

Abstract
Chromatin state maps were developed to elucidate sex differences in chromatin structure and their impact on sex-differential chromatin accessibility and sex-biased gene expression in mouse liver. Genes in active, inactive, and poised chromatin states exhibited differential responsiveness to ligand-activated nuclear receptors and distinct enrichments for functional gene categories. Sex-biased genes were clustered by chromatin environments and mapped to DNase-hypersensitive sites (DHS) classified by sex bias in chromatin accessibility and enhancer modifications. Results were integrated with genome-wide binding data for five transcription factors implicated in growth hormone-regulated, sex-biased liver gene expression, leading to the following findings. (i) Sex-biased DHS, but not sex-biased genes, are frequently characterized by sex-differential chromatin states, indicating distal regulation. (ii) Trimethylation of histone H3 at K27 (H3K27me3) is a major sex-biased repressive mark at highly female-biased but not at highly male-biased genes. (iii) FOXA factors are associated with sex-dependent chromatin opening at male-biased but not female-biased regulatory sites. (iv) Sex-biased STAT5 binding is enriched at sex-biased DHS marked as active enhancers and preferentially targets sex-biased genes with sex-differences in local chromatin marks. (v) The male-biased repressor BCL6 preferentially targets female-biased genes and regulatory sites in a sex-independent chromatin state. (vi) CUX2, a female-specific repressor of male-biased genes, also activates strongly female-biased genes, in association with loss of H3K27me3 marks. Chromatin states are thus a major determinant of sex-biased chromatin accessibility and gene expression, with FOXA pioneer factors proposed to confer sex-dependent chromatin opening and STAT5, but not BCL6, regulating sex-biased genes by binding to sites in a sex-biased chromatin state.
INTRODUCTION
The epigenetic environment is controlled by a variety of factors, most notably chromatin modifications, which regulate DNA methylation, chromatin accessibility, and transcription factor (TF) binding. Genomic regions active in a given cell type or regulatory state can be identified by the distinct and characteristic patterns of histone methylations, acetylations, and other chromatin modifications associated with transcribed genes, transcription start sites (TSS), and enhancers. Expressed genes can thus be distinguished from silenced genes and from genes that are poised for expression based on their unique epigenetic environment (1, 2). Enhancers exhibit cell-type-specific patterns of local chromatin modifications, which confer cell-type-specific activity and enable cell-type-specific regulation of distal target genes (3). These characteristic patterns of chromatin modifications can, in turn, be used to predict gene expression in multiple cell types (4).
Hypersensitivity to DNase I cleavage is commonly used to assay chromatin accessibility, which is a key feature of cell-type-specific regulation (5, 6). Regulation of chromatin opening is an important regulatory event in the binding of cell-type-specific TFs, which are invariably (>90%) bound at DNase-hypersensitive sites (DHS) (5). Factors regulating cell-type-specific chromatin opening and TF binding include differences in nucleosome occupancy and positioning, chromatin modifications, and other features of the chromatin environment (7–10). Thus, cell-type-specific gene expression involves a complex interplay between TF activators and repressors, chromatin modifiers, and the chromatin environment. Some TFs can bind to closed chromatin and facilitate nucleosome repositioning and/or chromatin opening (e.g., FOXA “pioneer factors”) (11, 12), while other factors preferentially bind open chromatin marked by cell-type- or condition-specific modifications (13–15). Comprehensive approaches, involving the integration of genome-wide chromatin state maps with binding site maps for TFs and other regulatory factors, are therefore needed to unravel the complexity of mammalian transcriptional networks (13, 14, 16).
Sex differences in gene expression are common and result from differences in genotypic sex as well as sex differences in circulating and local hormonal regulators (17). Sex differences in gene expression are best characterized in the liver, where ∼1,000 genes are expressed in a sex-biased manner, as seen in mice, rats, and humans (18–20). These sex differences affect diverse physiological processes, including hepatic steroid, lipid, and drug metabolism (21–23), and contribute to sex differences in cardiovascular disease risk, fatty liver disease, and the development of hepatocellular carcinoma (20, 24–26). Sex differences in liver gene expression are regulated by the temporal pattern of pituitary growth hormone (GH) secretion, which is sex dependent and programmed by androgen and estrogen exposure during the neonatal period (21). In males, GH is released from the pituitary gland in pulses at regular intervals, with little or no plasma GH detectable between GH secretory events, whereas the more frequent pituitary GH release in females gives a more continuous pattern of circulating hormone. Consequently, GH signaling to downstream transcriptional mediators is pulsatile in male liver and is persistent in female liver (27, 28).
Several TFs contribute to the sex-dependent actions of GH in the liver. STAT5, a major effector of the transcriptional actions of GH, is activated by GH-induced tyrosine phosphorylation, which leads to STAT5 dimerization and translocation into the nucleus (29). Liver nuclear STAT5 DNA-binding activity is pulsatile in males but persistent in females, mirroring the temporal plasma GH profiles of each sex (27, 28, 30). STAT5 is essential for sex-biased gene expression in mouse liver, where 75 to 82% of sex-differential expression is lost when Stat5b is inactivated (31). BCL6, a male-biased, GH-regulated transcriptional repressor, competes with STAT5 binding and antagonizes GH-stimulated STAT5 transcriptional activity (30, 32). CUX2 is a female-specific, continuous GH-inducible repressor (33) that downregulates many male-biased genes and activates a subset of female-biased genes in female liver (34). Other liver-expressed TFs, including HNF4A and HNF6, are also important for hepatic sex differences, as indicated by knockout studies and reporter gene analyses and by the enrichment of their motifs at GH-dependent regulatory sites (35–39).
Sex differences in gene expression have been linked to sex differences in GH-regulated chromatin accessibility, as revealed by genome-wide DHS mapping (39). Sex-biased STAT5 binding in mouse liver correlates with sex differences in DHS accessibility and the presence of activating chromatin marks and inversely correlates with the repressive mark trimethylation of histone H3 at K27 (H3K27me3) (30). GH-induced STAT5 DNA binding requires chromatin opening and activating histone modifications, as seen at several early GH response genes in rat liver (40). However, little is known about the chromatin states of sex-biased genes and their DHS regulatory sites and how the overall epigenetic environment might impact binding and regulation by STAT5 and other TFs required for sex-biased liver gene expression.
Here, we characterize chromatin environments on a global scale in both male and female mouse liver based on genome-wide data for four activating chromatin marks, two repressive chromatin marks, and global DHS maps. We identify genes in active, inactive, and poised chromatin states, and we establish the propensity of genes in each state for induction or repression by ligand-activated nuclear receptors, which mediate metabolic responses to myriad steroids, drugs, and environmental chemicals (41–43). Further, we elucidate sex-differential chromatin states at sex-biased genes and sex-biased DHS, and we integrate these chromatin state data with binding site data from chromatin immunoprecipitation with high-throughput sequencing (ChIP-seq) for the GH-regulated TFs STAT5 (30), BCL6 (30), and CUX2 (34) and for the pioneer factors FOXA1 and FOXA2 (11, 12), which impart male bias to hepatocellular carcinoma by facilitating androgen-mediated tumor promotion in males and estrogen-dependent resistance to tumorigenesis in females (44). Our findings reveal the importance of sex-dependent chromatin states in the control of liver gene expression. Strikingly, we find evidence for distinct sets of regulatory mechanisms in each sex. Many female-biased genes are associated with sex-independent chromatin marks and are preferentially repressed by the male-biased repressor BCL6, while a smaller subset, comprised of highly female-biased genes, shows sex differences in proximal chromatin marks and preferential activation by female-enriched STAT5 binding. Furthermore, we identify H3K27me3-based repression as an important mechanism regulating highly female-biased but not male-biased genes, and we identify sex differences in H3K4me1 (methylation of histone H3 at K4) profiles at male-biased but not female-biased DHS that suggest sex-dependent nucleosome repositioning facilitated by FOXA factors, in particular FOXA1. These findings are integrated into a model for GH-regulated transcriptional and epigenetic control of sex-biased gene expression.
MATERIALS AND METHODS
ChIP.
Chromatin isolation from formaldehyde cross-linked liver nuclei prepared from individual 7- to 8-week-old male and female CD-1 mouse livers (Charles River Laboratories, Kingston, NY) obtained using protocols approved by the Boston University Institutional Animal Care and Use Committee, followed by sonication and chromatin immunoprecipitation (ChIP), was carried out by E. Laz of this laboratory as reported previously (30). The following ChIP-validated antibodies were used: H3K27me3 (ab6002; Abcam) (2 μg per 10 μg of sonicated DNA), H3K9me3 (ab8898; Abcam) (0.8 μg per 10 μg of sonicated DNA), H3K4me3 (ab8580; Abcam) (0.8 μg per 10 μg of sonicated DNA), H3K4me1 (ab8895; Abcam) (0.8 μg per 10 μg of sonicated DNA), histone H3 acetylated at K27 (H3K27ac) (ab4729), H3K36me3 (ab9050), and normal rabbit IgG (sc-2027; Santa Cruz) (7.5 to 8 μg per 100 μg of sonicated DNA). ChIP DNA was analyzed by quantitative PCR (qPCR) (30) to interrogate genomic regions selected as positive controls and negative controls for each antibody based on initial ChIP-seq results.
High-throughput sequencing.
Liver genomic DNA isolated by ChIP was prepared for sequencing using a SPRI-TE Nucleic Acid Extractor for size selection (Beckman Coulter Genomics, Danvers, MA) followed by PCR enrichment with bar coding (30). Sample preparation and 35- to 41-nucleotide (nt) single-end read sequencing on an Illumina GAII or Illumina HiSeq2000 instrument, followed by mapping to the mouse genome build mm9 (NCBI 37) using Eland extended (Illumina, Inc.) or Bowtie (45), were carried out at the BioMicro Center at the Massachusetts Institute of Technology (MIT; Cambridge, MA). Raw sequencing reads were obtained for individual livers (n = 3 to 4 biological replicates/sex/mark, except for K9me3 and K36me3 at n = 2 replicates). All reads were trimmed to 35 nt to maintain a constant sequence length between samples. Replicates were evaluated by the percentage of reads in straight peaks (more than five identical reads that do not overlap any other reads and are therefore likely artifactual), by the degree of overlap between peaks detected in each replicate, and by correlation in read count between replicates (see Table S7 and “Analysis of data quality” in the supplemental material). Samples with >3% of the reads in straight peaks generally showed low overlap between biological replicates and were excluded. The final data sets for each chromatin mark ranged from 10.7 million (K9me3) to 52.5 million (K4me1) total reads (see Table S3A in the supplemental material).
Analysis of ChIP-seq data for chromatin modifications.
