

Abstract
One of the basic functions of insulin in the body is to inhibit lipolysis in adipocytes. Recently, we have found that insulin inhibits lipolysis and promotes triglyceride storage by decreasing transcription of adipose triglyceride lipase via the mTORC1-mediated pathway (P. Chakrabarti et al., Diabetes 59:775–781, 2010), although the mechanism of this effect remained unknown. Here, we used a genetic screen in Saccharomyces cerevisiae in order to identify a transcription factor that mediates the effect of Tor1 on the expression of the ATGL ortholog in yeast. This factor, Msn4p, has homologues in mammalian cells that form a family of early growth response transcription factors. One member of the family, Egr1, is induced by insulin and nutrients and directly inhibits activity of the ATGL promoter in vitro and expression of ATGL in cultured adipocytes. Feeding animals a high-fat diet increases the activity of mTORC1 and the expression of Egr1 while decreasing ATGL levels in epididymal fat. We suggest that the evolutionarily conserved mTORC1-Egr1-ATGL regulatory pathway represents an important component of the antilipolytic effect of insulin in the mammalian organism.
INTRODUCTION
Current epidemics of metabolic diseases, such as type 2 diabetes, cardiac dysfunction, hypertension, hepatic steatosis, etc., are largely caused by widespread obesity. Although obesity can affect human health via several different mechanisms, the best-established connection between obesity and metabolic disease is elevated and/or dysregulated levels of circulating free fatty acids (FFA). In addition to their direct pathological effects, superfluous FFA accumulate in the form of lipids, and their metabolic products in nonadipose peripheral tissues, such as liver, skeletal muscle, heart, and pancreas and cause detrimental effects on human health via mechanisms that are currently under intense investigation (1–5).
The levels of circulating FFA depend primarily on the rates of lipolysis in the adipose tissue. One of the key physiological functions of insulin as the major anabolic hormone in the body is to restrain lipolysis and to promote fat storage in adipose tissue in the postprandial state. The failure of insulin to suppress lipolysis in adipocytes has been long considered as a very serious metabolic defect and one of the most important if not the most important causative factor of insulin resistance and diabetes mellitus (6, 7).
Complete hydrolysis of triglycerides to glycerol and fatty acids is performed jointly by tri-, di-, and monoacylglyceride lipases (8–11). The recently discovered enzyme, adipose triglyceride lipase (ATGL; also known as desnutrin, PNPLA2, TTS2.2, and iPLA2ζ) (12–14), is responsible for the bulk of triacylglycerol hydrolase activity in various cells and represents the rate-limiting lipolytic enzyme. In every experimental model tested thus far, elevated ATGL expression increases, while attenuated ATGL expression decreases, both basal and cAMP-stimulated lipolysis (12–22). At the same time, ATGL has low affinity to di- and monoacylglycerides (8, 9). The major diacylglyceride lipase in adipocytes is hormone-sensitive lipase (HSL). Monoacylglyceride products of HSL are hydrolyzed by monoacylglyceride lipase (8, 9).
According to current views, lipolysis is regulated primarily at the posttranslational level with the cyclic AMP (cAMP)-mediated signaling pathway playing the key role in this process. Briefly, phosphorylation of perilipin and HSL by protein kinase A leads to the recruitment of HSL to the lipid droplet and activation of the enzyme. At the same time, a protein cofactor of ATGL, CGI-58, dissociates from phosphorylated perilipin and activates ATGL (10). Jointly, both processes rapidly and significantly stimulate lipolysis. Within this model, the inhibitory effect of insulin on lipolysis is attributed primarily to the inhibition of cAMP-mediated signaling to HSL via Akt-dependent (9, 23) and -independent (24) mechanisms.
However, in order to have a lasting effect on lipolysis, insulin has to suppress its rate-limiting enzyme, ATGL. Indeed, regulation of lipolysis in vivo by physiological stimuli, such as insulin, physical exercise, feeding, and fasting (13, 19, 25–29), are accompanied and likely to be mediated by changes in ATGL expression. Thus, not only posttranslational regulation of the enzymatic activity but also tight control of ATGL expression is necessary for the lipolytic control and FFA homeostasis. However, unlike posttranslational regulation that has been studied in much detail (8–11), very little is known about regulation of ATGL expression.
