

Abstract
In yeast, 25S rRNA makes up the major mass and shape of the 60S ribosomal subunit. During the last step of translation initiation, eukaryotic initiation factor 5B (eIF5B) promotes the 60S subunit joining with the 40S initiation complex (IC). Malfunctional 60S subunits produced by misfolding or mutation may disrupt the 40S IC stalling on the start codon, thereby altering the stringency of initiation. Using several point mutations isolated by random mutagenesis, here we studied the role of 25S rRNA in start codon selection. Three mutations changing bases near the ribosome surface had strong effects, allowing the initiating ribosomes to skip both AUG and non-AUG codons: C2879U and U2408C, altering the A loop and P loop, respectively, of the peptidyl transferase center, and G1735A, mapping near a Eukarya-specific bridge to the 40S subunit. Overexpression of eIF5B specifically suppressed the phenotype caused by C2879U, suggesting functional interaction between eIF5B and the A loop. In vitro reconstitution assays showed that C2879U decreased eIF5B-catalyzed 60S subunit joining with a 40S IC. Thus, eIF5B interaction with the peptidyl transferase center A loop increases the accuracy of initiation by stabilizing the overall conformation of the 80S initiation complex. This study provides an insight into the effect of ribosomal mutations on translation profiles in eukaryotes.
INTRODUCTION
Ribosomes catalyze mRNA-dependent protein synthesis with assistance from translation factors. While the basic mechanism of mRNA translation is similar in all the three domains of life on Earth, eukaryotic ribosomes bind more proteins in translation initiation and signal transduction and change translation profiles in response to distinct stimuli (1, 2). Despite extensive studies, however, the full repertoire of cellular regulation of mRNA translation has not been elucidated. For example, a group of genetic diseases called “ribosomopathies” occur due to defective ribosome biogenesis or function (3), but the mechanism by which the original genetic mutations change mRNA translation profile is not known. To understand the effect of ribosomal mutations on translation initiation, we have been studying yeast rRNA by genetics (4; N. Nemoto, T. Udagawa, D. Chowdhury, M. Kitabatake, H. Hiraishi, S. Wang, C. R. Singh, S. J. Brown, M. Ohno, and K. Asano, submitted for publication).
During translation initiation in eukaryotes, methionyl initiator tRNA (Met-tRNAiMet) is placed in the peptidyl-tRNA binding site (P site) of the small (40S) ribosomal subunit. Eukaryotic translation initiation factor 2 (eIF2) binds Met-tRNAiMet to form a ternary complex that then associates with the 40S subunit along with additional initiation factors, including eIF1 and eIF1A, to form a 43S preinitiation complex (PIC). This complex associates with an mRNA forming a 48S PIC and then scans to select a translation start site. Base-pairing interactions between the anticodon loop of Met-tRNAiMet in the 43S initiation complex (IC) and an AUG codon on the mRNA sets the translation start site. In addition to eIF2, eIF1, and eIF1A, the positioning of Met-tRNAiMet in the 48S PIC is influenced by the eukaryotic-specific initiation factors eIF3 and eIF5 (5, 6). After selection of the translation start site, eIF5B-mediated joining of the large (60S) ribosomal subunit generates an 80S ribosome that can then catalyze the synthesis of the encoded protein. Throughout these steps, the ribosome not only plays a passive role as the recipient of Met-tRNAiMet and mRNA but also plays an active role as the transducer of stringent start codon selection.
The mechanism by which start codon-anticodon pairing in the P site signals a conformational change of the 48S PIC has been studied extensively (7, 8). eIF1 plays a crucial role in this regulation: it inhibits the transition to the “closed” state of the 48S IC, which favors stable codon-anticodon binding to the P site. Initially during scanning, eIF1 found in a 48S PIC suppresses this transition, maintaining the open conformation in favor of scanning. Once a start codon is reached, eIF1 is released, and the 48S PIC transitions to the closed state (9–11). An important consequence of this transition is the release of translation factors eIF2 and eIF5, which otherwise block 60S subunit joining (10, 12, 13). The resulting 40S complex is termed the 40S initiation complex (IC). The factor eIF5B, which is an orthologue of bacterial IF2, promotes subunit joining by binding to the 40S IC, presumably in its closed conformation, producing the 80S IC that is ready to accept an aminoacyl tRNA (aa-tRNA) at the A site for peptide elongation (14).
In the bacterial 30S IC, fMet-tRNAfMet is proposed to position in a P/I hybrid state, such that codon-anticodon interactions take place in the P site and the methionyl moiety can interact with the peptidyl transferase center (PTC) of the 50S subunit after subunit joining (15). This P/I configuration is physically supported by bridging interactions between the G domain and domain II of IF2 and the 30S subunit and between domain IV of IF2 and fMet-tRNAfMet. The P/I state has not been demonstrated in eukaryotic complexes, but hydroxyl radical footprinting studies on human complexes and mutational studies on yeast eIF5B support the model that eIF5B binds to the 40S subunit via interactions homologous to those found in the bacterial 30S IC (16, 17).
In this communication, we examine the hypothesis that stable subunit joining involving the 60S subunit and eIF5B is a crucial step ensuring high stringency in start codon selection and that the 40S IC interaction with malfunctional 60S subunits, produced due to misfolding or mutation, changes the accuracy of translation initiation. For example, assume that an 80S IC forms between the malfunctional 60S subunit and the 40S IC. This could induce an open conformation in the 80S complex, followed by 60S dissociation and continued 40S scanning along mRNA attached. Alternatively, malfunctional 60S subunit may bind to a 40S IC more slowly, enabling a switch from a closed to an open 40S conformation, releasing the 60S subunit (without forming an 80S IC). We predict that if such an mRNA-40S (open) complex is formed, a 60S subunit mutation can cause the bypass of start codons. In agreement with the idea that this is an important checkpoint for 60S subunit quality, defective 80S IC formation or (initial) peptide elongation is coupled to various ribosome surveillance pathways (18–23).
Previously, we randomly mutated an RDN locus encoding all the rRNA components, isolated dozens of point mutations (rdn) that affected yeast cell growth and polysome profiles, and reported the characterization of 10 rdn mutations altering 18S rRNA making up the 40S subunit (4) and 7 rdn mutations altering 25S rRNA making up the 60S subunit (Nemoto et al., submitted). Here we studied the effects of six well-behaved 25S rRNA mutations on the accuracy of translation initiation. Interestingly, even though many 25S rRNA point mutants impaired translation initiation and allowed bypass of start codons, only one mutation, altering a base (C2879) near the peptidyl transferase center (PTC) A site, was suppressed by overexpression of eIF5B. Biochemical evidence that the C2879 mutation impairs 60S subunit joining to the 40S IC is provided. These results suggest that eIF5B functionally cooperates with the PTC to ensure stable and high-fidelity formation of an 80S IC on a start codon. Based on this study, we discuss the effect of ribosomal mutations on translation profiles in eukaryotes.
MATERIALS AND METHODS
Plasmids and yeast strains.
