

Abstract
We recently showed, in primary vascular smooth muscle cells (VSMCs), that the platelet-derived growth factor activates canonical store-operated Ca2+ entry and Ca2+ release-activated Ca2+ currents encoded by Orai1 and STIM1 genes. However, thrombin activates store-independent Ca2+ selective channels contributed by both Orai3 and Orai1. These store-independent Orai3/Orai1 channels are gated by cytosolic leukotriene C4 (LTC4) and require STIM1 downstream LTC4 action. However, the source of LTC4 and the signaling mechanisms of STIM1 in the activation of this LTC4-regulated Ca2+ (LRC) channel are unknown. Here, we show that upon thrombin stimulation, LTC4 is produced through the sequential activities of phospholipase C, diacylglycerol lipase, 5-lipo-oxygenease, and leukotriene C4 synthase. We show that the endoplasmic reticulum-resident STIM1 is necessary and sufficient for LRC channel activation by thrombin. STIM1 does not form sustained puncta and does not colocalize with Orai1 either under basal conditions or in response to thrombin. However, STIM1 is precoupled to Orai3 and Orai3/Orai1 channels under basal conditions as shown using Forster resonance energy transfer (FRET) imaging. The second coiled-coil domain of STIM1 is required for coupling to either Orai3 or Orai3/Orai1 channels and for LRC channel activation. We conclude that STIM1 employs distinct mechanisms in the activation of store-dependent and store-independent Ca2+ entry pathways.
INTRODUCTION
Ca2+ is a universal second messenger that controls a variety of cell functions in health and disease. In nonexcitable cells, most attention has focused on store-operated calcium entry (SOCE), first defined by Putney (1, 2). After more than 20 years of studies, SOCE is well recognized as a ubiquitous and physiologically important mode of receptor-regulated Ca2+ entry into most cells. Activation of a variety of phospholipase C (PLC)-coupled receptors leads to the hydrolysis of phosphatidylinositol-4,5-bisphosphate (PIP2) into 2 messengers, diacylglycerol (DAG) and inositol-1,4,5-trisphosphate (IP3) (3). The latter binds to IP3 receptors on the endoplasmic reticulum (ER), resulting in depletion of intracellular Ca2+ stores. The depletion of ER Ca2+ is sensed by stromal interaction molecule 1 (STIM1), which results in STIM1 aggregation and movement to areas where the ER comes close to the plasma membrane (PM) to physically interact with Orai1 channels and activate Ca2+ entry (4–8). In addition to SOCE, a store-independent pathway mediated by arachidonate-regulated Ca2+ (ARC) channels has been described and studied by Shuttleworth and colleagues during the past decade (9). ARC channels have been shown to be activated by exogenous arachidonic acid (AA) in HEK293 cells, contributed by both Orai1 and Orai3 subunits (10), and regulated by a specific pool of STIM1 located in the plasma membrane (11, 12). Subsequent data from the Shuttleworth group suggested that Orai3 confers sensitivity to AA via its N-terminal region (13). However, the precise gating of ARC channels by AA and the downstream signaling pathways and the cellular functions regulated by ARC channels remain largely unknown.
Vascular smooth muscle cells (VSMCs) are a major vascular cell type that forms the medial layer which is necessary for blood vessel integrity and for control of blood pressure (14). VSMCs are highly adaptable in nature and retain the ability to switch in vivo from a contractile excitable phenotype to a proliferative migratory nonexcitable phenotype (also called synthetic) (14, 15); the VSMC synthetic phenotype can be recapitulated by in vitro culture in the presence of serum. This physiological VSMC remodeling into a synthetic phenotype is essential for vascular development and repair. However, its dysfunction contributes to vascular diseases such as atherosclerosis, hypertension, restenosis, and leiomyosarcomas. We and others have previously shown that the migratory and proliferative VSMC agonist platelet-derived growth factor (PDGF) activates SOCE and corresponding Ca2+ release-activated Ca2+ (CRAC) currents in VSMCs (16–18). We further have shown that Orai1 is an important determinant for VSMC remodeling and development of neointimal hyperplasia in vivo upon vascular injury (19). Recently, we have identified a novel highly Ca2+-selective store-independent ARC-like channel in synthetic VSMCs that plays an important role in VSMC remodeling in vivo (17). This current is activated by thrombin, a pathophysiological VSMC agonist through cytosolic leukotriene C4 (LTC4)-produced downstream thrombin stimulation (17). We have shown that this LTC4-regulated Ca2+ (LRC) channel requires Orai3, Orai1, and STIM1. However, the source of LTC4 during receptor activation, the precise pool of STIM1 involved, and the mechanisms of STIM1 activation of LRC channels remain unknown.
In this study, we applied confocal and Forster resonance energy transfer (FRET) imaging, Fura2 Ca2+ imaging, and patch clamp electrophysiology coupled to pharmacology and “erase-and-replace” approaches to show that thrombin activates store-independent LRC channels through LTC4 produced via the sequential catalytic activities of PLC, DAG lipase, 5-lipo-oxygenase (5-LO), and LTC4 synthase (LTC4S). We provide evidence that ER-resident STIM1 is necessary and sufficient for LRC channel activation and that ER-STIM1 is precoupled to Orai3 under basal conditions through the second coiled-coil domain of STIM1.
MATERIALS AND METHODS
All Ca2+ imaging, patch clamp, confocal, and FRET imaging experiments were conducted at room temperature.
VSMC dispersion and cell culture.
The use of rats for these experiments has been reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) at the Albany Medical College Animal Resource Facility, which is licensed by the U.S. Department of Agriculture and the Division of Laboratories and Research of the New York State Department of Public Health and is accredited by the American Association for the Accreditation of Laboratory Animal Care. Male adult rats (150 g) were euthanized by suffocation in a CO2 chamber, and VSMCs from aortas were isolated, tested for expression of the smooth muscle marker SM22α, and cultured as previously described (17, 19). VSMCs are used in all experiments between passages 4 and 8. HEK293 cells were purchased from ATCC and cultured in standard Dulbecco modified Eagle medium (DMEM) containing 10% fetal bovine serum (FBS) and antibiotics.
Cell transfections.
Small interfering RNA (siRNA) sequences that induced significant decreases in their target mRNA (over 80%) without cross-effects on other mRNAs were used in Western blotting to confirm protein knockdown as described below. siRNA sequences targeting rat STIM1 were as follows: rSTIM1#1, UAAGGGAAGACCUCAAUU; rSTIM1#2, CAUCAGAAGUGUAUAACUG. All transfections in VSMCs were done using the Nucleofector device II (Amaxa Biosystems, Gaithersburg, MD) using program D033 according to the manufacturer's instructions. An 0.5-μg amount of green fluorescent protein (GFP) was cotransfected with siRNA for identification of successfully transfected cells. The control siRNA is a scrambled siRNA sequence. All transfections in HEK293 cells were done using the TransFectin reagent (Bio-Rad) according to the manufacturer's instructions.
Western blotting.
Cells were lysed using radioimmunoprecipitation assay (RIPA) lysis buffer, and 20 to 50 μg of proteins in denaturing conditions was subjected to SDS-PAGE and then electrotransferred onto polyvinylidene difluoride membranes. Blots were blocked with 5% nonfat dry milk (NFDM) dissolved in Tris-buffered saline containing 0.1% Tween 20 (TTBS) for 2 h at room temperature, incubated overnight at 4°C with specific primary antibodies in TTBS containing 2% NFDM, and incubated for 45 min at room temperature with the appropriate horseradish peroxidase-conjugated secondary antibody. Detection was performed using the enhanced chemiluminescence reagent (Amersham).