In order to fill this gap, we initiated a search for the pathways that regulate expression of ATGL by nutrients and insulin. We have found two novel pathways: the mTORC1-mediated pathway that inhibits lipolysis by decreasing transcription of ATGL (22) and the Sirt1/FoxO1-mediated pathway that activates lipolysis by increasing transcription of ATGL (30, 31). Since FoxO1 directly binds to and stimulates the activity of the ATGL promoter (30), the mechanism of its action seems evident. However, the mechanism of mTORC1 action on transcription of ATGL remained obscure. Importantly, a similar regulatory link between dTORC1 and the ATGL homologue, Brummer, exists in Drosophila (32, 33). Conservation of this regulatory pathway in the evolution suggests that it plays an essential role in animal physiology and makes it an attractive target for investigation. Here, we report that mTORC1 suppresses lipolysis in cultured adipocytes via the immediate-early response transcription factor Egr1 that directly inhibits ATGL gene expression.
MATERIALS AND METHODS
Antibodies.
Polyclonal antibodies against ATGL and perilipin were from Cell Signaling (Beverly, MA). Polyclonal antibody against Egr1 (sc110) was from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal anti-β-actin antibody was from Sigma (St. Louis, MO). A rabbit polyclonal antibody against cellugyrin was described previously (34). Polyclonal antibody against perilipin and all phospho-specific antibodies were from Cell Signaling (Beverly, MA).
Yeast strains and media.
The Saccharomyces cerevisiae wild-type strain BY4742 (MATα his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) and single gene deletion mutants (Invitrogen yeast gene deletion library) were cultured in YP medium (1% yeast extract and 2% peptone containing 2% glucose as a carbon source) at 30°C with rotational shaking at 200 rpm.
Cell culture.
3T3-L1 preadipocytes were cultured, differentiated, and maintained as described previously (34). HEK 293T cells and mouse embryonic fibroblasts (MEFs) were grown in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) in 2 mM l-glutamine, 100 U of penicillin/ml, and 100 μg of streptomycin/ml.
Animals and procedures.
Age-matched, 8-week-old C57/BL6 male mice were purchased from Jackson Laboratory, maintained at three mice per cage at a controlled temperature (22°C) and a 12-h light-dark cycle (light on from 7 a.m. to 5 p.m.), and allowed free access to food and water. At 10 weeks of age, the mice were fed under either high-fat (HF; 45% of calories from fat, mainly as lard) or low-fat (LF; 10% of calories from fat) diets for 14 weeks. Both diets were purchased from Research Diets, Inc. (New Brunswick, NJ). Body weights were recorded weekly. At the end of the LF/HF feeding and after a 4-h fast, the mice were decapitated after CO2 anesthesia, and the fat pads were dissected rapidly, divided into aliquots in ∼100-mg pieces, snap-frozen in liquid nitrogen, and stored at −80°C. All procedures were approved by the Boston University Medical Center Animal Care and Use Committee.
Transient transfections and reporter gene assays.
ATGL promoter luciferase constructs (25) and pcDNA3 expression plasmids for EGR1 and EGR2 (35) have been described earlier. Transient transfections with cDNA were performed using Lipofectamine 2000 (Invitrogen Life Technologies, Grand Island, NY) according to the manufacturer's instructions. Briefly, ca. 80% confluent HEK293T cells or ca. 50% confluent MEFs were transfected with 500 ng of luciferase reporter constructs, 500 ng of Egr1 cDNA, 500 ng of Egr2 cDNA, and 100 ng of enhanced green fluorescent protein (eGFP) cDNA in a six-well plate format. All experiments were performed in triplicate. After 48 h of transfection, the cells were harvested in reporter lysis buffer (Promega). The luciferase activity was determined in whole-cell lysates using a Promega luciferase assay kit and is expressed as relative light units. The expression of eGFP was measured fluorometrically. Firefly luciferase was normalized by eGFP fluorescence to correct for transfection efficiency.