Plasmids and yeast strains carrying rdn mutant 25S rRNA as the sole source are listed in Table 1. KAY171 (MATa ade2-1 his3-11 leu2-3,112 ura3-1 trp1-1 can1-100 rdnΔΔ::HIS3 his4-306 [TTG] p[2μ RDN URA3]) (Nemoto et al., submitted) was used to generate yeast rdn his4-306 strains by plasmid shuffling (Table 1). Other plasmids used include pC565-3 (high copy number [hc] FUN12 URA3), pD401-4 (hc FUN12 TRP1) (24), and the GCN4-lacZ reporter plasmid p180 and its modified version, pM226 (25, 26).
Table 1.
S. cerevisiae strains carrying 25S rdn mutations
| rdn allele | 25S rRNA base change | Escherichia coli nucleotide | Mutated domain | Plasmid introduced | Strain(s) |
|
|---|---|---|---|---|---|---|
| GCN2+a | his4-306b | |||||
| WT | WT | pNOY373 | NOY908, KAY761 | KAY165 | ||
| rdn40 | C2879U | C2510 | V (PTC, A loop) | pNOY373-40 | KAY968c | KAY427 |
| rdn47 | G1735A | C1507 | III | pNOY373-47 | KAY971c | KAY434 |
| rdn49 | A1446G | A1265 | II | pNOY373-49 | KAY964c | KAY436 |
| rdn56 | A1435U | A1254 | II | pNOY373-56 | KAY777 | KAY437 |
| rdn67b | U2408C | C2066 | V (PTC, P loop) | pNOY373-67b | KAY1016 | KAY460d |
| rdn69 | G651A | G577 | II | pNOY373-69 | KAY963c | KAY461 |
Derivatives of NOY908 (MATa ade2-1 his3-11 leu2-3,112 ura3-1 trp1-1 can1-100 rdnΔΔ::HIS3 pNOY373 [2μ RDN LEU2]) carrying the indicated mutations. The strains in this column were constructed previously (Nemoto et al., submitted).
Derivatives of KAY165 (MATa ade2-1 his3-11 leu2-3,112 ura3-1 trp1-1 can1-100 rdnΔΔ::HIS3 his4-306 [TTG] pNOY373 [2μ RDN LEU2]) carrying the indicated mutations.
pNOY373 derivatives used to generate these strains were prepared by reintroducing each mutation by site-directed mutagenesis (Nemoto et al., submitted).
The 25S rdn allele of KAY460 contains G1377A, a silent mutation, besides U2408C, since this strain was produced from transformants of KAY171 (4) carrying pNOY373-67, a plasmid isolated in the original conditionally lethal mutant screening (Nemoto et al., submitted).
80S complex formation assay.
Native gel assays monitoring 80S complex formation were performed as described previously (27). The final concentrations of the components in the 80S complex formation assay mixture were as follows: 800 nM eIF2, 200 μM GTP · Mg2+ (for ternary complex [TC] formation), 1.0 nM [35S]Met-tRNAiMet, 800 nM eIF1, 400 nM eIF1A, 1.0 μM mRNA, 400 nM 40S and 60S subunits, 800 nM eIF5, 500 nM (or 2.0 μM in the eIF5B excess condition) eIF5B, and 2 mM GTP · Mg2+ (or GDPNP · Mg2+) for chase. The limiting [35S]Met-tRNAiMet concentrations were used to achieve pseudo-first-order conditions and to ensure single-round turnover in the reactions. To calculate rate constants for 80S complex formation, fractions of [35S]Met-tRNAiMet in the 80S complex were fit using the program KaleidaGraph (Synergy) to the following equation: fraction of [35S]Met-tRNAiMet in 80S = A(1 − exp−kobst), where A is the amplitude and kobs is rate constant.
Other biochemical and molecular biology methods.
Standard molecular biology methods, including the β-galactosidase assay, were used throughout (26). To determine yeast titers on different agar plates, an overnight culture was diluted to an A600 of 0.15, and 5-μl portions of this mixture and 10-fold serial dilutions were spotted, incubated under specified conditions, and photographed (26).
RESULTS
25S rRNA mutations used in this study.
Taking advantage of the plasmid shuffling system for the yeast rDNA repeat locus (RDN) developed by M. Nomura (28), we previously isolated and characterized seven temperature-sensitive (Ts−) and slow-growth (Slg−) mutations altering yeast 25S rRNA (Nemoto et al., submitted). We chose six well-behaved mutants for this study. Figure 1 shows the locations of these mutation sites in the X-ray structure of the yeast 60S subunit (29). Three mutations, G651A (rdn69), A1435U (rdn56), and A1446G (rdn49), change a base involved in base triples that are crucial for structural integrity (slate spheres in Fig. 1). They are located in proximity to each other, disrupting a structural core in domain II located behind the PTC. Three other mutations change bases near the ribosome surface: C2879U (rdn40) and U2408C (rdn67b), which alter the A loop and P loop, respectively, of the PTC, and G1735A (rdn47) (green spheres in Fig. 1). The A loop and P loop contain G residues, which base pair with the CCA ends of aminoacyl and peptidyl tRNAs, respectively, during peptide elongation (30). G1735A (rdn47) maps near Rpl19e, making up eB12, a Eukarya-specific bridge to the 40S subunit, or nucleotides making up B4 (shown in red in Fig. 1). Thus, this mutation may directly impair subunit interaction. Polysome profile studies showed that all the mutants produced halfmers due to reduced 60S levels. The two PTC mutations, C2879U and U2408C, decreased polysome-to-monosome ratio, suggesting that they are defective in translation initiation, likely at the subunit joining step (Nemoto et al., submitted). In this study, we sought evidence that the six mutations show defects in start codon selection.
Fig 1.
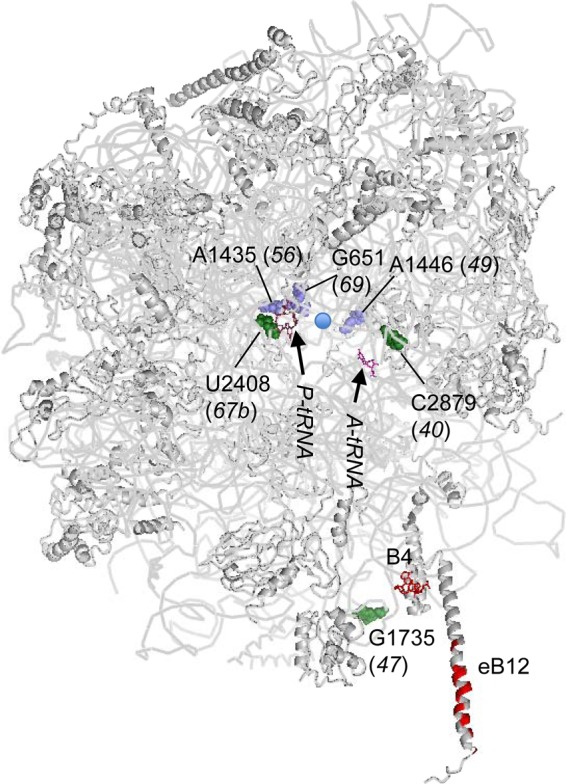
Locations of 25S rRNA point mutations in the yeast 60S subunit structure. The yeast 60S subunit (Protein Data Bank [PDB] 3O58) is shown with RNA and protein backbones in gray and viewed from the 40S interface side. 25S rRNA residues altered by rdn mutations (rdn allele numbers are in parentheses) are highlighted as green or slate spheres. Nucleotides in gray are located near the surface, whereas those in slate are located deep inside the ribosome structure. The blue circle indicates the location of the PTC (A2820 and C2821). Functional rRNA bases relevant to this study are shown as colored sticks; G2619 and G2620 are in raspberry (P loop), G2922 is in magenta (A loop), and A846 and A847 are in red (a 40S interface termed B4). Amino acids of Rpl19e involved in eB12, another 40S interface, are also in red. PYMOL was used to draw this figure.
