

Abstract
The yeast Saccharomyces cerevisiae possesses two distinct glycyl-tRNA synthetase (GlyRS) genes: GRS1 and GRS2. GRS1 is dually functional, encoding both cytoplasmic and mitochondrial activities, while GRS2 is dysfunctional and not required for growth. The protein products of these two genes, GlyRS1 and GlyRS2, are much alike but are distinguished by an insertion peptide of GlyRS1, which is absent from GlyRS2 and other eukaryotic homologues. We show that deletion or mutation of the insertion peptide modestly impaired the enzyme's catalytic efficiency in vitro (with a 2- to 3-fold increase in Km and a 5- to 8-fold decrease in kcat). Consistently, GRS2 can be conveniently converted to a functional gene via codon optimization, and the insertion peptide is dispensable for protein stability and the rescue activity of GRS1 at 30°C in vivo. A phylogenetic analysis further showed that GRS1 and GRS2 are paralogues that arose from a gene duplication event relatively recently, with GRS1 being the predecessor. These results indicate that GlyRS2 is an active enzyme essentially resembling the insertion peptide-deleted form of GlyRS1. Our study suggests that the insertion peptide represents a novel auxiliary domain, which facilitates both productive docking and catalysis of cognate tRNAs.
INTRODUCTION
Faithful decoding of mRNA depends on accurate aminoacylation of tRNA by aminoacyl-tRNA synthetases (aaRSs) and a specific readout of the codons by tRNAs. aaRSs are a group of structurally diverse enzymes, each of which catalyzes the ligation of a specific amino acid to its cognate tRNA. The resultant aminoacyl-tRNA is then delivered to ribosomes for protein translation. Typically, a full complement of aaRSs consists of 20 different enzymes in prokaryotes, one for each amino acid (1–4). In contrast, eukaryotes, such as yeast, contain two distinct sets of aaRSs, one localized to the cytoplasm and the other to mitochondria. With the exception of yeast glutaminyl-tRNA synthetase, which is distributed in both the cytoplasm and mitochondria (5), each set recognizes and aminoacylates cognate tRNAs within its respective cellular compartment and is sequestered from isoacceptors confined in other compartments. However, four Saccharomyces cerevisiae genes, ALA1 (which encodes alanyl-tRNA synthetase) (6, 7), GRS1 (which encodes glycyl-tRNA synthetase [GlyRS]) (8), HTS1 (which encodes histidyl-tRNA synthetase) (9), and VAS1 (which encodes valyl-tRNA synthetase) (10), specify both mitochondrial and cytoplasmic activities. This dually functional feature was found to be conserved in homologues of these genes in almost all yeast species studied (7, 11, 12).
Nearly all yeast cytoplasmic aaRSs possess an N- or C-terminal polypeptide extension (typically 80 to 200 amino acids long), known as an appended domain, which is absent from their prokaryotic counterparts (13). Many of these domains are rich in lysine residues and are involved in nonspecific tRNA binding, examples of which include glutaminyl-tRNA (14), arginyl-tRNA (15), and valyl-tRNA (16) synthetases. These domains act in cis as an auxiliary tRNA-binding domain and enhance the tRNA-binding affinity of the enzymes (17). In contrast, appended domains of some yeast cytoplasmic aaRSs participate in specific protein-protein interactions, examples of which include glutamyl-, methionyl-, and seryl-tRNA synthetases. Glutamyl- and methionyl-tRNA synthetases form a ternary complex with Arc1p, a nonspecific tRNA-binding protein encoded by ARC1, through their N-terminal appended domains (18), while seryl-tRNA synthetase forms a binary complex with the peroxisome biogenesis-related factor, Pex21p, through its C-terminal appended domain (19). These interactions were also shown to enhance tRNA binding and aminoacylation activities of the associated enzymes. In addition, Arc1p acts as a cytosolic sorting platform to regulate the subcellular distributions of glutamyl- and methionyl-tRNA synthetases (20).
Based on the conserved sequence motifs, quaternary structure, and aminoacylation function, aaRSs can be divided into two classes of 10 enzymes each (21, 22). Class I enzymes possess two conserved signature sequences, HIGH and KMSKS, while class II enzymes contain three conserved motifs, motifs 1, 2, and 3. In addition, class I enzymes first couple amino acids to the 2′-OH of the terminal adenylate residue of tRNA before transferring it to the 3′-OH, while class II enzymes directly couple it to the 3′-OH. Normally, orthologous enzymes that couple the same amino acid to isoaccepting tRNAs share high sequence similarities in their catalytic core domains and are grouped into the same class (I or II), which indicates that they shared a common ancestor. However, there are two exceptions to this rule: lysyl-tRNA synthetase and GlyRS. In the case of lysyl-tRNA synthetase, both class I- and II-type enzymes were found (23), while in the case of GlyRS, two oligomeric forms were identified: an α2β2 heterotetramer and an α2 homodimer (24, 25). Despite both forms of GlyRS containing a class II-defining architecture, they dramatically differ in size and sequence (26). As a result, they are believed to have evolved from different origins (27). To date, α2β2-type enzymes have been found only in bacteria and chloroplasts, while α2-type enzymes have been recovered from all three domains of life.
The yeast Saccharomyces cerevisiae possesses two distinct nuclear GlyRS genes, GRS1 and GRS2. GRS1 encodes both cytoplasmic and mitochondrial forms of GlyRS through alternative initiation of translation (8, 28), while GRS2 is defective in gene expression and dispensable for survival (29–31). GlyRS1 (encoded by GRS1) lacks an apparent N- or C-terminal appended domain but carries a lysine-rich insertion peptide, which is absent from GlyRS2 (encoded by GRS2) and other eukaryotic relatives (30). To provide further insights into the origin and function of this insertion peptide, various approaches were taken to characterize these two homologous genes.
MATERIALS AND METHODS
Plasmid construction.
Cloning of the wild-type (WT) GRS1 gene into pADH (a high-copy-number yeast shuttle vector with a constitutive ADH promoter, followed by multiple cloning sites and a short sequence coding for a His6 tag) or pRS315 (a low-copy-number yeast shuttle vector) followed a previously described protocol (30). To delete the insertion peptide (amino acid residues 126 to 158) from GlyRS1, DNA sequences flanking the insertion peptide of GRS1 were amplified by a PCR as two fragments, an EagI-SpeI fragment (containing bp −75 to +375) and a SpeI-SalI fragment (containing bp +475 to +2001). These two fragments were sequentially cloned into pADH, yielding GRS1(ΔIP) (pWYH184). Deletion of the K motif (amino acid residues 145 to 151) followed a similar strategy. Cloning of GRS1 mutants into pRS315 followed a similar protocol, except that the 5′ end was at position −300 instead.