Site-directed mutagenesis.
The consensus Egr1 binding site in the ATGL promoter was identified by using the MatInspector promoter analysis tool (Genomatix Software, Ann Arbor, MI), and conserved nucleotides −44, −42, −41, −40, and −36 were replaced by T's using a QuikChange II-XL site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA). Primers m1 and m2 for site-directed mutagenesis were synthesized by Eurofins MWG Operon, Huntsville, AL: m1, 5′-CAAAATTCTGAGCCAGGCGCCCtGtttTCAtCCGCACTAAAACACCTCCTC-3′; and m2, 5′-CTTACGCGTGCTAGCGCCCtGtttTCAtCCGCACTAAAACACCTCCTC-3′ (mutations are indicated in lowercase and underlined). The ATGL promoter constructs 2979/+21 and −48/+21 were used as templates for the PCR to produce mutations. The nucleotide sequence of all mutant constructs was confirmed by sequencing.
Adenoviral infection of 3T3-L1 adipocytes.
Egr1 cDNA in the adenoviral vector was kindly provided by E. Hofer (Medical University of Vienna, Vienna, Austria) (36). Differentiated 3T3-L1 adipocytes cultured in DMEM with 2% FBS in 6-cm plates were infected with AdEGR1 at a multiplicity of infection of 4,000. After 48 and 72 h, the cells were lysed and analyzed by Western blotting. Control cells underwent similar treatments with GFP cDNA in the adenoviral vector (kindly provided by A. S. Greenberg, Tufts University Medical School, Boston, MA).
RNA extraction and quantitative PCR.
Total RNA was extracted from the yeast cells by using acid phenol (Ambion) and from differentiated 3T3-L1 cells by using TRIzol reagent (Invitrogen). Reverse transcription of 500 ng of total RNA was performed using random decamers (RETROscript kit; Ambion, Austin, TX), and the gene expression was determined by quantitative PCR (MX4000 Multiplex qPCR system; Stratagene, La Jolla, CA). Reactions were performed in triplicate in a total volume of 25 μl containing 2.5 μl of 1:10-diluted cDNA, 1× SYBR green master mix (Brilliant II SYBR green qPCR Master Mix; Stratagene), and gene-specific primers. Gene expression was normalized by Act1 for yeast cells and by GAPDH (glyceraldehyde-3-phosphate dehydrogenase) or 36B4 for 3T3-L1 cells. Gene expression was measured by using the ΔΔCT method. DNase-treated samples and no-template controls were analyzed in parallel experiments to confirm specificity. Primer sequences are available upon request.
Lipolysis assay.
Differentiated 3T3-L1 adipocytes were incubated in phenol red-free DMEM with 2% fatty acid-free bovine serum albumin (BSA) for 1 h at 37°C in the presence or absence of 10 μM isoproterenol. The glycerol content in the medium was measured colorimetrically at 540 nm by using a triglyceride (GPO) reagent set (Pointe Scientific, Canton, MI) against a set of glycerol standards. The cells were then washed with cold phosphate-buffered saline (PBS) and lysed in 1% Triton X-100 buffer, and the protein concentration was determined and used to normalize glycerol release. All experiments were carried out in triplicate.
ChIP.
Chromatin immunoprecipitation (ChIP) studies were carried out in 3T3-L1 adipocytes using an EZ-chIP kit (Millipore) according to the manufacturer's instructions. Briefly, proteins were cross-linked to DNA by 18.5% formaldehyde, lysed in sodium dodecyl sulfate (SDS) lysis buffer, and sonicated seven times for 15 s. Egr1 proteins were then immunoprecipitated from the precleared lysates. Protein-DNA complexes were eluted, and the cross-links were reversed. Purified DNA was subjected to PCR to amplify the region between nucleotides −102 and +21 of the ATGL promoter using the following primers: 5′-CGGCGGAGGCGGAGACGCT-3′ and 5′-TCCCTGCTTGATCCAGTTGGAT-3′. For all PCRs, 10% input was used.
