

Abstract
Abnormal glucocorticoid and neurotrophin signaling has been implicated in numerous psychiatric disorders. However, the impact of neurotrophic signaling on glucocorticoid receptor (GR)-dependent gene expression is not understood. We therefore examined the impact of brain-derived neurotrophic factor (BDNF) signaling on GR transcriptional regulatory function by gene expression profiling in primary rat cortical neurons stimulated with the selective GR agonist dexamethasone (Dex) and BDNF, alone or in combination. Simultaneous treatment with BDNF and Dex elicited a unique set of GR-responsive genes associated with neuronal growth and differentiation and also enhanced the induction of a large number of Dex-sensitive genes. BDNF via its receptor TrkB enhanced the transcriptional activity of a synthetic GR reporter, suggesting a direct effect of BDNF signaling on GR function. Indeed, BDNF treatment induces the phosphorylation of GR at serine 155 (S155) and serine 287 (S287). Expression of a nonphosphorylatable mutant (GR S155A/S287A) impaired the induction of a subset of BDNF- and Dex-regulated genes. Mechanistically, BDNF-induced GR phosphorylation increased GR occupancy and cofactor recruitment at the promoter of a BDNF-enhanced gene. GR phosphorylation in vivo is sensitive to changes in the levels of BDNF and TrkB as well as stress. Therefore, BDNF signaling specifies and amplifies the GR transcriptome through a coordinated GR phosphorylation-dependent detection mechanism.
INTRODUCTION
The glucocorticoid receptor (GR) is a ligand-activated transcription factor that mediates many physiological processes, including the body's response to stress and circadian rhythms (1). Upon glucocorticoid binding, the receptor is released from a chaperone complex and translocates to the nucleus, where it activates and represses target gene transcription (2). GR-mediated transcription depends upon a number of factors, including the sequence and architecture of the glucocorticoid response elements (GREs), the availability and activity of interacting cofactors, and posttranslational modifications of the receptor (3–5). To date, multiple conserved (human, rat, and mouse) phosphorylated serine or threonine residues have been identified on the GR (6), yet only a select number of sites have been demonstrated to affect GR function (4, 7). For example, phosphorylation of serine 211, which is an agonist-dependent site, plays a role in transcriptional activation via its interaction with coactivator proteins (8), while serine 404 phosphorylation has been shown to decrease GR function by enhancing receptor degradation (9). Previously, our laboratory provided evidence for glucocorticoid-dependent phosphorylation at several N-terminal sites, including serines 203 (S203), 211 (S211), and 226 (S226) in the human GR numbering scheme, using phosphorylation-specific antibodies and site-specific GR mutants (10, 11). All three sites are hyperphosphorylated upon glucocorticoid treatment, yet S211 and S226 have a large impact on GR-mediated transcription in the cell culture models tested (10, 11).
Though the brain is a major site of glucocorticoid action, the impact of GR phosphorylation events in neurons remains largely uncharacterized. Aberrant glucocorticoid levels, GR expression, and GR genomic activity have been reported in the brains and peripheral blood of individuals presenting with neurodegenerative and neuropsychiatric disorders (12, 13). For example, GR expression is decreased in the hypothalamus and cerebral cortex of schizophrenic patients and depressed individuals (14). Also, single nucleotide polymorphisms in the GR gene (15) and in the glucocorticoid-synthetic enzyme 11β-HSD1 have been described in cognitive disorders (16, 17). Both circadian and stress-induced release of glucocorticoids is altered in posttraumatic stress disorders and depression (18, 19). A common feature shared by these conditions is reduced GR function in the early stage of disease development (18, 20). However, the molecular mechanisms underlying the modulation of GR function in the brain are largely unknown.
Neurotrophins, such as nerve growth factor (NGF) and brain-derived neurotrophic factor (BDNF), are peptide hormones that bind to specific Trk tyrosine kinase transmembrane receptors to promote survival, growth, and plasticity of neural networks (21). As with glucocorticoids, changes in the levels of BDNF are associated with a number of psychiatric maladies, including depression (22–24) and anxiety (25, 26). Recent evidence suggests that BDNF and glucocorticoid have converging, homeostatic effects in the nervous system (recently reviewed in references 27, 28, 29, and 30). For example, chronic stress or exogenous glucocorticoid administration suppresses hippocampal BDNF expression, resulting in neuronal atrophy and cognitive impairment (31–33).
We previously showed that glucocorticoid treatment could activate signaling of the receptor for BDNF, TrkB, and promote cell survival in neurons via a mechanism dependent on GR transcriptional activity (34). This prompted us to examine whether BDNF regulates GR transcriptional activity as a form of cell-autonomous, homeostatic feedback control of glucocorticoid action. In this report, we demonstrate that BDNF signaling modulates the effect of GR on gene expression in primary neurons in part through alterations in GR phosphorylation. This study portrays how neurotrophin signaling impacts GR function by modulating GR posttranslational modifications. Moreover, our findings provide a foundation for studying how GR activity is modulated in response to extracellular signaling pathways and the interplay between BDNF and glucocorticoid signaling in disorders featuring disrupted BDNF and glucocorticoid levels.
MATERIALS AND METHODS
Mice.
C57BL/6 mice (Charles River Laboratories), Sim1-Cre mice (Jackson Laboratories), GRFLOX mice (20), BDNFSTOP mice (35), and BDNF (36) and TrkB (37) knockouts were allowed ad libitum food access. For neurochemistry, the paraventricular nuclei (PVN) were punched out of frozen brain sections on glass slides using a tissue punch set (Stoelting). Before euthanasia, adult male mice either were exposed to systemic administration of dexamethasone 21-phosphate disodium salt (Sigma-Aldrich), 0.9% saline for 6 h or subjected to a forced swim test in room temperature water (25°C) for 10 min followed by a recovery period of 45 min in their home cage. The New York University School of Medicine Institutional Animal Care and Use Committee approved all procedures.
Cell culture.
Cultured neurons from embryonic day 18 rats were prepared from timed-pregnant Sprague-Dawley rats (Charles River Laboratories) as described previously (34). Primary neurons were cultured on poly-d-lysine-coated wells and maintained in neurobasal medium containing B27 supplement, 0.5 mM l-glutamine, 5-fluorouridine, and uridine (10 mM each). Experiments were conducted 7 to 10 days after plating. Lentiviruses expressing the wild-type rat GR and GR with the phosphorylation site mutations S155A and S287A were produced by transfecting expression and packaging plasmids into HEK-293FT cells as previously described (34). HEK-293 cells stably expressing TrkB (293TrkB) (38) were maintained in DMEM containing 10% fetal bovine serum supplemented with 200 μg/ml G418. Cells were serum starved overnight before experiments. Stable 293TrkB cell lines ectopically expressing rat GR (wild type and phosphorylation site S155A/S287A double mutants) were generated by infecting cells with lentivirus expressing GR and green fluorescent protein (GFP) and isolating individual GFP-positive clones via fluorescence-activated cell sorting. After 3 rounds of sorting, cells were assayed for GR expression by indirect immunofluorescence and immunoblotting with GR-specific antibodies. Clones homogeneously expressing GR were identified and maintained at 200 μg/ml G418.
Plasmids and reagents.
Transfections of HEK-293 and 293TrkB cells with TAT3-LUC reporter plasmid, pCMV-LacZ, pCMV-rat GR, or FCIV1-rat GR were performed using Lipofectamine 2000 (Invitrogen). Electroporations of neurons were performed using FCIV1-rat GR and GR S155A/S287A with an Amaxa rat neuron Nucleofector kit (Lonza) according to the manufacturer's instructions. Recombinant BDNF (450-02) was purchased from Pepro Tech, Inc. Small interfering RNA (siRNA) for human CREB1 (OnTarget Plus Smartpool L-003619-00), rat CREB1 (OnTarget Plus Smartpool L-092995-02), and siRNA control (OnTarget nontargeting pool) were obtained from ThermoFisher.
Western blotting and antibodies.
Western blotting was performed as previously described (11). Western blots were imaged and quantified using the Odyssey system (Li-Cor) or developed using ECL band intensity quantitated with Image J. Antibodies to S155∼P and S287∼P were generated by immunizing rabbits (Covance) with peptides corresponding to sequences in rat GR: 279DTGDTILS(pS)287PSSV291 and 147IANLNRST(pS)155VPEN159 (Anaspec, Inc.). High-titer antibodies were affinity purified (Covance). The rabbit polyclonal affinity purified phospho-TrkB antibody was developed against the phosphorylated residue Y816 (34). Commercially available antibodies used for Western blotting, immunohistochemistry, and immunoprecipitation were as follows: anti-GR (ab9568; Abcam), anti-GR (M-20 [Santa Cruz Biotechnology] and BuGr2 [EMD Millipore]), anti-pan Trk (C-14; Santa Cruz Biotechnology), anti TrkB (H-181, Santa Cruz Biotechnology), anti phosphoserine (p-Ser; 16B4; Santa Cruz Biotechnology), anti-phospho-CREB (S133∼P), anti-CREB (48H2; Cell Signaling), and anti-Hsp90 (BD Bioscience).
Microarray analysis.
Total RNA was isolated using the Trizol reagent (Invitrogen) as described by the manufacturer, and 3 μg of each sample was processed for hybridization to the rat genome 230 2.0 Affymetrix expression array by the Genomics Core Laboratory of Memorial Sloan-Kettering Cancer Center. Data are representative of four different conditions performed in duplicate (a total of eight) and were normalized using Robust Multichip Average (RMA) Express (39). The primary data were analyzed for changes of >2.0-fold when the logarithmic values for treated cells (Dex, BDNF, or BDNF plus Dex) and vehicle-treated cells were compared. DAVID (40, 41) was used to analyze the gene sets for Gene Ontology (GO), and Categorizer (http://www.animalgenome.org/tools/catego/) was utilized to determine occurrences of GO terms in each ancestral class of each treatment condition. DREME (discriminative regular expression motif elicitation) was used for transcription factor motif discovery in the BDNF- and Dex-coregulated gene sets (42).