To enhance the protein expression of GRS2, a codon-optimized GRS2 variant [here designated GRS2(CO)] was in vitro synthesized by Genscript USA (Piscataway, NJ) using a set of codons preferable for S. cerevisiae. To fuse the insertion peptide coding sequence of GRS1 into the corresponding position in GRS2(CO), a SpeI site was first created in GRS2(CO) between bp +369 and +370, resulting in GRS2(CO)-SpeI. The DNA sequence encoding the insertion peptide (bp +376 to +474) was amplified by PCR as a SpeI-SpeI fragment and cloned into the SpeI site of GRS2(CO)-SpeI, resulting in GRS2(CO)(IP) (pWYH167). To mutate the K motif of GRS1, the insertion peptide (a PCR-amplified SpeI-SpeI fragment) was cloned into pBluescript II KS(+/−) (Agilent, Santa Clara, CA), and the resultant construct was used as the template for mutagenesis. Mutagenesis was carried out according to standard protocols provided by the manufacturer (Stratagene, La Jolla, CA). After mutagenesis, the SpeI-SpeI fragment was retrieved from the plasmid and inserted into the SpeI site of GRS1(ΔIP), resulting in various GRS1(KMM) constructs. The K motif was mutated from KKKRKKK to SGSGSGT, SEEEEET, and SAAAAAT in GRS1(KMM1), GRS1(KMM2), and GRS1(KMM3), respectively.
Complementation assays for cytoplasmic GlyRS activity.
The yeast GRS1 knockout strain, RJT3/II-1, was previously described (29). Complementation assays for cytoplasmic GlyRS activity were carried out by introducing a test plasmid carrying the gene of interest and a LEU2 marker into RJT3/II-1, and the ability of the transformants to grow in the presence of 5-fluoroorotic acid (5-FOA) medium was determined. Starting from a cell density of an A600 of 4.0, cell cultures were 5-fold serially diluted, and 10-μl aliquots of each dilution were spotted onto the designated plates containing 5-FOA medium. Plates were incubated at 30°C for 3 to 5 days. The transformants evicted the maintenance plasmid with the URA3 marker in the presence of 5-FOA medium and thus could not grow on the selection medium unless a functional cytoplasmic GlyRS was encoded by the test plasmid.
Complementation assays for mitochondrial GlyRS activity.
RJT3/II-1 was cotransformed with a test plasmid (carrying a LEU2 marker) and a second maintenance plasmid (carrying a TRP1 marker) that expressed only the cytoplasmic form of GlyRS (due to a mutation in the initiator codon of the mitochondrial form). In the presence of 5-FOA medium, the first maintenance plasmid (carrying a URA3 marker) was evicted from the cotransformants, while the second maintenance plasmid was retained. Thus, all cotransformants survived 5-FOA selection, due to the presence of the cytoplasmic GlyRS derived from the second maintenance plasmid. The mitochondrial phenotypes of the cotransformants were further tested on yeast extract-peptone-glycerol (YPG) plates at 30°C, with results documented on day 3 following plating. Because a yeast cell cannot survive on glycerol without functional mitochondria, the cotransformants did not grow on the YPG plates unless a functional mitochondrial GlyRS was generated from the test plasmid.
Aminoacylation assay.
Aminoacylation reactions were carried out at 25°C in a buffer containing 50 mM HEPES (pH 7.5), 50 mM KCl, 15 mM MgCl2, 5 mM dithiothreitol, 10 mM ATP, 0.1 mg/ml bovine serum albumin (BSA), 100 μM unfractionated yeast tRNA (Boehringer Mannheim, Germany), and 20 μM glycine (2 μM [3H]glycine; Moravek Biochemicals, Brea, CA) (31, 32). The specific activity of [3H]glycine used was 35.0 Ci/mmol. Determination of active protein concentrations by active-site titration followed a previously described protocol (33). Reactions were quenched by spotting 10-μl aliquots of the reaction mixture onto Whatman filters (Maidstone, United Kingdom) soaked in 5% trichloroacetic acid and 1 mM glycine. Filters were washed three times for 15 min each in ice-cold 5% trichloroacetic acid before liquid scintillation counting. Data were obtained from three independent experiments and averaged. Error bars indicate ±2 times the standard deviation.
Kinetic parameters for aminoacylation of tRNA by the purified enzymes were determined by directly fitting the data points to the Michaelis-Menten equation. Initial rates of aminoacylation were determined at 25°C with tRNAGly concentrations ranging from 1 to 20 μM and enzyme concentrations ranging from 4 to 200 nM. tRNA used for the assay was unfractionated yeast tRNA or in vitro-transcribed yeast tRNAnGly. Km values of GlyRS variants for glycine were determined by an ATP-PPi exchange assay (34).
Miscellaneous methods.
Structure models of GlyRS1 (UniProt P38088) and GlyRS2 (UniProt Q06817) were taken from the Swiss-Model repository (35) and were prepared by superimposing to a crystal structure template of human GlyRS (PDB-ID 2zt5) (36) using the PyMOL program (37). The degradation assay and green fluorescence protein (GFP) assay followed previously described protocols (38). Western blotting used an anti-His6 tag antibody and followed a protocol described earlier (12). Purification of His6-tagged GlyRS enzymes was as previously described (39). Circular dichroism spectroscopy followed a protocol described earlier (40).
RESULTS
The insertion peptide folds into a discrete loop protruding from the main body of GlyRS1.
Yeast GlyRS1 and GlyRS2 are both α2-type enzymes and share ∼64% identity. Comparison of the predicted three-dimensional structures of these two proteins revealed that they are superimposable in most parts of their structures, with the exception of an insertion peptide of 33 amino acid residues (residues 126 to 158), which is present only in GlyRS1 (Fig. 1A). The catalytic domain, in particular the class II-defining motifs, is highly conserved in these two homologous enzymes (Fig. 1A and B) and in other α2-type GlyRS sequences (41). The insertion peptide protrudes from the main body of the protein as a discrete loop with two pairs of antiparallel β-pleated sheets interspersed with random coils (Fig. 1A). Sequence alignment among GlyRSs of various yeast species further showed that this peptide is conserved in all yeast GlyRS1 sequences (Fig. 1C) but absent from GlyRS2. This peptide is particularly enriched in lysine residues, which make up ∼25% of its amino acid composition. Notably, a lysine-rich cluster, KKKRKKK (here designated the K motif), is strictly conserved in the insertion peptide of all yeast GlyRS1 sequences (Fig. 1C) and was predicted to be folded into a β-pleated sheet close to the apex of the loop (Fig. 1A).