Ca2+ measurements.
Ca2+ was measured as described previously (17, 19). Briefly, coverslips with attached cells were mounted in a Teflon chamber and incubated at 37°C for 1 h in culture medium (DMEM with 10% FBS) containing 4 μM Fura2-AM (Molecular Probes, Eugene, OR). Cells were then washed and bathed in HEPES-buffered saline solution (140 mM NaCl, 1.13 mM MgCl2, 4.7 mM KCl, 2 mM CaCl2, 10 mM d-glucose, and 10 mM HEPES, with pH adjusted to 7.4 with NaOH) for ≥10 min before Ca2+ was measured. For Ca2+ measurements, fluorescence images of several cells were recorded and analyzed with a digital fluorescence imaging system (InCyt Im2; Intracellular Imaging, Cincinnati, OH). Fura2 fluorescence at an emission wavelength of 510 nm was induced by excitation of Fura2 alternately at 340 and 380 nm. The ratio of fluorescence at 340 nm to that at 380 nm was obtained on a pixel-by-pixel basis. The minimal and maximal values of Fura2 ratios, Rmin and Rmax, were 0.3 and 2.5, respectively. All experiments were conducted at room temperature. For pharmacological experiments reported in Fig. 1, cells were preincubated in the presence of either the vehicle or drugs for 10 min before recordings were initiated.
Fig 1.

Thrombin-activated Ca2+ entry requires the sequential catalytic activities of PLC, DAG lipase, 5-LO, and LTC4S. (A) Schematic representation of the pharmacological approach employed to study the pathway downstream thrombin receptor (crosses, drugs that inhibited thrombin-activated Ca2+ entry; checkmarks, drugs that had no effect; upward arrows, drugs that potentiated thrombin-activated Ca2+ entry). (B and C) Representative Ca2+ imaging traces from cells pretreated with either the vehicle control or various drugs at the concentrations indicated. (D) Statistical summary of effects of all drugs employed on thrombin-activated Ca2+ entry (data shown as x, y, where x = number of independent runs and y = total number of cells). Throughout the figures, *, **, and *** indicate P values of <0.05, 0.01, and 0.001, respectively.
Patch clamp electrophysiology.
Conventional whole-cell patch clamp recordings were carried out using an Axopatch 200B patch clamp and Digidata 1440A digitizer (Axon Instruments, NY) as previously published with a few important modifications (19–23). To reduce the noise to a minimum, we added in series a humbug noise eliminator that eliminates electrical interference such as simple 50-/60-Hz sine waves, mixtures of 50-/60-Hz harmonics, noise spikes from dimmers, and complex noise from fluorescent lamps. All experiments were performed at room temperature (20 to 25°C). Pipettes were pulled from borosilicate glass capillaries (World Precision Instruments, Inc., Sarasota, FL) with a P-97 flaming/brown micropipette puller (Sutter Instrument Company, Novato, CA) and polished with a DMF1000 microforge (World Precision Instruments). Resistances of filled glass pipettes were 2 to 3 MΩ. Series resistances were in the range of 2 to 10 MΩ. The liquid-junction potential offset was around 4.6 mV and was corrected. Only cells with tight seals (>16 GΩ) were selected for break-in. Immediately after establishing the whole-cell patch clamp configuration, we start the recording by running a 250-ms voltage ramp (from +100 mV to −140 mV) every 2 s and performing a first divalent-free (DVF) pulse when current development is minimal. The first I/V curves obtained immediately after break-in in Ca2+-containing bath solutions and divalent-free (DVF) bath solutions represent background currents that are subtracted from stimulus-activated Ca2+ currents and Na+ currents. After the currents are fully activated by different stimuli, I/V curves are obtained for Ca2+ currents (in Ca2+-containing bath solutions) and Na+ currents (in DVF bath solutions). Using OriginLab 7.5 software (OriginLab, Northampton, MA, USA), I/V curves corresponding to background currents obtained in Ca2+ and Na+ are subtracted from the I/V curves obtained in Ca2+ and Na+ after stimulus/agonist addition and maximal current activation. The subtracted I/V curves are represented as independent I/V curves in all figures. Cells were maintained at a 0-mV holding potential during experiments. Reverse ramps were designed to inhibit Na+ channels potentially expressed in VSMCs. High MgCl2 (8 mM) was included in the patch pipette to inhibit TRPM7 currents, and 3 μM nimodipine was added to the bath solution to generally stabilize membrane patches and reach better seals.
Solutions employed for whole-cell patch clamp electrophysiology. (i) Thrombin-activated currents (VSMCs).
Bath solution consisted of 135 mM sodium methanesulfonate, 10 mM CsCl, 1.2 mM MgSO4, 10 mM HEPES, 20 mM CaCl2, and 10 mM glucose (pH was adjusted to 7.4 with NaOH). Thrombin (100 nM) was added to the bath where indicated in the figures. Pipette solution consisted of 145 mM cesium methanesulfonate, 10 mM cesium-1,2-bis-(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (Cs-BAPTA), 5 mM CaCl2, 8 mM MgCl2, and 10 mM HEPES (pH adjusted to 7.2 with CsOH). Divalent-free (DVF) bath solution consisted of 155 mM sodium methanesulfonate, 10 mM HEDTA, 1 mM EDTA, and 10 mM HEPES (pH 7.4, adjusted with NaOH).
(ii) Store depletion-activated currents (in VSMCs).
Bath solution consisted of 135 mM sodium methanesulfonate, 10 mM CsCl, 1.2 mM MgSO4, 10 mM HEPES, 20 mM CaCl2, and 10 mM glucose (pH was adjusted to 7.4 with NaOH). Pipette solution consisted of 145 mM cesium methanesulfonate, 20 mM cesium-1,2-bis-(2-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid (Cs-BAPTA), 8 mM MgCl2, and 10 mM HEPES (pH adjusted to 7.2 with CsOH). Divalent-free (DVF) bath solution consisted of 155 mM sodium methanesulfonate, 10 mM HEDTA, 1 mM EDTA, and 10 mM HEPES (pH 7.4, adjusted with NaOH).
(iii) Store depletion-activated currents (in HEK293 cells).
Bath solution consisted of 145 mM NaCl, 5 mM CsCl, 1 mM MgCl2, 10 mM HEPES, 10 mM glucose, and 10 mM CaCl2 (pH was adjusted to 7.4 with NaOH). Pipette solution consisted of 145 mM cesium methanesulfonate, 8 mM NaCl, 3.5 mM MgCl2, 10 mM HEPES, and 20 mM EGTA (pH adjusted to 7.2 with CsOH).
Erase-and-replace experiments.
VSMCs were electroporated with 20 μg of either control siRNA or STIM1 siRNA and incubated for 72 h. Cells were then detached and electroporated again with siRNA along with siRNA-resistant plasmids encoding different versions of human STIM1. Cells were allowed to recover for an additional 36 h before Ca2+ measurements were performed. Protein expression of STIM1 constructs was verified by Western blotting and fluorescence (for enhanced yellow fluorescent protein [eYFP]-STIM1 plasmids). Western blotting assays reflect the total amount of proteins in transfected cell populations, while Ca2+ measurements were performed on individual cells that are positively transfected with plasmids (based on fluorescence of eYFP for tagged constructs and cotransfected GFP for untagged constructs).
Confocal imaging. (i) Confocal microscopy (eYFP-STIM1 punctum experiments).