Quantitative real-time PCR (qPCR).
Total RNA from primary neurons or 293TrkB cells was extracted with Trizol (Invitrogen) as described by the manufacturer. cDNA was synthesized from 1 μg of RNA using a First-Strand cDNA synthesis kit for real-time PCR (USB) and random primer mix (USB) following the manufacturer's instructions. cDNA was amplified with the SYBR green Taq Ready Mix (USB) using a MyiQ single-color real-time PCR detection system from Bio-Rad. The primers used for qPCR are listed in Table S3 in the supplemental material.
Mass spectrometry.
Primary rat cortical neurons were infected with a lentivirus expressing rat GR and, after 7 days, treated with BDNF (50 ng/ml) or vehicle for 30 min. The cells were lysed, GR was immunoprecipitated (anti-GR M-20; Santa Cruz Biotechnology), and the immunoprecipitated proteins were resolved by sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis. The Coomassie brilliant blue-stained GR band was excised, digested with trypsin, and analyzed by mass spectrometry (43, 44). For the enrichment of phosphopeptides, digests were dried, redissolved in 1% trifluoroacetic acid (TFA), 80% acetonitrile (AcN), and loaded onto a titanium dioxide tip (10 μl; Nutip; Glygen). The bound phosphopeptides were eluted using 500 mM ammonium hydroxide and then acidified by adding 5% formic acid (FA). The solutions were dried, redissolved (in 5 μl 0.1% FA, 2% AcN), and analyzed by ESI-QTOF nano-liquid chromatography-tandem mass spectrometry (LC-MS/MS) (Waters NanoAquity UPLC; the ionized electrospray flow rate of 200 nl/min coupled directly to a QTOF Premier mass spectrometer [Waters-Micromass, MA]) to identify the phosphorylation sites. Raw data were processed by Masslynx software and used to identify the phosphopeptides using MASCOT to search the Swiss-Prot Protein Knowledgebase (release 54.2), which contains 283,454 protein sequences and 104,030,551 residues, using “all entries” taxonomy.
Chromatin immunoprecipitation.
293TrkB cells stably expressing wild-type rat GR were analyzed by ChIP as described previously (45). Protein-DNA complexes were cross-linked by incubating cells in 1% formaldehyde for 10 min at room temperature, followed by incubation in 0.125 M glycine for 5 min to quench the cross-linking reaction. Cells were washed with PBS twice and lysed in Farnham lysis buffer [5 mM piperazine-N,N-bis(2-ethanesulfonic acid) (PIPES) at pH 8.0, 85 mM KCl, 0.5% NP-40] with added protease inhibitor (Roche). The cell lysate was then centrifuged at 2,000 rpm for 5 min at 4°C, and the crude nuclear pellets were collected. Chromatin was sonicated using the Bioruptor (Diagenode; Twin UCD-400) into 500-bp fragments. Immunoprecipitation of GR was performed with 6 μg of a GR antibody cocktail (MA1-510 [Thermo Scientific], PA1-511A [Thermo Scientific], and H-300 [Santa Cruz Biotechnology] for total GR) or with affinity-purified GR S155∼P and GR S287∼P antibodies. Immunoprecipitations for ChIP were also performed with 3 μg of anti-GRIP1 (M-343; Santa Cruz), anti-BRG1 (ab4081; Abcam), anti-p300 (C-20; Santa Cruz), and anti-CREB (48H2; Cell Signaling) or an equivalent amount of rabbit or mouse IgG (Sigma-Aldrich) overnight at 4°C. Protein A or G magnetic beads (Invitrogen) were used according to the manufacturer's instructions. After incubation, the beads were washed with LiCl wash buffer (100 mM Tris at pH 7.5, 500 mM LiCl, 1% NP-40, 1% sodium deoxycholate) and TE (10 mM Tris-HCl at pH 7.5, 0.1 mM Na2EDTA) at 4°C. Following reversal of cross-linking, recovered DNA was purified using a PrepEase DNA clean-up kit (USB). Cycle threshold values were normalized to percent input and IgG.
Immunocytochemistry.
Cells were serum starved for 5 h, fixed after treatments in paraformaldehyde (PFA), blocked in 5% bovine serum albumin (BSA)–0.1% Triton X-100–PBS for 30 min at 25°C, and incubated in blocking buffer overnight at 4°C with primary antibodies. Fluorescence-coupled secondary antibodies (Jackson Immuno Research) were incubated in blocking buffer for 30 min at 25°C. Cells were mounted in Vectashield containing DAPI (4′,6′-diamidino-2-phenylindole; Vector Laboratories). Epifluorescence was captured using a Nikon Eclipse E800 microscope and imaged with a Zeiss AxioCam HRc digital camera.
Immunohistochemistry.
Rats were perfused with 4% PFA, and brains were postfixed for 1 h and equilibrated in 30% sucrose. Free-floating coronal sections rinsed in PBS were blocked in 5% normal goat serum–5% normal horse serum–PBS–0.1% Triton X 100 for 1 h at 25°C. Primary antibodies were incubated overnight at 4°C with shaking. Alexa Fluor-conjugated secondary antibodies (Molecular Probes) were incubated for 90 min at 25°C. The p-TrkB (Y816∼P) rabbit polyclonal antibody was directly labeled with Alexa Fluor 555 according to the manufacturer's instructions (Molecular Probes) and was used in costaining experiments with the GR S287∼P affinity-purified rabbit antibody. Additional antibodies used include anti-CRH (T4037 and T5007; Peninsula Laboratories), anti-TrkB (H-181; Santa Cruz Biotechnology), and anti-GR (mouse monoclonal BUGR2; EDM Millipore). Sections mounted in Prolong Gold antifade reagent (Invitrogen) were imaged by using a Zeiss LSM 510 confocal microscope.
Statistical analysis.
Unless otherwise noted, all quantitative results are averages from at least three independent experiments, and each sample was analyzed in triplicate. Significance was determined using the two-tailed Student's t test.
RESULTS
BDNF signaling via TrkB affects GR-mediated transcriptional activation.
To assess the effects of BDNF signaling on GR-dependent transcriptional activity, we used a synthetic GR-sensitive promoter containing three tandem GREs from the tyrosine aminotransferase (TAT) gene fused to a minimal promoter driving the firefly luciferase gene (TAT3-Luc) (46) in HEK-293 cells ectopically expressing the BDNF receptor TrkB (here termed 293TrkB). Stimulation of 293TrkB cells with Dex induced TAT3-Luc promoter activity (Fig. 1A). BDNF treatment alone had no effect on the TAT3-Luc activity. Interestingly, costimulation of cells with BDNF and Dex induced the TAT3-Luc promoter activity 2-fold above that of Dex treatment alone (P < 0.05) (Fig. 1A). GR protein levels remained constant across treatments, indicating that enhanced GR activity upon BDNF treatment is not a result of increased GR expression (Fig. 1B). Importantly, BDNF-dependent enhancement of TAT3-Luc activity was dependent upon TrkB, since HEK-293 cells that do not express TrkB did not increase Dex-dependent activation of TAT3-Luc upon BDNF treatment (Fig. 1C). Again, GR protein levels remained constant across treatments (Fig. 1D).
Fig 1.

BDNF signaling enhances GR transcriptional activation. (A) HEK-293 cells ectopically expressing TrkB (293TrkB) were transiently transfected with pCMV-rat GR, the TAT3-Luc reporter construct, and pCMV-lacZ as an internal control. Twenty-four hours after transfection, cells were treated with ethanol (Veh), 100 nM dexamethasone (Dex) for 2 h, or 50 ng/ml BDNF for 2.5 h or treated with BDNF for 30 min followed by Dex for 2 h (BDNF+Dex). Luciferase activity was determined relative to β-galactosidase and is presented as relative (fold) induction, with the value for the vehicle-treated samples set to 1. (B) GR and TrkB protein expression was examined by Western blotting of 293TrkB lysates treated as described for panel A. (C) Parental HEK-293 cells that do not expression TrkB were assayed as described for panel A. (D) GR protein levels relative to tubulin were determined by Western blotting from HEK-293 lysates treated as described for panel B. (E) 293TrkB cells were transfected as described for panel A, treated with increasing concentrations of dexamethasone (Dex) for 24 h in the absence (−BDNF) or presence of 50 ng/ml BDNF (+BDNF), and luciferase activity was measured as described above. Error bars indicate standard deviations. Significance was determined using the two-tailed Student's t test (*, P < 0.05; **, P < 0.01). Western blots are representative of at least three independent experiments.
We next determined the minimal dose of Dex required to enhance TAT3-Luc promoter activity in the presence or absence of BDNF. We found that Dex at 0.1 nM induced TAT3-Luc activity to the same extent in the presence and absence of BDNF. In contrast, we observed a consistent 2-fold increase in GR transcriptional activity upon BDNF cotreatment with 1 nM, 10 nM, and 100 nM Dex (Fig. 1E). While the enhancement by BDNF of GR transcriptional activation was maintained, we also observed reduced luciferase activity at 10 nM and 100 nM Dex compared to 1 nM Dex consistent with “squelching” or titration of a limiting factor required for full response by the activated GR (47, 48). Thus, BDNF via TrkB increases GR transcriptional activity of a transfected reporter gene across a wide range of Dex concentrations, suggesting a direct effect of BDNF signaling on GR transcriptional activity.
BDNF and glucocorticoid cotreatment cooperatively regulate gene expression in neurons.
To address the relevance of BDNF and Dex transcriptional synergy, we examined the expression of TrkB and GR protein in the mouse cerebral cortex and hippocampus. We found that both TrkB and GR are concomitantly expressed in neurons throughout the cortex (Fig. 2A, top). GR is also expressed along with TrkB in neurons of the hippocampus (Fig. 2A, bottom). These data suggest that BDNF-TrkB signaling has the potential to regulate GR activity in cortical and hippocampal neurons.