Fig 1.

Insertion peptide of yeast GlyRS1. (A) Predicted three-dimensional structures of yeast GlyRS1 and GlyRS2. Left, GlyRS1; right, GlyRS2 (orange) superimposed onto GlyRS1 (blue). For distinction, the insertion peptide (IP), K motif (KM), anticodon-binding domain (ABD), and class II-defining motifs are highlighted in red, green, black, and magenta, respectively, in the GlyRS1 structure (left). (B) Relative positions of the functional domains of GlyRS1 and GlyRS2. The insertion peptide (IP) consists of the N-terminal amino acid residues 126 to 158. (C) Alignment of the insertion peptide of yeast GlyRS1s.
The insertion peptide is dispensable for the rescue activity of GRS1 in vivo.
The identity elements of tRNAGly include the discriminator base (N73), the first 3 bp of the acceptor stem (1:72, 2:71, and 3:70), and C35 and C36 in the anticodon loop (3). Despite the fact that the yeast mitochondrion-encoded tRNAGly, tRNAmGly, possesses an acceptor stem that appreciably diverges from that of its cytosolic counterpart, tRNAnGly (nucleus-encoded tRNAGly) (Fig. 2A), GlyRS1 can efficiently recognize both tRNAGly isoacceptors (8). To investigate whether the insertion peptide or the K motif is essential for recognition of these isoaccepting tRNAs, various deletions and mutations were introduced into the insertion peptide or the K motif therein, and the ability of the resultant constructs to rescue growth defects of a grs1− strain of S. cerevisiae on 5-FOA and YPG media was assayed.
Fig 2.
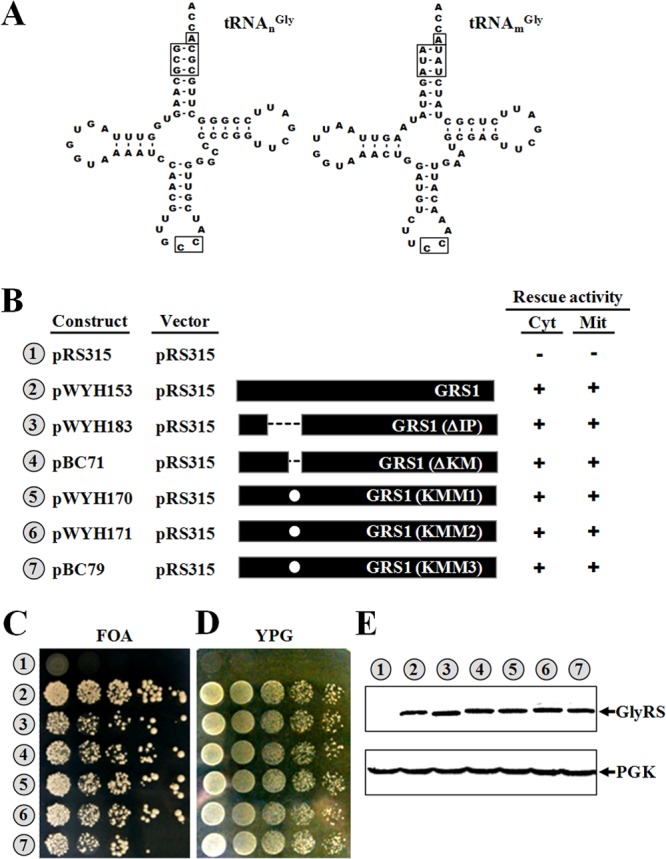
Functional assays of GRS1 variants. (A) Comparison of yeast cytoplasmic and mitochondrial tRNAGly isoacceptors. Secondary structures of tRNAsGly are shown in cloverleaf form. Nucleotides and base pairs that were shown to be important for recognition by GlyRS are boxed. (B) Summary of the constructs and their rescue activities. Constructs bearing wild-type or mutant GRS1 genes were transformed into a grs1− strain of Saccharomyces cerevisiae, and the ability of the transformants to grow on 5-FOA and YPG media was tested. The symbols + and − indicate positive and negative complementation, respectively. Mit, mitochondrial; Cyt, cytoplasmic. (C) Rescue of cytoplasmic GlyRS activity. (D) Rescue of mitochondrial GlyRS activity. (E) Western blotting. Top, GlyRS; bottom, phosphoglycerate kinase (PGK) (as a loading control). Indicated at the bottom of the Western blots are the amounts of protein extracts loaded into the gels. Numbers 1 to 7 (circled) in panels C to E represent the constructs shown in panel B. GlyRS and PGK were probed with an anti-His6 tag antibody and an anti-PGK antibody, respectively.
Figure 2 shows that deletion or mutation of the insertion peptide or the K motif had little effect on the enzyme's ability to restore the growth phenotypes of the knockout strain on both 5-FOA and YPG media, suggesting that the insertion peptide is dispensable for the enzyme's rescue activities in vivo [compare GRS1, GRS1(ΔIP), GRS1(ΔKM), GRS1(KMM1), GRS1(KMM2), and GRS1(KMM3) in Fig. 2B, C, and D]. GRS1(KMM1), GRS1(KMM2), and GRS1(KMM3) bear different mutations in the K motif. Western blotting using an anti-His6 tag antibody showed that all of the GRS1 constructs used were well expressed in cells (Fig. 2E), regardless of whether they contain mutations in the insertion peptide.
Deletion or mutation of the insertion peptide has little effect on the protein stability of GlyRS1 at 30°C.
To test whether deletion or mutation of the insertion peptide impairs the protein stability of GlyRS1, a cycloheximide (CHX) chase assay was carried out. GRS1, GRS1(ΔIP), GRS1(KM), and GRS1(ΔKMM1) were cloned into pGAL1, a high-copy-number yeast shuttle vector with an inducible GAL1 promoter and a short sequence coding for a His6 tag. Constructs bearing these GRS1 variants were transformed into INVSc1, and cultures of the transformants were induced with galactose for 2 h, followed by the addition of CHX to terminate protein synthesis. Cells were harvested at various time points following CHX treatment and prepared for Western blot analyses using an anti-His6 tag antibody. As shown in Fig. 3A, the WT enzyme was quite stable, and its protein level remained almost constant throughout the time period tested (up to 16 h). A similar scenario was observed for the mutants, suggesting that deletion or mutation of the insertion peptide did not perturb the overall folding or stability of GlyRS1. This outcome is not unexpected, considering the fact that this peptide protrudes from the main body of the protein as a discrete loop (Fig. 1A).
