As shown in Fig. S4 in the supplemental material, WT and Foxa3-null mice have comparable amounts of adipocyte precursors, suggesting that Foxa3 selectively affects the differentiation capabilities of preadipocytes rather than the size of the precursor pool. We next assessed whether the phenotype of 2-week-old Foxa3-null mice would be recapitulated in adult mice. Although we found no differences in the adipose depot amounts in adult WT and Foxa3-null mice fed a normal chow diet (Fig. 4D), as previously reported (17), we detected a reduction in PPARγ, aP2, and adiponectin mRNA levels only and selectively in the epididymal depot of Foxa3-null mice (Fig. 4E). These data suggest that in the absence of Foxa3, PPARγ gene expression, and that of its direct downstream targets aP2 and adiponectin, is affected in a depot-specific manner. To determine the effects of Foxa3 ablation after challenge with a HFD, we subsequently exposed 8-week-old mice to an obesogenic high-fat diet. Our analysis revealed a reduction in body weight in Foxa3-null mice on a HFD (Fig. F) and a selective decrease in the amount of epididymal fat depot in Foxa3-null mice compared to WT mice (Fig. 4G), while no differences between WT and Foxa3-null mice were observed in the amounts of other fat depots. These data suggest that ablation of Foxa3 can cause a decrease in epididymal depot adiposity in adult mice exposed to a HFD, which is consistent with our observations of reduced epididymal fat in preweaning Foxa3-null mice. Interestingly, we did not record any differences in total fat mass by nuclear magnetic resonance (NMR) analysis (see Fig. S5 in the supplemental material) or in food intake (see Fig. S6 in the supplemental material), and energy expenditure measurements appeared to be similar between the WT and Foxa3-null mice (see Fig. S7 in the supplemental material).
Fig 4.

Foxa3-null mice have decreased visceral adiposity but no reduction in inguinal fat depot compared to WT mice. (A) Epididymal (eWAT) and inguinal (iWAT) fat depot morphology of 2-week-old WT and Foxa3-null (KO) mice. (B) Weights of eWAT and iWAT depots of 2-week-old WT (n = 6) and Foxa3-null (KO) (n = 6) mice. B.W., body weight. (C) Relative mRNA levels of adipocyte markers in epididymal (eWAT) and inguinal (iWAT) depots of 2-week-old WT (n = 6) and Foxa3-null (KO) (n = 6) mice. (D) Weights of epididymal (eWAT) and inguinal (iWAT) fat depots in 8-week-old, adult WT (n = 5) and Foxa3-null (KO) (n = 5) mice. (E) Relative mRNA levels of adipocyte markers in epididymal (eWAT) and inguinal (iWAT) depots of 8-week-old WT (n = 5) and Foxa3-null (KO) (n = 5) mice. (F) Body weight of WT (n = 5) and Foxa-null (KO) (n = 5) mice after HFD regimen. (G) Weights of epididymal (eWAT), inguinal (iWAT), mesenteric (mWAT), retroperitoneal (rWAT), and brown fat (BAT) depots in adult WT (n = 5) and Foxa3-null (KO) (n = 5) mice fed a HFD. (H) Hematoxylin and eosin (H&E) staining of epididymal (eWAT) and inguinal (iWAT) fat depots of adult WT and Foxa3-null (KO) mice after HFD regimen. (I) Size measurements of adipocytes in the epididymal and inguinal depots of adult WT (n = 5) and Foxa3-null (KO) (n = 5) mice fed a HFD. (J) Relative mRNA levels of adipocyte differentiation markers in epididymal and inguinal fat depots of adult WT (n = 5) and Foxa3-null (KO) (n = 5) mice after HFD regimen. (K) Relative mRNA levels of adipocyte markers at day 6 after induction of adipocyte differentiation in stromal-vascular fractions (SVF) of cells obtained from visceral (visc) and inguinal (ing) fat depots of WT and Foxa3-null (KO) mice. Data represent means ± SEM (***, P < 0.001; **, P < 0.01; *, P < 0.05).