To identify peaks and regions of chromatin mark enrichment, each data set, after combining data for biological replicates, was analyzed separately for male and female liver, as follows. K4me1, K27ac, and K4me3 form focal (localized) peaks and were analyzed using MACS, version 1.4.1 (46), with default parameters. K27me3, K9me3, and K36me3 were analyzed using SICER, version 1.1 (47), which identifies broad genomic regions (i.e., broad domains or islands). For K27me3, a window size of 400 bp was used, and a gap size of 2,400 was chosen as most appropriate (see Fig. S12 in the supplemental material). The same parameters were used for K9me3. For K36me3, whose peak regions were not as broad (see Table S3A), a window size of 200 bp and a gap size of 800 bp were used. Sex-enriched peaks and regions were identified for each chromatin mark, as follows. For each MACS peak and SICER region, the number of male sequence reads was compared to the number of female sequence reads after normalization by the total number of reads in common peaks—peaks (or regions) that were detected by MACS (or by SICER) in both male and female liver—in order to account for any bias between male and female livers in the percentage of reads in peaks/regions. The log2 male/female ratio (M value) was then calculated, along with a P value based on a Bayesian model (48) as implemented by Shao et al. (49). Each chromatin mark (focal peak or broad genomic region) was classified as male enriched (M > 1 and P < 0.001), female enriched (M < −1 and P < 0.001), or sex independent (−1 < M < 1 or P > 0.001). Table S3 in the supplemental material lists the numbers of peaks and domains identified for each chromatin mark along with their full data sets and genomic coordinates.
Chromatin states in mouse liver.
The six chromatin mark data sets along with liver DHS data reported previously (39) were analyzed using ChromHMM (1) to learn a hidden Markov model and to assign chromatin states across the mouse genome. ChromHMM was run using an IgG control and with default parameters. A single joint model was learned using data from both male and female liver. A 15-state model was previously found to be biologically meaningful and consistently recovered across cell types for the human genome (1). We therefore started with 20 states and used the ChromHMM CompareModels module to compare decreasing numbers of states to the 20-state model. We then calculated, for each of the 20 states, the similarity—i.e., the correlation between emission parameters—to its closest state in smaller models (see Fig. S3B in the supplemental material). A 14-state model was chosen as the point after which any further decrease in the number of states in the model resulted in states from the 20-state model being recovered with decreasing similarity (see Fig. S3B and C and “Chromatin states in mouse liver” in the supplemental material).
Clustering of genes by chromatin mark densities around TSS and TES.
Analysis using high-throughput sequencing of RNA transcripts of total mouse liver (RNA-seq) was carried out using two independent pools of male and two independent pools of female mouse liver RNA (n = 6 livers/pool). Poly(A)-enriched RNA was used to prepare RNA strand-specific libraries (50) for Illumina sequencing at the BioMicro Center at MIT (Cambridge, MA). Forty-nucleotide sequence tags were mapped to the mm9 genome using Bowtie (45) and mapped to Mouse Genome Informatics (MGI) genes (www.informatics.jax.org/) using SeqMonk (Babraham Bioinformatics, Cambridge, United Kingdom [www.bioinformatics.bbsrc.ac.uk/projects/seqmonk/]). The set of all genes represented in this data set was clustered by their read densities in the 2 kb surrounding the TSS and 2 kb surrounding the transcription end sites (TES), with clustering carried out separately in male and female liver. A data set of 15,533 genes was obtained after the exclusion of short genes (<5 kb in length) to eliminate genes containing TSS-associated sequences that overlap the TES-associated region; 7,864 of the 15,533 genes were defined as liver expressed based on log2 reads per kilobase per million mapped (RPKM) was ≥1 in either male or female liver. At this RPKM but not at lower RPKM values, ∼80% of genes contained a K4me3 peak and ∼90% of genes overlapped with a K27ac island. For each gene, the region of the TSS ± 1 kb and of the TES ± 1 kb was subdivided into a total of 20 200-bp windows. Read counts were obtained for these 20 windows for seven features—six chromatin marks and DNase hypersensitivity—yielding 140 columns of data. Read counts were normalized by total sequencing library size for each ChIP-seq data set, and log transformed. The 15,533 genes were then clustered by their read densities by k-means clustering using Cluster (51), with a k value of 6. The six clusters of genes were visualized as a heat map (see Fig. S5A in the supplemental material) using Java Treeview (52). Each cluster was characterized as active or inactive based on the relative density of activating chromatin marks (K4me1, K27ac, K4me3, and K36me3) and repressive chromatin marks (K27me3 and K9me3). The frequencies with which each 200-bp bin in the TSS and TES regions, expanded to ±2 kb, were assigned 1 of the 14 chromatin states were calculated for genes in each cluster. Three of the six clusters were identified as active based on high read density of K27ac, K4me3, K36me3, and DNase hypersensitivity. Two clusters were designated poised based on the presence of K27me3 together with K4me1, with the poised chromatin state 12 at the TSS. The sixth cluster was inactive, characterized by K27me3 and low levels of activating marks.
In other analyses, sex-biased genes were clustered by their chromatin mark densities. Sex-biased genes were identified from the RNA-seq data set using edgeR (53) for differential expression analysis, as implemented by P.-Y. Hao and T. Melia of this laboratory. A total of 423 male-biased and 477 female-biased genes (listed in Table S1B and C in the supplemental material) were selected based on the following criteria: adjusted P value of <0.01 for sex difference, corresponding to a minimum sex differences in expression of 1.4-fold; liver expressed at a level of log2 RPKM of ≥1 in either sex; genes located on autosomes; and gene length of ≥2 kb. Each set of sex-biased genes was clustered by read densities in male liver and separately in female liver for the 2-kb regions surrounding the TSS and the TES, as described above, by k-means clustering using a k value of 3. Each of the clusters was defined by its chromatin activity—high, medium, and low—as indicated by the relative abundance of activating and repressive chromatin marks (see Fig. S6A in the supplemental material). Genes were then classified into six classes according to the cluster they fell into in male liver and in female liver (see Table S4A in the supplemental material). Each of the six gene classes was then characterized by mapping each gene to chromatin modifications identified by MACS (46) or SICER (47), which were subjected to a direct male-to-female comparison as described above (“Analysis of ChIP-seq data for chromatin modifications”). K4me3 marks were mapped to the promoter (TSS ± 500 bp), and K27ac marks were mapped to the promoter (TSS ± 500 bp) and separately to the gene body (from TSS up to TES); the other four marks were mapped to the gene body alone. Genes that contained both a male-enriched mark and a female-enriched mark for a particular modification were assigned to the sex-independent mark class. Box plots were generated using the box plot function of R with default parameters.
Gene enrichment analysis.
Gene sets that respond to activators of the nuclear receptors constitutive androstane receptor (CAR) (54, 55), pregnane X receptor (PXR) (54), and peroxisome proliferator-activated receptor alpha (PPARα) and PPARβ/δ (56) and to activators of AhR (57) were obtained from the indicated references. For CAR, the union of CAR-upregulated genes from Tojima et al. (54) and Tian et al. (55) was considered, with a corresponding set used for CAR downregulated genes. Genes up- or downregulated by each of the receptors were tested for enrichment for one of the six clusters (see Fig. S5 in the supplemental material), compared to the background set of all 15,533 genes (as defined above), using Fisher's exact test. For enrichment analysis of sex-biased genes shown in Fig. 5, genes were mapped to ChIP-seq-determined binding sites for STAT5, BCL6 (30), CUX2 (34), FOXA1, or FOXA2 (44). For each set of TFBS, the nearest gene body within 10 kb of a TFBS were defined as the target of the corresponding TF. Gene targets of these TFs were then tested using Fisher's exact test for enrichment for belonging to each of the classes of sex-biased genes (Fig. 5A and B; see also Table S4B and C in the supplemental material) compared to a background set comprised of 8,764 liver-expressed genes (log2 PRKM of >1 in either sex) that were >2 kb in length. For each TF the fold enrichment was calculated as (number of genes in each sex-biased gene class that are a TF target/total number of sex-biased genes in the gene class)/(number of all other liver-expressed genes that are a TF target/number of all other liver-expressed genes). Functional term enrichment analysis was performed using DAVID (58, 59). FOXA1 and FOXA2 ChIP-seq data from Li et al. (44) were analyzed by G. Bonilla of this laboratory to identify sex-biased FOXA binding sites using methods described elsewhere (30).
Fig 5.
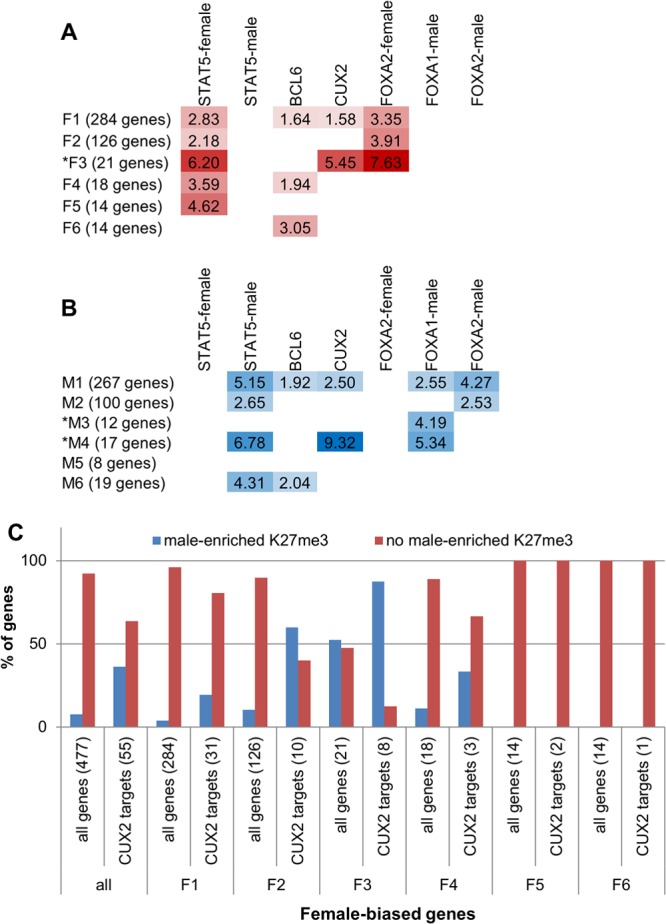
Enrichment of TF targets within each sex-biased gene class. (A and B) Fold enrichments for the genes in each class being a target of the specified TF (i.e., nearest gene within 10 kb of a TF binding site), compared to the background of all liver-expressed genes (see Materials and Methods). For both female-biased gene classes (A) and male-biased gene classes (B), data are shown for enrichments that have a P value of <0.05 and that contain at least five genes. Asterisks mark gene classes with sex-biased local chromatin marks (cf. Fig. 4A). For STAT5, FOXA1, and FOXA2, the enrichments shown are for targets of TF binding sites enriched in male or female liver, as indicated. No enrichments were seen for female-enriched FOXA1 binding. See Table S4B and C in the supplemental material for P values and numbers of genes represented by each enrichment score. (C) Percentage of CUX2 target genes in each female-biased gene class that overlap a male-enriched K27me3 mark compared to all female-biased genes in the class. Numbers of genes are shown in parentheses.
Correlation between TF binding and DHS/chromatin mark sex ratios.
The set of 72,862 merged liver DHS identified previously (39) was ranked by the male-female ratio after normalization by reads in male-female liver common peaks. The DHS genomic regions were ranked separately by their male-female ratio in DNase hypersensitivity and in K4me1, K27ac, and K27me3 read density over the entire peak region. Overlaps between DHS and TF binding sites (TFBSs) (male-enriched STAT5, female-enriched STAT5, male-enriched FOXA1 and FOXA2, and female-enriched FOXA2 binding sites) were also computed. Female-enriched FOXA1 binding sites did not follow the patterns exhibited by the other sex-biased TF binding sites examined (see Fig. S8F and S9B in the supplemental material) and were excluded from subsequent analyses. A TF binding site was considered to overlap a DHS if the ChIP-seq-identified peak region for the TF overlapped the DHS peak region by at least one base pair. Gene set enrichment analysis (60) was used to calculate a running enrichment score for each of the five types of TF binding sites for each of the four ranked lists: sites ranked by sex ratio in DHS, K4me1, K27ac, and K27me3.