Fig 3.

Degradation and stability assays for GlyRS1 variants. (A) Cycloheximide chase assay. Transformants harboring various GRS1 constructs were grown in a raffinose-containing medium to a cell density of an A600 of ∼1.0 and then induced with galactose for 2 h before the addition of cycloheximide. Cells were harvested at various time periods following treatment with cycloheximide and lysed. T0, T0.5, T1, T2, T4, T8, and T16 denote 0, 0.5, 1, 2, 4, 8, and 16 h postinduction, respectively. PGK served as a loading control for each assay. Quantitative data for relative levels of GlyRSs are shown in a separate diagram below the Western blots. (B) The melting curves of the WT and mutant GlyRS1 enzymes. The melting curves of GlyRS1 and its mutants were determined via circular dichroism spectroscopy at 222 nm. The final concentration of the proteins was 2.4 μM. Spectra were recorded from 10°C to 80°C in a 1-mm-path-length cell on a Jasco J-810 spectropolarimeter using a scan speed of 50 nm per min, a time constant of 1 s, and a bandwidth of 1 nm. Three scans were accumulated and averaged for each protein tested.
To investigate whether the deletion or mutation affects the protein folding of GlyRS1 in vitro, the melting curves of GlyRS1 and its derivatives were determined via circular dichroism (CD) spectroscopy at 222 nm. As shown in Fig. 3B, all GlyRS1 variants tested possessed a molar ellipticity (θ) similar to that of the WT enzyme at 30°C or a lower temperature. At 30°C, the GlyRS1 protein contained ∼35% α-helix and ∼18% β-sheet as estimated by the K2D3 software (42). Thus, the deletion or mutation did not significantly alter the conformation of GlyRS1 at 30°C, which is largely consistent with the finding of the cycloheximide chase assay shown in Fig. 3A. However, the deletion mutants GlyRS1(ΔIP) and GlyRS1(ΔKM) appeared to have a melting temperature slightly lower than that of the WT enzyme. The melting curves of these two enzymes slightly shifted to the left (∼5°C) relative to that of their WT counterpart. As for GlyRS2, it had a melting temperature ∼5°C higher than that of GlyRS1. This result suggests that deletion of the insertion peptide did not alter the enzyme's conformation at a temperature below 30°C but did impair its stability at a higher temperature.
GRS2 can be converted into a functional gene via codon optimization.
Since the main difference between GlyRS1 and GlyRS2 is the insertion peptide, we then asked whether GRS2 can be converted to a functional gene by fusing the insertion peptide coding sequence of GRS1 at its matching position. Given that the native GRS2 gene is very poor at both transcription and translation (30, 31), a constitutive ADH promoter and a codon-optimized GRS2 variant [here designated GRS2(CO)] were used instead to improve the expression efficiency. For this purpose, GRS2(CO) was first in vitro synthesized using a set of codons preferable for S. cerevisiae and then cloned into pADH. Unexpectedly, GRS2(CO) per se rescued the growth defect of the GRS1 knockout strain on 5-FOA medium with an appreciably high efficiency. On the other hand, fusion of the insertion peptide coding sequence to GRS2(CO), resulting in GRS2(CO)(IP), did not further enhance its efficiency (compare pWYH166 and pWYH167 in Fig. 4). As a matter of fact, GRS2(CO)(IP) could barely support the growth of the strain with the null allele on 5-FOA medium. Perhaps, fusion of the insertion peptide somehow destabilizes the protein structure of GlyRS2. In addition, fusion of a sequence encoding a mitochondrion-targeted signal (MTS) 5′ to GRS2(CO) or GRS2(CO)(IP) failed to confer positive mitochondrial activity to either of the two constructs on YPG medium (see pWYH186 and pWYH187 in Fig. 4). To provide direct evidence that the MTS used actually guided GlyRS2(CO) to mitochondria, cellular distributions of two GFP fusion constructs, GRS2(CO)-GFP and MTS-GRS2(CO)-GFP, were analyzed by fluorescence microscopy. MitoTracker and 4′,6-diamidino-2-phenylindole (DAPI) were used to label mitochondria and nuclei, respectively. Figure 4F shows that MTS-GlyRS2(CO) was indeed localized in mitochondria, while GlyRS2(CO) was restricted in the cytoplasm.
Fig 4.

Functional assays of GRS2 variants. Constructs bearing GRS2 or its derivatives were transformed into a grs1− strain of Saccharomyces cerevisiae, and the ability of the transformants to grow on 5-FOA and YPG media was tested. (A) Summary of the constructs and their rescue activities. Mit, mitochondrial; Cyt, cytoplasmic; MTS, mitochondrial targeting signal; IP, insertion peptide; GRS2(CO), a codon-optimized GRS2. (B) Rescue of cytoplasmic GlyRS activity. (C) Rescue of mitochondrial GlyRS activity. (D) Western blotting. Top, GlyRS; bottom, PGK (as a loading control). (E) Relative protein levels of GlyRS1, GlyRS2, and GlyRS2(CO). (F) Fluorescence microscopy. MitoTracker and 4′,6-diamidino-2-phenylindole (DAPI) were used to label mitochondria and nuclei, respectively. Indicated at the bottom of the Western blots are the amounts of protein extracts loaded into the gels. Numbers 1 to 8 (circled) in panels B to E represent constructs shown in panel A.
Western blotting using an anti-His6 tag antibody showed that all of the GRS2 constructs used were properly expressed in the knockout strain, but their expression levels were much lower than that of GRS1 (Fig. 4D). To get more quantitative data, protein extracts of GRS1 and GRS2(CO) were 2-fold serially diluted before being loaded into the gel. As shown in Fig. 4E, GRS2 had a protein expression level ∼25-fold lower than that of GRS1 under the conditions used (compare GRS1 and GRS2). However, the protein expression level of GRS2 was modestly enhanced (by ∼3-fold) by codon optimization [compare GRS2 and GRS2(CO)], which might account for the positive rescue activity of GRS2(CO) on 5-FOA medium. As suspected, fusion of the insertion peptide coding sequence to GRS2(CO) slightly reduced its protein expression level [compare GRS2(CO) and GRS2(CO)(IP) in Fig. 4D]. Taking into account the fact that GRS2(CO)(IP) had a protein expression level almost equivalent to that of GRS2, it is possible that insertion of the peptide to GlyRS2 somehow reduces its protein stability but enhances its aminoacylation activity (Fig. 4B).