VSMCs were transfected by electroporation with eYFP-STIM1 (0.75 μg plasmid DNA per 106 cells) and seeded on glass-bottom petri dishes (MatTek) for live confocal imaging using a Zeiss LSM 510 Meta confocal microscope. Images were collected using an ×63 oil immersion lens. LSM software was used to drive the hardware and image acquisition. Vertical sections were collected at 0.5-μm intervals, with pinhole settings at 1 Airy unit. A z-stack was acquired every 20 s for 20 min. The first 5 stacks before addition of stimuli were used as control. Treatments with stimuli (thapsigargin, PDGF, and thrombin) were performed between the 5th and 6th acquisition time points. ImageJ software was used to provide a z-projection (maximum intensity) for better visualization of protein distribution.
(ii) Colocalization studies: Orai1/STIM1 and Orai3/STIM1 (in VSMCs).
VSMCs were cotransfected by electroporation with eYFP-STIM1 (0.5 μg plasmid DNA per 106 cells) and CFP-Orais (either CFP-Orai1 or CFP-Orai3; 3 μg). Data acquisition was achieved as described above with pinhole settings automatically optimized by the LSM software. Cyan fluorescent protein (CFP)-Orai1/3 and eYFP-STIM1 were individually stimulated by using 458 and 514 argon laser lines, respectively. Time series of single-plane images of STIM1/Orai1 cotransfection were acquired every 20 s after stimulation with either thapsigargin or thrombin. The first 5 images without stimulation were used as controls. ImageJ was employed to adjust the image brightness and contrast for better visualization. Plot profiles for the CFP and YFP intensities were obtained by using the plot profile feature from ImageJ. For the three-dimensional (3-D) reconstruction of STIM1/Orai colocalization under basal conditions, z-stack images were acquired as described above and transferred to Imaris 7.2.3 software. This software was used for generation of 3-D surfaces based on a size/intensity threshold. A colocalization feature was employed to calculate the extent of STIM1/Orai specific surface overlap.
(iii) Fluorescence microscopy (Orai1/STIM1, Orai3/STIM1, Orai3-Orai1/STIM1, Orai1/STIM1-A376K, Orai3/STIM1-A376K, Orai3-Orai1/STIM1-A376K [in HEK293 cells]) and FRET (Orai1/STIM1 and Orai3/STIM1 [in HEK293 cells]).
Confocal FRET microscopy was performed as previously described (24). In brief, a QLC100 real-time confocal system (VisiTech Int., United Kingdom) was used for recording fluorescence images connected to two Photometrics CoolSNAPHQ monochrome cameras (Roper Scientific) and a dual port adapter (dichroic, 505lp; cyan emission filter, 485/30; yellow emission filter, 535/50; Chroma Technology Corp.). This system was attached to an Axiovert 200 M microscope (Zeiss, Germany) in conjunction with an argon ion multiwavelength (457-, 488-, and 514-nm) laser (Spectra Physics). The wavelengths were selected by an Acousto optical tuneable filter (VisiTech Int., United Kingdom). Image acquisition and control of the confocal system were performed with MetaMorph 6.1 software (Universal Imaging Corp.). CFP, YFP, and overlay images were consecutively recorded with a minimum delay. Image analysis was performed with custom-made software (25) integrated in MatLab 7.0.4 according to the method previously published (26). Scale bars are 5 μm throughout unless otherwise noted.
Statistics.
For a patch clamp where n is less than 10, data are expressed as mean/range (instead of standard errors of the means [SEM]), and statistical analyses comparing two experimental groups were performed using the two-tailed t test with OriginLab 7.5 software (OriginLab, Northampton, MA). Differences were considered significant when P was <0.05. For studies with bigger sample sizes, including Ca2+ imaging, data are represented as means ± SEM. n in the representative Ca2+ imaging traces represents the number of cells analyzed simultaneously in the same coverslip and averaged. The two numbers between parentheses (x, y) next to each data point in bar graphs represent the following: x = number of independent experiments and y = total number of cells from independent experiments/transfections. For comparison between control and one experimental condition, the two-tailed t test was used. For multiple comparisons such as in the erase-and-replace experiments, one-way analysis of variance (ANOVA) was performed. Differences were considered significant when P was <0.05.
RESULTS
LRC channel activation by thrombin requires the sequential catalytic activities of PLC, DAG lipase, 5-LO, and LTC4S.
To determine the source of LTC4 during LRC channel activation by thrombin in primary synthetic rat aortic VSMCs, we used pharmacological blockers of enzymes involved in AA synthesis and metabolism (Fig. 1A) and determined their effect on thrombin-mediated Ca2+ entry by using Fura2 imaging. We used a standard Fura2 Ca2+ imaging protocol with agonist stimulation in a nominally Ca2+-free solution followed by restoration of Ca2+ (2 mM) to the extracellular milieu. While inhibitors of PLC, DAG lipase, and 5-lipooxygensase (5-LO) inhibited thrombin-activated Ca2+ entry (Fig. 1B), inhibitors of G protein-associated phospholipase A2 (PLA2), cyclooxygenases (Cox1/2), and leukotriene A4 (LTA4) hydrolase had no effect (Fig. 1D; see also Fig. S1A in the supplemental material). With the exception of the PLC inhibitor U73122, all inhibitors had no statistically significant effect on thrombin-mediated Ca2+ release. We previously showed that LTC4 is the signal required for LRC channel activation (17). Inhibition of the transporter multidrug resistance-associated protein 1 (MRP1) with high concentrations of montelukast (1 μM) potentiated thrombin-activated Ca2+ entry (Fig. 1C and D), suggesting a role for LTC4 from inside the cell. However, inhibition of the cysteinyl leukotriene G protein-coupled receptors 1/2 (CysLTR1/2) with three independent drugs (montelukast, MK-571, and Bay-U9773) had no effect on thrombin-activated Ca2+ entry, ruling out a role for LTC4 from outside the cell (Fig. 1D). Statistical analysis of all data from several independent experiments is depicted in Fig. 1D; representative Fura2 traces for all drugs that showed no effect on thrombin-activated Ca2+ entry are shown in Fig. S1A. Figure S1B and C shows that nordihydroguaiaretic acid (NDGA), which affected thrombin-activated Ca2+ entry, has no effect on SOCE activated by platelet-derived growth factor (PDGF).
ER-resident STIM1 is required and sufficient for activation of thrombin-activated Ca2+ entry.
We then sought to determine which pool of STIM1 is required for this store-independent thrombin-activated Ca2+ entry. Given the striking similarity between this pathway and the ARC pathway (dependence on STIM1, Orai1, and Orai3), we considered the possibility that thrombin-activated Ca2+ entry depends on the pool of STIM1 (∼10 to 15% of total) that is located in the plasma membrane, as was shown for ARC channels (11). Therefore, we performed erase-and-replace experiments where knockdown of endogenous rat STIM1 in VSMCs was achieved using specific siRNA followed by subsequent rescue with different versions of human siRNA-resistant STIM1 constructs to near-endogenous levels (Fig. 2A). These are human wild-type untagged STIM1 that is expressed at both ER and plasma membrane and two versions of STIM1 that were proposed to be unable to traffic to the plasma membrane and were expressed exclusively in ER, eYFP-STIM1 (27) and an untagged glycosylation mutant of STIM1 (N131Q and N171Q) (11). All these STIM1 constructs were expressed in VSMCs (Fig. 2B); please note that while Western blotting assays represent total protein expression in a mixed population of cells, Ca2+ imaging experiments were conducted only in cells that were successfully transfected as evidenced by eYFP-STIM1 fluorescence or cotransfected GFP fluorescence (for untagged plasmids). Surprisingly, all versions of STIM1 were capable of rescuing thrombin-activated Ca2+ entry (Fig. 2C and D), suggesting that ER-resident STIM1 is necessary and sufficient to support thrombin-activated Ca2+ entry.