Characterization of DHS by enhancer modifications and enrichment of TF binding.
Male-biased, female-biased, and sex-independent DHS (39) were characterized by the presence or absence of the two enhancer-associated chromatin marks, K4me1 and K27ac, and by whether the marks were classified as male enriched or female enriched (|M| > 1 and P < 0.001) (see above) or sex independent. For each DHS, the DHS peak region was defined as a 1-kb window centered at the DHS peak summit. Each DHS was considered to contain a K4me1 or K27ac peak if at least 200 bp of the 1-kb DHS peak region overlapped a K4me1 or a K27ac peak. The requirement for a 200-bp overlap was chosen based on the K4me1 and K27ac read profiles at DHS, which peak at ∼300 bp on either side of the DHS summit (see Fig. 2E).
Fig 2.

Chromatin states in mouse liver and chromatin environments surrounding TSS and DHS summits. (A) Emission probabilities for the 14 chromatin states. A darker shade of blue corresponds to greater emission frequency of a chromatin modification in that state. (B) Chromatin states surrounding TSS of liver-expressed and non-liver-expressed genes. The y axis shows the frequency at which each 200-bp bin in a 4-kb region surrounding the TSS is assigned each of the 14 chromatin states shown in panel A. (C) Average ChIP-seq read densities for DHS and each of six chromatin marks in a 4-kb region surrounding TSS of liver-expressed and non-liver-expressed genes. (D) Distributions of 4 of the 14 chromatin states in male and female liver surrounding male-biased and female-biased DHS peak summits. Shown are the following: state 6, active enhancer state (frequency is significantly different between male and female liver at male-biased DHS [P = 4e−7] and also at female-biased DHS [P = 1e−5]); state 1, containing K27me3 alone (significantly different at male-biased DHS [P = 6e−4] and at female-biased DHS [P = 1e−8]); state 12, containing K27me3 and K4me1 (not significant [P > 0.05] at male-biased DHS or at female-biased DHS); state 2, another inactive state (significant at male-biased DHS [P = 4e−7] and at female-biased DHS [P = 2e−6]). See Fig. S4B in the supplemental material for distributions of all 14 states. (E and F) Chromatin mark read densities surrounding DHS peak summits (E) and TSS (F) in male and female liver for male-biased and female-biased DHS and genes, respectively.
Since K27ac marks identify active enhancers and since K4me1 marks without K27ac marks identify enhancers that may not be active (61), DHS were assigned enhancer categories in a hierarchical manner, in the following order (see Table S6A in the supplemental material): (i) sex-biased K27ac, (ii) sex-biased K4me1, (iii) sex-independent K27ac, (iv) sex-independent K4me1, and (v) no K27ac or K4me1 mark. Any DHS that overlapped both a male-biased mark and a female-biased mark for a particular modification was considered to overlap a sex-independent mark. Each of these categories of DHS was compared to the same background set of sex-independent DHS (58,087 DHS in male liver; 52,187 DHS in female liver). Background sex-independent DHS were those whose nearest gene TSS was not sex biased, and additionally, they were required to be >500 kb from the nearest sex-biased TSS. DHS were considered to contain a STAT5, BCL6, CUX2, FOXA1, or FOXA2 binding site (identified by ChIP-seq) if the DHS overlapped the TF's binding site by at least 1 bp. For each category of DHS by enhancer status, the enrichment for a particular TF was calculated as follows: (number of DHS in a category that overlap a TFBS/total number of DHS in that category)/(number of background sex-independent DHS that overlap a TFBS/total number of sex-independent DHS). The Fisher exact test was used to calculate a P value for each enrichment or depletion. Enrichments having a P value of <0.001 are shown in Fig. 6B.
Fig 6.

Relationship between TF binding and sex bias in DHS and chromatin marks at regulatory sites. (A) Gene set enrichment analysis for DHS ranked by male/female (M/F) ratio in normalized sequence reads for each of the following (from left to right): DNase hypersensitivity, K4me1, K27ac, and K27me3. x axis, DHS ranked by M/F ratio in each mark, from male bias on left to female bias on right; y axis, running enrichment score for each TF. Male-enriched STAT5, FOXA1, and FOXA2 binding are enriched toward the male-biased (i.e., left) end of the ranked lists for DHS, K4me1, and K27ac and toward the female-biased (i.e., right) end of the ranked list for K27me3. The opposite pattern is seen for female-enriched STAT5 and FOXA2 binding. (B) Enrichment of ChIP-seq binding sites at male-biased, female-biased, and sex-independent DHS categorized by their patterns of enhancer-associated modifications (K27ac and K4me1). DHS are assigned enhancer categories in a hierarchical manner (see Materials and Methods). Sex-independent DHS shown are limited to those whose nearest gene TSS within 250 kb is sex biased in expression. Shown are enrichments and depletions at a P value of <0.001, and enriched groups were additionally required to contain at least five DHS. Green, enrichment; purple, depletion. Numbers of DHS and enrichment P values associated with these enrichment scores are shown in the upper sections of Table S6B to D in the supplemental material. Mapping of these DHS to the nearest gene TSS within 250 kb (to include more distal regulatory sites compared to the 10-kb distance used to map TF targets shown in Fig. 5) yielded enrichments for enhancer categories further broken down by the gene class of their target genes (see Table S6B to D).
Microarray data accession numbers.
Raw sequence reads for the six sets of chromatin marks in both male and female mouse liver are available at the Gene Expression Omnibus (GEO) website as series GSE44571 (samples GSM1087069 to GSM1087105 and GSM1181495), and sequence reads for RNA-seq data for gene expression in male and female mouse liver are available as series GSE48109 (samples GSM1168535 to GSM1168538). Mouse liver DNase-seq data sets (39) are available as series GSE21777, and ChIP-seq data sets are available for STAT5 (GSE31578; male and female liver, STAT5 high-activity status), BCL6 (GSE31578; male liver) (30), CUX2 (GSE35985; female liver) (34), and FOXA1 and FOXA2 (44) (untreated male and female liver; E-MTAB-805) in ArrayExpress (www.ebi.ac.uk/arrayexpress/).
RESULTS
Chromatin states and distributions around genomic features.
Six chromatin marks were mapped genome-wide in male and female mouse liver. All four activating marks (histone H3 K4me1, K4me3, K27ac, and K36me3) showed the anticipated positive correlations with each other and with expression of associated genes, and both repressive marks (histone H3 K9me3 and K27me3) were negatively correlated with gene expression (Fig. 1A; see also Fig. S1A in the supplemental material). Genomic localizations and distributions across gene bodies (see Fig. S2 in the supplemental material) are consistent with previously reported associations of each mark: K4me3 with active promoters (62–64), K36me3 with actively transcribed regions (62, 63, 65), K4me1 and K27ac with enhancers (61, 66), K27me3 with polycomb-mediated gene silencing, and K9me3 with constitutive heterochromatin and nongenic regions (67). K27me3 and K9me3 were enriched in female liver on chromosome X (ChrX) (Fig. 1B; see also Fig. S1B), consistent with their role in X inactivation (68). Conversely, K4me1 and K27ac are enriched in male liver on ChrX, consistent with depletion of K4 methylation from the inactive X chromosome in female cells (69). ChrX was excluded from further analysis to eliminate bias in genome-wide male-female comparisons.
Fig 1.
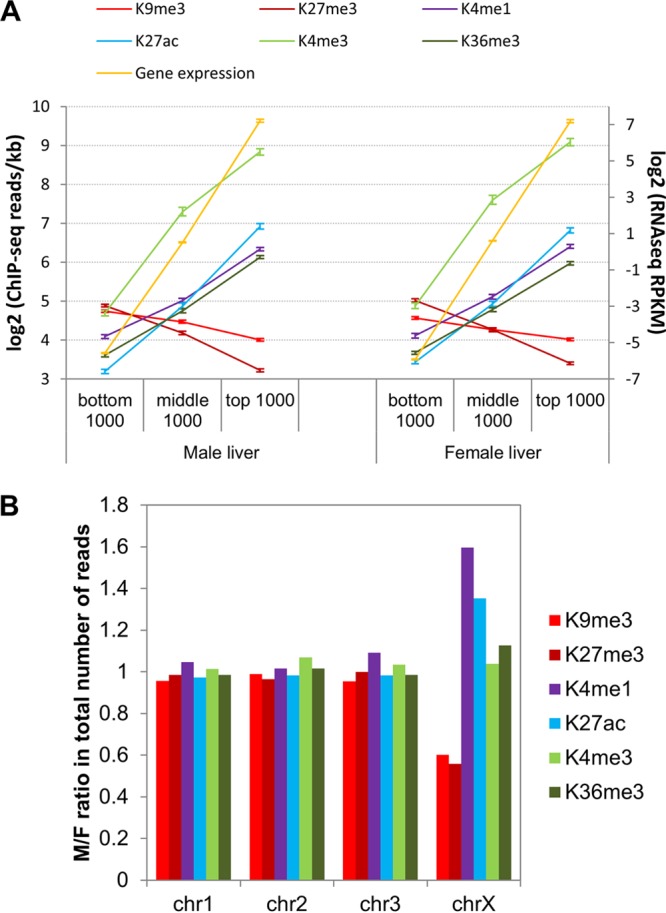
Chromatin mark read densities in gene bodies and across chromosomes. (A) Mean ± 95% confidence intervals for ChIP-seq read densities (left y axis) in TSS ± 500 bp (K4me3) or within gene bodies (all other marks) of top, middle, and bottom 1,000 genes (full set of 21,828 genes ranked by expression level) in male and female liver. Average gene expression for each category of genes (right y axis) is shown in yellow. (B) Male/female (M/F) ratio in total ChIP-seq reads for each chromatin modification on Chr1, Chr2, Chr3, and ChrX. Other chromosomes are shown in Fig. S1B in the supplemental material.
ChromHMM (1) was used to learn 14 chromatin states in male and female liver based on DHS data (39) combined with the above six chromatin marks (Fig. 2A, and B; see also Fig. S3A in the supplemental material). These include inactive states (marked by K27me3 [state 1] or K9me3 [state 3]), a bivalent promoter-associated state (K27me3 plus K4me1 [state 12]), a transcribed state (K36me3 [state 14]), promoter states, which contain K4me3 (states 7 and 8), and several enhancer states, which contain K27ac or K4me1 but lack K4me3 (states 5, 6, 9, 10, and 11). Liver-expressed genes show active promoter states at the transcription start site (TSS) (Fig. 2B, left), with an asymmetric pattern reflecting the asymmetric distribution of DHS and K4me1, K4me3, and K27ac marks at promoters (Fig. 2C, left) (5). Active chromatin states occur more frequently in one sex than the other at GH-regulated DHS whose accessibility differs significantly between male and female liver (sex-biased, GH-responsive DHS [39]) (Fig. 2D; see also Fig. S4B in the supplemental material) but not at sex-independent DHS (see Fig. S4A). K27me3 states are elevated in male compared to female liver at female-biased DHS (cf. male prominence of K27me3-containing states 1 and 12), whereas we did not find a corresponding female bias in K27me3 states at male-biased DHS (Fig. 2D and E). This indicates a role for K27me3 in sex-biased gene silencing in male but not female liver. Overall, sex differences in chromatin environment are much less pronounced at sex-biased TSS than at sex-biased DHS (Fig. 2F versus E; see also Fig. S4C), suggesting that many sex-biased genes are controlled by distal regulatory sites.