Fig 2.

Transcriptional response of cortical neurons to BDNF and glucocorticoid cotreatment reveals a unique transcriptome associated with neuronal differentiation. (A) TrkB and GR expression in the cortex. Immunostaining of GR (BuGr2; EDM Millipore) (green) and TrkB (H-181; Santa Cruz Biotechnology) (red) from sections of the mouse cortex (layers LI to LVI) and hippocampus [stratum oriens (S.O.), stratum pyramidale (S.Py), stratum radiatum (S.R.), stratum lacunosum (S.L.M.), molecular layer (Mol.), and granule cell layer (GCL)] are shown. Enlarged areas at the right show GR and TrkB colocalization in neurons from layer LV of the cortex and the S.Py region of the hippocampus. (B) Primary cortical neurons cultured from E18 rats were infected with a lentivirus expressing rat GR. After 7 days in culture, the cells were treated with ethanol (Veh), 100 nM dexamethasone for 3 h (Dex), or 50 ng/ml BDNF for 3 h or cotreated with BDNF and Dex for 3 h (BDNF and Dex). Total RNA was isolated, and gene expression was assessed using the rat genome 230 2.0 Affymetrix expression array. (C) Probe sets were analyzed for significance (P < 0.05) using ANOVA. Genes were considered significant if they passed the ANOVA test and displayed a change of >2.0-fold when the logarithmic values between treated and control samples were compared. Significantly regulated genes in each treatment were graphed. (D) Venn diagram of the >2-fold-responsive genes (induced and repressed) from each treatment. (E) GO-Slim annotations of the ontology associations from the uniquely induced genes upon treatment with Dex alone and with BDNF and Dex. The number of detailed annotations associated with the designated parent annotation is indicated.
Given the communication between glucocorticoid and BDNF signaling in neuronal physiology and stress-associated pathophysiology (27–29), as well as the coexpression of GR and TrkB in neurons, we sought to understand the genes regulated by both signaling pathways in neurons. We used a microarray approach to determine the genes that were regulated at least 2-fold in primary rat cortical neurons upon treatments with Dex and BDNF alone or in combination (Fig. 2B). Since primary neurons when cultured ex vivo lose GR expression (49), cells were transduced with a lentivirus expressing rat GR to enrich for receptor expression prior to treatments. The levels of GR do not change with the various treatments (see Fig. S1 in the supplemental material).
The analysis identified 435 genes sensitive to Dex treatment, 456 genes responsive to BDNF, and 933 genes regulated by BDNF and Dex cotreatment after a 3 h stimulation (Fig. 2C). Our analysis revealed a set of Dex-responsive genes that were enhanced by BDNF treatment, representing 27% of the Dex-induced genes. We also found a small cohort (8%) of BDNF-responsive genes that were increased upon Dex treatment. Although we expected an additive representation of responsive genes from BDNF and Dex cotreatment, this was not the case. Surprisingly, nearly half (455 of 933 genes; 49%) of all genes in the BDNF and Dex cohort were uniquely induced or repressed above 2-fold (45% and 55%, respectively) by cotreatment relative to Dex or BDNF alone (Fig. 2D). Thus, cotreatment of neurons with BDNF and Dex results in a unique set of genes being expressed compared to either treatment alone, and this indicates that a distinct glucocorticoid-responsive transcriptome is evoked upon BDNF signaling.
We next performed gene ontology (GO) analysis using the Database for Annotation, Visualization and Integrated Discovery (DAVID) to classify the functional relationships among responsive genes (40, 50). As expected, genes responsive to BDNF and Dex cotreatment were significantly associated with receptor signaling (e.g., TrkB) and response to glucocorticoid stimulus (see Table S1 in the supplemental material). In addition, using GO Slim analysis, which generates a more focused view of the GO classes, we found that upon cotreatment, the coresponsive genes associate more with neuronal communication and differentiation than with metabolism, as elicited by individual treatments (Fig. 2E; also, see Fig. S2 in the supplemental material). Indeed, regulation of synaptic plasticity and neurotransmission as well as axon guidance shows a more significant association in the cotreated genes than with BDNF or Dex stimulation alone. For example, the upregulation of the DUSP1 gene expression by glucocorticoid and BDNF (Fig. 3A) is consistent with such GO class predictions. Moreover, the protein encoded by DUSP1, MKP-1, has been shown to directly regulate BDNF-induced axon branching (51).
Fig 3.

BDNF and glucocorticoid signaling regulate GR target genes. qPCR validation of the induced (A) and repressed (B) genes identified from the microarray analysis upon Dex, BDNF, and BDNF+Dex treatment. Cells were treated as described for Fig. 2. Error bars indicate standard deviations. (C) Heat maps of normalized ΔCT values of validated genes in the absence and presence of cycloheximide (CHX) upon Dex and Dex+BDNF stimulation. Asterisks indicate genes that upon cycloheximide treatment are no longer modulated by Dex treatment and are likely indirect targets of GR action.
Upon analysis of the uniquely coinduced gene sets, we found a greater enrichment with functions associated with differentiation, cell organization, and biogenesis (Fig. 2E; also, see Table S1 in the supplemental material). Surprisingly, a majority of the annotations for biological processes represented in the uniquely coinduced cluster are similar to those associated with BDNF induction (see Table S1 and Fig. S2 in the supplemental material). Similarly, the top annotations that arose from uniquely corepressed genes were similar to the most significant Dex-repressed subset (see Table S2 in the supplemental material). The predominant annotations in this subset were genes associated with the regulation of transcription and RNA metabolic processes. These predictions suggest that BDNF and glucocorticoid signaling pathways converge in specific gene expression profiles similar to those regulated by single treatments. Importantly, within the induced unique gene profiles are some GO classes that were not represented by separate treatments, such as zinc ion transport and angiogenic processes (see Table S1). Taken together, these results suggest that the GR transcriptome is reshaped by BDNF signaling, which in turn can lead to the expression of functionally distinct classes of genes.
Cotreatment with BDNF and Dex amplifies a subset of GR neuronal target genes.
Next, we validated by real-time qPCR using distinct sets of RNA a selection of genes from the microarray regulated upon cotreatment with BDNF and Dex. This includes many of the highly induced (PRL3C1, FGF8, MYOM2, NR4A1 [NUR77], AGTR1A, SRGN, and ANGPTL1) and repressed (PCDH20 and CRHR1) genes, as well as established GR targets (DUSP1) and genes uniquely induced upon cotreatment (IRX2) (Fig. 3A). Interestingly, IRX2, a homeobox transcription factor responsible for cerebellum formation, is induced by fibroblast growth factor 8 (FGF8) signaling (52). Expression of genes such as PRL3C1, FGF8, and MYOM2 was increased more than 6-fold by BDNF plus Dex above Dex treatment alone. A number of known neuronal GR target genes were revealed from this analysis, such as CRH, MYCN, and DUSP1, while others are less characterized in a neuronal context, such as FGF8 and SRGN. FGF8, not previously linked to GR, is a diffusible morphogen that patterns the neocortex during embryonic development (53). Of note, prenatal exposure to Dex retards the migration of cortical neuron progenitors throughout the embryonic cortical plate (54). SRGN (serglycin) is a proteoglycan critical for maintaining storage of secretory granule proteases. More interestingly, proteoglycans have been implicated as modulators of axonal growth and plasticity via their chondroitin sulfate chains, a function regulated by both BDNF and glucocorticoids in the brain (55, 56).
To investigate if the BDNF-dependent effects on Dex responsive genes are a primary event of GR-mediated activation or a result of a secondary effect via new protein synthesis, we treated cells for 30 min with the protein synthesis inhibitor cycloheximide and then reassessed gene expression. Nine of the 11 representative genes, namely, PRL3C1, FGF8, MYOM2, AGTR1A, SRGN, ANGPTL1, NR4A1, DUSP1, and CRH, demonstrated similar induction profiles in the presence of cycloheximide (Fig. 3C), indicating that these genes do not require new protein synthesis to modulate gene expression by GR and therefore are considered primary GR target genes.
BDNF signaling induces GR phosphorylation at two evolutionarily conserved sites.
We next tested the possibility that BDNF-dependent enhancement of GR activity requires the posttranslational modification of GR. We found increased phosphoserine immunoreactivity in GR immunoprecipitated from HEK-293 cell extracts cotransfected with TrkB and stimulated with BDNF (Fig. 4A). In contrast, transfection of GFP, TrkA, or TrkC in HEK-293 cells and stimulation with cognate ligands NGF for TrkA and NT3 for TrkC resulted in little phosphoserine immunoreactivity in GR immunoprecipitates. These results indicate that GR could be phosphorylated upon TrkB activation with BDNF in this system.
Fig 4.

BDNF signaling induces GR phosphorylation at S155 and S287. (A) Phosphoserine immunoblot (p-Ser; 16B4 mouse monoclonal antibody; Santa Cruz Biotechnology) of GR immunoprecipitates (α-GR M-20; Santa Cruz Biotechnology) from HEK-293 cells cotransfected with rat GR and TrkA, TrkB, TrkC, or GFP and treated with cognate neurotrophins (50 ng/ml) for 30 min. Expression of the Trk and GR receptors was determined as a control. (B) Primary neurons were infected with rat GR-expressing lentivirus and treated with BDNF as described for Fig. 2. Cells were lysed, GR was immunoprecipitated, resolved by SDS-PAGE, and stained, and the GR band was excised and digested with trypsin. Western blots are representative of at least three independent experiments. (C and D) Tryptic phosphopeptides from BDNF-treated GR were enriched using a titanium dioxide tip, eluted, and analyzed by nanoflow LC-ESI Q-TOF mass spectrometry. The spectra indicate peptides present in the mixture that were phosphorylated at S155 (C) and S287 (D). The loss of phosphoric acid (H3PO4; molecular weight [MW] = 98) from b3 in the Ser155 phosphopeptide and y11 in the Ser287 phosphopeptide indicates that Ser155 and Ser287 were phosphorylated (boxes).