Gel electrophoresis and Western blotting.
Proteins were separated in SDS-polyacrylamide gels and transferred to Immobilon-P membranes (Millipore Corp., Bedford, MA) in 25 mM Tris and 192 mM glycine. After transfer, the membrane was blocked with 10% nonfat milk in PBS with 0.5% Tween 20 for 2 h. The blots were probed overnight with specific primary antibodies at 4°C, followed by 1 h of incubation at room temperature with horseradish peroxidase-conjugated secondary antibodies (Sigma). Protein bands were detected with an enhanced chemiluminescence substrate kit (Perkin-Elmer Life Sciences, Boston, MA) using a Kodak Image Station 440CF (Eastman Kodak, Rochester, NY).
Statistics.
A Student paired two-tailed t test was used to evaluate the statistical significance of the results.
RESULTS AND DISCUSSION
Previously, we found that the activation of mTORC1 suppresses ATGL gene expression and lipolysis, whereas the inhibition of mTORC1 has the opposite effect (22). In order to dissect the mechanism of mTORC1 action, we expressed several truncated mutants of the ATGL promoter linked to the luciferase cDNA (25) in wild-type and TSC2−/− MEFs that have hyperactive mTORC1 (37). In agreement with our previous observations (22), the expression of luciferase driven by the full-size ATGL promoter is suppressed in TSC2−/− MEFs (Fig. 1). Interestingly, the negative effect of mTORC1 on transcription of the reporter gene is maintained even in the case of the shortest ATGL promoter that includes only the proximal 48 bp (Fig. 1). This suggests that neither of the transcription factors that are known to control ATGL expression, namely, peroxisome proliferator-activated receptor γ (PPARγ) (25, 38–40), FoxO1 (30), and interferon regulatory factor 4 (41), play a major role in mediating the effect of mTORC1 because their binding sites are located between 1 and 3 kb upstream of the transcription initiation site (data not shown).
Fig 1.
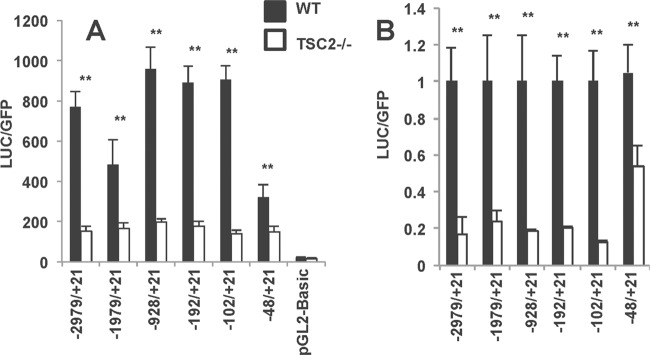
mTORC1 suppresses ATGL promoter activity. Wild-type (WT) and TSC2−/− MEFs were transiently transfected with different lengths of mouse ATGL luciferase promoter constructs, as indicated, together with eGFP. After 48 h, the cells were washed three times in cold PBS and harvested in the reporter lysis buffer. The absolute luciferase activity (A) and the relative luciferase activity (B) in cell lysates was assayed as described in Materials and Methods and normalized based on the eGFP fluorescence. Experiments were repeated four times, and data are presented for triplicate samples as means ± the standard deviations (SD). **, P < 0.01.
In order to search for a mechanism of mTORC1 action, we turned to yeast. ATGL has a functional ortholog in S. cerevisiae, a triglyceride lipase called Tgl4p (21). In order to determine whether or not the expression of Tgl4 is regulated via the Tor1-mediated pathway, we grew S. cerevisiae in the absence and in the presence of the specific Tor1/mTORC1 inhibitor, rapamycin, for 30 and 90 min and determined the expression levels of mRNA for Tgl4p and two related lipases, Tgl3p and Tgl5p, by qPCR. As shown in Fig. 2A, rapamycin strongly and specifically increased expression of Tgl4p mRNA. Thus, the transcriptional control of ATGL expression by mTORC1 should be essential for regulation of metabolism, or it would not be conserved in evolution from yeast (Fig. 2A) through Drosophila (32, 33) to mammals (22).