Clustering of genes by local chromatin environment.
Genes were clustered by chromatin mark and DHS read densities surrounding the TSS and transcript end site (TES) (see Fig. S5A and Table S1 [for the full data set for 15,533 genes] in the supplemental material). Chromatin state distributions for the resultant six gene clusters are shown in Fig. 3A. Clusters 1 to 3 were characterized by high levels of DHS and the presence of all four activating marks and by low levels of repressive marks and are primarily in chromatin state 7 at the TSS (Fig. 3A). Genes in these active chromatin state clusters show high expression (≥80% at log2 RPKM ≥ 1) (Fig. 3B; see also Fig. S5B). Cluster 1 differs from clusters 2 and 3 in its active enhancer states around the TES (Fig. 3A). Cluster 1 genes also have the highest coverage by broad domains of DNase hypersensitivity, with 70% of the genes being at least half covered by an extended DHS island (39), compared to 16 to 23% for the other two active clusters (see Fig. S5C and D). Genes in clusters 4 to 6 have high K27me3 read densities and, correspondingly, show low expression (Fig. 3B; see also Fig. S5B). Genes in clusters 4 and 5 are characterized by K4me1 in combination with K27me3 (chromatin state 12 at the TSS) (Fig. 3A), i.e., the signature of a poised gene (70), while cluster 6 genes have K27me3 without K4me1 or other activating marks (state 1) (Fig. 3A). The poised-gene clusters show significantly higher mean expression levels than cluster 6 genes (Fig. 3B).
Fig 3.

Genes clustered by chromatin densities around TSS and TES. (A) Chromatin state environments, as described in the legend of Fig. 2B, surrounding the TSS (left) and TES (right) for six clusters of 15,533 liver-expressed and non-liver-expressed genes clustered by chromatin read densities in 200-bp nonoverlapping windows within 2-kb regions surrounding the TSS and TES. See Fig. S5A in the supplemental material for a heat map of read intensities in six gene clusters. (B) Box plots of gene expression distributions for each cluster in female liver, with similar results in male liver (not shown). Clusters 1 to 3 differ significantly from clusters 4 to 6 (P < e−221), and clusters 4 and 5 differ significantly from cluster 6 (P = 2e−113 and P = 9e−117, respectively, Wilcoxon rank sum test). (C and D) CAR-, AhR-, PXR-, and PPAR-responsive genes mapped to the six gene clusters. For each category of upregulated (C) or downregulated (D) genes, the percentage of genes in each of the six clusters is shown. Significant enrichments and depletions for each gene set belonging to a given cluster compared to a background set of all 15,533 genes are indicated by black and gray asterisks, respectively. *, P < 0.05; **, P < 0.01. PPARa, PPARα; PPARbd, PPARβ/δ.
To test the hypothesis that the basal chromatin state informs responsiveness to transcriptional regulators, genes in all six chromatin clusters were compared to the sets of genes that respond to four ligand-activated TFs of the nuclear receptor superfamily: CAR, PXR, PPARα, and PPARβ/δ (71). Genes regulated by these TFs were enriched for one or more active chromatin clusters (Fig. 3C and D; see also Table S1 in the supplemental material). Further, genes downregulated by PXR and PPARβ/δ were enriched for poised chromatin clusters, suggesting that ligands that activate these two TFs induce removal of K4me1 while maintaining K27me3 at target genes. In contrast, genes upregulated by another ligand-activated TF, AhR, were enriched for genes in a poised chromatin state, indicating loss of K27me3 upon ligand activation. Consistent with the above preference for activation of genes already in an active chromatin state, the upregulated gene targets of all four nuclear receptors were significantly depleted from the inactive chromatin state (cluster 6) (Fig. 3C). Thus, the basal chromatin state is apparently a determinant of responsiveness to these ligand-activated TFs.
Functional term enrichment analysis (see Table S2 in the supplemental material) indicated enrichment for liver-specific terms such as drug, lipid, and steroid metabolism in cluster 1, the most active gene cluster. The other two active clusters were enriched for a broad range of non-liver-specific terms, e.g., mitochondrion, RNA processing, protein transport, and cell cycle. The two poised clusters were enriched for terms such as cell adhesion, cell junction, mesenchymal cell differentiation, and extracellular matrix, while the inactive cluster 6 was enriched for genes not expressed in liver (e.g., neurotransmission, muscle, vision, and memory).
Classes of sex-biased genes.
Genes that show sex-biased expression (423 male-biased genes and 477 female-biased genes) were clustered by their chromatin mark and DHS densities around the TSS and TES in male liver and, separately, in female liver (see “Classification of sex-biased genes” and also Fig. S6A to C in the supplemental material). Six female-biased and six male-biased gene classes (F1 to F6 and M1 to M6, respectively) were identified based on their chromatin activity classifications in each sex (see Table S4A and Fig. S6A in the supplemental material). Next, sex differences in the chromatin environment of each gene class were characterized by comparing normalized densities of each of the six marks on a genome-wide basis in male versus female liver. Genomic regions that showed significant male enrichment or female enrichment were thus identified for each chromatin mark (see Table S3 in the supplemental material). The frequency of sex-biased and sex-independent chromatin marks was then determined for the genes in each class. Figure 4A presents these data for the two largest classes of sex-biased genes, which, as shown, generally lack sex differences in proximal chromatin marks (classes F1 and M1), as do a majority of all sex-biased genes. In contrast, genes in sex-biased gene classes F3, M3, and M4 show sex differences in proximal chromatin marks at comparatively high frequencies (Fig. 4A; see the full data set in Fig. S6E and Table S1B and C in the supplemental material). Classes F3 and M3 also showed the largest sex differences in expression (Fig. 4B), while classes F3 and M4 showed the highest level of expression (see Fig. S6G). Heat maps of chromatin mark sex ratios are shown for classes F3, M3, and M4 in Fig. 4C. These three classes (F3, M3, and M4) represent only 5.6% of all sex-biased genes, consistent with the conclusion that many sex-biased genes are not in a sex-biased chromatin environment (cf. Fig. 2F). Indeed, taking into account distal chromatin marks, which may represent distal regulatory sites, less than half of sex-biased genes have sex-biased chromatin marks within 10 kb, and only 66 to 69% have them within 100 kb (see Fig. S6F).
Fig 4.

Characteristics of sex-biased genes classified by chromatin cluster. (A) Sex dependence of the chromatin marks identified using MACS or SICER associated with the following sets of genes: all male- and female-biased genes, the two largest classes of sex-biased genes that lack sex differences in local chromatin marks (F1 and M1), and three classes of sex-biased genes that exhibit sex differences in local chromatin marks (F3, M3, and M4). none, no MACS- or SICER-identified mark associated with the promoter (K4me3) or gene body (all other marks); *, chromatin mark for which the distribution (male, female, sex-independent [sex-indep], or none) for that category of genes is significantly different from that for the set of all male-biased or all female-biased genes (P < 0.05; chi-square test). See Fig. S6E in the supplemental material for a complete data set for all six classes of male-biased genes and all six classes of female-biased genes. (B) Gene expression log2 sex ratios for female-biased and male-biased gene classes. The dashed horizontal line indicates median log2 sex ratio for all female-biased genes or all male-biased genes. (C) Heat map of sex ratios in chromatin marks in the 2-kb regions surrounding the TSS and TES for gene classes F3, M3, and M4. Red, female bias; blue, male bias.
Importantly, the highly female-biased class F3 genes are characterized by female-biased activating marks and male-biased repressive marks (note the high frequency of male-enriched K27me3), whereas class M3 and M4 genes frequently show male-biased activating marks but not female-biased repressive marks (Fig. 4A). Our finding of male-enriched K27me3 marks at highly female-biased genes but not the corresponding female-enriched K27me3 marks at highly male-biased genes is supported by a separate analysis in which sex-biased genes were divided into four equal groups based on their ranking in sex ratio in expression (see Fig. S7A in the supplemental material). As shown in Fig. S7A, 30 out of the 118 most highly female-biased genes (25%) have male-enriched K27me3 marks, compared to ≤4% for each of the other quarters. In contrast, none of the male-biased genes have female-enriched K27me3 marks.
Enrichment of TF targets among sex-biased gene classes.
Next, we investigated whether sex-biased genes in different chromatin states utilize the same regulatory factors and mechanisms. To identify putative targets of such regulatory factors, the sex-biased genes in each class were mapped to nearby (within 10 kb) binding sites for five TFs implicated in GH-regulated, sex-dependent liver gene expression. Enrichment scores were then calculated for each TF compared to a background set of all liver-expressed genes. Figure 5A and B present these data for three GH-regulated TFs: STAT5 (male-enriched, female-enriched, and sex-independent STAT5 binding sites) (30), BCL6, a male-biased repressor (30), and CUX2, a highly female-specific repressor (34). Data are also shown for FOXA1 and FOXA2, which contribute to the sex-biased incidence of hepatocellular carcinoma (44). Several of the sex-biased gene classes were enriched for being targets of sex-biased STAT5 binding, with the highest enrichments seen for genes in classes F3 and M4, whose proximal chromatin marks show sex bias (Fig. 4A and C). In contrast, BCL6 targets were enriched among female-biased genes that do not show sex differences in chromatin marks (classes F1, F4, and F6), suggesting that the repression of female-biased genes in male liver by BCL6 (30) does not introduce (or require) a sex-biased chromatin state. BCL6 targets were also enriched at a subset of male-biased genes, reflecting the STAT5 competition mechanism described earlier (30).
CUX2 targets were highly enriched among male-biased genes in class M4 and, to a lesser extent, in class M1 (Fig. 5A), consistent with the proposed role for CUX2 as a repressor of many male-biased genes in female liver (34). CUX2 targets were also enriched among class F3 genes, consistent with CUX2 directly activating a subset of female-biased genes (34). Female-biased CUX2 targets are more likely than other female-biased genes to have a male-enriched K27me3 mark, especially the target genes in class F3 (Fig. 5C), which show the greatest enrichment of CUX2 binding (Fig. 5A) and the highest frequency of male-biased K27me3 marks (Fig. 4A). Since class F3 genes are also highly enriched for being targets of STAT5 and FOXA2 (Fig. 5A), these three factors may cooperatively regulate the most highly female-biased genes. Overall, male-enriched K27me3 domains at the gene body of female-biased genes coassociate with CUX2 binding (within 10 kb) more than with any other TF examined, and, correspondingly, CUX2 binding is more likely to target female-biased genes with male-enriched K27me3 domains than any other sex-biased mark (see Table S4D and E in the supplemental material). FOXA2 shows female-biased binding enriched at female-biased genes with male-enriched K27me3 domains, and this enrichment is almost as high as the corresponding enrichment for CUX2 (see Table S4D and E), suggesting that CUX2 and FOXA2 cooperate to derepress these genes in female liver.