Fig 2.
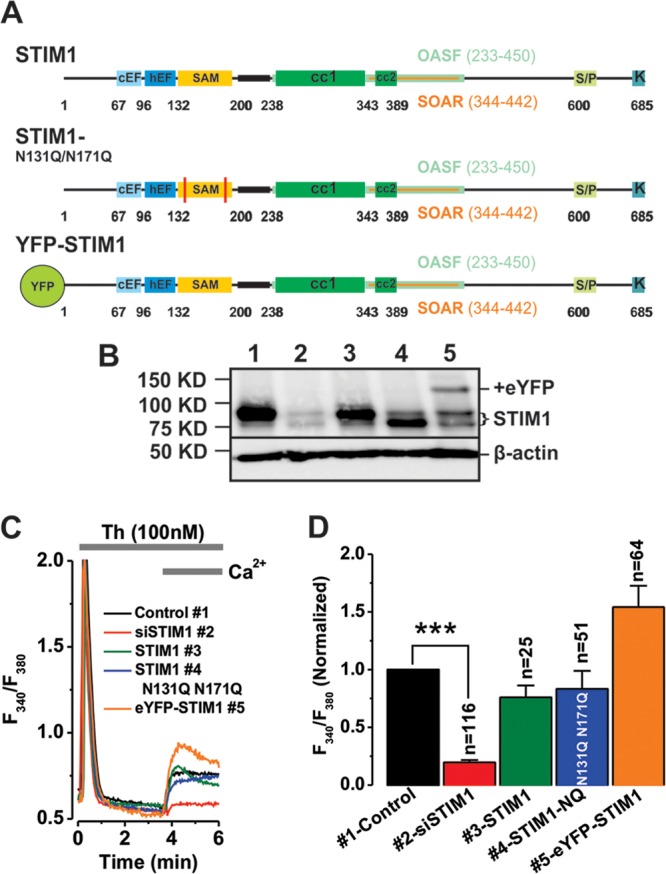
ER-resident STIM1 is required for activation of thrombin-activated Ca2+ entry. (A) Schematic representation of various versions of STIM1 employed in erase-and-replace experiments (see Materials and Methods). (B) Western blot analysis confirmed successful endogenous STIM1 knockdown and validated the erase-and-replace approach. Endogenous STIM1 is detected by Western blotting (lane 1), and knockdown was achieved using specific siRNA against STIM1 (lane 2). STIM1 rescue was achieved by transfecting 1.5 μg of plasmid DNA per 106 cells. Plasmids expressing full-length STIM1 (lane 3), STIM1 N131Q N171Q glycosylation mutant (lane 4; note the lower molecular mass of this mutant), and eYFP-STIM1 (lane 5). Please note that while Western blotting assays represent total protein expression in a mixed population of cells, Ca2+ imaging experiments were conducted only in cells that were successfully transfected as evidenced by eYFP-STIM1 or cotransfected GFP (for untagged plasmids) fluorescence. Positions of protein molecular mass markers are shown on the left. (C) Representative Ca2+ imaging traces from cells subjected to the erase-and-replace approach showing rescue of thrombin-activated Ca2+ entry with all STIM1 constructs. (D) Summary of Ca2+ imaging data is shown.
Thrombin-activated Ca2+ entry is not blocked by drugs that inhibit STIM1 punctum formation.
We previously showed that although it requires STIM1, thrombin-activated Ca2+ entry is store independent and that thrombin does not cause sustained Ca2+ store depletion (17), suggesting that store depletion and subsequent STIM1 punctum formation are likely not required for thrombin-activated Ca2+ entry. Therefore, we sought to determine the effects on thrombin-activated Ca2+ entry of SOCE blockers that inhibit STIM1 puncta and interfere with Orai-STIM1 interactions, namely, 2-aminoethyldiphenyl borate (2-APB) and ML9 (28). As expected, Fura2 Ca2+ imaging revealed that the thrombin-activated Ca2+ entry pathway in VSMCs is insensitive to 2-APB and ML9 (Fig. 3A), suggesting that thrombin-activated Ca2+ entry does not require STIM1 reorganization into puncta. SOCE activated by thapsigargin was consistently blocked by both 2-APB and ML9 (Fig. 3B). Similar results were obtained with whole-cell current measurements. As shown previously (17), whole-cell patch clamp recordings using a pipette solution where Ca2+ was buffered to 150 nM showed that thrombin activated an inwardly rectifying Ca2+-selective current that was recorded in Ca2+-containing (20 mM) bath solutions and amplified in divalent-free (DVF; Na+ as the charge carrier) solutions (Fig. 3C). However, this current was insensitive to 2-APB (Fig. 3D and E); 2-APB at the same concentration blocked CRAC currents activated through Ca2+ store depletion by dialysis of the cell with a pipette solution containing 20 mM BAPTA (Fig. 3F to H). Of note, while CRAC showed the typical depotentiation in DVF solutions (29), thrombin-activated Ca2+ currents did not depotentiate in DVF solutions (see Fig. S2 in the supplemental material). These results are consistent with ectopically expressed Orai3 currents that show less depotentiation in DVF solutions (30).
Fig 3.
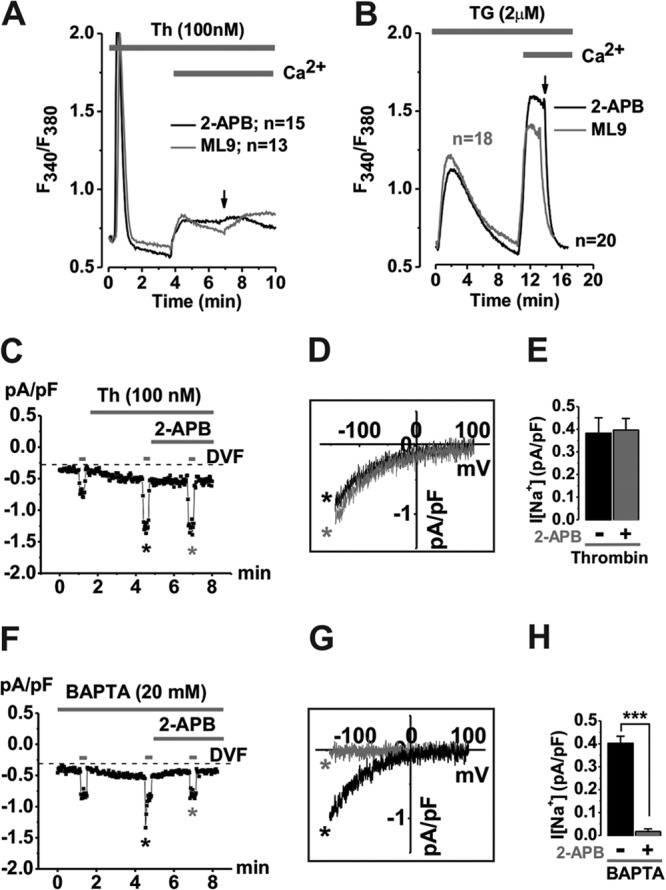
Thrombin-activated Ca2+ entry and currents are not blocked by drugs that inhibit STIM1 punctum formation. Ca2+ signals in VSMCs were measured in response to either thrombin (Th, 100 nM) (A) or thapsigargin (TG, 2 μM) (B). Pharmacological agents known to inhibit SOCE (50 μM 2-APB and 50 μM ML-9) completely abrogated thapsigargin-induced Ca2+ entry (B) with no effect in thrombin-activated Ca2+ signals (A). Whole-cell patch clamp electrophysiology confirmed that thrombin-activated Ca2+ (measured in Ca2+-containing bath solutions) and Na+ (measured in DVF bath solutions) currents (C) are not sensitive to 2-APB (Na+ I/V depicted; n = 5) (D). However, store depletion (20 mM BAPTA in pipette)-activated Ca2+ and Na+ CRAC currents (F) are sensitive to 2-APB (Na+ I/V depicted; n = 5) (G). Statistics on LRC and CRAC current data are shown in panels E and H, respectively, as mean/range.