Histological analysis of epididymal adipose tissues revealed the presence of smaller adipocytes in Foxa3-null mice (Fig. 4H), as confirmed by cell size measurements (Fig. 4I). Further molecular analysis of adipocyte markers showed a decrease in PPARγ, aP2, and adiponectin mRNA levels in the epididymal fat depot of Foxa3-null mice compared to WT mice, with no differences detected in the inguinal compartments of WT and Foxa3-null mice (Fig. 4J). To determine whether the decreased visceral adiposity in Foxa3-null mice was due to a cell-autonomous defect, we differentiated cells obtained from the visceral and inguinal stromal-vascular fractions (SVF) of cells from WT and Foxa3-null mice and examined the levels of adipocyte differentiation markers. Figure 4K shows that Foxa3-null visceral cells have decreased levels of PPARγ compared to WT cells, while no reduction in PPARγ mRNAs was detected in the inguinal stromal-vascular fraction of cells obtained from either WT and Foxa3-null mice. In addition, analysis of other adipocyte markers revealed a significant reduction in Foxa3-null visceral fat cells compared to WT cells. These data suggest that the selective decrease in visceral fat mass observed in Foxa3-null mice may be due to an intrinsic defect of visceral adipocytes.
Foxa3 levels are regulated depot selectively during HFD.
To better understand the basis for the selective effects of Foxa3 ablation specifically on the epididymal fat depot, we examined whether Foxa3 mRNA levels were differentially regulated by a HFD in different fat depots. As shown in Fig. 5A, Foxa3 levels appeared to be preferentially induced in the epididymal fat of mice fed a HFD, suggesting a depot-selective regulation of Foxa3. To better define whether the increase in Foxa3 levels during HFD occurred in the SVF of cells or in the adipocytes, we fractionated cells obtained from the epididymal depot of mice exposed to a HFD. As shown in Fig. 5B, Foxa3 mRNA levels increased selectively in the SVF of HFD-exposed mice. We next measured FOXA3 mRNA levels in fat depots of obese subjects. Our analysis of biopsy specimens obtained from intra-abdominal and subcutaneous adipose tissues of 14 individuals with body mass index ranging from 36.1 to 55.6 revealed higher levels of FOXA3 mRNA in the visceral depot of these obese subjects compared to their subcutaneous adipose tissue (Fig. 5C). These data support the notion that Foxa3 may play a role in the expansion of visceral adipose tissues in obese states in humans.
Fig 5.
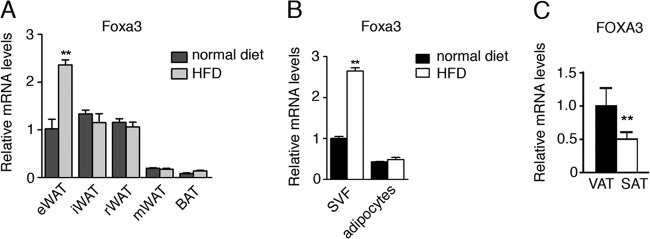
Foxa3 mRNA levels are regulated by a HFD. (A) Relative Foxa3 mRNA levels in fat depots of WT mice fed a normal diet (n = 4) or a HFD (n = 4) for 14 weeks. eWAT, epididymal WAT; iWAT, inguinal WAT; mWAT, mesenteric WAT; rWAT, retroperitoneal WAT; BAT, brown adipose tissue. (B) Foxa3 mRNA levels in SVF of cells and adipocytes obtained from epididymal fat depot of mice fed a normal diet (n = 4) or a HFD (n = 4) for 14 weeks. (C) Human FOXA3 mRNA levels in visceral (VAT) and subcutaneous (SAT) adipose tissues in obese women (n = 14). Data represent means ± SEM (**, P < 0.01).
Foxa3-null mice have increased insulin sensitivity.
Diet-induced obesity in animal models is generally accompanied by glucose intolerance and insulin resistance. To determine the effects of high fat intake on glucose homeostasis and insulin sensitivity in WT and Foxa3-null mice exposed to a HFD, we performed glucose and insulin tolerance tests (GTT and ITT). (Figure 6A through D shows that Foxa3-null mice were more insulin sensitive than WT mice, with increased serum adiponectin and decreased insulin and cholesterol levels, consistent with an improved metabolic profile (Fig. 6E).
Fig 6.

Foxa3-null mice are more insulin sensitive than WT mice. (A to D) GTT (A) and ITT (C) assays in adult WT (n = 5) and Foxa3-null (KO) mice (n = 5) fed a HFD and quantification of area under the curve (AUC) (B and D). (E) Serum parameters in WT (n = 5) and Foxa3-null (KO) mice (n = 5) after HFD regimen. Data represent means ± SEM (**, P < 0.01; *, P < 0.05).