Among female-biased genes, the preference for STAT5 and FOXA2 to target genes that have proximal sex-biased marks and for BCL6 to target genes that lack sex-biased marks is further supported by enrichment analysis of female-biased genes ranked by gene expression sex ratio (see Fig. S7B in the supplemental material). The enrichment for being a target of female-enriched STAT5 or FOXA2 binding increases with an increasing sex ratio in gene expression. In contrast, BCL6 targets are enriched only among the female-biased genes with the lowest sex ratio in expression. Thus, female-enriched STAT5 and FOXA2 binding sites preferentially map to the most highly female-biased genes (Fig. 5A; see also Fig. S7B), which in turn exhibit sex differences in proximal chromatin marks (Fig. 4A and B), while BCL6 targets weakly female-biased genes, which lack sex differences in proximal chromatin marks.
Relationship between sex bias in DHS, chromatin modifications, and TF binding at regulatory sites.
Female-biased DHS (39) are highly enriched (18-fold), compared to sex-independent DHS, for the presence of female-biased K27ac or K4me1 marks; correspondingly, a 16-fold enrichment of a male-enriched K27ac or K4me1 mark is seen at male-biased DHS (see Table S5A in the supplemental material). Thus, many sex-biased DHS have the marks of sex-dependent enhancers. Moreover, sex-biased gene classes F3 and M3, which comprise the most highly sex-biased genes, are enriched for association (within 250 kb) with sex-biased DHS that have sex-biased K27ac (see Table S5B and C), the mark of an active enhancer (61, 66).
Next, we investigated the relationship between sex bias in chromatin marks and DNase hypersensitivity and sex-biased binding of the five TFs discussed above. Gene set enrichment analysis showed positive correlations between the sex-biased binding of STAT5, FOXA1, and FOXA2 and the ranking by sex ratio of each enhancer mark (K27ac and K4me1) and of DNase hypersensitivity and inverse correlations in the case of K27me3 (Fig. 6A). These findings are supported by our analysis of TF binding site frequency in DHS ranked by sex ratio (see Fig. S8A to E in the supplemental material) and by the correlation between sex ratio in TF binding and sex ratio in DHS and in activating chromatin marks (see Fig. S9A to C in the supplemental material). BCL6 is most frequently bound at sex-independent DHS (see Fig. S8G), and BCL6 binding intensity does not correlate with sex ratio in chromatin marks (see Fig. S9D), consistent with its preferred sex-biased target genes (Fig. 5A) having sex-independent chromatin marks (Fig. 4A). CUX2 binding is enriched at male-biased DHS, consistent with the study of Conforto et al. (34), but is not associated with or correlated with sex differences in chromatin marks at the sites where it is bound (see Fig. S8H and S9E).
To examine more closely the relationship between chromatin modifications and regulation by liver TFs, we integrated TF binding data with the enhancer status of DHS and the chromatin class of their respective gene targets, considering genes within 250 kb to allow for distal regulation. For each TF, we identified significant enrichments (P < 0.001) of its binding sites at subsets of DHS categorized by their enhancer modification patterns (Fig. 6B). Sex-biased DHS that have sex-biased enhancer marks are most highly enriched for being GH responsive, linking sexually dimorphic plasma GH profiles and sex differences in enhancer modifications. Consistent with the results above, female-biased binding of STAT5 and of FOXA2 is most highly enriched at female-biased DHS that have female-biased K27ac marks. Similarly, male-biased STAT5, FOXA1, and FOXA2 binding is most highly enriched at male-biased DHS with male-biased K27ac. This preference for sites with sex-biased K27ac is also seen at sex-independent DHS but with lower enrichments (Fig. 6B). Enrichments of TF binding sites were also determined for subsets of DHS that target (within 250 kb) genes in individual sex-biased gene classes (see Table S6B to D in the supplemental material). Female-biased STAT5 binding was most strongly enriched (22-fold) at DHS mapping to class F3 genes, consistent with the results shown in Fig. 5A. In contrast, the strongest enrichment for male-biased STAT5 binding was at DHS that map to class M1 genes (47-fold), which lack proximal sex-biased chromatin marks (Fig. 4A). Thus, distal regulation by male-enriched STAT5 binding is associated with male-biased genes that lack nearby sex differences at the chromatin level.
BCL6 and CUX2 binding are enriched at both male-biased and sex-independent DHS, in particular for sites with a sex-independent K27ac mark (Fig. 6B), in agreement with the lack of correlation between binding sites for these factors and sex differences in chromatin marks described above. Among DHS with sex-independent K27ac, BCL6 binding is enriched in association with female-biased genes that lack nearby sex-biased chromatin marks (classes F1, F2, F4, and F6), while CUX2 binding is most highly enriched in association with male-biased class M4 genes (7.8-fold) (see Table S6C and D in the supplemental material), in agreement with the results shown in Fig. 5A and B. Bcl6 itself is a target of male-biased STAT5 binding (see Table S1C in the supplemental material) and is an example of a male-biased TF that binds to sex-independent regulatory sites, which enables it to regulate secondary target genes that do not exhibit sex differences in chromatin marks. CUX2 also shows enrichment for binding at sex-independent DHS with female-biased K27ac marks (Fig. 6B). The highest CUX2 enrichment was associated with female class F2 genes (which includes Cux2 itself) and class F3 genes (see Table S6B and D). CUX2 is thus a female-specific TF that regulates female-specific genes via sex-independent DHS regulatory elements.
Sex difference in K4me1 distribution surrounding sex-biased DHS summits.
FOXA1 acts as a liver-enriched pioneer factor (11, 12) that opens chromatin at cell-type-specific enhancers by binding compacted chromatin via a core histone binding motif, resulting in the generation of open chromatin regions (DHS) (72, 73). Given the strong enrichment of sex-biased binding of FOXA1 and of the related FOXA2 at DHS showing a sex bias in DNase hypersensitivity and K27ac and K4me1 marks (Fig. 6), we hypothesized that FOXA1 and/or FOXA2 mediates sex-dependent nucleosome repositioning and sex-differential chromatin opening at sex-biased DHS. Supporting this hypothesis, male-biased DHS show a sex-dependent distribution of K4me1 marks surrounding the DHS summit, with a bimodal peak in male liver and a monomodal peak in female liver (Fig. 7A). This pattern is consistent with sex-dependent nucleosome positioning (13) and is most pronounced at DHS that bind FOXA1 or FOXA2 in a male-enriched manner (cf. the greater dip in K4me1 reads at the summit of male-biased DHS bound by FOXA1 or FOXA2 compared to male-biased DHS not bound by these factors, as shown in Fig. 7B). Moreover, in contrast to the monomodal K4me1 peak seen in female liver at DHS with male-enriched FOXA1 or FOXA2 binding, a trough in K4me1 reads is discernible in female liver at DHS with sex-independent FOXA1 or FOXA2 binding. Further support for sex-dependent nucleosome repositioning at male-biased DHS by FOXA1/FOXA2 is provided by the deeper K4me1 trough at male-biased DHS in male liver when FOXA1 or FOXA2 binding occurs in combination with male-biased STAT5 binding than at sites bound by STAT5 alone (Fig. 7C; see also Fig. S10A and B in the supplemental material).
Fig 7.

K4me1 read profiles at DHS sites. K4me1 read density in male (blue) and female liver (red). Read counts are normalized to the total number of DHS in each panel. (A) Male-biased, female-biased, and sex-independent DHS. While male-biased DHS and female-biased DHS both show sex differences in K4me1 intensity (normalized read density), only male-biased DHS show a sex-dependent K4me1 distribution, with a monomodal peak in female liver and bimodal peak in male liver. (B) Male-biased DHS and FOXA1 or FOXA2 binding. (C) Male-biased DHS with male-enriched STAT5 binding, with and without FOXA1 binding. (D) Female-biased DHS and FOXA2 binding. The effect of STAT5 binding is shown in Fig. S10A to C in the supplemental material, and trough depths and sex differences in K4me1 profiles are quantified in Fig. S10D and E. The difference between the K4me1 profiles at FOXA-binding male-biased DHS and non-FOXA-binding DHS is retained when samples of non-FOXA-binding sites are chosen to match FOXA-binding sites by either DHS intensity or DHS sex ratio (see Fig. S10F to H).
In contrast to male-biased DHS, DHS that are female biased or sex independent have bimodal K4me1 peaks in both male and female liver (Fig. 7A). Female-biased DHS show no sex difference in K4me1 profiles at sites of female-enriched FOXA2 binding (Fig. 7D), despite the strong enrichment of female-biased FOXA2 binding at female-biased DHS (Fig. 6) and at female-biased genes (Fig. 5A; see also Fig. S8D and S9C and Table S6B in the supplemental material). Thus, while FOXA2 is likely involved in female-biased gene regulation, it apparently does not confer sex-dependent nucleosome repositioning at female-biased DHS. The finding that the FOXA factor(s) is apparently active in nucleosome repositioning at male-biased but not female-biased DHS can be explained if FOXA1, rather than FOXA2, is the key factor in sex-dependent chromatin opening, as suggested by the deficiency of female-enriched compared to male-enriched FOXA1 binding sites (577 and 3,280 sites, respectively) and by the more similar numbers of female-enriched and male-enriched FOXA2 binding sites (780 and 976 sites, respectively). Supporting this proposal, 71% of the male-enriched FOXA2 binding sites found at male-biased DHS cooccur with a male-enriched FOXA1 binding site, whereas only 6% of female-enriched FOXA2 binding sites cooccur with a female-enriched FOXA1 binding site. Moreover, the female-enriched FOXA1 binding sites that are present are unusual insofar as they do not preferentially overlap, nor do they correlate with the magnitude of the sex bias in female-biased DHS and chromatin modifications (see Fig. S8F and S9B). To summarize, the pronounced sex-dependent trough of K4me1 reads at male-biased DHS is indicative of nucleosome repositioning occurring in male but not female liver. This sex-differential pattern of marks is associated with, and may be regulated by, male-enriched FOXA1 binding, although it often coincides with male-enriched FOXA2 binding. The relative deficiency of female-enriched FOXA1 binding sites may account for the absence of a corresponding strong, female-specific trough of K4me1 reads at female-biased DHS.
DISCUSSION
We have developed genome-wide chromatin state maps for male and female mouse liver based on DHS profiles and patterns of six chromatin marks and then used these maps to elucidate chromatin environments at DHS and across gene bodies, in particular, those associated with sex differences in gene expression, a characteristic of ∼1,000 genes in mouse liver. By integrating chromatin maps with TF binding data, we show that the mechanisms of sex-biased gene regulation differ between male and female liver, with male-biased DHS but not female-biased DHS being characterized by a sexually dimorphic K4me1 profile that is most closely associated with male-enriched binding of FOXA pioneer factors (Fig. 8). These FOXA factors, in turn, are proposed to facilitate male-enriched binding of STAT5, which activates male-biased genes by mechanisms that include distal regulation of the male-biased genes that lack nearby sex differences in chromatin marks. Genes that show a weak female bias in expression and are associated with sex-independent chromatin marks are preferentially repressed in male liver by the male-biased repressor BCL6, while other, more highly female-biased genes show sex differences in proximal chromatin marks and are preferentially activated by female-enriched STAT5 binding. Many of the most strongly female-biased genes are regulated by K27me3-based repression in male liver, which may be countered by the stimulatory actions of CUX2 in female liver; however, strongly male-biased genes are not repressed by a corresponding K27me3-based mechanism in female liver (Fig. 8). These insights evidence the power of functional genomic studies and epigenetic analysis for elucidating transcriptional regulatory networks in a complex mammalian tissue.