Thrombin causes neither sustained STIM1 puncta nor colocalization with Orai1.
Next, we sought to determine whether, in response to thrombin stimulation, STIM1 patterns within VSMC change and whether STIM1 colocalizes with Orai1 upon thrombin stimulation. Expression of enhanced yellow fluorescent protein (eYFP)-tagged STIM1 in VSMCs and confocal microscopy showed that stimulation of VSMCs with a SOCE activator, thapsigargin (22) or PDGF (16, 17), caused eYFP-STIM1 punctum formation that was apparent within seconds (30 s for PDGF; 60 s for thapsigargin) and sustained over the duration of the experiments (up to 20 min) (Fig. 4A, panels 1 to 6; see also Movies S1 and S2 in the supplemental material). Please note that all stimuli were added at t = 120 s and that images were collected every 30 s. However, thrombin caused only a transient redistribution of eYFP-STIM1 into puncta; these puncta were maximal 30 s after stimulation and completely reversed 60 s after stimulation (Fig. 4A, panels 7 to 9; see also Movie S3 in the supplemental material), consistent with the timing and duration of the Ca2+ release peak observed in Fura2 experiments after addition of thrombin in nominally Ca2+-free solution (Fig. 1B and C and Fig. 3A). Coexpression of eYFP-STIM1 with cyan fluorescent protein (CFP)-tagged Orai1 in VSMCs showed that eYFP-STIM1 and CFP-Orai1 colocalized near the plasma membrane (PM) after addition of thapsigargin but failed to colocalize in response to thrombin (Fig. 4B and C; see also Movies S4 and S5 in the supplemental material).
Fig 4.

Thrombin stimulation failed to cause sustained STIM1 puncta and STIM1/Orai1 colocalization. (A) Representative confocal time-lapse images of VSMCs expressing 0.75 μg of eYFP-STIM1 and nonstimulated (Control; panels 1, 4, and 7) or upon stimulation with maximal concentrations of thapsigargin (TG, 4 μM; panels 2 and 3; n = 5) (see also Movie S1 in the supplemental material), PDGF (100 ng/ml; panels 5 and 6; n = 3) (see also Movie S2 in the supplemental material), and thrombin (100 nM; panels 8 and 9; n = 8) (see also Movie S3 in the supplemental material) showing sustained puncta only in response to thapsigargin and PDGF. Images were acquired every 30 s, and stimuli were added at t = 120 s. (B and C) Confocal time-lapse images of VSMCs cotransfected with eYFP-STIM1 (0.5 μg) and CFP-Orai1 (3 μg) before and after stimulation with thapsigargin (B) (TG, 2 μM; n = 3) (see also Movie S4 in the supplemental material) or thrombin (C) (100 nM; n = 3) (see also Movie S5 in the supplemental material). Images were acquired every 20 s. Intensity profiles for STIM1 (green) and Orai1 (red) showed that while thapsigargin stimulation caused these proteins to colocalize in areas close to the plasma membrane, thrombin stimulation failed to cause STIM1/Orai1 colocalization. Scale bars for panels B and C are 15 μm.
Orai3/STIM1 precoupling is required for thrombin-activated Ca2+ entry.
The requirement of ER-resident STIM1 coupled with lack of sustained STIM1 punctum formation and lack of STIM1/Orai1 colocalization upon thrombin stimulation was puzzling. This prompted us to consider the possibility that the eYFP-STIM1 requirement is mediated through its known interaction with microtubules via the plus-end binding protein EB1 (31). However, this possibility was ruled out as a mutant of STIM1 unable to bind EB1 (640TRIP→TRNN) fully rescued thrombin-activated Ca2+ entry in VSMCs using the erase-and-replace strategy (see Fig. S3 in the supplemental material).
A fallacy in our studies thus far is the assumption that Orai1 and Orai3 interact in similar fashions with STIM1. The results obtained forced us to consider the possibility that ER-resident STIM1 is required for thrombin-mediated LRC channel activation through interactions with Orai3 (rather than Orai1). We therefore hypothesized that ER-resident STIM1 interacts with LRC channels through Orai3 upon thrombin-mediated production of LTC4. Surprisingly, confocal colocalization experiments in VSMCs using z-stacks with eYFP-STIM1 and CFP-Orai3 showed a substantial degree of STIM1/Orai3 colocalization at room temperature which was apparent under basal unstimulated conditions (Pearson coefficient of colocalized volume [PCCV], 0.56 ± 0.07 versus 0.01 ± 0 for STIM1-Orai1 interactions) (Fig. 5A and B; see also Movies S6 and S7 in the supplemental material depicting 3-dimensional reconstructions). These results were confirmed in the HEK293 cell expression system (Fig. 6A, panels 1 to 6), and a CFP-tagged Orai1-Orai3 tandem construct also showed basal colocalization with eYFP-STIM1, although to a lesser extent than that with STIM1/Orai3 (Fig. 6A, panels 7 to 9); STIM1/Orai1 showed very little to no basal colocalization. Remarkably, this basal STIM1/Orai3 and STIM1/Orai1-Orai3 tandem interaction does not translate into channel activation; channel activation was observed, however, after store depletion with 1,4-dihydroxy-2,5-di-tert-butylbenzene (BHQ) with little or no increase in STIM1/Orai3 and STIM1/Orai1-Orai3 tandem colocalization (see Fig. S4 in the supplemental material). FRET experiments using a C-terminally tagged STIM1 construct showed that strong STIM1/Orai1 interaction was seen only after store depletion with BHQ while a much stronger STIM1/Orai3 interaction was readily detected under basal conditions and marginally increased after store depletion (Fig. 6B to D). Importantly, similar coexpression experiments in HEK293 cells employing a STIM1 construct where the second coiled-coil domain was rendered less canonical through mutagenesis (32) (A376K-eYFP-STIM1) showed impaired basal colocalization when coexpressed with either Orai3 or the Orai1-Orai3 tandem (Fig. 6E) as well as impaired BHQ-induced colocalization and CRAC currents (see Fig. S4).
Fig 5.