DISCUSSION
As worldwide rates of obesity and related metabolic disorders increase, we are compelled to better understand the fundamental mechanisms underlying adipocyte formation and fat tissue expansion. Given that Forkhead transcription factors serve as critical regulators of developmental and differentiation decisions in a number of organs and the current incomplete understanding of their function in adipose tissue, we systematically investigated the role of Forkhead factors in adipocyte and lipid metabolism. Using a siRNA screen, we identified the winged helix protein Foxa3 as a novel positive regulator of fat differentiation. Our data showed that Foxa3 is an early modulator that cooperates with well-established upstream regulators of PPARγ. Furthermore, we demonstrated that mice with Foxa3 ablation have a depot-selective decrease in fat tissue. The differential levels of expression of FOXA3 in fat depots in obese patients also highlighted the potential importance of Foxa3 in contributing to human visceral obesity.
Our finding that Foxa3 is an early modulator of adipocyte differentiation provides novel evidence regarding how early regulators, such as C/EBPβ and -δ, cooperate synergistically to induce PPARγ expression. Interestingly, although Foxa3 is sufficient to enhance C/EBP-mediated induction of PPARγ, as shown through gain-of-function experiments, it does not appear to be fully required for the regulation of PPARγ basal expression levels, since in the absence of Foxa3 in vivo, PPARγ mRNA is reduced but not ablated. These data implicate Foxa3 as a modulator of PPARγ levels.
Our data in mice show that ablation of Foxa3 reduces epididymal adipose tissue expansion in response to a high-fat diet. Interestingly, the decrease in fat weight occurred selectively in the epididymal adipose tissue of Foxa3-null mice, not in the inguinal depots, suggesting that Foxa3 contributes specifically to the regulation of depots involved in visceral fat expansion. This depot-selective role of Foxa3 is substantiated by two lines of evidence: first, our ex vivo experiments show that only SVF of cells derived from visceral adipose tissue of Foxa3-null mice, and not of those derived from the inguinal depot, have an intrinsic defect in their ability to induce PPARγ and to differentiate; and second, Foxa3 levels are elevated selectively in the visceral adipose tissue in response to a HFD. Interestingly, this study appears to be the first to have demonstrated a selective role of a transcription factor in visceral obesity.
The reduction in the epididymal fat amount of mice with genetic ablation of Foxa3 did not appear to be caused by differences between WT and Foxa3-null mice in food intake or in energy expenditure but rather seemed to be due to a defect intrinsic to the visceral Foxa3-null preadipocytes, as demonstrated by our ex vivo studies. Given that NMR analysis showed no statistical differences between the two genotypes in fat mass, it is plausible that lipid redistribution and ectopic deposition to other tissues may have occurred.
Our data suggest that Foxa3 ablation in vivo leads to reduced visceral depots due to decreased PPARγ levels and possibly reduced lipid storage capacity. This scenario is supported by the ex vivo experiments suggesting that the decrease in epididymal adipose tissue levels observed in Foxa3-null mice may be due to a cell-autonomous defect of the visceral adipocytes, pointing to a direct role of Foxa3 in fat tissue. Further genetic and physiological studies employing conditional Foxa3 knockout systems will define the role of Foxa3 tissue specifically in physiology and metabolism under conditions of high dietary lipid intake.
In conclusion, through a systematic siRNA knockdown of all Fox factors, we identified Foxa3 as a novel positive regulator of adipocyte differentiation. Our data show that Foxa3 functions early in the adipocyte differentiation program by cooperating with well-established regulators of PPARγ. Furthermore, we demonstrated a novel role for Foxa3 in visceral adipose tissue expansion through the control of PPARγ levels during high-fat feeding and in the regulation of insulin sensitivity. Our findings also highlight the potential importance of Foxa3 in contributing to human intra-abdominal obesity. Overall, our data implicate Foxa3 as a novel critical modulator of adipocyte differentiation and of lipid accumulation in the visceral fat depot.
Supplementary Material
ACKNOWLEDGMENTS
We thank Richard Proia for comments on the manuscript and Pasha Sarraf for critical discussions throughout the project. We are grateful to Sam Cushman (National Institutes of Health), Gerald Reaven, and Philip Tsao (Stanford University, Stanford, CA) for providing patient samples and thank Laura Allende for help in fluorescence-activated cell sorter (FACS) analysis and William Jou and Tatyana Chanturiya for their technical assistance.