Next, we sought to identify the phosphorylation sites of GR that are induced by BDNF signaling in primary cortical neurons. To do so, we purified GR from neuronal extracts by immunoprecipitation followed by SDS-PAGE (Fig. 4B, left) and mass spectrometry. The GR bands were detected by Coomassie staining and then excised from the gel for matrix-assisted laser desorption ionization–quantitative time of flight (MALDI-QTOF) analysis (Fig. 4B right). GR proteins were trypsinized and phosphopeptides were enriched in a titanium column, and LC-electrospray ionization (ESI)-QTOF MS/MS was used to sequence the phosphopeptides. The spectra of two phosphopeptides are presented in Fig. 4C and D. We found that phosphorylation of the rat GR protein upon BDNF treatment occurred at serines 155 and 287 (Fig. 4C and D), which are conserved in mouse (S143 and S275) and human GR (S134 and S267). These sites have not been extensively studied.
To validate that GR is phosphorylated by BDNF at S155 and S287, we generated GR phosphorylation site-specific antibodies (Fig. 5A) and tested GR phosphorylation upon BDNF treatment in 293TrkB cells expressing rat GR or the corresponding GR nonphosphorylatable mutants S155A and S287A. Basal GR phosphorylation at both S155 (Fig. 5B) and S287 (Fig. 5C) was low but highly inducible by BDNF treatment but not in the respective GR serine-to-alanine mutants. GR was also phosphorylated in primary cortical neurons (Fig. 5D) and in the neuroendocrine cell line PC12, which harbors endogenous GR in response to BDNF treatment (see Fig. S3A in the supplemental material).
Fig 5.

Characterization of BDNF-induced GR phosphorylation sites. (A) Schematic diagram of the functional domains and some major phosphorylation sites of rat GR. NTD, amino-terminal domain; AF-1, activation function 1; DBD, DNA-binding domain; LBD, ligand-binding domain; AF-2, activation function 2. (B and C) Specificity of S155∼P and S287∼P antibodies tested in whole-cell extracts of 293TrkB cells transfected with rat GR (WT) or phosphorylation site-deficient S155A or S287A mutants and stimulated with 50 ng/ml BDNF for 30 min. Activated, phosphorylated TrkB (TrkB∼P) and total TrkB protein expression upon BDNF treatment is also shown. (D) Western blot analysis of S155∼P, S287∼P, and total GR from primary rat cortical neurons treated with BDNF (50 ng/ml, 30 min), Dex (100 nM, 30 min), or BDNF+Dex (30 min). The blots is representative of at least three independent experiments. (E and F) Kinetics of GR S155 and S287 phosphorylation in response to BDNF or Dex treatment, respectively. Western blot analysis for S155∼P, S287∼P, and total GR was performed from cells treated with either 50 ng/ml BDNF or 100 nM Dex at the indicated times. GR phosphoisoforms were quantified relative to total GR levels, and values are expressed as fold change relative to vehicle-treated cells (means ± standard errors of the means [SEM] from 3 independent experiments). (G) BDNF-dependent phosphorylation at additional GR phosphorylation sites (S224∼P, S246∼P, and S232∼P) not recovered in the mass spectrometry analysis highlights a >2-fold signal detection technical limit. Data (means ± SEM) were collected for whole-cell extracts from 293TrkB cells treated with 50 ng/ml BDNF for 30 min. Western blots are representative of at least three independent experiments.
We next examined the kinetics of GR phosphorylation upon BDNF or Dex treatment at S155 and S287. Phosphorylation at S155 and S287 in 293TrkB cells increased rapidly within 15 min of BDNF treatment and remained elevated for 60 min, after which the signal decreased quickly from 90 to 120 min back to basal levels. Phosphorylation of S155 followed a pattern similar to that of S287 throughout the time course of BDNF treatment (Fig. 5E). Phosphorylation at S287 also increased after 30 min of Dex treatment, remained elevated for 60 min, and progressively declined after 90 min. In contrast, S155 phosphorylation was not induced upon Dex treatment at any of time points examined (Fig. 5F). Thus, S287 phosphorylation is induced by both BDNF and Dex, albeit with different kinetics, whereas S155 phosphorylation responds only to BDNF.
We also examined whether BDNF treatment would induce phosphorylation at other previously characterized rat GR phosphorylation sites, S224, S232, and S246 (human GR S203, S211, and S226). BDNF stimulation increased GR phosphorylation at S224, S232, and S246 only 1.5-fold (Fig. 5G), whereas S155 and S287 showed the greatest induction in response to BDNF (3.5-fold and 3-fold, respectively) compared to untreated 293TrkB cells. Together, these data indicate that (57) BDNF treatment induces GR phosphorylation in a rapid manner at multiple sites (22), S155 is BDNF dependent but GC independent (58), and S287 is a GC- and BDNF-dependent site, with cotreatment resulting in S287 hyperphosphorylation of GR.
S155 and S287 phosphorylation mediate BDNF-dependent induction of GR target genes.
To assess the role of BDNF-induced GR phosphorylation, we generated stable cell lines expressing wild-type GR and the S155A/S287A mutant in a 293TrkB background and selected single clones expressing similar protein levels of GR and TrkB (data not shown). We next examined the transcription of two well-characterized direct GR transcriptional targets, namely, serum- and glucocorticoid-regulated kinase 1 (SGK1), a gene product that inhibits apoptosis (59), and glucocorticoid-induced leucine zipper protein (GILZ), a protein that regulates multiple signal transduction pathways involved in cell growth, cell differentiation, and cell survival (58). The expression of both SGK1 and GILZ were induced by Dex stimulation. However, BDNF treatment increased Dex-mediated SGK1 induction but did not enhance Dex-mediated GILZ expression (Fig. 6B). When GR phosphorylation sites were mutated, BDNF treatment failed to augment Dex-mediated SGK1 induction, whereas Dex-mediated expression of GILZ was insensitive to the mutations (Fig. 6B). Moreover, the expression of another GR target gene FKBP5 resembles that of SGK1 in that it is enhanced upon BDNF and Dex cotreatment and sensitive to GR phosphorylation (see Fig. S4A in the supplemental material). We also observed a small amount of BDNF-dependent stimulation of SGK1 (Fig. 6B) and FKBP5 (see Fig. S4A) expression in the absence of Dex treatment that is not observed with the GR S155A/S287A mutant. This is likely a result of BDNF treatment affecting a preexisting nuclear pool of GR present in the absence of added glucocorticoids to induce gene expression in a GR phosphorylation-dependent manner. Consistent with this idea, GR occupancy was observed at the SGK1 regulatory region upon BDNF treatment in the absence of Dex, and ∼20% of the cells exhibited some level of nuclear GR in untreated and BDNF-treated cells (see Fig. S5 in the supplemental material). In contrast, the Dex-mediated repression of C-MYC by GR, like GILZ induction, is insensitive to BDNF-induced GR phosphorylation (see Fig. S4A). Although these experiments were performed using a single clone of 293TrkB cells expressing either wild-type GR or the GR S155A/S287A mutant, we also observed in transient-transfection assays GR phosphorylation-dependent activation of SGK1 by Dex and BDNF (see Fig. S4C in the supplemental material), indicating that the stable GR 293TrkB lines accurately reflect GR and BDNF cross talk. Therefore, GR phosphorylation at S155 and S287 contributes to BDNF-mediated, Dex-dependent induction of selected GR target genes.
Fig 6.
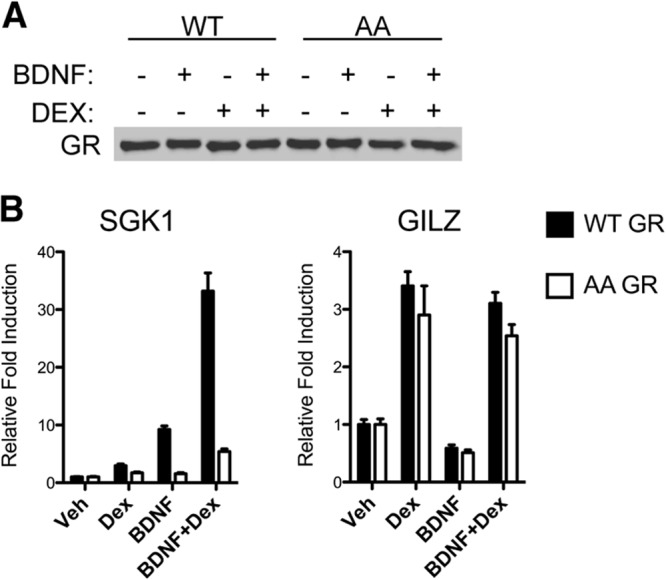
S155- and S287-dependent induction of GR target genes by BDNF. (A) Comparison of stable expression of rat GR (WT) or a S155A/S287A double mutant (AA) in transfected 293TrkB clones after treatment with 0.1% ethanol (Veh), 100 nM Dex, 50 ng/ml BDNF, or BDNF and Dex for 1 h and Western blot analysis for total GR. The Western blot is representative of at least three independent experiments. (B) 293TrkB cells stably expressing rat GR WT or AA were serum starved overnight and treated with the indicated agents for 3 h. Total RNA was prepared, and mRNA expression for SGK1 and GILZ was assessed by qPCR (means ± SEM from 3 independent experiments).