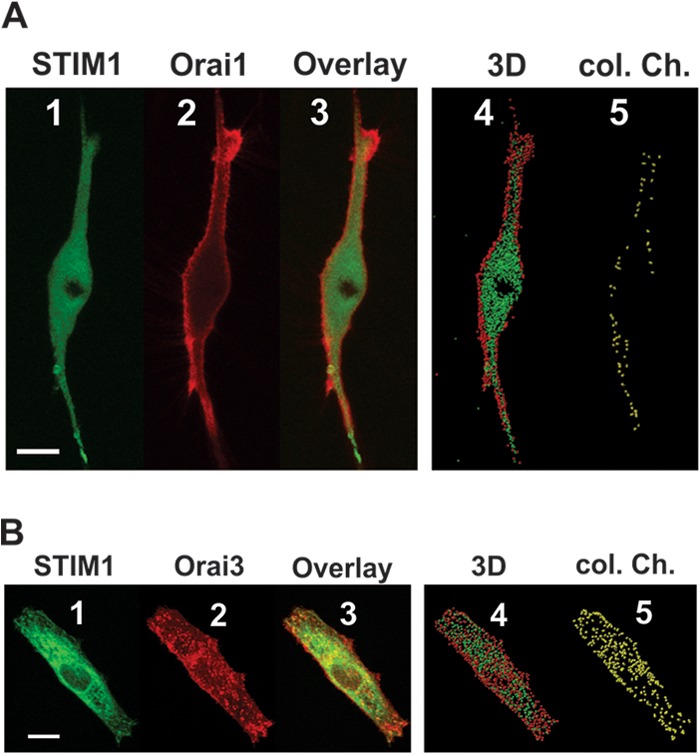
STIM1 colocalizes with Orai3, not Orai1, under basal conditions. (A) Representative confocal z-stack images of VSMCs coexpressing eYFP-STIM1 (green; 0.5 μg) (panel 1) and CFP-Orai1 (red; 3 μg) (panel 2) acquired under basal conditions (in Hanks balanced salt solution [HBSS] containing 2 mM Ca2+; n = 4) show no significant colocalization (panel 3). Three-dimensional surface generation for visualization of protein distribution (see Movie S6 in the supplemental material) shows minimal stochastic basal colocalization (panels 4 and 5). (B) However, coexpression of eYFP-STIM1 (green; 0.5 μg) (panel 1) and CFP-Orai3 (red; 3 μg) (panel 2) shows significant basal colocalization (panel 3) that is confirmed by three-dimensional surface generation (panel 4) and STIM1/Orai3 surface interactions (colocalization channel) (panel 5) (see also Movie S7 in the supplemental material) (PCCV = 0.56 ± 0.07; n = 10). All images were acquired under basal conditions in the absence of agonists. Scale bars for panels A and B are 15 μm.
Fig 6.

Orai3/STIM1 basal precoupling is not associated with constitutive current activity. (A) Representative confocal images of HEK293 cells overexpressing eYFP-STIM1/CFP-Orai1 (panels 1 to 3; n = 15), eYFP-STIM1/CFP-Orai3 (panels 4 to 6; n = 3), and eYFP-STIM1/CFP-Orai3-Orai1 tandem (panels 7 to 9; n = 6) acquired under basal conditions. (B) Confocal images showing colocalization of STIM1 (green) and Orai1 (red) protein coexpressed in HEK293 cells acquired under basal conditions (panels 1 to 3) and upon store depletion with 60 μM BHQ (panels 5 to 7). Net FRET values confirmed the minimal basal interaction of Orai1/STIM1 (panel 4) that is induced in response to store depletion (60 μM BHQ for 5 min) (panel 8). (C) However, confocal images showing colocalization of STIM1 (green) and Orai3 (red) protein coexpressed in HEK293 cells showed marked colocalization (panel 3) without store depletion. Net FRET values confirm strong interaction of Orai3/STIM1 (panel 4) that is marginally enhanced in response to store depletion (panel 8). (D) Statistical analysis of FRET experiments from several experiments similar to those in panels B (n = 5) and C (n = 4). (E) Representative confocal images of HEK293 cells coexpressing STIM1-A376K/Orai1 (panels 1 to 3; n = 15), STIM1-A376K/Orai3 (panels 4 to 6; n = 3), and STIM1-A376K/Orai3-Orai1 tandem (panels 7 to 9; n = 6) confirmed loss of basal colocalization (panels 6 and 9). Scale bars for panels A to C and E represent 5 μm.
The STIM1 coiled-coil domain is required for thrombin-activated Ca2+ entry.
The fact that the A376K STIM1 coiled-coil domain mutant resulted in loss of STIM1/Orai3 interaction prompted us to investigate whether this interaction was functionally required for thrombin-activated Ca2+ entry in VSMCs. To test this hypothesis, we utilized erase-and-replace experiments using two versions of eYFP-STIM1 with mutations in their coiled-coil domain (Fig. 7A), A376K (32) and L373S/A376S (33), shown previously to be deficient in supporting interactions with Orai1 and mediating SOCE and CRAC currents. Both STIM1 mutant constructs were expressed in VSMCs (Fig. 7B) but failed to rescue thrombin-activated Ca2+ entry (Fig. 7C and D).
Fig 7.
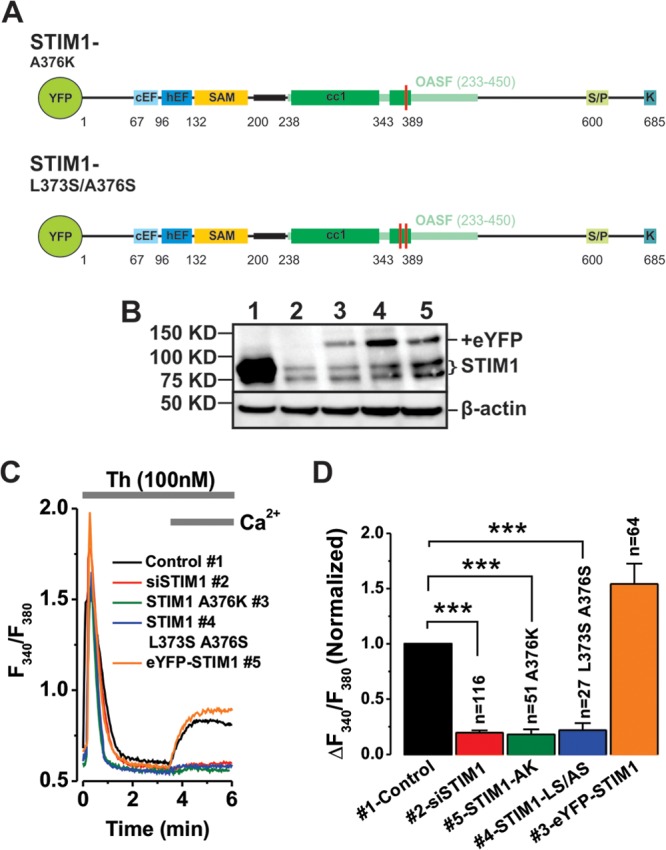
The second coiled-coil domain of ER-STIM1 is important for activation of thrombin-activated Ca2+ entry. (A) Schematic representations of mutant versions of eYFP-STIM1 employed in erase-and-replace assays. (B) Western blot shows STIM1 expression in siRNA control-transfected VSMC (lane 1) and successful endogenous STIM1 knockdown with siRNA (lane 2). Rescue of STIM1 expression was achieved using wild-type human eYFP-STIM1 (lane 5) and two eYFP-STIM1 coiled-coil mutants (lanes 3 and 4). EYFP-STIM1 plasmids were transfected at 1.5 μg per 106 cells. Positions of protein molecular mass markers are shown on the left. (C) Among plasmids expressing either STIM1-A376K (lane 3), STIM1-L373S A376S (lane 4), or eYFP-STIM1 (lane 5), only wild-type eYFP-STIM1 could rescue thrombin-activated Ca2+ entry. (D) Statistical analysis of the Ca2+ imaging data from several independent runs similar to those for panel C.