C2879U and U2408C, altering the PTC A loop and P loop, respectively, and G1735A, mapping near a subunit bridge, facilitate skipping of start codons.
If the mutant 60S subunits disrupt the 40S IC, 80S IC, or A-site-loaded 80S elongation complex, following the subunit joining, we anticipated that an mRNA-40S (open) complex is produced at a higher frequency in the mutant cells, allowing bypass of the original start codons (see the introduction). To address this point, we examined specifically whether the rdn mutations display phenotypes related to altered start codon selection. We focused on two phenotypes, Gcn and Ssu. Yeast can overcome growth inhibition by 3-aminotrizole (3AT), an inhibitor of His3p enzyme, by activating translation of the mRNA encoding the transcription factor Gcn4p. This translational regulation is dependent on upstream open reading frames (uORFs) present in the GCN4 mRNA leader: after translating uORF1, ribosomes resume scanning and translate one of the subsequent uORFs (uORF2 to -4), leading to the ribosome disengaging from the mRNA under normal, noninducing conditions. However, under starvation conditions (such as one induced by 3AT), Gcn2p phosphorylates eIF2 and inhibits its GTP binding, thereby decreasing eIF2/GTP/Met-tRNAiMet ternary complex (TC) levels. Accordingly, ribosomes that have translated uORF1 bypass uORF2 to -4 without binding the TC and instead reinitiate translation at the GCN4 start codon, eliciting a general control response governed by Gcn4p (Fig. 2A [top] shows the GCN4 mRNA leader structure).
Fig 2.

C2879U (rdn40), G1735A (rdn47), and U2408C (rdn67b) allow bypass of AUG codons. (A) Gcn− phenotype test. Fixed amounts (5 μl of culture diluted to an A600 of 0.15) of NOY908 (WT), KAY968 (rdn40), KAY971 (rdn47), KAY964 (rdn49), KAY777 (rdn56), KAY1016 (rdn67b), and KAY963 (rdn69) (Table 1) and their 10-fold serial dilutions were spotted onto synthetic complete (SC) medium lacking histidine but supplemented with 40 mM leucine and with (panel 2) or without (panel 1) 50 mM 3AT and incubated for 3 days at 36°C. The schematic at the top depicts the structure of the GCN4-lacZ leader region. (B) Transformants of designated strains in panel A carrying p180 (GCN4-lacZ) were grown in the absence (−) or presence (+) of 30 mM 3AT for 6 h following preculturing for 2 h and assayed for β-galactosidase (26) at 36°C (panel 1) or 34°C (panel 2). The graphs show the averages using 2 or more independent transformants, with bars indicating standard errors (SE) (n = 4 to 10). (C) Leaky scanning of the uORF1 start codon. The graph presents β-galactosidase activities expressed from pM226 in transformants of rdn strains listed to the left in panel A that were grown at 30°C and subjected to the assay, as described previously (26). The schematic at the top depicts the structure of the modified GCN4-lacZ leader carried on the plasmid used. Bars indicate SE from 4 reactions using two independent transformants. Each of the rdn mutations tested increased expression from pM226 compared to the wild type (P < 0.002).
Translation initiation defects that cause leaky scanning of uORF1 and/or the GCN4 start codon prevent translational induction of Gcn4p, resulting in a general control nonderepressible (Gcn−) phenotype. When spotted on 3AT medium agar plates and incubated at a restrictive temperature (36°C), the six 25S rdn mutants grew worse than the WT RDN control (Fig. 2A). Interestingly, C2879U (rdn40), G1735A (rdn47), and U2408C (rdn67b), changing bases near the 60S subunit surface (raspberry spheres in Fig. 1A), and A1435U (rdn56), disrupting an internal core behind the PTC, showed the strongest 3AT sensitivity. Reporter assays with a GCN4-lacZ plasmid (p180) (31) confirmed that the four mutants are defective in inducing GCN4 in response to 3AT treatment at 36°C (Fig. 2B, panel 1). The assays at a semipermissive temperature (34°C) indicated a stronger defect in GCN4 induction in the two PTC mutants, carrying C2879U and U2408C (Fig. 2B, panel 2). Interestingly, the uninduced GCN4-lacZ level was 2-fold higher than the wild-type level in the C2879U mutant (Fig. 2B, panel 2, columns 1 and 3) (general control derepressed [Gcd−]), as observed previously with a mutant deleted for the large ribosomal protein (RP) Rpl16p (32). This Gcd− phenotype happens when the inhibitory uORF2 to -4 in the GCN4 leader are bypassed. Thus, C2879U appears to increase the frequency of ribosome bypass of uORF2 to -4, besides increasing the bypass of the uORF1 (and/or GCN4) start codon causing the Gcn− phenotype. In agreement with the strong Gcn− phenotypes, the three surface mutations C2879U, U2408C, and G1735A (Fig. 1A, raspberry) displayed the strongest leaky scanning of the uORF1 start codon (Fig. 2C): expression of GCN4 from a reporter in which uORF1 was elongated such that it overlapped the GCN4 ORF in an alternate reading frame (Fig. 2C [top] shows the modified GCN4 leader structure) was enhanced in these mutants, consistent with the notion that these mutants promoted leaky scanning of the uORF1 start codon. Since the 25S rdn mutations displayed similar 60S/40S ratios (Nemoto et al., submitted) and yet showed distinct levels of leaky scanning (Fig. 2C), the stronger leaky scanning by the three ribosome surface mutations results from functional defects and not merely from reduced 60S levels.