We also tested whether regulation of GR gene expression by BDNF-induced GR phosphorylation applied in primary cortical neurons. Real time qPCR of a set of microarray targets yielded similar results. Genes highly responsive to BDNF and Dex cotreatment, like NR4A1, PRL3C1, and SRGN required GR phosphorylation. A gene responsive to BDNF and Dex cotreatment, like MYOM2, was unaffected by the mutation of GR phosphorylation sites (see Fig. S4B in the supplemental material). Taken together, these data illustrate that BDNF enhances the expression of a subset of Dex-responsive genes that depend on GR phosphorylation.
Impact of BDNF signaling on GR recruitment to DNA.
To address whether BDNF signaling affects GR recruitment to chromatin, we performed a series of chromatin immunoprecipitation (ChIP) experiments from 293TrkB cells at the GREs upstream of SGK1 and GILZ (Fig. 7A). Since BDNF alone does not affect translocation of GR from the cytoplasm to the nucleus (see Fig. S5 in the supplemental material), we focused on cells stimulated with Dex in the absence or presence of BDNF. We found that Dex alone increased GR recruitment severalfold at both SGK1 (Fig. 7B)- and GILZ (Fig. 7C)-proximal promoter regions harboring the GREs. In the presence of BDNF, GR occupancy significantly increased at the SGK1 (Fig. 7B) but not GILZ GR-responsive regulatory regions (Fig. 7C), consistent with the enhanced mRNA induction of SGK1 and the lack of an effect on expression of GILZ by GR upon BDNF treatment. Thus, BDNF treatment results in increased GR occupancy at SGK1.
Fig 7.

BDNF-induced GR phosphorylation does not affect GR recruitment to the sgk1 regulatory region. (A) Schematic representations of SGK1 and GILZ loci. Arrows indicate the transcription start site. Gray box, CRE; black boxes, predicted GREs; thick arrows, regions amplified by qPCR to determine the level of occupancy of GR and cofactors. (B and C) Chromatin immunoprecipitations were performed with total GR or GR S155∼P S287∼P antibodies at the SGK1 (B) and GILZ (C) regulatory regions amplified with the primers used for panel A. 293TrkB cells stably expressing rat GR were treated for 1 h with Veh, Dex, or BDNF and Dex. (D) Chromatin immunoprecipitations were performed from 293TrkB cells expressing wild-type GR (WT) or GR S155A/S287A (AA) with antibodies to total GR at the SGK1 GR-responsive regulatory region. Error bars indicate standard deviations. Significance was determined using the two-tailed Student's t test (*, P < 0.05).
Next we investigated whether the phosphorylated forms of GR were differentially recruited to DNA as a potential mechanism of GR phosphorylation-dependent gene expression. Occupancy of GR S155∼P was minimal at SGK1 and GILZ under vehicle and Dex treatment, consistent with the lack of basal and Dex-induced GR phosphorylation at this site (Fig. 7B and C). GR S287∼P occupancy at SGK1 and GILZ regulatory regions was enhanced upon cotreatment with BDNF and Dex (Fig. 7B and C), reflecting the fact that cotreatment of BDNF and Dex has a cumulative effect on GR phosphorylation at S287 (see Fig. S6 in the supplemental material). Therefore, enrichment of phospho-GR isoforms at the representative SGK1 and GILZ regulatory regions reflected the level of GR phosphorylation and did not correlate with the transcription of these genes. This indicates that phosphoisoform recruitment does not distinguish activity between phosphorylation-sensitive and -insensitive genes. Consistent with this finding is the observation that GR recruitment to the SGK1 regulatory region upon BDNF and Dex treatment was similar in cells expressing the phosphorylated GR WT and GR S155A/S287A mutant (Fig. 7D). Thus, selective phosphoisoform recruitment per se does not appear to be the mechanism underlying differential effects of BDNF enhancement on GR activity.
BDNF signaling facilitates the recruitment of GR cofactors at selected promoters.
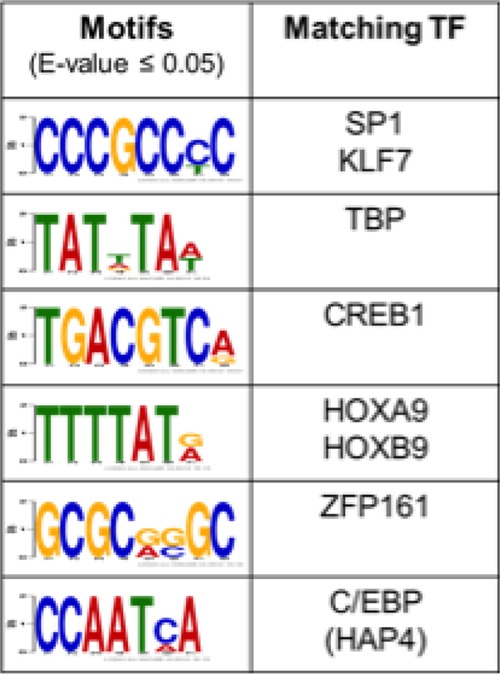
Given the likelihood that transcription factors downstream of BDNF signaling cooperate with GR to regulate BDNF- and Dex-sensitive genes, we searched for transcription factor-binding motifs within 500 bp upstream and 100 bp downstream of the transcription start sites of the BDNF- and Dex-coresponsive genes using DREME, an algorithm designed to find short DNA-binding motifs of eukaryotic transcription factors from large data sets (42). We found that binding sites for CREB, SP1, TBP, HOXA9, ZFP161, and C/EBP were enriched within the upstream regulatory regions of genes regulated by BDNF and Dex (P < 0.05) (Table 1). The validity of the bioinformatic analysis is emphasized by the identification of established GR cooperative transcription factors, including CREB (45, 60, 61) and SP1 (62). Although GREs were not recovered using this approach, this is not surprising given that the vast majority of GREs are not in promoter-proximal regions but are located >5 kb upstream or downstream of the transcription start site of most GR-responsive genes (45, 63). This suggests that combinatorial control of GR upon BDNF-induced phosphorylation with other transcription factors and coregulators promotes the expression of this unique cohort of genes induced by BDNF and Dex treatment.
Table 1.
Identification of transcription factors motifs enriched at the promoters of the uniquely BDNF+Dex induced genes
This prompted us to investigate whether specific GR cofactors are enriched at the prototypical SGK1 and GILZ promoters in response to cotreatment of 293TrkB cells with BDNF and Dex. We found that CREB, p300 (Fig. 8A), and GRIP1 (see Fig. S7 in the supplemental material) were enriched together with GR at the SGK1 promoter, whereas only GR, p300, and GRIP1 occupied the GILZ promoter (Fig. 8B; also, see Fig. S7). Also, the Brahma-related gene, BRG1, was not enriched at these promoters, suggesting some level of specificity (see Fig. S7). Therefore, CREB binding is correlated with the transcription of BDNF- and Dex-coresponsive genes.
Fig 8.

BDNF-induced GR phosphorylation stabilizes recruitment of p300 and CREB to the SGK1 promoter. (A and B) Chromatin immunoprecipitations were performed with 293TrkB cells expressing wild type GR (WT) or GR S155A/S287A (AA) with antibodies to GR, p300, or CREB at the SGK1 or GILZ GR-responsive regulatory regions. (C) BDNF-dependent activation of CREB by phosphorylation at S133∼P in the WT GR- and GR AA-expressing cells. The Western blot is representative of at least three independent experiments. (D) Coimmunoprecipitation of CREB with GR from nuclear extracts of 293TrkB cells depends upon BDNF and Dex cotreatment (D+B) and BDNF-sensitive phosphorylation sites. Raw data analysis (data are means ± SEM; *, P < 0.05 [t test]) of CREB recovered in two independent GR immunoprecipitates that compared the effect of cotreatment (50 ng/ml BDNF and 1 μM Dex) with 0.1% ethanol (Veh) and wild type with mutant GR. (E) 293TrkB cells were transfected with control scramble siRNA (siSCR) or a CREB siRNA pool (siCREB) and allowed to recover for 24 h. Cells were then serum starved, and RNA was assessed as described for Fig. 6. (F) Primary cortical neurons were transfected with control scramble siRNA (siSCR) or a CREB siRNA pool (siCREB), and expression of NR4A1 was measured upon BDNF+Dex treatment. Error bars indicate standard deviations. Significance was determined using the two-tailed Student's t test (*, P < 0.05; **, P < 0.01).
BDNF-induced GR phosphorylation regulates the recruitment of CREB to select promoters.
We next investigated whether recruitment of cofactors, notably CREB, are sensitive to BDNF-induced GR phosphorylation from 293TrkB cells expressing the GR wild type or the GR AA mutant. We found that the occupancy of CREB at the SGK1 promoter was reduced when the phosphodeficient GR mutant was expressed (Fig. 8A). In contrast, basal occupancy of CREB to the GILZ promoter was insensitive to the alanine mutations of GR (Fig. 8B). Recruitment of GR and GRIP1 was not significantly altered between wild-type GR and the phosphodeficient GR mutant (see Fig. S7 in the supplemental material). Phosphorylation of CREB at serine 133, which induces p300 binding (64), was not sensitive to Dex treatment or to GR phosphorylation (Fig. 8C). Therefore, combinatorial binding of GR with CREB may explain how BDNF-induced GR phosphorylation affects the transcription of a select group of genes.
BDNF- and Dex-sensitive binding of CREB to GR modulates transcription at selected genes.
We next tested by coimmunoprecipitation the possibility that GR and CREB associate in a GR phosphorylation-dependent manner. We prepared nuclear extracts from 293TrkB cells expressing the wild-type GR or the GR phosphorylation site mutant. We observed an association of GR with CREB upon Dex and BDNF cotreatment that was reduced with the GR phosphorylation site mutant (Fig. 8D).