DISCUSSION
Our previous work showed that different agonists in primary cells can couple to distinct endogenous Ca2+ selective channels contributed by different combinations of Orai isoforms (16, 17). Here, using pharmacological tools, we show that the store-independent LRC pathway activated by thrombin requires the production of LTC4 through the sequential catalytic activities of PLC, DAG lipase, 5-LO, and LTC4S. This is the first difference noted between this pathway in VSMCs and the ARC pathway in HEK293 cells, which has been suggested to require AA produced through PLA2 action with no subsequent downstream AA metabolism (34). The second difference is related to STIM1; whereas ARC required plasma membrane STIM1 (11, 12), ER-STIM1 is sufficient for LRC channel activation. The differences between these two conductances might be related to the existence of two distinct channels, one regulated by AA and plasma membrane STIM1 and the other by LTC4 and ER-STIM1. Alternatively, these two conductances might be mediated by the same channel that shows promiscuity for activation by AA and LTC4 and a requirement for both pools of STIM1. Additional studies are needed to further understand the diversity of these store-independent Ca2+ entry pathways mediated by Orai proteins. While thrombin activates the production of AA and LTC4 in VSMCs through sequential activation of PLC and DAG lipase, previous studies in immune cells, including mast cells, identified an important role for the Ca2+-sensitive cytosolic phospholipase A2 (cPLA2) activated by Ca2+ influx through CRAC channels in the production of AA and LTC4 (35, 36). These studies suggest that different stimuli in different cell types might use different routes for AA and LTC4 synthesis and further highlight the complexity of the AA signaling pathway.
Both the N terminus and C terminus of Orai channels are required for binding to STIM1 during SOCE activation (37). Unlike STIM1/Orai1 coexpression in VSMCs and HEK293 cells, coexpression of STIM1 and Orai3 leads to constitutive coupling between STIM1 and Orai3 through the STIM1 C-terminal coiled-coil domain, which is consistent with the store independence and STIM1 punctum independence of LRC channel activation. Remarkably, the precoupling between STIM1 and Orai3 does not translate into channel activation and requires stimulation to elicit membrane currents. Recent work from our lab showed that activation of LRC currents in VSMCs with direct dialysis of LTC4 through the patch pipette was abrogated upon STIM1 knockdown, suggesting that STIM1 is required downstream from LTC4 action (17). Presumably, the constitutive STIM1/Orai3 interaction in LRC channels requires LTC4 to trigger channel activation, and further studies are needed to determine precisely how this occurs. STIM1/Orai3 basal precoupling might have a role in fast physiological responses of VSMCs to thrombin. In fact, findings in skeletal muscle show that STIM1 and Orai1 are precoupled under resting conditions within the triad junction (38, 39). This is likely an evolutionarily acquired characteristic that helps in providing fast Ca2+ entry from the outside in skeletal muscle to prevent decline in contractility during long periods of activity. The important question that remains to be answered by structural studies is how LTC4 triggers LRC channel activation and whether this is a direct action. LTC4 could interact directly with Orai3 or could interact with combinations of STIM1/Orai3 and/or Orai1. One way to answer this question is to use erase-and-replace strategies (as performed for STIM1 in this study) with various chimeras of Orai1/Orai3 where the cytosolic N terminus, 2d loop, and C terminus of Orai3 alone or in combination are replaced by the equivalent domains of Orai1. ARC channels were proposed to form pentamers between two Orai3 and three Orai1 molecules (40). However, the crystal structure of Drosophila melanogaster Orai was resolved as a hexamer (41). Future studies aimed at determining the oligomeric state and the subunit requirement of LRC channels are needed to help understand the exact mechanisms of activation of LRC channels by LTC4. For instance, a series of concatenated multimers of Orai1 and Orai3 coupled with erase-and-replace experiments in VSMCs would help determine the subunit composition of LRC channels. Future studies will be also required to determine the transcriptional programs activated by Ca2+ signals through LRC channels and how these programs impact cell function in vascular smooth muscle and other tissues.
Supplementary Material
ACKNOWLEDGMENTS
This work was mainly supported by Public Health Service grant HL097111 from the NIH to M.T. and in part by NIH grant HL095566 to K.M. and Austrian Science Fund (FWF) grants P22747 to R.S. and P22565 to C.R.
Footnotes
Published ahead of print 22 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00554-13.
REFERENCES
- 1.Putney JW., Jr 1986. A model for receptor-regulated calcium entry. Cell Calcium 7:1–12 [DOI] [PubMed] [Google Scholar]
- 2.Parekh AB, Putney JW., Jr 2005. Store-operated calcium channels. Physiol. Rev. 85:757–810 [DOI] [PubMed] [Google Scholar]
- 3.Berridge MJ. 1993. Inositol trisphosphate and calcium signalling. Nature 361:315–325 [DOI] [PubMed] [Google Scholar]
- 4.Liou J, Kim ML, Heo WD, Jones JT, Myers JW, Ferrell JE, Jr, Meyer T. 2005. STIM is a Ca2+ sensor essential for Ca2+-store-depletion-triggered Ca2+ influx. Curr. Biol. 15:1235–1241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Roos J, DiGregorio PJ, Yeromin AV, Ohlsen K, Lioudyno M, Zhang S, Safrina O, Kozak JA, Wagner SL, Cahalan MD, Velicelebi G, Stauderman KA. 2005. STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 169:435–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vig M, Beck A, Billingsley JM, Lis A, Parvez S, Peinelt C, Koomoa DL, Soboloff J, Gill DL, Fleig A, Kinet JP, Penner R. 2006. CRACM1 multimers form the ion-selective pore of the CRAC channel. Curr. Biol. 16:2073–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feske S, Gwack Y, Prakriya M, Srikanth S, Puppel SH, Tanasa B, Hogan PG, Lewis RS, Daly M, Rao A. 2006. A mutation in Orai1 causes immune deficiency by abrogating CRAC channel function. Nature 441:179–185 [DOI] [PubMed] [Google Scholar]
- 8.Zhang SL, Yeromin AV, Zhang XH, Yu Y, Safrina O, Penna A, Roos J, Stauderman KA, Cahalan MD. 2006. Genome-wide RNAi screen of Ca(2+) influx identifies genes that regulate Ca(2+) release-activated Ca(2+) channel activity. Proc. Natl. Acad. Sci. U. S. A. 103:9357–9362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mignen O, Shuttleworth TJ. 2000. I(ARC), a novel arachidonate-regulated, noncapacitative Ca(2+) entry channel. J. Biol. Chem. 275:9114–9119 [DOI] [PubMed] [Google Scholar]