This research was supported by P01-DK049210 (to K.H.K.) and by the Intramural Research Program of the National Institutes of Health and National Institute of Diabetes and Digestive and Kidney Diseases (to E.M.).
Footnotes
Published ahead of print 24 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00244-13.
REFERENCES
- 1.Anghel SI, Wahli W. 2007. Fat poetry: a kingdom for PPAR gamma. Cell Res. 17:486–511 [DOI] [PubMed] [Google Scholar]
- 2.Farmer SR. 2006. Transcriptional control of adipocyte formation. Cell Metab. 4:263–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scherer PE. 2006. Adipose tissue: from lipid storage compartment to endocrine organ. Diabetes 55:1537–1545 [DOI] [PubMed] [Google Scholar]
- 4.Wajchenberg BL. 2000. Subcutaneous and visceral adipose tissue: their relation to the metabolic syndrome. Endocr. Rev. 21:697–738 [DOI] [PubMed] [Google Scholar]
- 5.Tontonoz P, Hu E, Spiegelman BM. 1994. Stimulation of adipogenesis in fibroblasts by PPAR gamma 2, a lipid-activated transcription factor. Cell 79:1147–1156 [DOI] [PubMed] [Google Scholar]
- 6.Cristancho AG, Lazar MA. 2011. Forming functional fat: a growing understanding of adipocyte differentiation. Nat. Rev. Mol. Cell Biol. 12:722–734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hannenhalli S, Kaestner KH. 2009. The evolution of Fox genes and their role in development and disease. Nat. Rev. Genet. 10:233–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sund NJ, Ang SL, Sackett SD, Shen W, Daigle N, Magnuson MA, Kaestner KH. 2000. Hepatocyte nuclear factor 3beta (Foxa2) is dispensable for maintaining the differentiated state of the adult hepatocyte. Mol. Cell. Biol. 20:5175–5183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee CS, Friedman JR, Fulmer JT, Kaestner KH. 2005. The initiation of liver development is dependent on Foxa transcription factors. Nature 435:944–947 [DOI] [PubMed] [Google Scholar]
- 10.Gao N, LeLay J, Vatamaniuk MZ, Rieck S, Friedman JR, Kaestner KH. 2008. Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev. 22:3435–3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakae J, Kitamura T, Kitamura Y, Biggs WH, III, Arden KC, Accili D. 2003. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Dev. Cell 4:119–129 [DOI] [PubMed] [Google Scholar]
- 12.Wolfrum C, Shih DQ, Kuwajima S, Norris AW, Kahn CR, Stoffel M. 2003. Role of Foxa-2 in adipocyte metabolism and differentiation. J. Clin. Invest. 112:345–356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Davis KE, Moldes M, Farmer SR. 2004. The forkhead transcription factor FoxC2 inhibits white adipocyte differentiation. J. Biol. Chem. 279:42453–42461 [DOI] [PubMed] [Google Scholar]
- 14.Friedman JR, Kaestner KH. 2006. The Foxa family of transcription factors in development and metabolism. Cell. Mol. Life Sci. 63:2317–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meruvu S, Hugendubler L, Mueller E. 2011. Regulation of adipocyte differentiation by the zinc finger protein ZNF638. J. Biol. Chem. 286:26516–26523 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu A, McLaughlin T, Liu T, Sherman A, Yee G, Abbasi F, Lamendola C, Morton J, Cushman SW, Reaven GM, Tsao PS. 2009. Differential intra-abdominal adipose tissue profiling in obese, insulin-resistant women. Obes. Surg. 19:1564–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaestner KH, Hiemisch H, Schutz G. 1998. Targeted disruption of the gene encoding hepatocyte nuclear factor 3gamma results in reduced transcription of hepatocyte-specific genes. Mol. Cell. Biol. 18:4245–4251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shen W, Scearce LM, Brestelli JE, Sund NJ, Kaestner KH. 2001. Foxa3 (hepatocyte nuclear factor 3gamma) is required for the regulation of hepatic GLUT2 expression and the maintenance of glucose homeostasis during a prolonged fast. J. Biol. Chem. 276:42812–42817 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.