Stringent AUG selection depends on translation factor-regulated transition of the PIC to the closed conformation of the 40S subunit, which then becomes the substrate for 60S subunit joining. Mutational disruption of the closed state increases the accuracy of initiation by preventing translation from a noncanonical start codon UUG (7). A 25S rRNA mutation can increase the accuracy of initiation by disrupting the 80S IC or switching the 40S IC back to the open conformation during an improper subunit joining at a non-AUG codon. To test whether the rdn mutations increase the accuracy of translation initiation, we generated yeast 25S rdn strains bearing his4-306, which alters the HIS4 start codon to UUG. A wild-type RDN strain bearing his-306 is His− due to the initiation codon mutation of the histidine synthesis enzyme (Fig. 3, row 1). The introduction of a dominant allele, SUI3-2 (eIF2β-S254Y), conferred a His+ phenotype, consistent with the mutation enhancing translation of His4p from the UUG codon (a Sui− [suppressor of initiation codon mutation] phenotype) (Fig. 3, row 2). We found that C2879U (rdn40), G1735A (rdn47), and U2408C (rdn67) mutations suppressed the SUI3-2-induced His+ phenotype (Fig. 3, rows 4, 6, and 12), suggesting that these mutations increase the accuracy of initiation and thereby inhibit the relaxed initiation from the UUG codon (suppressor of Sui [Ssu−] phenotype). These data are congruent with the results in Fig. 2 and together suggest that the three rdn mutations allow ribosomes to strongly bypass start codons, probably by dissociating the 40S initiation complex during subunit joining.
Fig 3.

C2879U (rdn40), G1735A (rdn47), and U2408C (rdn67b) increase the initiation accuracy by bypassing UUG codons (Ssu− phenotype test). Fixed amounts of transformants of KAY165 (his4-306 RDN+) and its 25S rdn derivatives (Table 1) carrying YDpU-SUI3 (SUI3) or YDpU-SUI3-2 (SUI3-2) (44) and their 10-fold serial dilutions were spotted onto synthetic complete (SC) medium lacking uracil (+His) or lacking uracil and histidine (−His) and incubated for 4 or 6 days, respectively. The column to the right summarizes the Ssu phenotypes of the rdn mutants.
eIF5B interaction with the 60S subunit PTC A loop is important for normal control of accurate initiation in vivo.
Since the docking studies of the bacterial 30S IC suggest that domain IV of IF2 (eIF5B orthologue) touches the A loop structure on the 50S subunit (15), it was conceivable that the phenotypes observed with C2879U (rdn40) result at least in part from defective interaction with eIF5B. If this is the case, the C2879U phenotypes might be suppressed by eIF5B overexpression (mass action effect). We therefore examined whether expression of eIF5B from a high-copy-number (hc) plasmid could suppress the phenotypes in C2879U mutants. As expected, we found that the slow-growth and 3AT-sensitive phenotypes in the C2879U mutant were partially suppressed by eIF5B overexpression (Fig. 4A). hc eIF5B did not suppress similar phenotypes caused by other 25S rdn mutants (data not shown), indicating that the genetic interaction with eIF5B is specific to C2879U, altering the 25S rRNA A loop.
Fig 4.
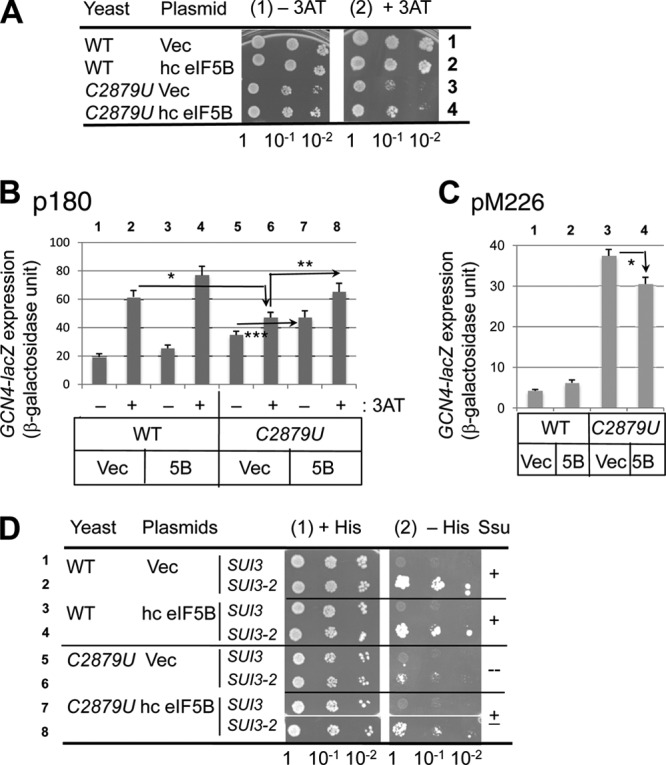
C2879U (rdn40) is suppressed by overexpression of eIF5B. (A) Effect on Gcn− phenotype. Cultures of transformants of NOY908 (RDN+; WT) and KAY968 (C2879U) bearing pC565-3 (hc eIF5B) (24) or a vector control (Vec) and their 10-fold serial dilutions were spotted onto SC medium plates lacking histidine and uracil with (panel 2) or without (panel 1) 50 mM 3AT. The plates were incubated for 3 (panel 1) or 7 (panel 2) days at 34°C. (B and C) Effect on GCN4 expression and leaky scanning of the uORF1 start codon. Transformants of NOY908 (WT) or KAY968 (C2879U) carrying p180 (B) or pM226 (C) and pD401-4 (hc eIF5B TRP1) (5B) or a vector control (Vec) were assayed for β-galactosidase as for Fig. 2B and C. (B) Transformants were grown at 34°C in SC medium lacking His, Trp, and uracil (SC-His-ura) supplemented (+) or not supplemented (−) with 30 mM 3AT. The β-galactosidase units presented are the averages from 10 reactions using 5 independent transformants. *, P = 0.02; **, P = 0.0003; ***, P = 0.03. (C) Transformants were grown at 30°C in SC-Trp-ura. Shown are the averages from 16 reactions using 4 independent transformants. *, P = 0.04. (D) Effect on Ssu− phenotype. Transformants of KAY165 (WT) and KAY427 (C2879U) carrying pD401-4 (hc eIF5B) or a vector control (Vec) and YDpU-SUI3 (SUI3) or YDpU-SUI3-2 (SUI3-2) were assayed except as follows, and the results are presented as for panel A. We used SC medium lacking uracil and tryptophan (+His) or lacking uracil, tryptophan, and histidine supplemented with 0.3 μM histidine (−His), and the plates were incubated for 4 and 7 days, respectively.
Reporter assays with the GCN4-lacZ plasmid showed that hc eIF5B significantly increased the lowered GCN4 expression in the C2879U mutant under the starvation conditions (P < 0.02; compare columns 2, 6 and 8 in Fig. 4B), in support of the idea that hc eIF5B partially suppressed the 3AT-sensitive phenotype through GCN4 regulation. Since hc eIF5B decreased leaky scanning of the uORF1 start codon (Fig. 4C) (P = 0.04), the increase in GCN4 translation is partly due to the suppression of leaky scanning of the uORF1 start codon. Second, hc eIF5B also increased GCN4 expression in the C2879U mutant, even under the nonstarvation conditions (Fig. 4B, column 5 versus 7). Thus, higher eIF5B abundance can restore defective GCN4 initiation regardless of TC levels (regulated by eIF2 phosphorylation).