Fig 8.

Model for role of GH-regulated TFs and chromatin states in sex-biased gene expression in male liver and female liver. The model describes the two largest classes of sex-biased genes, which lack local sex-biased chromatin marks (class M1 with 267 genes and class F1 with 284 genes), and two classes of sex-biased genes with sex-biased local chromatin marks (class M4 with 17 genes and class F3 with 21 genes). Male-biased chromatin marks, genes, and TF binding sites are indicated in blue text; female-biased features are in red text; and sex-independent chromatin marks are in green text. Orange curves represent the distribution of K4me1 marks around DHS summits. (A) Male-biased DHS and genes. In male liver, male-biased DHS are marked by a bimodal distribution in K4me1 marks and by male-enriched K4me1/K27ac marks and are bound in a male-enriched manner by STAT5 and/or FOXA1/FOXA2. These DHS activate male-biased genes with local sex differences in chromatin marks (class M4) as well as genes that lack local sex differences in chromatin marks (class M1). In female liver, male-biased DHS exhibit a monomodal K4me1 distribution and are deficient in binding of STAT5 and in the binding of FOXA1/FOXA2, which facilitate male-biased DHS opening. CUX2 binding, which is restricted to female liver, is enriched at sites with sex-independent DHS/enhancer modifications and male-enriched STAT5 binding (34) and preferentially targets for repression class M4 male-biased genes and, to a lesser extent, the larger class M1 male-biased genes. (B) Female-biased DHS and genes. Female-biased DHS exhibit a bimodal K4me1 distribution in both male and female liver, have female-enriched enhancer modifications, and show female-enriched binding by STAT5, FOXA2, and CUX2. These three TFs preferentially target female-biased genes with sex differences in local chromatin marks (class F3 genes) and could act in a cooperative manner. CUX2 activation of class F3 and other female-biased genes may be facilitated by removal of K27me3 marks, which are enriched at these genes in male liver. (C) Sex-independent DHS. The male-biased repressor BCL6 shows enriched binding at sex-independent DHS that have sex-independent enhancer modifications, and it preferentially represses female-biased genes that lack local sex differences in chromatin marks.
Impact of chromatin state on functional gene enrichments and nuclear receptor responsiveness.
Clustering of genes by local chromatin mark intensities and DNase hypersensitivity identified genes in three distinct chromatin environments: active (clusters 1 to 3), poised (K27me3 plus K4me1 state at the TSS; clusters 4 and 5), and inactive (low activating marks, high K27me3 levels; cluster 6) (Fig. 3). Cluster 1 genes are commonly involved in liver-specific tasks, such as drug, steroid, and lipid metabolism; genes in clusters 2 and 3 perform many non-liver-specific functions, and poised cluster 4 and 5 genes encompass biological processes activated in response to specific stimuli. Genes responsive to nuclear receptors/TFs that regulate many liver metabolic functions (CAR, PXR, PPARα, and PPARβ/δ) (41–43) and genes responsive to AhR were depleted of inactive cluster genes, indicating that these TFs do not target genes in an inactive chromatin state. A poised chromatin state is preferentially associated with genes downregulated by PXR and PPARβ/δ and with genes upregulated by AhR, indicating that poised chromatin can resolve into either an inactive or an active state, depending on the TF and depending on the target gene. In contrast, many genes upregulated by PXR and PPARβ/δ, as well as genes responsive to CAR and PPARα, are already in an active chromatin state in unstimulated liver.
Regulation of sex-biased genes.
Sex differences in chromatin environment were commonly seen at male-biased and female-biased DHS but not at sex-biased genes, a majority of which lack sex differences in local chromatin marks. Consistently, genes differentially expressed between cell types show cell-type-specific chromatin states at regulatory sites but common chromatin states at the promoter (3, 6, 74). However, some sex-biased genes showing large differences in expression between sexes do show notable sex differences in local chromatin states (e.g., Cyp2b9 and Hsd3b5) (see Fig. S11 in the supplemental material) while others do not (Sult2a6 and Cabyr) (see Fig. S11). Genes with sex differences in local chromatin environment show the strongest enrichment for local sex-biased STAT5 binding (Fig. 5A and B). Distal regulation is most likely for class M1 genes (the largest class of male-biased genes), which generally lack sex differences in local chromatin marks but show 47-fold enrichment for distal (within 250 kb) male-biased STAT5 binding sites at male-biased DHS with male-biased K27ac marks (see Table S6C in the supplemental material). Globally, sex-biased STAT5 binding at sex-biased DHS is 3.6-fold more common in male than female liver (male-biased STAT5 binding at 624 of 2,714 male-biased DHS versus female-biased STAT5 binding at 173 of 1,333 female-biased DHS), which may enable STAT5 to distally regulate a larger number of sex-biased genes in male liver. Finally, sex-biased STAT5 binding is most highly enriched in association with sex-biased K27ac, the mark of an active enhancer, both at sex-biased and at sex-independent DHS (Fig. 6B). This finding suggests that many of the sex-dependent actions of STAT5 action proceed via sex-biased enhancer sequences and highlights the importance of sex differences in chromatin state for sex-dependent gene expression. A failure to recapitulate a sex-dependent chromatin environment may explain the inability of some sex-biased DHS sequences to confer sex-differential reporter gene expression when they are assayed for enhancer activity in liver in vivo (39).
Our analysis of sex-differentiated chromatin states in combination with binding data for TFs implicated in sex-biased liver gene regulation supports the model depicted in Fig. 8, which shows proposed regulatory events and relationships for the largest classes of sex-biased genes with (F3 and M4) or without (F1 and M1) sex differences in proximal chromatin marks. Plasma GH profiles, which are sexually dimorphic, impart sex-differential patterns of STAT5 activation (75, 76) and sex-differential expression of Bcl6 (32) and Cux2 (33) and regulate DHS opening and closing in a sex-biased manner (39). This sex-dependent and plasma GH pattern-dependent remodeling of regulatory sites involves the enhancer-associated modifications K4me1 and K27ac, as evidenced by the strong enrichment for GH responsiveness among DHS that show sex bias in these two activating marks (Fig. 6B). The pioneer factor FOXA1, which initiates chromatin opening that activates enhancers during liver development (77), and the related FOXA2 show sex-dependent binding that correlates closely with sex bias in DHS and in K4me1 and K27ac marks (Fig. 6; see also Fig. S8 and S9 in the supplemental material) and are proposed to induce the observed sex-differential opening at male-biased DHS in mouse liver. Supporting this proposal, male-biased DHS show a sexually dimorphic K4me1 distribution around the DHS summits that is FOXA dependent (Fig. 7). This pattern is reminiscent of unstimulated LNCaP cells, where androgen receptor binding sites lacking FOXA1 binding have a broad K4me2 peak compared to a bimodal peak surrounding the summit of androgen receptor binding sites associated with FOXA1 binding (8). Furthermore, cell-type-specific binding by FOXA1 is enriched at sites with cell-type-specific K4me1 and K4me2 marks, which are required for FOXA1 binding at enhancers (72). Thus, FOXA1 is recruited to sites marked by cell-type-specific K4 methylation, which it utilizes as an epigenetic signature for chromatin opening. By extension, our finding that FOXA1 and FOXA2 are recruited in a male-biased manner to sites with male-enriched K4me1 marks in a bimodal distribution suggests that FOXA induces repositioning of nucleosomes to flank the site, thereby increasing DNase hypersensitivity in male liver at the peak center. Since 71% of the male-enriched FOXA2 binding sites at male-biased DHS overlap a male-enriched FOXA1 binding site, FOXA1 could be the key factor for sex-dependent chromatin opening. Indeed, at female-biased DHS, where FOXA2, but not FOXA1, commonly exhibits female-enriched binding, sex differences in the pattern of K4me1 read density are not apparent (Fig. 7D).
Both bimodal and monomodal patterns of K4me1 marks are seen at FOXA2 and HNF4A binding sites in liver, but the bimodal loci are associated with more highly expressed genes and are more likely to confer liver specificity (13). Further, K4me1 peaks that are bimodal in liver and monomodal in islets are associated with liver-specific genes, and a corresponding relationship holds for bimodal peaks in islets and islet-specific genes (13). The bimodal K4me1 peak is associated with nucleosome depletion at the peak center, as confirmed by nucleosome positioning analysis at a few representative loci (13). In liver, bimodal K4me1 loci occupied by FOXA2 are most likely to be cooccupied by HNF4A (13), and STAT5 binding sites in female liver show significant cooccupation by HNF4A, FOXA1, and FOXA2 (78). HNF4A is required for sex-biased gene expression and may act cooperatively with STAT5 to influence gene expression (35, 36). Thus, FOXA1, and perhaps FOXA2, when recruited to male-biased DHS marked by male-biased K4me1, may act in cooperation with HNF4A and/or STAT5 to regulate sex-biased genes in male liver. Of note, about half of all FOXA2 binding sites in mouse liver occur at nucleosome-bound sites, including some sites that exhibit bimodal K4me1 distribution (79). Consequently, the bimodal distribution of K4me1 cannot be taken as a definitive indication of nucleosome repositioning at every such site (79). Further studies are needed to identify sites of sex-dependent, GH-regulated nucleosome remodeling and to elucidate the individual roles of FOXA1 and FOXA2 in these remodeling events.
Sex-dependent nucleosome positioning at male-biased DHS but not female-biased DHS may explain our previous finding that male-enriched STAT5 binding sites are characterized by open chromatin in male liver versus relatively closed chromatin in female liver (30), which suggests that there is a specific, sex-dependent mechanism to open these sites in male liver. In contrast, female-enriched liver STAT5 binding sites are relatively open in both males and females (30) and therefore may not require sex-dependent chromatin opening by FOXA or other factors. Other mechanisms, such as the persistence of STAT5 binding activity in female liver (30), may be sufficient to confer female-biased gene expression for the target genes of female-enriched STAT5 binding sites.
BCL6 and CUX2: chromatin environments of binding sites and at target genes.
BCL6, which is expressed in a male-biased manner subject to regulation by GH and STAT5, is a potent transcriptional repressor that competes with STAT5 for binding to chromatin and is associated with repression of many female-biased genes in mouse liver (30, 32). BCL6 preferentially binds sex-independent DHS with sex-independent chromatin marks (see Fig. S8G in the supplemental material), and the female-biased targets of BCL6 are enriched for genes that lack local sex-biased chromatin marks (Fig. 5A and 8B and C; see also Table S6C and D in the supplemental material) and show weak sex bias (see Fig. S7B in the supplemental material). These findings support our earlier proposal that sex-biased genes deficient in nearby sex-biased DHS are in part regulated by TFs whose expression or activity is sex dependent (e.g., BCL6), without a need for sex bias in the chromatin environment at the TF binding site (39). The enrichment that BCL6 shows for weakly but not strongly female-biased targets may reflect the modest sex difference that characterizes BCL6 expression in mouse liver (32).