Fig 2.
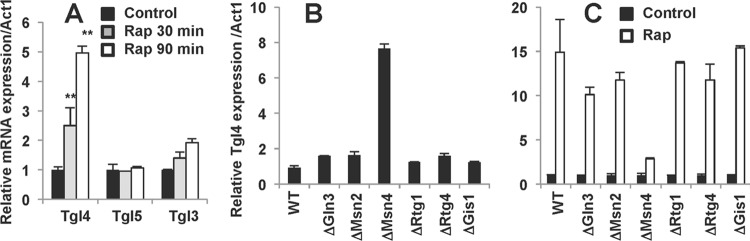
Yeast TGL4 expression is regulated by the Tor1-Msn4 pathway. (A) Wild-type yeast cells were incubated with 50 nM rapamycin (Rap) for the indicated times. mRNA expression levels for Tgl3, Tgl4, and Tgl5 were determined in triplicate samples by quantitative PCR and normalized based on the Act1 mRNA levels. The experiment was repeated three times. Data are presented relative to the expression levels in control cells and expressed for triplicate samples as means ± the SD. **, P < 0.01. (B) mRNA expression of Tgl4 in the wild type (WT) and in the indicated mutant yeast cells were determined in triplicate samples by quantitative PCR. (C) Wild-type and mutant yeast cells were incubated with or without (Control) 50 nM rapamycin (Rap) for 60 min, and the mRNA expression of Tgl4 was determined and normalized based on the Tgl4 expression levels in the respective controls. Experiments were repeated three times, and data are presented for triplicate samples as means ± the SD.
To determine which transcription factors specifically control the expression of Tgl4, we screened the S. cerevisiae deletion library. It turned out that the deletion of Msn4 not only elevated the basal expression level of Tgl4 (Fig. 2B) but also dramatically inhibited the effect of rapamycin on Tgl4 expression (Fig. 2C). Still, rapamycin had a small positive effect on Tgl4 expression even in the absence of Msn4 (Fig. 2C), suggesting that not all effects of TOR1 on Tgl4 are mediated by Msn4.
Mammalian homologues of Msn4p form a family of early growth response factors with the best-studied members called Egr1 (Krox24) and Egr2 (Krox20) (42). Although specific targets of the Egr transcription factors remain largely unknown, this family has recently been implicated in adipogenesis (43–45), insulin resistance (46), and cholesterol biosynthesis (47). Furthermore, two polymorphisms in Egr1 have been associated with impaired lipid metabolism in humans (48). In order to determine whether or not mammalian Egr1 and Egr2 can regulate expression of ATGL, we analyzed their activity in vitro toward ATGL promoter deletion constructs shown in Fig. 1. We found that Egr1 is a potent inhibitor of ATGL transcription in vitro, whereas Egr2 is significantly less effective (Fig. 3A and B). Note that Egr1 exerts a negative effect on the “minimal” 48-bp ATGL promoter (Fig. 3A) that has a consensus Egr1 binding site (Fig. 3C) (see also references 35 and 49). Indeed, the replacement of five nucleotides in this site for T's strongly decreases the inhibitory effect of Egr1 on the activity of both full-size and minimal ATGL promoters but does not change the effect of Egr2 (Fig. 3D).
Fig 3.

Egr1 suppresses ATGL promoter activity. 293T cells were transfected with different lengths of mouse ATGL luciferase promoter constructs, together with eGFP. Cells were cotransfected with either Egr1 cDNA (A), Egr2 cDNA (B), or empty vector (EV). The luciferase activity in cell lysates was assayed as described in Materials and Methods and normalized based on the GFP fluorescence. Experiments were repeated three times, and data are presented for triplicate samples as means ± the SD. (C) Schematic representation of the proximal region of ATGL promoter with the consensus Egr1 binding site. Nucleotides that have been chosen for the site-directed mutagenesis are underlined. (D) Conserved nucleotides −44, −42, −41, −40, and −36 in the ATGL promoter (underlined in panel C) were substituted by T's as described in Materials and Methods. HEK293T cells were transfected with wild-type (wt) or mutant (mt) ATGL luciferase promoter constructs, together with eGFP. Cells were cotransfected with Egr1 or Egr2 or empty vector (EV) and analyzed as described for panels A and B. (E) Wild-type and TSC2−/− MEFs were treated with 100 nM rapamycin (Rap) for 48 h. Total cell lysates were analyzed by Western blotting for Egr1. Actin served as a loading control. *, P < 0.05; **, P < 0.01 (all panels).