Next, we examined the effect of hc eIF5B on the potential Ssu− phenotype in the C2879U mutant. As shown in Fig. 4D, hc eIF5B partially suppressed the Ssu− phenotype observed in the C2879U (rdn40) strain expressing SUI3-2 (panel 2, compare rows 6 and 8), even though the growth of the two transformants compared here is identical in the presence of histidine (panel 1). Thus, both the Gcn− and Ssu− phenotypes in C2879U mutants are suppressed by hc eIF5B, in support of functional interaction between eIF5B and the 25S rRNA A loop.
C2879, located near the PTC A loop, is involved in eIF5B-dependent 60S subunit joining.
To delineate the mechanism whereby the C2879U mutation impairs 60S subunit joining in vitro, we purified 60S subunits from the mutant strain as well as from the isogenic wild-type control. Primer extension analysis of the purified mutant ribosomes confirmed that >90% of the ribosomes from the C2879U mutant contained the expected mutation (Fig. 5). The activity of these purified ribosomes was then analyzed in a 60S subunit joining assay. A simplified 48S complex was formed using purified 40S subunits, mRNA, eIF1, eIF1A, and a preformed eIF2/GTP/[35S]Met-tRNAi ternary complex. The 60S subunit was then added together with eIF5, eIF5B, and GTP. Formation of 80S ICs was monitored by native gel electrophoresis (Fig. 6A and B). In the absence of eIF5B, only 48S complexes were observed due to impairment of the subunit joining step (Fig. 6B, panel a), confirming activity of the eIF5B. The 80S IC formation was also confirmed to depend on GTP hydrolysis, because the addition of GDPNP prevented release of eIF2, resulting in stable 48S PICs and inhibition of 80S ICs (Fig. 6C).
Fig 5.
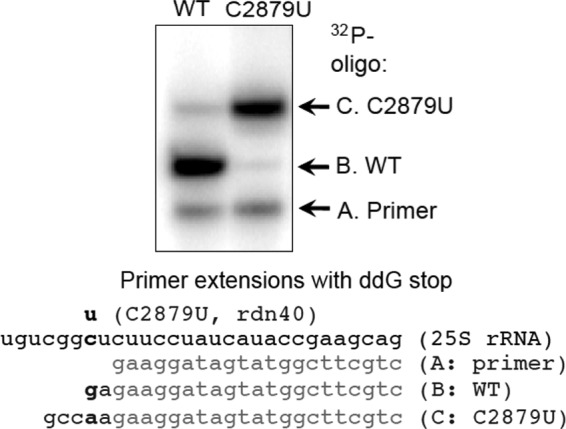
Primer extension analysis of purified 40S subunits. The homogeneity of the C2879U mutant 40S subunit was confirmed by primer extension using 25S rRNA isolated from the purified sample (right lane of the gel). Wild-type (WT) 40S subunit was used as a control (left lane). The mutation site for C2879U, the primer, and expected cDNAs obtained from primer extensions are shown below the gel.
Fig 6.

The C2879U (rdn40) mutation in 25S rRNA impairs 80S complex formation. (A) Scheme of 80S complex formation assay. A final concentration of 1 nM [35S]Met-tRNAiMet was used as a limiting component for the assay. (B) 80S complex formation assay. 48S complexes were formed using a limited amount of 35S-labeled Met-tRNAiMet, and, with GTP chase, subunit joining was performed according to the scheme described in panel A. The reactions were quenched at the indicated time by loading onto a 4% acrylamide native gel, and subunit joining was monitored by electrophoresis. The results of experiments done in the absence of eIF5B or in the presence of 0.5 or 2 μM eIF5B are presented. *, smearing bands below the 80S complex. (C) 80S complex formation assay in the presence of two different concentrations of eIF5B with GDPNP chase, as shown in the scheme in panel A. (D) Kinetics of subunit joining with 0.5 μM eIF5B. Amounts of [35S]Met-tRNAiMet that were free or bound to 80S complexes at the indicated times were quantified, and the fraction of [35S]Met-tRNAiMet in the 80S complexes was calculated. The fitting curve shown here is from one of two independent experiments.
As expected, the C2879U mutation impaired 80S IC formation with 0.5 μM eIF5B (Fig. 6B panel b). As quantified in Fig. 6D, C2879U decreased the rate of 80S complex formation to 40% compared to that of the wild type (0.29 ± 0.06 min−1 for the C2879U mutant and 0.73 ± 0.28 min−1 for the WT). More importantly, this defect was not observed when eIF5B was added in excess at 2 μM (Fig. 6B, panel c), analogous to its in vivo defects suppressed by hc eIF5B (Fig. 4) (note that eIF5B is substoichiometric to eIF1, eIF1A, and eIF2 in vivo [33]). These results support the idea that the C2879U mutation in 25S rRNA impairs 60S subunit joining due to defective interaction with eIF5B in vivo and in vitro.
One interesting observation was the smearing of the 80S complex bands in the reaction mixtures containing the C2879U 60S subunits (box with asterisk in Fig. 6B), which was not evident for reactions performed with wild-type 60S subunits. This smearing might reflect unstable binding of initiator Met-tRNAiMet to the 80S complex, consistent with the location of C2879 near the PTC. Alternatively, the smearing might be due to unstable subunit joining due to impaired interaction of eIF5B and the mutant 60S subunit. In any case, the results support the idea that C2879 contributes to anchoring Met-tRNAiMet in the 80S IC, depending on eIF5B either directly or indirectly. Together, the results presented in Fig. 6 suggest that eIF5B functionally cooperates with the PTC during subunit joining to ensure tight 80S IC formation and stringency in start codon selection.
DISCUSSION
25S rRNA mutagenesis: implication for 60S subunit structure and function in initiation.
In this study, we found that all six rdn mutants tested significantly increased leaky scanning of the uORF1 start codon on the GCN4 mRNA (Fig. 2C) (P < 0.002) and obtained genetic evidence that C2879U (rdn40), U2408C (rdn67b), and G1735A (rdn47), altering a base near the PTC or 40S subunit-binding surfaces, bypass a non-AUG start codon, UUG (Fig. 3). The 60S subunit joins the initiation complex at the last step of the initiation pathway. Thus, a simple explanation for the mechanism of the observed initiation defects caused by the 25S rRNA mutations is that each mutation impairs an interface with the 40S IC and thereby decreases the efficiency of 60S subunit joining, as demonstrated for the C2879U (rdn40) mutation altering a base near the PTC A loop (Fig. 6).
All six rdn mutations tested increased leaky scanning of the uORF1 start codon (Fig. 2C), but the Gcn− phenotype of only C2879U (rdn40) was suppressed by hc eIF5B (Fig. 4A). This specific effect suggests that eIF5B might form an interface to a limited part of the 60S subunit, including the PTC near the A loop. This is in agreement with the current model of the 80S IC structure, in which the PTC P site interacts with Met-tRNAiMet (34) and numerous intersubunit bridges form directly between the 60S and 40S subunits (29). U2408C (rdn67b) and G1735A (rdn47), altering a base near the P loop and eB12, respectively, showed a strong bypass of start codons (Fig. 2 and 3), similar to the case for C2879U. These former mutations probably weaken the interaction with Met-tRNAiMet (at the P site) and the 40S subunit, respectively. We propose that the failure of proper alignment between the 40S IC (with eIF5B) and the 60S subunit destabilizes the former and produces at a certain frequency mRNA attached to the 40S subunit in an open conformation, thereby allowing reinitiation at downstream start codons (see below) (Fig. 7).