The high female specificity of CUX2 expression (∼100-fold) (33) effectively restricts its binding to female liver chromatin, where CUX2 binding is enriched at male-biased DHS and at sites with male-enriched STAT5 binding, and is associated with repression of ∼30% of male-biased genes (34). Unlike the female-biased targets of BCL6, male-biased genes that show the highest enrichment for being a proximal target of CUX2 (class M4 genes) are characterized by sex differences in local chromatin marks and also show the highest enrichment of male-biased binding of STAT5 and FOXA1 (Fig. 5B and 8A). The male-gene-repressive actions of CUX2 in female liver are also associated with CUX2 binding at male-biased DHS and at sites with male-enriched STAT5 binding (34). This suggests that CUX2 may keep male-biased DHS closed in female liver and thereby contribute to the male bias in chromatin accessibility and STAT5 binding seen at those sites, which may be opened by FOXA1 in male liver. CUX2 can also activate female-biased genes in female mouse liver (34), and, consistently, CUX2 binding was enriched at both female-biased and sex-independent DHS that map to several classes of female-biased genes (see Table S6B and D in the supplemental material). Furthermore, female-biased targets of CUX2 are frequently marked by enrichment for K27me3 at the gene body in male liver (Fig. 5C). Thus, the activating role of CUX2 may involve chromatin remodeling at target genes—in this case, derepression of female-biased genes by removal of K27me3 repressive marks across the gene body (Fig. 8B). Presumably, the male bias in K27me3 marks at these genes is introduced at puberty, when the female-specific expression of CUX2 and a majority of its targets first emerge (80).
Conclusions.
Comparison of the epigenetic environment and chromatin states that characterize male and female mouse liver and integration of these data with gene expression and TF binding data revealed distinct mechanisms of sex-biased gene regulation in male and female liver. Sex-dependent K27me3-mediated repression is shown to be an important mechanism of repression of highly female-biased but not of male-biased genes, and a sexually dimorphic K4me1 profile, which likely indicates sex-dependent nucleosome repositioning by FOXA pioneer factors, is seen at male-biased but not female-biased regulatory sites. TFs implicated in sex-biased gene expression were found to exhibit distinct relationships with epigenetic marks: STAT5-mediated activation was associated with sex-biased chromatin modifications, while BCL6-mediated repression was primarily associated with sex-independent chromatin modifications, both at BCL6 binding sites and at its target genes. Finally, the female-specific CUX2 may repress male-biased genes by interfering with STAT5 binding, while CUX2 activation of highly female-biased genes may involve removal of K27me3 repressive marks.
Supplementary Material
ACKNOWLEDGMENTS
We thank the following lab members for their contributions to this study: Ekaterina Laz for ChIP sample preparation, Andy Rampersaud for confirmatory qPCR analysis of ChIP samples, Peng-Ying Hao and Tisha Melia for providing RNA-seq data, and Gracia Bonilla for initial analysis of sex-biased FOXA1 and FOXA2 binding sites.
Author contributions were as follows: A.S. and D.J.W. conceived and designed the study; A.S. carried out all of the computational studies, including analysis of ChIP-seq and DNase-seq data sets, and chromatin state analysis and prepared figures and tables for publication; A.S. and D.J.W. jointly wrote and revised the manuscript.
This work was supported in part by NIH grant DK33765 (to D.J.W.).
Footnotes
Published ahead of print 8 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00280-13.
REFERENCES
- 1.Ernst J, Kheradpour P, Mikkelsen TS, Shoresh N, Ward LD, Epstein CB, Zhang X, Wang L, Issner R, Coyne M, Ku M, Durham T, Kellis M, Bernstein BE. 2011. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature 473:43–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Heintzman ND, Stuart RK, Hon G, Fu Y, Ching CW, Hawkins RD, Barrera LO, Van Calcar S, Qu C, Ching KA, Wang W, Weng Z, Green RD, Crawford GE, Ren B. 2007. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome. Nat. Genet. 39:311–318 [DOI] [PubMed] [Google Scholar]
- 3.Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, Zhang X, Green RD, Lobanenkov VV, Stewart R, Thomson JA, Crawford GE, Kellis M, Ren B. 2009. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature 459:108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dong X, Greven MC, Kundaje A, Djebali S, Brown JB, Cheng C, Gingeras TR, Gerstein M, Guigo R, Birney E, Weng Z. 2012. Modeling gene expression using chromatin features in various cellular contexts. Genome Biol. 13:R53. 10.1186/gb-2012-13-9-r53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, Garg K, John S, Sandstrom R, Bates D, Boatman L, Canfield TK, Diegel M, Dunn D, Ebersol AK, Frum T, Giste E, Johnson AK, Johnson EM, Kutyavin T, Lajoie B, Lee BK, Lee K, London D, Lotakis D, Neph S, Neri F, Nguyen ED, Qu H, Reynolds AP, Roach V, Safi A, Sanchez ME, Sanyal A, Shafer A, Simon JM, Song L, Vong S, Weaver M, Yan Y, Zhang Z, Lenhard B, Tewari M, Dorschner MO, Hansen RS, Navas PA, Stamatoyannopoulos G, Iyer VR, Lieb JD, Sunyaev SR, Akey JM, Sabo PJ, Kaul R, Furey TS, Dekker J, Crawford GE, Stamatoyannopoulos JA. 2012. The accessible chromatin landscape of the human genome. Nature 489:75–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Natarajan A, Yardimci GG, Sheffield NC, Crawford GE, Ohler U. 2012. Predicting cell-type-specific gene expression from regions of open chromatin. Genome Res. 22:1711–1722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arvey A, Agius P, Noble WS, Leslie C. 2012. Sequence and chromatin determinants of cell-type-specific transcription factor binding. Genome Res. 22:1723–1734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He HH, Meyer CA, Chen MW, Jordan VC, Brown M, Liu XS. 2012. Differential DNase I hypersensitivity reveals factor-dependent chromatin dynamics. Genome Res. 22:1015–1025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kundaje A, Kyriazopoulou-Panagiotopoulou S, Libbrecht M, Smith CL, Raha D, Winters EE, Johnson SM, Snyder M, Batzoglou S, Sidow A. 2012. Ubiquitous heterogeneity and asymmetry of the chromatin environment at regulatory elements. Genome Res. 22:1735–1747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang J, Zhuang J, Iyer S, Lin X, Whitfield TW, Greven MC, Pierce BG, Dong X, Kundaje A, Cheng Y, Rando OJ, Birney E, Myers RM, Noble WS, Snyder M, Weng Z. 2012. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 22:1798–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zaret KS, Carroll JS. 2011. Pioneer transcription factors: establishing competence for gene expression. Genes Dev. 25:2227–2241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Friedman JR, Kaestner KH. 2006. The Foxa family of transcription factors in development and metabolism. Cell. Mol. Life Sci. 63:2317–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hoffman BG, Robertson G, Zavaglia B, Beach M, Cullum R, Lee S, Soukhatcheva G, Li L, Wederell ED, Thiessen N, Bilenky M, Cezard T, Tam A, Kamoh B, Birol I, Dai D, Zhao Y, Hirst M, Verchere CB, Helgason CD, Marra MA, Jones SJ, Hoodless PA. 2010. Locus co-occupancy, nucleosome positioning, and H3K4me1 regulate the functionality of FOXA2-, HNF4A-, and PDX1-bound loci in islets and liver. Genome Res. 20:1037–1051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. 2010. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38:576–589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng C, Alexander R, Min R, Leng J, Yip KY, Rozowsky J, Yan KK, Dong X, Djebali S, Ruan Y, Davis CA, Carninci P, Lassman T, Gingeras TR, Guigo R, Birney E, Weng Z, Snyder M, Gerstein M. 2012. Understanding transcriptional regulation by integrative analysis of transcription factor binding data. Genome Res. 22:1658–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Garber M, Yosef N, Goren A, Raychowdhury R, Thielke A, Guttman M, Robinson J, Minie B, Chevrier N, Itzhaki Z, Blecher-Gonen R, Bornstein C, Amann-Zalcenstein D, Weiner A, Friedrich D, Meldrim J, Ram O, Cheng C, Gnirke A, Fisher S, Friedman N, Wong B, Bernstein BE, Nusbaum C, Hacohen N, Regev A, Amit I. 2012. A high-throughput chromatin immunoprecipitation approach reveals principles of dynamic gene regulation in mammals. Mol. Cell 47:810–822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rinn JL, Snyder M. 2005. Sexual dimorphism in mammalian gene expression. Trends Genet. 21:298–305 [DOI] [PubMed] [Google Scholar]
- 18.Wauthier V, Sugathan A, Meyer RD, Dombkowski AA, Waxman DJ. 2010. Intrinsic sex differences in the early growth hormone responsiveness of sex-specific genes in mouse liver. Mol. Endocrinol. 24:667–678 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wauthier V, Waxman DJ. 2008. Sex-specific early growth hormone response genes in rat liver. Mol. Endocrinol. 22:1962–1974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang Y, Klein K, Sugathan A, Nassery N, Dombkowski A, Zanger UM, Waxman DJ. 2011. Transcriptional profiling of human liver identifies sex-biased genes associated with polygenic dyslipidemia and coronary artery disease. PLoS One 6:e23506. 10.1371/journal.pone.0023506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Waxman DJ, Holloway MG. 2009. Sex differences in the expression of hepatic drug metabolizing enzymes. Mol. Pharmacol. 76:215–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Scandlyn MJ, Stuart EC, Rosengren RJ. 2008. Sex-specific differences in CYP450 isoforms in humans. Expert Opin. Drug Metab. Toxicol. 4:413–424 [DOI] [PubMed] [Google Scholar]
- 23.Schwartz JB. 2007. The current state of knowledge on age, sex, and their interactions on clinical pharmacology. Clin. Pharmacol. Ther. 82:87–96 [DOI] [PubMed] [Google Scholar]
- 24.Yokoyama Y, Nimura Y, Nagino M, Bland KI, Chaudry IH. 2005. Current understanding of gender dimorphism in hepatic pathophysiology. J. Surg. Res. 128:147–156 [DOI] [PubMed] [Google Scholar]
- 25.Baik M, Yu JH, Hennighausen L. 2011. Growth hormone-STAT5 regulation of growth, hepatocellular carcinoma, and liver metabolism. Ann. N. Y. Acad. Sci. 1229:29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ruggieri A, Barbati C, Malorni W. 2010. Cellular and molecular mechanisms involved in hepatocellular carcinoma gender disparity. Int. J. Cancer 127:499–504 [DOI] [PubMed] [Google Scholar]
- 27.Choi HK, Waxman DJ. 2000. Plasma growth hormone pulse activation of hepatic JAK-STAT5 signaling: developmental regulation and role in male-specific liver gene expression. Endocrinology 141:3245–3255 [DOI] [PubMed] [Google Scholar]
- 28.Choi HK, Waxman DJ. 1999. Growth hormone, but not prolactin, maintains, low-level activation of STAT5a and STAT5b in female rat liver. Endocrinology 140:5126–5135 [DOI] [PubMed] [Google Scholar]
- 29.Lanning NJ, Carter-Su C. 2006. Recent advances in growth hormone signaling. Rev. Endocr Metab. Disord. 7:225–235 [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y, Laz EV, Waxman DJ. 2012. Dynamic, sex-differential STAT5 and BCL6 binding to sex-biased, growth hormone-regulated genes in adult mouse liver. Mol. Cell. Biol. 32:880–896 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clodfelter KH, Holloway MG, Hodor P, Park SH, Ray WJ, Waxman DJ. 2006. Sex-dependent liver gene expression is extensive and largely dependent upon signal transducer and activator of transcription 5b (STAT5b): STAT5b-dependent activation of male genes and repression of female genes revealed by microarray analysis. Mol. Endocrinol. 20:1333–1351 [DOI] [PubMed] [Google Scholar]