The insertion peptide facilitates both productive tRNA binding and catalysis in vitro.
To examine whether the insertion peptide contributes to the enzymatic activity of GlyRS1, aminoacylation activities of the WT and mutant GlyRS1 enzymes were assayed in vitro. To this end, the WT and mutant GRS1 genes were cloned in pADH and then transformed into a yeast strain, INVSc1. Recombinant His6-tagged GlyRS enzymes were purified from the transformants to homogeneity using nickel-nitrilotriacetic acid (Ni-NTA) column chromatography. As shown in Fig. 5, deletion of the insertion peptide appreciably reduced the enzyme's aminoacylation activity (by ∼3-fold) [compare GlyRS1 and GlyRS1(ΔIP)]. Moreover, deletion or mutation of the K motif had a similar effect on aminoacylation (with a 3- to 4-fold decrease) [see GlyRS1(ΔKM) and GlyRS1(KMM1)], suggesting that the effect of the insertion peptide on aminoacylation is probably mediated through the positively charged K motif. On the other hand, the deletion or mutation had no significant effect on the Km values of the GlyRS1 variants for glycine (∼135 μM) as determined by the ATP-PPi exchange assay shown in Table 1, suggesting that the insertion peptide contributes to tRNA binding or catalysis rather than cognate amino acid binding. In addition, GlyRS2 had a Km value for glycine (∼145 μM) very close to that of GlyRS1.
Fig 5.

Aminoacylation assays for GlyRS variants. Aminoacylation activities of the purified recombinant GlyRS enzymes were determined in vitro by measuring relative amounts of [3H]glycine that were incorporated into tRNA using a liquid scintillation counter. The final concentration of the enzymes used in the reaction mixtures was 20 nM.
Table 1.
Km values of GlyRS variants for Gly determined by the ATP-PPi exchange
| GlyRS variant | Gly Km (μM) |
|---|---|
| GlyRS1 | 135 |
| GlyRS1(ΔIP) | 145 |
| GlyRS1(ΔKM) | 140 |
| GlyRS1(KMM1) | 130 |
| GlyRS2 | 145 |
To gain further insights into the effect of the insertion peptide on aminoacylation, kinetic parameters for aminoacylation of tRNAGly by GlyRS enzymes were subsequently determined using unfractionated yeast tRNA as the substrate. As shown in Table 2, the WT GlyRS1 enzyme had a Km value of 0.33 μM for tRNAGly and a kcat value of 0.38 s−1. In contrast, the mutant enzymes, including GlyRS1(ΔIP), GlyRS1(ΔKM), and GlyRS1(KMM1), had Km values for tRNAGly ranging from 0.65 to 0.91 μM and kcat values ranging from 0.05 to 0.08 s−1. Thus, deletion or mutation of the insertion peptide or the K motif increased the enzyme's Km value for tRNAGly by 2- to 3-fold and decreased the enzyme's kcat value by 5- to 8-fold. Overall, the catalytic efficiency (kcat/Km) of the glycine enzyme was reduced by 10- to 24-fold upon deletion or mutation of the insertion peptide or the K motif. It is also noted that GlyRS2, which inherently lacks the insertion peptide, possessed kinetic parameters comparable to those of GlyRS1(ΔIP). These data suggest that the insertion peptide, while dispensable for rescue activity and protein stability, plays a significant role in both tRNA binding and catalysis, and this effect on aminoacylation appears to be mediated through the K motif.
Table 2.
Kinetic parameters for aminoacylation of yeast tRNAGly by GlyRS variants
| GlyRS variant | Unfractionated yeast tRNA |
In vitro-transcribed yeast tRNAnGly |
||||
|---|---|---|---|---|---|---|
| Km (μM) | kcat (s−1) | kcat/Km (M−1 s−1) | Km (μM) | kcat (s−1) | kcat/Km (M−1 s−1) | |
| GlyRS1 | 0.33 ± 0.03 | 0.38 ± 0.15 | 11.4 × 105 | 0.28 ± 0.03 | 0.33 ± 0.03 | 12.3 × 105 |
| GlyRS1(ΔIP) | 0.89 ± 0.39 | 0.08 ± 0.01 | 1.0 × 105 | 1.37 ± 0.23 | 0.19 ± 0.02 | 1.4 × 105 |
| GlyRS1(ΔKM) | 0.91 ± 0.45 | 0.05 ± 0.02 | 0.5 × 105 | NDa | ND | ND |
| GlyRS1(KMM1) | 0.65 ± 0.13 | 0.08 ± 0.03 | 1.2 × 105 | ND | ND | ND |
| GlyRS2 | 0.53 ± 0.08 | 0.10 ± 0.02 | 1.8 × 105 | 0.73 ± 0.27 | 0.15 ± 0.03 | 2.2 × 105 |
ND, not determined.
To eliminate the effect of competition by noncognate tRNAs, we next used in vitro-transcribed tRNAnGly as the substrate for determination of the kinetic parameters. As shown in Table 2, deletion of the insertion peptide increased the enzyme's Km value for tRNAnGly by 5-fold and decreased the enzyme's kcat value by 2-fold. Overall, the catalytic efficiency (kcat/Km) of the glycine enzyme was reduced by ∼9-fold upon deletion of the insertion peptide. These data reinforce the hypothesis that the insertion peptide plays an important role in both tRNA binding and catalysis.
GRS1 and GRS2 arose from a gene duplication event.
As GRS2 of S. cerevisiae appreciably diverged from its GRS1 counterpart, we analyzed the possible historical relationships between GRS2 and its yeast homologues using the neighbor-joining (NJ) method (43). To get a more-complete picture, representative α2-dimeric GlyRS sequences from all three of the major branches of life (Bacteria, Archaea, and Eukarya) were retrieved from databases. Bias in the alignments of these sequences was minimized by taking away all major extensions and insertions. Thus, the portion used for analysis comprised only the core active site and the anticodon-binding domain, which accounts for ∼64% of the sequence of GlyRS2.