Fig 7.
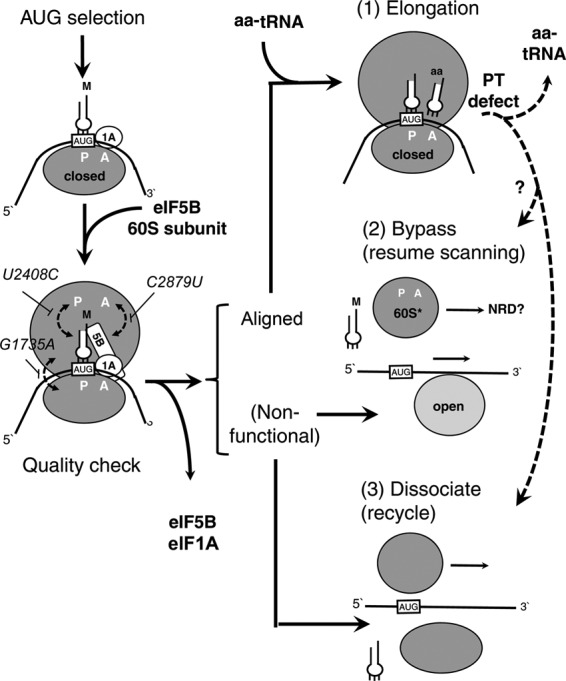
Effect of the 25S rRNA mutations on translation initiation and quality control coupled to ribosome bypass and recycling. The left column describes translation initiation steps until the subunit joining. The gray oval represents the 40S subunit with the AUG codon (box AUG) of the mRNA (line), loaded on its P site (P) and base-paired to the anticodon of Met (M)-tRNAiMet (a plug). eIF1A (circle 1A) is bound to the 40S A site (indicated by A), while eIF5B (rectangle 5B) forms a bridge between the 40S subunit and Met-tRNAiMet. When a wild-type 60S subunit (a large gray circle with A and P denoting the A site and P site, respectively) joins, eIF5B and eIF1A are released to produce the 80S IC, which binds aa-tRNA and undergoes elongation (right column, step 1). However, when a mutant 60S subunit (a gray circle labeled 60S*) joins, the 40S IC components (eIF5B, Met-tRNAiMet, or 40S subunit) sense the misalignment (dashed arrows for each rdn mutation indicated) and reject the mutant 60S subunit (right column, steps 2 and 3). The 40S subunit may remain bound to the mRNA for ribosome bypass and reinitiation (step 2) or become dissociated from it for recycling (step 3). In this way, the 25S rdn mutations increase the frequency of reinitiation or recycling, bypassing AUG or UUG codons as observed in this study. Mutant 60S subunits released from misaligned complexes may be targeted for degradation (? in steps 2 and 3). Alternatively, they are degraded after stalling the elongation complex (dashed arrows).
How does the C2879U mutation affect the interaction with eIF5B? C2879 is located in helix 90, which lies directly behind the helix 92 A loop that interacts with the CCA end of the A-site tRNA (29, 35). When a 50S subunit is docked onto the bacterial 30S IC, domain IV of IF2 clashes with helix 92 on the 50S subunit (15). Thus, C2879, or an A loop structure disrupted by C2879U, may physically interact with eIF5B domain IV. In this model, part of eIF5B would occupy the A loop, blocking it from binding the methionyl moiety of Met-tRNAiMet. If this is the case, eIF5B may play a role in guiding Met-tRNAiMet binding to the PTC, besides supporting the P/I configuration of the tRNA.
Subunit joining is the checkpoint for initiation accuracy and ribosome quality control.
Based on the finding that hc eIF5B suppressed the yeast phenotypes suggestive of ribosome bypass of start codons (Fig. 4), we propose that eIF5B-dependent 60S subunit joining is a crucial checkpoint for the decision to initiate, bypass, or dissociate (Fig. 7). In wild-type cells, most of the 60S subunits are normal. Therefore, subunit joining actively produces 80S ICs ready for elongation (Fig. 7, step 1). In the 25S rdn mutant cells, a significant proportion (up to ∼10%) of the 60S subunits are defective. This results in misalignment between the mutant 60S subunit and the 40S IC, thereby disrupting the entire 80S preinitiation complex. After the release from 80S ICs, the 40S IC is either completely dissociated to recycle Met-tRNAiMet, mRNA, and the 40S subunit (Fig. 7, step 3) or partially dissociated, such that the 40S subunit remains bound to the mRNA in an open conformation (step 2). The latter pathway allows bypass of the original start codon and reinitiation at a downstream start codon, as observed in this study.
Biogenesis of ribosomal 60S subunits is coupled to the cellular surveillance pathways that trigger nuclear or cytoplasmic degradation of the mutant rRNA (18–23). At least a part of the nonfunctional 60S subunits released as the result of the misalignment may be targeted for proteasome-dependent degradation by the Mms1p-dependent NRD, as its substrate is suggested to be cytoplasmic 80S complexes (20) (Fig. 7, steps 2 and 3). It is entirely possible, however, that the misalignment at the initiation stage dissociates 80S complexes without targeting the mutant 60S subunits for degradation. The mutant 80S complexes can be monitored at the next step of elongation after aa-tRNA is delivered to the A site (Fig. 7, step 1). The defect in peptidyl transferase activity due to 25S rRNA mutations may stall the ribosome on the start codon or a subsequent codon, allowing a surveillance system to pull the mutant 60S subunit off for degradation (dashed arrows in Fig. 7) (21).
Effect of ribosomal mutations on mRNA translation profile in eukaryotes.
25S rRNA mutations C2879U, U2408C, and G1735A allowed the stalled PIC to skip both AUG and UUG codons (Fig. 2 and 3). What is the effect of these mutations on the overall accuracy of translation initiation? The AUG codon of uORF1 that was skipped by these mutations is in the good context AAAAUG (the good context in yeast is AA[A/G]AUG [36]). However, the frequency of 40S IC skipping versus initiating at this codon is only <10%, since the fully induced level of GCN4-lacZ expression is ∼500 to 1,000 units (the value from pM199 with only an intact uORF1 preceding GCN4 ORF). In contrast, a near elimination of the SUI3-2-induced His+ phenotype by these mutations (Fig. 3) suggests that the majority of the UUG codons are skipped. Thus, we believe that the 25S rRNA mutations generally increase the accuracy of initiation at the expense of missing some initiation at strong start codons. Likewise, 18S rRNA mutations A1193U, altering the P site, and G875A, altering a core of the eIF1- and eIF2-binding platform, allowed the IC to similarly skip both AUG and UUG codons (4). Thus, it could be proposed that the general consequence of rRNA mutations is to increase the accuracy of initiation by destabilizing the closed state of the PIC stalling on the start codon. (Theoretically, the effect of 18S rRNA mutations depends on the balance between the effects on the open versus the closed conformation. See reference 37 for an example of 18S rRNA mutations that appear to be selected [by Gcd− screening] for destabilizing the open PIC more strongly.)