- 32.Meyer RD, Laz EV, Su T, Waxman DJ. 2009. Male-specific hepatic Bcl6: growth hormone-induced block of transcription elongation in females and binding to target genes inversely coordinated with STAT5. Mol. Endocrinol. 23:1914–1926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Laz EV, Holloway MG, Chen CS, Waxman DJ. 2007. Characterization of three growth hormone-responsive transcription factors preferentially expressed in adult female liver. Endocrinology 148:3327–3337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Conforto TL, Zhang Y, Sherman J, Waxman DJ. 2012. Impact of CUX2 on the female mouse liver transcriptome: activation of female-biased genes and repression of male-biased genes. Mol. Cell. Biol. 32:4611–4627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holloway MG, Laz EV, Waxman DJ. 2006. Codependence of growth hormone-responsive, sexually dimorphic hepatic gene expression on signal transducer and activator of transcription 5b and hepatic nuclear factor 4α. Mol. Endocrinol. 20:647–660 [DOI] [PubMed] [Google Scholar]
- 36.Holloway MG, Miles GD, Dombkowski AA, Waxman DJ. 2008. Liver-specific hepatocyte nuclear factor-4α deficiency: greater impact on gene expression in male than in female mouse liver. Mol. Endocrinol. 22:1274–1286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Delesque-Touchard N, Park SH, Waxman DJ. 2000. Synergistic action of hepatocyte nuclear factors 3 and 6 on CYP2C12 gene expression and suppression by growth hormone-activated STAT5b. Proposed model for female specific expression of CYP2C12 in adult rat liver. J. Biol. Chem. 275:34173–34182 [DOI] [PubMed] [Google Scholar]
- 38.Lahuna O, Fernandez L, Karlsson H, Maiter D, Lemaigre FP, Rousseau GG, Gustafsson J, Mode A. 1997. Expression of hepatocyte nuclear factor 6 in rat liver is sex-dependent and regulated by growth hormone. Proc. Natl. Acad. Sci. U. S. A. 94:12309–12313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ling G, Sugathan A, Mazor T, Fraenkel E, Waxman DJ. 2010. Unbiased, genome-wide in vivo mapping of transcriptional regulatory elements reveals sex differences in chromatin structure associated with sex-specific liver gene expression. Mol. Cell. Biol. 30:5531–5544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chia DJ, Rotwein P. 2010. Defining the epigenetic actions of growth hormone: acute chromatin changes accompany GH-activated gene transcription. Mol. Endocrinol. 24:2038–2049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chai X, Zeng S, Xie W. 2013. Nuclear receptors PXR and CAR: implications for drug metabolism regulation, pharmacogenomics and beyond. Expert Opin. Drug Metab. Toxicol. 9:253–266 [DOI] [PubMed] [Google Scholar]
- 42.Kakizaki S, Yamazaki Y, Takizawa D, Negishi M. 2008. New insights on the xenobiotic-sensing nuclear receptors in liver diseases—CAR and PXR. Curr. Drug Metab. 9:614–621 [DOI] [PubMed] [Google Scholar]
- 43.Hernandez JP, Mota LC, Baldwin WS. 2009. Activation of CAR and PXR by dietary, environmental and occupational chemicals alters drug metabolism, intermediary metabolism, and cell proliferation. Curr. Pharmacogenomics Person Med. 7:81–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li Z, Tuteja G, Schug J, Kaestner KH. 2012. Foxa1 and Foxa2 are essential for sexual dimorphism in liver cancer. Cell 148:72–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Langmead B, Trapnell C, Pop M, Salzberg SL. 2009. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10:R25. 10.1186/gb-2009-10-3-r25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. 2008. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9:R137. 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zang C, Schones DE, Zeng C, Cui K, Zhao K, Peng W. 2009. A clustering approach for identification of enriched domains from histone modification ChIP-Seq data. Bioinformatics 25:1952–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Audic S, Claverie JM. 1997. The significance of digital gene expression profiles. Genome Res. 7:986–995 [DOI] [PubMed] [Google Scholar]
- 49.Shao Z, Zhang Y, Yuan GC, Orkin SH, Waxman DJ. 2012. MAnorm: a robust model for quantitative comparison of ChIP-Seq data sets. Genome Biol. 13:R16. 10.1186/gb-2012-13-3-r16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Parkhomchuk D, Borodina T, Amstislavskiy V, Banaru M, Hallen L, Krobitsch S, Lehrach H, Soldatov A. 2009. Transcriptome analysis by strand-specific sequencing of complementary DNA. Nucleic Acids Res. 37:e123. 10.1093/nar/gkp596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Eisen MB, Spellman PT, Brown PO, Botstein D. 1998. Cluster analysis and display of genome-wide expression patterns. Proc. Natl. Acad. Sci. U. S. A. 95:14863–14868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Saldanha AJ. 2004. Java Treeview–extensible visualization of microarray data. Bioinformatics 20:3246–3248 [DOI] [PubMed] [Google Scholar]
- 53.Robinson MD, McCarthy DJ, Smyth GK. 2010. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26:139–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tojima H, Kakizaki S, Yamazaki Y, Takizawa D, Horiguchi N, Sato K, Mori M. 2012. Ligand dependent hepatic gene expression profiles of nuclear receptors CAR and PXR. Toxicol. Lett. 212:288–297 [DOI] [PubMed] [Google Scholar]
- 55.Tian J, Huang H, Hoffman B, Liebermann DA, Ledda-Columbano GM, Columbano A, Locker J. 2011. Gadd45β is an inducible coactivator of transcription that facilitates rapid liver growth in mice. J. Clin. Invest. 121:4491–4502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sanderson LM, Boekschoten MV, Desvergne B, Muller M, Kersten S. 2010. Transcriptional profiling reveals divergent roles of PPARα and PPARβ/δ in regulation of gene expression in mouse liver. Physiol. Genomics 41:42–52 [DOI] [PubMed] [Google Scholar]
- 57.Boverhof DR, Burgoon LD, Tashiro C, Chittim B, Harkema JR, Jump DB, Zacharewski TR. 2005. Temporal and dose-dependent hepatic gene expression patterns in mice provide new insights into TCDD-mediated hepatotoxicity. Toxicol. Sci. 85:1048–1063 [DOI] [PubMed] [Google Scholar]
- 58.Huang da W, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4:44–57 [DOI] [PubMed] [Google Scholar]
- 59.Huang da W, Sherman BT, Lempicki RA. 2009. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102:15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R. 2010. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. U. S. A. 107:21931–21936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. 2007. High-resolution profiling of histone methylations in the human genome. Cell 129:823–837 [DOI] [PubMed] [Google Scholar]
- 63.Mikkelsen TS, Ku M, Jaffe DB, Issac B, Lieberman E, Giannoukos G, Alvarez P, Brockman W, Kim TK, Koche RP, Lee W, Mendenhall E, O'Donovan A, Presser A, Russ C, Xie X, Meissner A, Wernig M, Jaenisch R, Nusbaum C, Lander ES, Bernstein BE. 2007. Genome-wide maps of chromatin state in pluripotent and lineage-committed cells. Nature 448:553–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bernstein BE, Humphrey EL, Erlich RL, Schneider R, Bouman P, Liu JS, Kouzarides T, Schreiber SL. 2002. Methylation of histone H3 Lys 4 in coding regions of active genes. Proc. Natl. Acad. Sci. U. S. A. 99:8695–8700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bannister AJ, Schneider R, Myers FA, Thorne AW, Crane-Robinson C, Kouzarides T. 2005. Spatial distribution of di- and tri-methyl lysine 36 of histone H3 at active genes. J. Biol. Chem. 280:17732–17736 [DOI] [PubMed] [Google Scholar]
- 66.Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. 2011. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470:279–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rosenfeld JA, Wang Z, Schones DE, Zhao K, DeSalle R, Zhang MQ. 2009. Determination of enriched histone modifications in non-genic portions of the human genome. BMC Genomics 10:143. 10.1186/1471-2164-10-143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Silva J, Mak W, Zvetkova I, Appanah R, Nesterova TB, Webster Z, Peters AH, Jenuwein T, Otte AP, Brockdorff N. 2003. Establishment of histone h3 methylation on the inactive X chromosome requires transient recruitment of Eed-Enx1 polycomb group complexes. Dev. Cell 4:481–495 [DOI] [PubMed] [Google Scholar]
- 69.Boggs BA, Cheung P, Heard E, Spector DL, Chinault AC, Allis CD. 2002. Differentially methylated forms of histone H3 show unique association patterns with inactive human X chromosomes. Nat. Genet. 30:73–76 [DOI] [PubMed] [Google Scholar]
- 70.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, Jaenisch R, Wagschal A, Feil R, Schreiber SL, Lander ES. 2006. A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125:315–326 [DOI] [PubMed] [Google Scholar]
- 71.Waxman DJ. 1999. P450 gene induction by structurally diverse xenochemicals: central role of nuclear receptors CAR, PXR, and PPAR. Arch. Biochem. Biophys. 369:11–23 [DOI] [PubMed] [Google Scholar]
- 72.Lupien M, Eeckhoute J, Meyer CA, Wang Q, Zhang Y, Li W, Carroll JS, Liu XS, Brown M. 2008. FoxA1 translates epigenetic signatures into enhancer-driven lineage-specific transcription. Cell 132:958–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. 2002. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol. Cell 9:279–289 [DOI] [PubMed] [Google Scholar]
- 74.Song L, Zhang Z, Grasfeder LL, Boyle AP, Giresi PG, Lee BK, Sheffield NC, Graf S, Huss M, Keefe D, Liu Z, London D, McDaniell RM, Shibata Y, Showers KA, Simon JM, Vales T, Wang T, Winter D, Clarke ND, Birney E, Iyer VR, Crawford GE, Lieb JD, Furey TS. 2011. Open chromatin defined by DNase I and FAIRE identifies regulatory elements that shape cell-type identity. Genome Res. 21:1757–1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gebert CA, Park SH, Waxman DJ. 1999. Down-regulation of liver JAK2-STAT5b signaling by the female plasma pattern of continuous growth hormone stimulation. Mol. Endocrinol. 13:213–227 [DOI] [PubMed] [Google Scholar]
- 76.Gebert CA, Park SH, Waxman DJ. 1999. Termination of growth hormone pulse-induced STAT5b signaling. Mol. Endocrinol. 13:38–56 [DOI] [PubMed] [Google Scholar]
- 77.Zaret K. 1999. Developmental competence of the gut endoderm: genetic potentiation by GATA and HNF3/fork head proteins. Dev. Biol. 209:1–10 [DOI] [PubMed] [Google Scholar]
- 78.Kang K, Robinson GW, Hennighausen L. 2013. Comprehensive meta-analysis of Signal Transducers and Activators of Transcription (STAT) genomic binding patterns discerns cell-specific cis-regulatory modules. BMC Genomics 14:4. 10.1186/1471-2164-14-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Li Z, Schug J, Tuteja G, White P, Kaestner KH. 2011. The nucleosome map of the mammalian liver. Nat. Struct. Mol. Biol. 18:742–746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Conforto TL, Waxman DJ. 2012. Sex-specific mouse liver gene expression: genome-wide analysis of developmental changes from pre-pubertal period to young adulthood. Biol. Sex Differ. 3:9. 10.1186/2042-6410-3-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.