- 10.Mignen O, Thompson JL, Shuttleworth TJ. 2008. Both Orai1 and Orai3 are essential components of the arachidonate-regulated Ca2+-selective (ARC) channels. J. Physiol. 586:185–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mignen O, Thompson JL, Shuttleworth TJ. 2007. STIM1 regulates Ca2+ entry via arachidonate-regulated Ca2+-selective (ARC) channels without store depletion or translocation to the plasma membrane. J. Physiol. 579:703–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thompson JL, Shuttleworth TJ. 2012. A plasma membrane-targeted cytosolic domain of STIM1 selectively activates ARC channels, an arachidonate-regulated store-independent Orai channel. Channels (Austin) 6:370–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Thompson J, Mignen O, Shuttleworth TJ. 2010. The N-terminal domain of Orai3 determines selectivity for activation of the store-independent ARC channel by arachidonic acid. Channels (Austin) 4:398–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.House SJ, Potier M, Bisaillon J, Singer HA, Trebak M. 2008. The non-excitable smooth muscle: calcium signaling and phenotypic switching during vascular disease. Pflugers Arch. 456:769–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang W, Trebak M. 2011. STIM1 and Orai1: novel targets for vascular diseases? Sci. China Life Sci. 54:780–785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bisaillon JM, Motiani RK, Gonzalez-Cobos JC, Potier M, Halligan KE, Alzawahra WF, Barroso M, Singer HA, Jourd'heuil D, Trebak M. 2010. Essential role for STIM1/Orai1-mediated calcium influx in PDGF-induced smooth muscle migration. Am. J. Physiol. Cell Physiol. 298:C993–C1005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gonzalez-Cobos JC, Zhang X, Zhang W, Ruhle BC, Motiani RK, Schindl R, Muik M, Spinelli AM, Bisaillon JM, Shinde AV, Fahrner M, Singer HA, Matrougui K, Barroso M, Romanin C, Trebak M. 2013. Store-independent Orai1/3 channels activated by intracrine leukotriene C4: role in neointimal hyperplasia. Circ. Res. 112:1013–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McKeown L, Moss NK, Turner P, Li J, Heath N, Burke D, O'Regan D, Gilthorpe MS, Porter KE, Beech DJ. 2012. Platelet-derived growth factor maintains stored calcium through a nonclustering Orai1 mechanism but evokes clustering if the endoplasmic reticulum is stressed by store depletion. Circ. Res. 111:66–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang W, Halligan KE, Zhang X, Bisaillon JM, Gonzalez-Cobos JC, Motiani RK, Hu G, Vincent PA, Zhou J, Barroso M, Singer HA, Matrougui K, Trebak M. 2011. Orai1-mediated I (CRAC) is essential for neointima formation after vascular injury. Circ. Res. 109:534–542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Abdullaev IF, Bisaillon JM, Potier M, Gonzalez JC, Motiani RK, Trebak M. 2008. Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 103:1289–1299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Motiani RK, Zhang X, Harmon KE, Keller RS, Matrougui K, Bennett JA, Trebak M. 2013. Orai3 is an estrogen receptor alpha-regulated Ca2+ channel that promotes tumorigenesis. FASEB J. 27:63–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Potier M, Gonzalez JC, Motiani RK, Abdullaev IF, Bisaillon JM, Singer HA, Trebak M. 2009. Evidence for STIM1- and Orai1-dependent store-operated calcium influx through ICRAC in vascular smooth muscle cells: role in proliferation and migration. FASEB J. 23:2425–2437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shinde AV, Motiani RK, Zhang X, Abdullaev IF, Adam AP, Gonzalez-Cobos JC, Zhang W, Matrougui K, Vincent PA, Trebak M. 2013. STIM1 controls endothelial barrier function independently of Orai1 and Ca2+ entry. Sci. Signal. 6:ra18. 10.1126/scisignal.2003425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Muik M, Fahrner M, Schindl R, Stathopulos P, Frischauf I, Derler I, Plenk P, Lackner B, Groschner K, Ikura M, Romanin C. 2011. STIM1 couples to ORAI1 via an intramolecular transition into an extended conformation. EMBO J. 30:1678–1689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Derler I, Hofbauer M, Kahr H, Fritsch R, Muik M, Kepplinger K, Hack ME, Moritz S, Schindl R, Groschner K, Romanin C. 2006. Dynamic but not constitutive association of calmodulin with rat TRPV6 channels enables fine tuning of Ca2+-dependent inactivation. J. Physiol. 577:31–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xia Z, Liu Y. 2001. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys. J. 81:2395–2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mercer JC, Dehaven WI, Smyth JT, Wedel B, Boyles RR, Bird GS, Putney JW., Jr 2006. Large store-operated calcium selective currents due to co-expression of Orai1 or Orai2 with the intracellular calcium sensor, Stim1. J. Biol. Chem. 281:24979–24990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Smyth JT, Dehaven WI, Bird GS, Putney JW., Jr 2008. Ca2+-store-dependent and -independent reversal of Stim1 localization and function. J. Cell Sci. 121:762–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Prakriya M, Lewis RS. 2002. Separation and characterization of currents through store-operated CRAC channels and Mg2+-inhibited cation (MIC) channels. J. Gen. Physiol. 119:487–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DeHaven WI, Smyth JT, Boyles RR, Putney JW., Jr 2007. Calcium inhibition and calcium potentiation of Orai1, Orai2, and Orai3 calcium release-activated calcium channels. J. Biol. Chem. 282:17548–17556 [DOI] [PubMed] [Google Scholar]
- 31.Grigoriev I, Gouveia SM, van der Vaart B, Demmers J, Smyth JT, Honnappa S, Splinter D, Steinmetz MO, Putney JW, Jr, Hoogenraad CC, Akhmanova A. 2008. STIM1 is a MT-plus-end-tracking protein involved in remodeling of the ER. Curr. Biol. 18:177–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Covington ED, Wu MM, Lewis RS. 2010. Essential role for the CRAC activation domain in store-dependent oligomerization of STIM1. Mol. Biol. Cell 21:1897–1907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Frischauf I, Muik M, Derler I, Bergsmann J, Fahrner M, Schindl R, Groschner K, Romanin C. 2009. Molecular determinants of the coupling between STIM1 and Orai channels: differential activation of Orai1-3 channels by a STIM1 coiled-coil mutant. J. Biol. Chem. 284:21696–21706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Osterhout JL, Shuttleworth TJ. 2000. A Ca(2+)-independent activation of a type IV cytosolic phospholipase A(2) underlies the receptor stimulation of arachidonic acid-dependent noncapacitative calcium entry. J. Biol. Chem. 275:8248–8254 [DOI] [PubMed] [Google Scholar]
- 35.Currie S, Roberts EF, Spaethe SM, Roehm NW, Kramer RM. 1994. Phosphorylation and activation of Ca(2+)-sensitive cytosolic phospholipase A2 in MCII mast cells mediated by high-affinity Fc receptor for IgE. Biochem. J. 304:923–928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chang WC, Nelson C, Parekh AB. 2006. Ca2+ influx through CRAC channels activates cytosolic phospholipase A2, leukotriene C4 secretion, and expression of c-fos through ERK-dependent and -independent pathways in mast cells. FASEB J. 20:2381–2383 [DOI] [PubMed] [Google Scholar]
- 37.Park CY, Hoover PJ, Mullins FM, Bachhawat P, Covington ED, Raunser S, Walz T, Garcia KC, Dolmetsch RE, Lewis RS. 2009. STIM1 clusters and activates CRAC channels via direct binding of a cytosolic domain to Orai1. Cell 136:876–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dirksen RT. 2009. Checking your SOCCs and feet: the molecular mechanisms of Ca2+ entry in skeletal muscle. J. Physiol. 587:3139–3147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Darbellay B, Arnaudeau S, Bader CR, Konig S, Bernheim L. 2011. STIM1L is a new actin-binding splice variant involved in fast repetitive Ca2+ release. J. Cell Biol. 194:335–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mignen O, Thompson JL, Shuttleworth TJ. 2009. The molecular architecture of the arachidonate-regulated Ca2+-selective ARC channel is a pentameric assembly of Orai1 and Orai3 subunits. J. Physiol. 587:4181–4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hou X, Pedi L, Diver MM, Long SB. 2012. Crystal structure of the calcium release-activated calcium channel Orai. Science 338:1308–1313 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.