Ribosome profiling of mouse embryonic stem cells showed that translation of the 5′ untranslated region (UTR) involving non-AUG codons is upregulated in these cells and suppressed when differentiation is induced (38). Thus, ribosomal mutations are expected to suppress 5′ UTR translation by increasing initiation accuracy, leading to a trend in favor of cell differentiation. Can this model explain the disease expression of “ribosomopathies”?
Diamond-Blackfan anemia and 5q− syndrome, founding members of ribosomopathies, arise by various large or small ribosomal protein (RP) mutations and display complex symptoms, including macrocytic anemia and congenital disorders, while other ribosomopathies with related symptoms arise due indirectly to defective ribosome biogenesis (3). Ribosomopathies are also associated with an increased risk of cancer (3), and in zebrafish, various large and small RP genes were isolated as sites of insertional mutations causing cancer (39). Similar to the case for yeast rRNA mutations, the RP mutations lead to a decrease in the corresponding ribosomal subunits both in HeLa cells (40) and in zebrafish (39). The loss of RP is expected to destabilize ribosome conformations, thereby compromising its function at least partially. Thus, the molecular basis for altered ribosome function would be similar, although the effect of heterozygous disease-causing mutations might be smaller than that of the ribosome mutations characterized in haploid yeast.
Interestingly, the ribosomopathies are often associated with the MDM2-regulated activation of the tumor suppressor p53, causing cell cycle arrest and apoptosis and thereby leading to the disease states (3). Furthermore, all the zebrafish tumors that arose from RP insertions lost p53 protein without losing p53 mRNA (41), indicating that the elimination of the antiproliferation effect by p53 with translational control is required for tumor formation. If the ribosomal defects increase the initiation accuracy and suppress 5′ UTR translation that may be required for cell proliferation, this effect would enhance the antiproliferation effects caused by p53 activation, in favor of disease development. In addition, the p53 mRNA contains an ∼200-base-long 5′ UTR that binds Rpl26 for its translational activation in response to DNA damage (42, 43). Thus, it would be intriguing to learn whether the RP mutations silence 5′ UTR translation for p53 mRNA and thereby contribute to translational control of p53, and if so, reveal the mechanism by which tumor-initiating stimuli inhibit this translational activation.
ACKNOWLEDGMENTS
We are greatly indebted to Tom Dever, Ranjit Singh, and Makoto Kitabatake for discussions and comments on the manuscript. We thank Angela Carpenter for technical assistance.
This work was supported by NIH R01 grant GM64781 and its ARRA supplement (K.A.), a Kansas COBRE PSF pilot grant (K.A.), an Innovative Award from KSU Terry Johnson Cancer Center (K.A.), an internal grant from the KSU Division of Biology (K.A.), and grants from NCRR (5P20RR016475) and NIGMS (8P20GM103418) (S.J.B.) and, in part, by the Intramural Research Program of the National Institutes of Health (B.-S.S. and Thomas E. Dever).
Footnotes
Published ahead of print 8 July 2013
REFERENCES
- 1.Jackson RJ, Hellen CUT, Pestova TV. 2010. The mechanism of eukaryotic translation initiation and principles of its regulation. Nat. Rev. Mol. Cell Biol. 10:113–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sonenberg N, Hinnebusch AG. 2009. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell 136:731–745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Narla A, Ebert BL. 2010. Ribosomopathies: human disorders of ribosome dysfunction. Blood 115:3196–3205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nemoto N, Udagawa T, Singh CR, Wang S, Thorson E, Winter ZA, Ohira T, Brown SJ, Asano K. 2010. Yeast 18S rRNA is directly involved in the ribosomal response to stringent AUG selection during translation initiation. J. Biol. Chem. 285:32200–32212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Algire MA, Maag D, Lorsch JR. 2005. Pi release from eIF2, not GTP hydrolysis, is the step controlled by start-site selection during eukaryotic translation initiation. Mol. Cell 20:251–262 [DOI] [PubMed] [Google Scholar]
- 6.Asano K, Clayton J, Shalev A, Hinnebusch AG. 2000. A multifactor complex of eukaryotic initiation factors eIF1, eIF2, eIF3, eIF5, and initiator tRNAMet is an important translation initiation intermediate in vivo. Genes Dev. 14:2534–2546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Asano K, Sachs MS. 2007. Translation factor control of ribosome conformation during start codon selection. Genes Dev. 21:1280–1287 [DOI] [PubMed] [Google Scholar]
- 8.Lorsch JR, Dever TE. 2010. Molecular view of 43S complex formation and start site selecion in eukaryotic translation initiation. J. Biol. Chem. 285:21203–21207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cheung Y-N, Maag D, Mitchell SF, Fekete CA, Algire MA, Takacs JE, Shirokikh N, Pestova T, Lorsch JR, Hinnebusch A. 2007. Dissociation of eIF1 from the 40S ribosomal subunit is a key step in start codon selection in vivo. Genes Dev. 21:1217–1230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luna RE, Arthanari H, Hiraishi H, Nanda J, Martin-Marcos P, Markus M, Arabayov B, Milbradt A, Hyperts S, Luna LE, Reibarkh M, Miles D, Farmy A, Seo H-C, Marintchev A, Hinnebusch AG, Lorsch JR, Asano K, Wagner G. 2012. The C-terminal domain of eukaryotic initiation factor 5 promotes start codon recognition by its dynamic interplay with eIF1 and eIF2β Cell Rep. 1:689–702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Singh CR, Watanabe R, Chowdhury D, Hiraishi H, Murai MJ, Yamamoto Y, Miles D, Ikeda Y, Asano M, Asano K. 2012. Sequential eIF5 binding to the charged disordered segments of eIF4G and eIF2β stabilizes the 48S pre-initiation complex and promotes its shift to the initiation mode. Mol. Cell. Biol. 32:3978–3989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rabl J, Leibundgut M, Ataide SF, Haag A, Ban N. 2011. Crystal structure of the eukaryotic 40S ribosomal subunit in complex with initiaiton factor 1. Science 331:730–736 [DOI] [PubMed] [Google Scholar]
- 13.Shin B-S, Kim J-R, Walker SE, Dong J, Lorsch JR, Dever TE. 2011. Initiation factor eIF2gamma promotes eIF2-GTP-tRNAiMet ternary complex binding to the 40S ribosome. Nat. Struct. Mol. Biol. 18:1227–1234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shin BS, Maag D, Roll-Mecak A, Arefin MS, Burley SK, Lorsch JR, Dever TE. 2002. Uncoupling of initiation factor eIF5B/IF2 GTPase and translational activities by mutations that lower ribosome affinity. Cell 111:1015–1025 [DOI] [PubMed] [Google Scholar]