If the recruitment of CREB is required to facilitate the transcription of select Dex-sensitive genes, then loss of function of CREB should prevent the induction of SGK1 in response to BDNF and Dex cotreatment. We found that knockdown of CREB1 by RNAi in 293TrkB cells diminished the induction of SGK1 following BDNF and Dex cotreatment compared to a control siRNA (Fig. 8E; also, see Fig. S8A in the supplemental material). The induction of GILZ expression was similar whether cells were transfected with the scrambled or specific CREB RNAi. Similarly, in primary neurons the expression of the BDNF- and Dex-sensitive gene NR4A1 was reduced upon CREB1 knockdown (Fig. 8F; also, see Fig. S8B). Taken together, our data indicate that BDNF-induced GR phosphorylation and CREB activation are required for full expression in the context of BDNF and glucocorticoid cosignaling.
GR phosphorylation at S287 is sensitive to stress and genetic manipulation of the BDNF-TrkB signaling pathway.
To assess GR phosphorylation at S287 in vivo as a function of stress and BDNF signaling, GR S287∼P reactivity in the paraventricular nucleus (PVN) of the hypothalamus was determined by Western blotting of tissue biopsy specimens and by immunostaining of brain sections (Fig. 9A and B). Importantly, the specificity of the GR S287∼P antibody was confirmed by a reduction in GR S287∼P immunoreactivity upon conditional inactivation of GR in the PVN through crossing of GR-floxed mice (20) with the PVN-selective Sim1-Cre transgenic mice (65) (see Fig. S9A in the supplemental material).
Fig 9.

GR phosphorylation at S287 is sensitive to BDNF and TrkB dosage and to stress in the hypothalamus. (A) Quantification of hypothalamic GR S287 phosphorylation by Western blotting of brain lysates from adult mice subjected to a single systemic injection of vehicle (0.9% saline), Dex (10 mg/kg for 6 h), or an acute 10-min forced swim stress. Data are expressed as means ± SEM of the optical density (OD) of GR S287∼P normalized to total GR (5 mice/group). The Western blot is representative of at least three independent experiments. (B) Localization of phospho-TrkB and GR S287∼P in hypothalamic CRH-producing neurons of a representative adult resting mouse is shown. Blue, CRH; green, GR S287∼P; red, TrkB∼P. Bar, 10 μm. (C) Representative GR S287∼P immunostaining in the PVN of adult BDNF and TrkB heterozygotes, as well as conditional hypothalamic BDNF overexpression mice, using the Sim1-Cre line [Tg(BDNF)Cre]. Bar, 50 μm. (D) Number of GR S287∼P-positive cells (mean ± SEM; *, P < 0.05 [t test]) in the PVN from littermates of the BDNF knockout line (5 +/+ and 4 +/−) or TrkB knockout line (6 +/+ and 6 +/−) and transgenic BDNF-overexpressing Tg(BDNF)×Sim1-Cre (7 Cre+ and 7 Cre−) mice is shown. At least 600 GR S287∼P-positive cells were counted per group. Error bars indicate standard deviations. Significance was determined using the two-tailed Student's t test (*, P < 0.05).
Because CRH neurons of the PVN are highly sensitive to stress and glucocorticoid feedback, we examined GR S287∼P expression as a function of increased glucocorticoid levels in vivo either by exogenous Dex administration or by elevation of endogenous glucocorticoids via stress. Both treatments result in an increase in GR S287∼P in hypothalamic extracts by Western blot (Fig. 9A).
We also found that CRH-expressing neurons of the PVN display GR S287∼P immunoreactivity that colocalizes with active, phospho-TrkB-expressing cells, suggesting that these neurons are responsive to TrkB signaling (Fig. 9B).
To further determine the requirement of BDNF and TrkB signaling in GR phosphorylation in vivo, we compared GR S287∼P immunostaining in tissue sections from wild-type mice and from heterozygous BDNF (BDNF+/−) and TrkB (TrkB+/−) mice. The number of cells in the PVN expressing GR S287∼P was decreased in both BDNF+/− and TrkB+/− mice compared to wild-type controls (Fig. 9C and D). Conversely, using a transgenic mouse that conditionally overexpresses BDNF in the PVN by Sim1-Cre-mediated deletion of a floxed-STOP cassette [referred to as Tg(BDNF)Cre mice] (20), we found an increase in GR S287∼P in the PVN neurons compared to levels in littermate controls (Fig. 9C and D). This demonstrates that changes in BDNF and TrkB levels in the hypothalamus modulate GR S287∼P in vivo. Although we detected expression of GR S155∼P in neurons of the PVN (see Fig. S9B in the supplemental material), we have yet to investigate its regulation in the hypothalamus as a function of BDNF signaling. This demonstrates that changes in BDNF, TrkB, or glucocorticoid levels modulate GR S287∼P in vivo.
DISCUSSION
This study explored the mechanism whereby BDNF affects the transcriptional activity of GR. We demonstrate that BDNF not only alters the GR transcriptome by affecting the regulation of known GR-responsive genes but also facilitates the expression of a new repertoire of genes. This is mediated in part through changes in GR phosphorylation at S155 and S287 upon BDNF signaling via TrkB. Interestingly, these events not only were observed in primary cortical neurons harboring endogenous TrkB, in PVN neurons rich in endogenous GR and TrkB, but also were observed in 293 cells reconstituted with TrkB, suggesting that cell signaling events mediated via BDNF-TrkB are sufficient to modulate GR phosphorylation and transcriptional regulatory functions. In fact, in both cell contexts, select GR targets induced by BDNF treatment were no longer sensitive to BDNF stimulation when the GR S155A/S287A mutant was expressed. BDNF treatment also enhanced the ability of GR to repress gene expression, although in some cases this appears to be a secondary effect of GR regulation of gene expression that requires new protein synthesis. We suggest that BDNF-dependent phosphorylation of GR modulates GR cofactor recruitment to affect GR transcriptional activation and repression.
How might BDNF treatment lead to a new repertoire of genes upon Dex stimulation? The most likely explanation is that BDNF-TrkB signaling affects the activity of key transcription factors that cooperate with phospho-GR to drive gene expression or that BDNF acts through epigenetic mechanisms that open chromatin to allow GR to bind and regulate new gene targets. Such mechanisms are not mutually exclusive. Our comparative genomic analysis of conserved motifs in gene promoters supports the hypothesis that cooperating transcription factors upon BDNF treatment align GR activity to shape the GR transcriptome. If this is true, then one would predict that GR would associate with new binding sites genomewide upon BDNF and Dex treatment compared to Dex alone. In fact, elegant work from the Stunnenberg laboratory found that in HeLa cells, coactivation of GR in the context of NF-κB activation via tumor necrosis factor alpha alters expression of genes regulated by activation of each factor separately and results in GR associating with novel sites in the genome (66). Whether BDNF signaling elicits such effects upon GR binding genomewide remains to be elucidated.
For some genes, BDNF-induced phosphorylation enhanced transcriptional activation (PRL3C1), while for other genes, enhanced transcription appears to be insensitive to phosphorylation at S155 and S287 (MYOM2). This could be a function of the chromatin architecture of different gene-regulatory elements. Genes like GILZ that are GR phosphorylation state independent mitigate the need for phosphorylation-dependent coregulators, whereas genes sensitive to BDNF treatment and GR phosphorylation would require a GR N-terminal AF-1 domain cofactor to assemble a productive transcription initiation complex, resulting in greater occupancy of GR at the promoter (10). Interestingly, we observed higher GR occupancy at the phosphorylation-sensitive SGK1 promoter under Dex and BDNF conditions relative to Dex treatment alone. Given that both wild-type GR and GR S155A/S287A are recruited to DNA with the same magnitude, this suggests that BDNF-induced GR phosphorylation affects a postrecruitment step. We further demonstrate that CREB, likely through enhanced p300 recruitment, contributes to GR-mediated induction of SGK1 and NR4A1 in a GR phosphorylation-dependent manner. Thus, induction of gene expression by BDNF and Dex is dependent upon (i) BDNF-induced GR phosphorylation at S155 and S287, (ii) recruitment of BDNF-activated CREB to target composite promoters, and (iii) BDNF-enhanced GR recruitment to DNA. Recently, CREB DNA binding motifs were found to be enriched near GR binding sites in A549 human lung adenocarcinoma cells (60). We propose a model whereby upon BDNF stimulation, other transcription factors (SP1, TBP, CREB, HOXA9, ZFP161, and C/EBP) act to cooperatively recruit GR as well as phosphorylation-sensitive coactivators at selected genes, thus changing the repertoire of genes controlled upon signaling by glucocorticoids and BDNF.
Work from Galliher-Beckley et al. (67) has demonstrated that GR S155∼P (S134 human GR) is a site that is phosphorylated upon H2O2-induced cellular stress and glucose deprivation but not by glucocorticoids, as was found here (Fig. 5F). This site was shown to recruit 14-3-3 proteins and when mutated to a nonphosphorylated residue impacts gene expression in U2OS cells (67). Consistent with their conclusion that S155 is a sensor, we have observed this phosphorylation to be a readout of BDNF activation in cortical neurons.
Future investigations will be necessary to define both the role of GR phosphorylation in the central nervous system and the physiological consequences of BDNF signaling in modulating phosphorylation and GR transcriptional activity in neuronal function. We have shown previously that genetic disruption of GR signaling in the paraventricular nucleus results in disinhibition of the HPA axis, increased CRH expression, and upregulation of hypothalamic levels of BDNF (20). It would be interesting to explore the requirement for GR phosphorylation in hypothalamic gene expression. Furthermore, in tissues such as the prefrontal cortex and the hippocampus, modulation of GR phosphorylation may be important for glucocorticoid function in the brain. Recent work from the Pariante laboratory demonstrated a role for SGK1 in neurogenesis via changes in GR phosphorylation and suggested that BDNF-dependent GR enhancement of SGK1 expression could influence neural progenitor cell formation (57).