As shown in Fig. 6, all eukaryotic GlyRSs were clustered within a monophyletic branch. In contrast, the archaeal and bacterial sequences were clustered into two paraphyletic branches, with the archaeal branch having higher affinity with the eukaryotic branch. This result argues that all prevailing eukaryotic α2-dimeric GlyRS genes originated from a eukaryotic source. Thus, yeast GRS1 and GRS2 are paralogues that arose from a gene duplication event relatively recently. Since the lysine-rich insertion peptide exists only in yeast GlyRS1, it appears that this insert was added to the enzyme after the yeast branch separated from other eukaryotic branches. Moreover, it is likely that GlyRS2 once possessed this insert, but the insert was later deleted during evolution.
Fig 6.

Phylogenetic analysis of α2-dimeric GlyRS proteins. Sequences comprising the core active site and anticodon-binding domain of α2-dimeric GlyRS proteins were aligned using CLUSTAL W (51) and analyzed with the neighbor-joining method (43). Numbers at the nodes denote bootstrapping frequencies (shown as percent) calculated from 1,000 trees. The accession number of the GlyRS sequence is shown in the parentheses.
DISCUSSION
The Thermus thermophilus GlyRS enzyme possesses an insertion peptide located at a position analogous to that of the insertion peptide of yeast GlyRS1. This peptide was predicted to interact with the acceptor arm of its cognate tRNA (44). Consistent with this hypothesis, deletion or mutation of the insertion peptide of yeast GlyRS1 impairs its apparent affinity for tRNAGly and catalytic rate (Table 2). Moreover, this effect of the insertion peptide on aminoacylation appears to be mediated through its K motif. However, even with such a low catalytic efficiency (5% relative to that of the WT GlyRS1 enzyme), these mutants effectively rescued growth defects of the knockout strain on both 5-FOA and YPG media (Fig. 2). A similar scenario was previously observed in yeast ALA1 (6, 12), GLN4 (16), and VAS1 (11, 45) genes, indicative of a common feature of the ability to rescue a knockout strain with a considerably low level of aaRS activity. It is noteworthy that the fragment containing the amino acid residues 122 to 165 of GlyRS1 was previously considered to be the insertion peptide solely on the basis of sequence alignment, but deletion of such a peptide considerably destabilizes the protein structure of GlyRS1 (30). In contrast, the insertion peptide described here was predicted on the basis of structure, and deletion of such a peptide had little effect on protein stability at 30°C. It thus appears that the insertion peptide specified here is more representative of its location and size.
With the exception of two yeast species, Saccharomyces cerevisiae and Vanderwaltozyma polyspora, which contain a second GlyRS homologue (GRS2), all other yeast species studied thus far possess a single, dually functional GRS1 homologue (31). GRS2 is poor at both transcription and translation, and thus, it cannot substitute for GRS1 even under the control of a constitutive ADH promoter (29, 30). As a result, this redundant gene was once thought to be an evolutionary relic. Despite that, a recent report argued that expression of GRS2 can be drastically induced by various stresses such as heat, alkali, hydrogen peroxide, and ethanol, suggesting that it may have a purpose under certain special conditions (31). Evidence supporting this hypothesis came from the discovery that the purified recombinant GlyRS2 enzyme is fairly active in vitro (30) (Fig. 5; Table 2). In addition, GRS2 can be conveniently converted to a functional copy by fusion to ARC1 (30) or via codon optimization (Fig. 4). More efforts are under way to elucidate its tRNA specificity and bona fide biological functions in vivo.
The major identity elements of tRNAGly reside on the discriminator base (N73), the first 3 bp of the acceptor stem (1:72, 2:71, and 3:70), and C35 and C36 in the anticodon loop (3). These identity elements determine the efficiency of tRNAGly aminoacylation by its cognate enzyme (46). The most striking difference between bacterial and eukaryotic tRNAGly isoacceptors is the discriminator base, which is nearly always a “U” in bacteria and an “A” in eukaryotic cytoplasm. For example, in Escherichia coli, N73 is a U, while in Arabidopsis thaliana, N73 is a U in the mitochondrion-encoded tRNAGly and an A in nucleus-encoded cytosolic tRNAsGly (47). Normally, tRNAGly bearing U73 is recognized by an α2β2-heterotetrameric GlyRS enzyme, while tRNAGly bearing A73 is recognized by an α2-homodimeric GlyRS enzyme (46). However, the discovery that a homodimeric GlyRS enzyme from Thermus thermophilus can efficiently recognize tRNAGly bearing U73 destroys the once-tight relationship between the type of discriminator base and the oligomeric structure of its cognate enzyme (46). Despite that, it was still amusing to find that both yeast nucleus- and mitochondrion-encoded tRNAGly isoacceptors possessed the same discriminator base, A73. Such an unusual feature might account for the dually functional phenotype of GRS1 (30, 31).
Escherichia coli glutaminyl-tRNA synthetase per se cannot substitute for its yeast homologue in vivo, but it can be converted into a functional yeast enzyme when fused to a nonspecific tRNA-binding domain, such as Arc1p or the appended domain of yeast glutaminyl- or valyl-tRNA synthetase (16, 38, 48). Similarly, E. coli valyl-tRNA synthetase can be converted to a functional yeast enzyme through a similar approach (45). In these instances, Arc1p and the appended domains act in cis as an auxiliary tRNA-binding domain to facilitate nonspecific tRNA binding of the enzyme. It thus appears that acquiring such a domain is a prerequisite for proper functioning of yeast cytoplasmic aaRSs. In that sense, results presented here are of particular interest. Our study suggests that the relatively small insertion peptide of yeast GlyRS1 facilitates both productive docking and catalysis of cognate tRNAs (Table 2). Conceivably, yeast aaRSs have acquired various auxiliary domains during evolution to optimize their performance in nonspecific tRNA binding (as in the cases of glutaminyl- and valyl-tRNA synthetases), protein sorting (as in the cases of glutamyl- and methionyl-tRNA synthetases), editing (as in the case of mitochondrial leucyl-tRNA synthetase) (49), intron splicing (as in the case of yeast mitochondrial leucyl-tRNA synthetase) (50), and catalysis (as in the case of GlyRS1).
ACKNOWLEDGMENT
This work was supported by a grant (NSC101-3113-B-008-001 to C.-C.W.) from the National Science Council (Taipei, Taiwan).