- 15.Simonetti A, Marzi S, Myasnikov AG, Fabbretti A, Yusupov M, Gualerzi CO, Klaholz B. 2008. Structure of the 30S translation initiation complex. Nature 455:416–420 [DOI] [PubMed] [Google Scholar]
- 16.Shin B-S, Kim J-R, Acker MG, Maher KN, Lorsch JR, Dever TE. 2009. rRNA suppressor of a eukaryotic translation initiation factor 5B/initiation factor 2 mutant reveals a binding site for translational GTPases on the small ribosomal subunit. Mol. Cell. Biol. 29:808–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Unbehaun A, Marintchev A, Lomakin IB, Didenko T, Wagner G, Hellen CUT, Pestova TV. 2007. Position of eukaryotic initiation factor eIF5B on the 80S ribosome mapped by directed hydroxyl radical probing. EMBO J. 26:3109–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bussiere C, Hashem Y, Arora S, Frank J, Johnson AW. 2012. Integrity of the P-site is probed during maturation of the 60S ribosomal subunit. J. Cell Biol. 197:747–759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dez C, Houseley J, Tollervey D. 2006. Surveillance of nuclear-restricted pre-ribosomes within a subnucleolar region of Saccharomyces cerevisiae. EMBO J. 25:1534–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fujii K, Kitabatake M, Sakata T, Miyata A, Ohno M. 2009. A role for ubiquitin in the clearance of nonfunctional rRNAs. Genes Dev. 23:963–974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujii K, Kitabatake M, Sakata T, Ohno M. 2012. 40S subunit dissocation and proteasome-dependent RNA degradation in nonfunctional 25S rRNA decay. EMBO J. 31:2579–2589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hage AE, Tollervey D. 2004. A surfeit of factors: why is ribosome assembly so much more complicated in eukaryotes than bacteria? RNA Biol. 1:10–15 [PubMed] [Google Scholar]
- 23.LaRiviere FJ, Cole SE, Ferullo DJ, Moore MJ. 2006. A late-acting quality control process for mature eukaryotic rRNAs. Mol. Cell 24:619–626 [DOI] [PubMed] [Google Scholar]
- 24.Choi SK, Olsen SD, Roll-Mecak A, Martung A, Remo KL, Burley SK, Hinnebusch AG, Dever TE. 2000. Physical and functional interaction between the eukaryotic orthologs of prokaryotic translation initiation factors IF1 and IF2. Mol. Cell. Biol. 20:7183–7191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grant CM, Miller PF, Hinnebusch AG. 1994. Requirements for intercistronic distance and level of eIF-2 activity in reinitiation on GCN4 mRNA varies with the downstream cistron. Mol. Cell. Biol. 14:2616–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee B, Udagawa T, Singh CS, Asano K. 2007. Yeast phenotypic assays on translational control. Methods Enzymol. 429:105–137 [DOI] [PubMed] [Google Scholar]
- 27.Acker MG, Kolitz SE, Mitchell SF, Nanda JS, Lorsch JR. 2007. Reconstitution of yeast translation initiation. Methods Enzymol. 430:111–145 [DOI] [PubMed] [Google Scholar]
- 28.Wai HH, Vu L, Oakes M, Nomura M. 2000. Complete deletion of yeast chromosomal rDNA repeats and integration of a new rDNA repeat: use of rDNA deletion strains for functional analysis of rDNA promoter elements in vivo. Nucleic Acids Res. 28:3524–3534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ben-Shem A, Jenner L, Yusupova G, Yusupov M. 2010. Crystal structure of the eukaryotic ribosome. Science 330:1203–1209 [DOI] [PubMed] [Google Scholar]
- 30.Moore PB, Steitz TA. 2002. The involvement of RNA in ribosome funcion. Nature 418:229–235 [DOI] [PubMed] [Google Scholar]
- 31.Mueller PP, Hinnebusch AG. 1986. Multiple upstream AUG codons mediate translational control of GCN4. Cell 45:201–207 [DOI] [PubMed] [Google Scholar]
- 32.Foiani M, Cigan AM, Paddon CJ, Harashima S, Hinnebusch AG. 1991. GCD2, a translational repressor of the GCN4 gene, has a general function in the initiation of protein synthesis in Saccharomyces cerevisiae. Mol. Cell. Biol. 11:3203–3216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Singh CR, Udagawa T, Lee B, Wassink S, He H, Yamamoto Y, Anderson JT, Pavitt GD, Asano K. 2007. Change in nutritional status modulates the abundance of critical pre-initiation intermediate complexes during translation initiation in vivo. J. Mol. Biol. 370:315–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Allen GS, Frank J. 2007. Structural insights on the translation initiation complex: ghosts of a universal initiation complex. Mol. Microbiol. 63:941–950 [DOI] [PubMed] [Google Scholar]
- 35.Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. 2000. The structural basis of ribosome activity in peptide bond synthesis. Science 289:920–930 [DOI] [PubMed] [Google Scholar]
- 36.Martin-Marcos P, Cheung Y-N, Hinnebusch AG. 2011. Functional elements in initiation factors 1, 1A, and 2β discriminate against poor AUG Context and Non-AUG Start Codons. Mol. Cell. Biol. 31:4814–4831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Dong J, Nanda JS, Rahman H, Pruitt MR, Shin B-S, Wong C-M, Lorsch JR, Hinnebusch AG. 2008. Genetic idenfication of yeast 18S rRNA residues required for efficient recruitment of initiator tRNAMet and AUG selection. Genes Dev. 22:2242–2255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ingolia NT, Lareau LF, Weissman JS. 2011. Ribosome profiling of mouse embryonic stem cells reveals the complexity of mammalian proteomes. Cell 147:789–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Amsterdam A, Sadler KC, Lai K, Farrington S, Bronson RT, Lees JA, Hopkins N. 2004. Many ribosomal protein genes are cancer genes in zebrafish. PLoS Biol. 2:E139. 10.1371/journal.pbio.0020139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Robledo S, Idol R, Crimmins D, Ladenson J, Mason P, Bessler M. 2008. The role of human ribosomal proteins in the maturation of rRNA and ribosome production. RNA 14:1918–1929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.MacInnes AW, Amsterdam A, Whittaker CA, Hopkins N, Lees J. 2008. Loss of p53 synthesis in zebrafish tumors with ribosomal protein gene mutations. Proc. Natl. Acad. Sci. U. S. A. 105:10408–10413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chen J, Kastan MB. 2010. 5′-3′-UTR interactions regulate p53 mRNA translation and provide a target for modulating p53 induction after DNA damage. Genes Dev. 24:2146–2156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Takagi M, Absalon MJ, McLure KG, Kastan MB. 2005. Regulation of p53 translation and induction after DNA damage by ribosomal protein L26 and nucleolin. Cell 123:49–63 [DOI] [PubMed] [Google Scholar]
- 44.Watanabe R, Murai MJ, Singh CR, Fox S, Ii M, Asano K. 2010. The eIF4G HEAT domain promotes translation re-initiation in yeast both dependent on and independent of eIF4A mRNA helicase. J. Biol. Chem. 285:21922–21933 [DOI] [PMC free article] [PubMed] [Google Scholar]