Factors regulating GR-dependent gene expression are not limited to BDNF. Norepinephrine is capable of increasing ligand-induced GR transcriptional activity and DNA binding (68). Adrenergic receptor agonists synergistically enhance GR-dependent transcription, and cAMP-elevating drugs increase the potency of GR activity (69). Consequently, the effect of BDNF upon GR transcriptional activity via receptor phosphorylation likely represents a general mechanism shared by others neurotransmitter systems to tailor transcriptional responses of glucocorticoids. Although GR S155 and S287 are targets for BDNF-induced phosphorylation, other trophic or hormonal pathways do not result in GR phosphorylation at these sites in neurons, demonstrating a level of specificity (F. Jeanneteau, unpublished observation).
We have previously shown that glucocorticoids, via a GR transcriptional mechanism, can activate TrkB signaling independently of neurotrophins (34). It is conceivable that a reciprocal homeostatic sensing mechanism exists between GR and BDNF-TrkB signaling to adjust GR transcriptional output. Consistent with this idea is the rapid feed-forward kinetics of BDNF-induced GR phosphorylation, which occurs within minutes, and the slower feedback phase of the Dex-induced TrkB phosphorylation, which occurs over hours. Synchronizing these events is key to balancing GR and TrkB activity in response to corticosteroid and neurotrophic signals, and disruption of this process may lead to neurological disorders in animal models and humans.
Supplementary Material
ACKNOWLEDGMENTS
This research was supported by the Brain and Behavior Foundation and an Inserm ATIP/AVENIR grant (to F.D.J.) and by grants MH086651 and NS21072 (to M.V.C. and M.J.G.), P30 NS050276 to T.A.N., and 5T32 AI07180-29 (to W.M.L.) from the National Institutes of Health.
We thank Maryem Hussein, Elina Shrestha, Naoko Tanese, and Inez Rogatsky for critically reading the manuscript.
Footnotes
Published ahead of print 22 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00150-13.
REFERENCES
- 1.de Kloet ER, Vreugdenhil E, Oitzl MS, Joels M. 1998. Brain corticosteroid receptor balance in health and disease. Endocrine Rev. 19:269–301 [DOI] [PubMed] [Google Scholar]
- 2.Yamamoto KR. 1995. Multilayered control of intracellular receptor function. Harvey Lect. 91:1–19 [PubMed] [Google Scholar]
- 3.Hittelman AB, Burakov D, Iniguez-Lluhi JA, Freedman LP, Garabedian MJ. 1999. Differential regulation of glucocorticoid receptor transcriptional activation via AF-1-associated proteins. EMBO J. 18:5380–5388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ismaili N, Garabedian MJ. 2004. Modulation of glucocorticoid receptor function via phosphorylation. Ann. N. Y. Acad. Sci. 1024:86–101 [DOI] [PubMed] [Google Scholar]
- 5.Rogatsky I, Wang JC, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq CM, Darimont BD, Garabedian MJ, Yamamoto KR. 2003. Target-specific utilization of transcriptional regulatory surfaces by the glucocorticoid receptor. Proc. Natl. Acad. Sci. U. S. A. 100:13845–13850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Webster JC, Jewell CM, Bodwell JE, Munck A, Sar M, Cidlowski JA. 1997. Mouse glucocorticoid receptor phosphorylation status influences multiple functions of the receptor protein. J. Biol. Chem. 272:9287–9293 [DOI] [PubMed] [Google Scholar]
- 7.Galliher-Beckley AJ, Cidlowski JA. 2009. Emerging roles of glucocorticoid receptor phosphorylation in modulating glucocorticoid hormone action in health and disease. IUBMB Life 61:979–986 [DOI] [PubMed] [Google Scholar]
- 8.Chen W, Rogatsky I, Garabedian MJ. 2006. MED14 and MED1 differentially regulate target-specific gene activation by the glucocorticoid receptor. Mol. Endocrinol. 20:560–572 [DOI] [PubMed] [Google Scholar]
- 9.Galliher-Beckley AJ, Williams JG, Collins JB, Cidlowski JA. 2008. Glycogen synthase kinase 3β-mediated serine phosphorylation of the human glucocorticoid receptor redirects gene expression profiles. Mol. Cell. Biol. 28:7309–7322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen W, Dang T, Blind RD, Wang Z, Cavasotto CN, Hittelman AB, Rogatsky I, Logan SK, Garabedian MJ. 2008. Glucocorticoid receptor phosphorylation differentially affects target gene expression. Mol. Endocrinol. 22:1754–1766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang Z, Frederick J, Garabedian MJ. 2002. Deciphering the phosphorylation “code” of the glucocorticoid receptor in vivo. J. Biol. Chem. 277:26573–26580 [DOI] [PubMed] [Google Scholar]
- 12.Holsboer F. 2000. The corticosteroid receptor hypothesis of depression. Neuropsychopharmacology 23:477–501 [DOI] [PubMed] [Google Scholar]
- 13.Pariante CM. 2003. Depression, stress and the adrenal axis. J. Neuroendocrinol. 15:811–812 [DOI] [PubMed] [Google Scholar]
- 14.Webster MJ, Knable MB, O'Grady J, Orthmann J, Weickert CS. 2002. Regional specificity of brain glucocorticoid receptor mRNA alterations in subjects with schizophrenia and mood disorders. Mol. Psychiatry 7:985–994 [DOI] [PubMed] [Google Scholar]
- 15.van Rossum EF, Lamberts SW. 2004. Polymorphisms in the glucocorticoid receptor gene and their associations with metabolic parameters and body composition. Recent Prog. Horm Res. 59:333–357 [DOI] [PubMed] [Google Scholar]
- 16.de Quervain DJ, Poirier R, Wollmer MA, Grimaldi LM, Tsolaki M, Streffer JR, Hock C, Nitsch RM, Mohajeri MH, Papassotiropoulos A. 2004. Glucocorticoid-related genetic susceptibility for Alzheimer's disease. Hum. Mol. Genet. 13:47–52 [DOI] [PubMed] [Google Scholar]
- 17.Green KN, Billings LM, Roozendaal B, McGaugh JL, LaFerla FM. 2006. Glucocorticoids increase amyloid-beta and tau pathology in a mouse model of Alzheimer's disease. J. Neurosci. 26:9047–9056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holsboer F, Ising M. 2010. Stress hormone regulation: biological role and translation into therapy. Annu. Rev. Psychol 61:81–109 [DOI] [PubMed] [Google Scholar]
- 19.Yehuda R, Halligan SL, Grossman R, Golier JA, Wong C. 2002. The cortisol and glucocorticoid receptor response to low dose dexamethasone administration in aging combat veterans and holocaust survivors with and without posttraumatic stress disorder. Biol. Psychiatry 52:393–403 [DOI] [PubMed] [Google Scholar]
- 20.Jeanneteau FD, Lambert WM, Ismaili N, Bath KG, Lee FS, Garabedian MJ, Chao MV. 2012. BDNF and glucocorticoids regulate corticotrophin-releasing hormone (CRH) homeostasis in the hypothalamus. Proc. Natl. Acad. Sci. U. S. A. 109:1305–1310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chao MV. 2003. Neurotrophins and their receptors: a convergence point for many signalling pathways. Nat. Rev. Neurosci. 4:299–309 [DOI] [PubMed] [Google Scholar]
- 22.Angelucci F, Brene S, Mathe AA. 2005. BDNF in schizophrenia, depression and corresponding animal models. Mol. Psychiatry 10:345–352 [DOI] [PubMed] [Google Scholar]
- 23.Schmidt HD, Duman RS. 2010. Peripheral BDNF produces antidepressant-like effects in cellular and behavioral models. Neuropsychopharmacology 35:2378–2391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Shirayama Y, Chen AC, Nakagawa S, Russell DS, Duman RS. 2002. Brain-derived neurotrophic factor produces antidepressant effects in behavioral models of depression. J. Neurosci. 22:3251–3261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen ZY, Jing D, Bath KG, Ieraci A, Khan T, Siao CJ, Herrera DG, Toth M, Yang C, McEwen BS, Hempstead BL, Lee FS. 2006. Genetic variant BDNF (Val66Met) polymorphism alters anxiety-related behavior. Science 314:140–143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Soliman F, Glatt CE, Bath KG, Levita L, Jones RM, Pattwell SS, Jing D, Tottenham N, Amso D, Somerville LH, Voss HU, Glover G, Ballon DJ, Liston C, Teslovich T, Van Kempen T, Lee FS, Casey BJ. 2010. A genetic variant BDNF polymorphism alters extinction learning in both mouse and human. Science 327:863–866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gray JD, Milner TA, McEwen BS. 2013. Dynamic plasticity: the role of glucocorticoids, brain-derived neurotrophic factor and other trophic factors. Neuroscience 239:214–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jeanneteau F, Chao MV. 2013. Are BDNF and glucocorticoid activities calibrated? Neuroscience 239:173–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Numakawa T, Adachi N, Richards M, Chiba S, Kunugi H. 2013. Brain-derived neurotrophic factor and glucocorticoids: reciprocal influence on the central nervous system. Neuroscience 239:157–172 [DOI] [PubMed] [Google Scholar]
- 30.Suri D, Vaidya VA. 2013. Glucocorticoid regulation of brain-derived neurotrophic factor: relevance to hippocampal structural and functional plasticity. Neuroscience 239:196–213 [DOI] [PubMed] [Google Scholar]
- 31.McEwen BS. 2005. Glucocorticoids, depression, and mood disorders: structural remodeling in the brain. Metabolism 54:20–23 [DOI] [PubMed] [Google Scholar]