Footnotes
Published ahead of print 1 July 2013
REFERENCES
- 1.Burbaum JJ, Schimmel P. 1991. Structural relationships and the classification of aminoacyl-tRNA synthetases. J. Biol. Chem. 266:16965–16968 [PubMed] [Google Scholar]
- 2.Carter CW., Jr 1993. Cognition, mechanism, and evolutionary relationships in aminoacyl-tRNA synthetases. Annu. Rev. Biochem. 62:715–748 [DOI] [PubMed] [Google Scholar]
- 3.Giege R, Sissler M, Florentz C. 1998. Universal rules and idiosyncratic features in tRNA identity. Nucleic Acids Res. 26:5017–5035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giege R. 2006. The early history of tRNA recognition by aminoacyl-tRNA synthetases. J. Biosci. 31:477–488 [DOI] [PubMed] [Google Scholar]
- 5.Rinehart J, Krett B, Rubio MA, Alfonzo JD, Söll D. 2005. Saccharomyces cerevisiae imports the cytosolic pathway for Gln-tRNA synthesis into the mitochondrion. Genes Dev. 19:583–592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tang HL, Yeh LS, Chen NK, Ripmaster T, Schimmel P, Wang CC. 2004. Translation of a yeast mitochondrial tRNA synthetase initiated at redundant non-AUG codons. J. Biol. Chem. 279:49656–49663 [DOI] [PubMed] [Google Scholar]
- 7.Huang HY, Tang HL, Chao HY, Yeh LS, Wang CC. 2006. An unusual pattern of protein expression and localization of yeast alanyl-tRNA synthetase isoforms. Mol. Microbiol. 60:189–198 [DOI] [PubMed] [Google Scholar]
- 8.Chang KJ, Wang CC. 2004. Translation initiation from a naturally occurring non-AUG codon in Saccharomyces cerevisiae. J. Biol. Chem. 279:13778–13785 [DOI] [PubMed] [Google Scholar]
- 9.Natsoulis G, Hilger F, Fink GR. 1986. The HTS1 gene encodes both the cytoplasmic and mitochondrial histidine tRNA synthetases of S. cerevisiae. Cell 46:235–243 [DOI] [PubMed] [Google Scholar]
- 10.Chatton B, Walter P, Ebel JP, Lacroute F, Fasiolo F. 1988. The yeast VAS1 gene encodes both mitochondrial and cytoplasmic valyl-tRNA synthetases. J. Biol. Chem. 263:52–57 [PubMed] [Google Scholar]
- 11.Chiu WC, Chang CP, Wen WL, Wang SW, Wang CC. 2010. Schizosaccharomyces pombe possesses two paralogous valyl-tRNA synthetase genes of mitochondrial origin. Mol. Biol. Evol. 27:1415–1424 [DOI] [PubMed] [Google Scholar]
- 12.Chang CP, Tseng YK, Ko CY, Wang CC. 2012. Alanyl-tRNA synthetase genes of Vanderwaltozyma polyspora arose from duplication of a dual-functional predecessor of mitochondrial origin. Nucleic Acids Res. 40:314–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mirande M. 2010. Processivity of translation in the eukaryote cell: role of aminoacyl-tRNA synthetases. FEBS Lett. 584:443–447 [DOI] [PubMed] [Google Scholar]
- 14.Wang CC, Schimmel P. 1999. Species barrier to RNA recognition overcome with nonspecific RNA binding domains. J. Biol. Chem. 274:16508–16512 [DOI] [PubMed] [Google Scholar]
- 15.Frugier M, Moulinier L, Giege R. 2000. A domain in the N-terminal extension of class IIb eukaryotic aminoacyl-tRNA synthetases is important for tRNA binding. EMBO J. 19:2371–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chang CP, Lin G, Chen SJ, Chiu WC, Chen WH, Wang CC. 2008. Promoting the formation of an active synthetase/tRNA complex by a nonspecific tRNA-binding domain. J. Biol. Chem. 283:30699–30706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grant TD, Snell EH, Luft JR, Quartley E, Corretore S, Wolfley JR, Snell ME, Hadd A, Perona JJ, Phizicky EM, Grayhack EJ. 2012. Structural conservation of an ancient tRNA sensor in eukaryotic glutaminyl-tRNA synthetase. Nucleic Acids Res. 40:3723–3731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simos G, Segref A, Fasiolo F, Hellmuth K, Shevchenko A, Mann M, Hurt EC. 1996. The yeast protein Arc1p binds to tRNA and functions as a cofactor for the methionyl- and glutamyl-tRNA synthetases. EMBO J. 15:5437–5448 [PMC free article] [PubMed] [Google Scholar]
- 19.Godinic V, Mocibob M, Rocak S, Ibba M, Weygand-Durasevic I. 2007. Peroxin Pex21p interacts with the C-terminal noncatalytic domain of yeast seryl-tRNA synthetase and forms a specific ternary complex with tRNA(Ser). FEBS J. 274:2788–2799 [DOI] [PubMed] [Google Scholar]
- 20.Frechin M, Senger B, Braye M, Kern D, Martin RP, Becker HD. 2009. Yeast mitochondrial Gln-tRNA(Gln) is generated by a GatFAB-mediated transamidation pathway involving Arc1p-controlled subcellular sorting of cytosolic GluRS. Genes Dev. 23:1119–1130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schimmel P. 1987. Aminoacyl tRNA synthetases: general scheme of structure-function relationships in the polypeptides and recognition of transfer RNAs. Annu. Rev. Biochem. 56:125–158 [DOI] [PubMed] [Google Scholar]
- 22.Eriani G, Delarue M, Poch O, Gangloff J, Moras D. 1990. Partition of tRNA synthetases into two classes based on mutually exclusive sets of sequence motifs. Nature 347:203–206 [DOI] [PubMed] [Google Scholar]
- 23.Ibba M, Morgan S, Curnow AW, Pridmore DR, Vothknecht UC, Gardner W, Lin W, Woese CR, Söll D. 1997. A euryarchaeal lysyl-tRNA synthetase: resemblance to class I synthetases. Science 278:1119–1122 [DOI] [PubMed] [Google Scholar]
- 24.Ostrem DL, Berg P. 1970. Glycyl-tRNA synthetase: an oligomeric protein containing dissimilar subunits. Proc. Natl. Acad. Sci. U. S. A. 67:1967–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mazauric MH, Reinbolt J, Lorber B, Ebel C, Keith G, Giege R, Kern D. 1996. An example of non-conservation of oligomeric structure in prokaryotic aminoacyl-tRNA synthetases. Biochemical and structural properties of glycyl-tRNA synthetase from Thermus thermophilus. Eur. J. Biochem. 241:814–826 [DOI] [PubMed] [Google Scholar]
- 26.Shiba K, Schimmel P, Motegi H, Noda T. 1994. Human glycyl-tRNA synthetase. Wide divergence of primary structure from bacterial counterpart and species-specific aminoacylation. J. Biol. Chem. 269:30049–30055 [PubMed] [Google Scholar]
- 27.Nada S, Chang PK, Dignam JD. 1993. Primary structure of the gene for glycyl-tRNA synthetase from Bombyx mori. J. Biol. Chem. 268:7660–7667 [PubMed] [Google Scholar]
- 28.Chen SJ, Lin G, Chang KJ, Yeh LS, Wang CC. 2008. Translational efficiency of a non-AUG initiation codon is significantly affected by its sequence context in yeast. J. Biol. Chem. 283:3173–3180 [DOI] [PubMed] [Google Scholar]
- 29.Turner RJ, Lovato M, Schimmel P. 2000. One of two genes encoding glycyl-tRNA synthetase in Saccharomyces cerevisiae provides mitochondrial and cytoplasmic functions. J. Biol. Chem. 275:27681–27688 [DOI] [PubMed] [Google Scholar]
- 30.Chen SJ, Lee CY, Lin ST, Wang CC. 2011. Rescuing a dysfunctional homologue of a yeast glycyl-tRNA synthetase gene. ACS Chem. Biol. 6:1182–1187 [DOI] [PubMed] [Google Scholar]
- 31.Chen SJ, Wu YH, Huang HY, Wang CC. 2012. Saccharomyces cerevisiae possesses a stress-inducible glycyl-tRNA synthetase gene. PLoS One 7:e33363. 10.1371/journal.pone.0033363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nameki N, Tamura K, Asahara H, Hasegawa T. 1997. Recognition of tRNA(Gly) by three widely diverged glycyl-tRNA synthetases. J. Mol. Biol. 268:640–647 [DOI] [PubMed] [Google Scholar]
- 33.Fersht AR, Ashford JS, Bruton CJ, Jakes R, Koch GL, Hartley BS. 1975. Active site titration and aminoacyl adenylate binding stoichiometry of aminoacyl-tRNA synthetases. Biochemistry 14:1–4 [DOI] [PubMed] [Google Scholar]
- 34.Simlot MM, Pfaender P. 1973. Amino acid dependent ATP-32PPi exchange measurement. A filter paper disk method. FEBS Lett. 35:201–203 [DOI] [PubMed] [Google Scholar]
- 35.Schwede T, Kopp J, Guex N, Peitsch MC. 2003. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 31:3381–3385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Nangle LA, Zhang W, Xie W, Yang XL, Schimmel P. 2007. Charcot-Marie-Tooth disease-associated mutant tRNA synthetases linked to altered dimer interface and neurite distribution defect. Proc. Natl. Acad. Sci. U. S. A. 104:11239–11244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schrodinger LLC. 2010. The PyMOL molecular graphics system, version 1.3r1. Schrodinger LLC, New York, NY. [Google Scholar]
- 38.Liao CC, Lin CH, Chen SJ, Wang CC. 2012. Trans-kingdom rescue of Gln-tRNAGln synthesis in yeast cytoplasm and mitochondria. Nucleic Acids Res. 40:9171–9181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen SJ, Ko CY, Yen CW, Wang CC. 2009. Translational efficiency of redundant ACG initiator codons is enhanced by a favorable sequence context and remedial initiation. J. Biol. Chem. 284:818–827 [DOI] [PubMed] [Google Scholar]
- 40.Kelly SM, Jess TJ, Price NC. 2005. How to study proteins by circular dichroism? Biochim. Biophys. Acta 1751:119–139 [DOI] [PubMed] [Google Scholar]
- 41.Mazauric MH, Keith G, Logan D, Kreutzer R, Giege R, Kern D. 1998. Glycyl-tRNA synthetase from Thermus thermophilus—wide structural divergence with other prokaryotic glycyl-tRNA synthetases and functional inter-relation with prokaryotic and eukaryotic glycylation systems. Eur. J. Biochem. 251:744–757 [DOI] [PubMed] [Google Scholar]
- 42.Louis-Jeune C, Andrade-Navarro MA, Perez-Iratxeta C. 2012. Prediction of protein secondary structure from circular dichroism using theoretically derived spectra. Proteins 80:374–381 [DOI] [PubMed] [Google Scholar]
- 43.Saitou N, Nei M. 1987. The neighbor-joining method: a new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 4:406–425 [DOI] [PubMed] [Google Scholar]
- 44.Logan DT, Mazauric MH, Kern D, Moras D. 1995. Crystal structure of glycyl-tRNA synthetase from Thermus thermophilus. EMBO J. 14:4156–4167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chiu WC, Chang CP, Wang CC. 2009. Evolutionary basis of converting a bacterial tRNA synthetase into a yeast cytoplasmic or mitochondrial enzyme. J. Biol. Chem. 284:23954–23960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Mazauric MH, Roy H, Kern D. 1999. tRNA glycylation system from Thermus thermophilus. tRNAGly identity and functional interrelation with the glycylation systems from other phylae. Biochemistry 38:13094–13105 [DOI] [PubMed] [Google Scholar]
- 47.Marechal-Drouard L, Small I, Weil JH, Dietrich A. 1995. Transfer RNA import into plant mitochondria. Methods Enzymol. 260:310–327 [DOI] [PubMed] [Google Scholar]
- 48.Whelihan EF, Schimmel P. 1997. Rescuing an essential enzyme-RNA complex with a non-essential appended domain. EMBO J. 16:2968–2974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nawaz MH, Pang YL, Martinis SA. 2007. Molecular and functional dissection of a putative RNA-binding region in yeast mitochondrial leucyl-tRNA synthetase. J. Mol. Biol. 367:384–394 [DOI] [PubMed] [Google Scholar]
- 50.Sarkar J, Poruri K, Boniecki MT, McTavish KK, Martinis SA. 2012. Yeast mitochondrial leucyl-tRNA synthetase CP1 domain has functionally diverged to accommodate RNA splicing at expense of hydrolytic editing. J. Biol. Chem. 287:14772–14781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Thompson JD, Higgins DG, Gibson TJ. 1994. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22:4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]