- 32.Tata DA, Marciano VA, Anderson BJ. 2006. Synapse loss from chronically elevated glucocorticoids: relationship to neuropil volume and cell number in hippocampal area CA3. J. Comp. Neurol. 498:363–374 [DOI] [PubMed] [Google Scholar]
- 33.Watanabe Y, Gould E, McEwen BS. 1992. Stress induces atrophy of apical dendrites of hippocampal CA3 pyramidal neurons. Brain Res. 588:341–345 [DOI] [PubMed] [Google Scholar]
- 34.Jeanneteau F, Garabedian MJ, Chao MV. 2008. Activation of Trk neurotrophin receptors by glucocorticoids provides a neuroprotective effect. Proc. Natl. Acad. Sci. U. S. A. 105:4862–4867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chang Q, Khare G, Dani V, Nelson S, Jaenisch R. 2006. The disease progression of Mecp2 mutant mice is affected by the level of BDNF expression. Neuron 49:341–348 [DOI] [PubMed] [Google Scholar]
- 36.Ernfors P, Lee KF, Jaenisch R. 1994. Mice lacking brain-derived neurotrophic factor develop with sensory deficits. Nature 368:147–150 [DOI] [PubMed] [Google Scholar]
- 37.Rohrer B, Korenbrot JI, LaVail MM, Reichardt LF, Xu B. 1999. Role of neurotrophin receptor TrkB in the maturation of rod photoreceptors and establishment of synaptic transmission to the inner retina. J. Neurosci. 19:8919–8930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Narisawa-Saito M, Iwakura Y, Kawamura M, Araki K, Kozaki S, Takei N, Nawa H. 2002. Brain-derived neurotrophic factor regulates surface expression of alpha-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid receptors by enhancing the N-ethylmaleimide-sensitive factor/GluR2 interaction in developing neocortical neurons. J. Biol. Chem. 277:40901–40910 [DOI] [PubMed] [Google Scholar]
- 39.Bolstad BM, Irizarry RA, Astrand M, Speed TP. 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19:185–193 [DOI] [PubMed] [Google Scholar]
- 40.Huang DW, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4:44–57 [DOI] [PubMed] [Google Scholar]
- 41.Huang DW, Sherman BT, Lempicki RA. 2009. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bailey TL. 2011. DREME: motif discovery in transcription factor ChIP-seq data. Bioinformatics 27:1653–1659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ma Y, Lu Y, Zeng H, Ron D, Mo W, Neubert TA. 2001. Characterization of phosphopeptides from protein digests using matrix-assisted laser desorption/ionization time-of-flight mass spectrometry and nanoelectrospray quadrupole time-of-flight mass spectrometry. Rapid Commun. Mass Spectrom. 15:1693–1700 [DOI] [PubMed] [Google Scholar]
- 44.Xu CF, Lu Y, Ma J, Mohammadi M, Neubert TA. 2005. Identification of phosphopeptides by MALDI Q.-TOF MS in positive and negative ion modes after methyl esterification. Mol. Cell. Proteomics 4:809–818 [DOI] [PubMed] [Google Scholar]
- 45.Reddy TE, Pauli F, Sprouse RO, Neff NF, Newberry KM, Garabedian MJ, Myers RM. 2009. Genomic determination of the glucocorticoid response reveals unexpected mechanisms of gene regulation. Genome Res. 19:2163–2171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vivanco MD, Johnson R, Galante PE, Hanahan D, Yamamoto KR. 1995. A transition in transcriptional activation by the glucocorticoid and retinoic acid receptors at the tumor stage of dermal fibrosarcoma development. EMBO J. 14:2217–2228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lefstin JA, Thomas JR, Yamamoto KR. 1994. Influence of a steroid receptor DNA-binding domain on transcriptional regulatory functions. Genes Dev. 8:2842–2856 [DOI] [PubMed] [Google Scholar]
- 48.Wright AP, McEwan IJ, Dahlman-Wright K, Gustafsson JA. 1991. High level expression of the major transactivation domain of the human glucocorticoid receptor in yeast cells inhibits endogenous gene expression and cell growth. Mol. Endocrinol. 5:1366–1372 [DOI] [PubMed] [Google Scholar]
- 49.Xiao L, Qi A, Chen Y. 2005. Cultured embryonic hippocampal neurons deficient in glucocorticoid (GC) receptor: a novel model for studying nongenomic effects of GC in the neural system. Endocrinology 146:4036–4041 [DOI] [PubMed] [Google Scholar]
- 50.Dennis G, Jr, Sherman BT, Hosack DA, Yang J, Gao W, Lane HC, Lempicki RA. 2003. DAVID: database for annotation, visualization, and integrated discovery. Genome Biol. 4:P3. 10.1186/gb-2003-4-5-p3 [DOI] [PubMed] [Google Scholar]
- 51.Jeanneteau F, Deinhardt K, Miyoshi G, Bennett AM, Chao MV. 2010. The MAP kinase phosphatase MKP-1 regulates BDNF-induced axon branching. Nat. Neurosci. 13:1373–1379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Matsumoto K, Nishihara S, Kamimura M, Shiraishi T, Otoguro T, Uehara M, Maeda Y, Ogura K, Lumsden A, Ogura T. 2004. The prepattern transcription factor Irx2, a target of the FGF8/MAP kinase cascade, is involved in cerebellum formation. Nat. Neurosci. 7:605–612 [DOI] [PubMed] [Google Scholar]
- 53.Toyoda R, Assimacopoulos S, Wilcoxon J, Taylor A, Feldman P, Suzuki-Hirano A, Shimogori T, Grove EA. 2010. FGF8 acts as a classic diffusible morphogen to pattern the neocortex. Development 137:3439–3448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fukumoto K, Morita T, Mayanagi T, Tanokashira D, Yoshida T, Sakai A, Sobue K. 2009. Detrimental effects of glucocorticoids on neuronal migration during brain development. Mol. Psychiatry 14:1119–1131 [DOI] [PubMed] [Google Scholar]
- 55.Kwok JC, Dick G, Wang D, Fawcett JW. 2011. Extracellular matrix and perineuronal nets in CNS repair. Dev. Neurobiol. 71:1073–1089 [DOI] [PubMed] [Google Scholar]
- 56.Pejler G, Abrink M, Wernersson S. 2009. Serglycin proteoglycan: regulating the storage and activities of hematopoietic proteases. Biofactors 35:61–68 [DOI] [PubMed] [Google Scholar]
- 57.Anacker C, Cattaneo A, Musaelyan K, Zunszain PA, Horowitz M, Molteni R, Luoni A, Calabrese F, Tansey K, Gennarelli M, Thuret S, Price J, Uher R, Riva MA, Pariante CM. 2013. Role for the kinase SGK1 in stress, depression, and glucocorticoid effects on hippocampal neurogenesis. Proc. Natl. Acad. Sci. U. S. A. 110:8708–8713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ayroldi E, Riccardi C. 2009. Glucocorticoid-induced leucine zipper (GILZ): a new important mediator of glucocorticoid action. FASEB J. 23:3649–3658 [DOI] [PubMed] [Google Scholar]
- 59.Mikosz CA, Brickley DR, Sharkey MS, Moran TW, Conzen SD. 2001. Glucocorticoid receptor-mediated protection from apoptosis is associated with induction of the serine/threonine survival kinase gene, sgk-1. J. Biol. Chem. 276:16649–16654 [DOI] [PubMed] [Google Scholar]
- 60.Reddy TE, Gertz J, Crawford GE, Garabedian MJ, Myers RM. 2012. The hypersensitive glucocorticoid response specifically regulates period 1 and expression of circadian genes. Mol. Cell. Biol. 32:3756–3767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Stauber C, Altschmied J, Akerblom IE, Marron JL, Mellon PL. 1992. Mutual cross-interference between glucocorticoid receptor and CREB inhibits transactivation in placental cells. New Biol. 4:527–540 [PubMed] [Google Scholar]
- 62.Strahle U, Schmid W, Schutz G. 1988. Synergistic action of the glucocorticoid receptor with transcription factors. EMBO J. 7:3389–3395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.John S, Sabo PJ, Johnson TA, Sung MH, Biddie SC, Lightman SL, Voss TC, Davis SR, Meltzer PS, Stamatoyannopoulos JA, Hager GL. 2008. Interaction of the glucocorticoid receptor with the chromatin landscape. Mol. Cell 29:611–624 [DOI] [PubMed] [Google Scholar]
- 64.Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. 1993. Phosphorylated CREB binds specifically to the nuclear protein CBP. Nature 365:855–859 [DOI] [PubMed] [Google Scholar]
- 65.Balthasar N, Dalgaard LT, Lee CE, Yu J, Funahashi H, Williams T, Ferreira M, Tang V, McGovern RA, Kenny CD, Christiansen LM, Edelstein E, Choi B, Boss O, Aschkenasi C, Zhang CY, Mountjoy K, Kishi T, Elmquist JK, Lowell BB. 2005. Divergence of melanocortin pathways in the control of food intake and energy expenditure. Cell 123:493–505 [DOI] [PubMed] [Google Scholar]
- 66.Rao NAS, McCalman MT, Moulos P, Francoijs K-J, Chatziioannou A, Kolisis FN, Alexis MN, Mitsiou DJ, Stunnenberg HG. 2011. Coactivation of GR and NFKB alters the repertoire of their binding sites and target genes. Genome Res. 21:1404–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Galliher-Beckley AJ, Williams JG, Cidlowski JA. 2011. Ligand-independent phosphorylation of the glucocorticoid receptor integrates cellular stress pathways with nuclear receptor signaling. Mol. Cell. Biol. 31:4663–4675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Schmidt P, Holsboer F, Spengler D. 2001. Beta(2)-adrenergic receptors potentiate glucocorticoid receptor transactivation via G protein beta gamma-subunits and the phosphoinositide 3-kinase pathway. Mol. Endocrinol. 15:553–564 [DOI] [PubMed] [Google Scholar]
- 69.Kaur M, Chivers JE, Giembycz MA, Newton R. 2008. Long-acting beta2-adrenoceptor agonists synergistically enhance glucocorticoid-dependent transcription in human airway epithelial and smooth muscle cells. Mol. Pharmacol. 73:203–214 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.