Since the same proximal region of the ATGL promoter is responsible for the transcriptional inhibition by mTORC1 (Fig. 1) and Egr1 (Fig. 3A), we hypothesize that mTORC1 and Egr1 are engaged in the same regulatory pathway. In support of this idea, we found that the expression of Egr1 is very low in wild-type MEFs but is significant in TSC2−/− MEFs (Fig. 3E), which may explain the results shown in Fig. 1. Also, rapamycin inhibits the expression of Egr1 in TSC2−/− MEFs (Fig. 3E).
In order to test the newly discovered regulatory link between mTORC1, Egr1, and ATGL in physiologically relevant cells and conditions, we analyzed the effect of insulin in cultured adipocytes. We have found that, in basal 3T3-L1 adipocytes, the expression of Egr1 is very low and is virtually undetectable. Insulin administration causes dramatic but transient induction of Egr1 in these cells (Fig. 4A) (see also reference 50). In agreement with several previously published reports (19, 25, 51), we have found that insulin inhibits ATGL expression (Fig. 4A). Note that suppression of ATGL mRNA and protein by insulin takes place after induction of Egr1 (Fig. 4A) and is completely preventable by the transcriptional inhibitor actinomycin D (Fig. 4B), suggesting that inhibition of ATGL expression is mediated by the de novo-synthesized Egr1. Indeed, infection of 3T3-L1 adipocytes with adenovirus carrying the Egr1 gene inhibits the expression of ATGL (Fig. 4C), as well as basal and isoproterenol-stimulated lipolysis (Fig. 4D). In order to confirm the direct interaction between Egr1 and the ATGL promoter, we carried out a ChIP experiment. We have found that, after insulin stimulation, endogenous Egr1 specifically interacts with the endogenous ATGL promoter in cultured adipocytes (Fig. 4E).
Fig 4.

Insulin-dependent Egr1 expression controls ATGL expression in 3T3-L1 adipocytes. (A) Differentiated 3T3-L1 adipocytes were treated with 100 nM insulin for the indicated periods of time. (Top panel) The levels of ATGL and Egr1 mRNA were determined in triplicate by quantitative PCR and normalized based on 36B4 mRNA. (Bottom panel) Total cell lysates were analyzed by Western blotting for Egr1 and ATGL. Actin served as a loading control. (B) 3T3-L1 adipocytes were treated with 100 nM insulin in the presence or absence of actinomycin D (AcD; 0.2 μg/ml) for 16 h. Total cell lysates were analyzed by Western blotting for ATGL. Perilipin served as a control for AcD action, and cellugyrin and actin served as loading controls. (C) 3T3-L1 adipocytes were infected with adenovirus expressing Egr1 (AdEgr1) and GFP (AdGFP) and cultured for 48 and 72 h. Total cell lysates were analyzed by Western blotting for Egr1 and ATGL. Actin served as a loading control. (D) 3T3-L1 adipocytes infected with adenovirus expressing Egr1 (AdEgr1) and GFP (AdGFP) and cultured for 48 h. Cells were then incubated in phenol red-free DMEM with 2% fatty acid-free BSA without (white bars) or with (black bars) 10 μM isoproterenol (Iso) for 2 h. Glycerol was measured in medium aliquots in triplicate and normalized by protein concentration in whole-cell lysates. Data are expressed as means ± the SD. (E) ChIP assays were performed in 3T3-L1 adipocytes treated with 100 nM insulin for 4 h. Genomic fragments were immunoprecipitated with antibody against Egr1 or rabbit IgG, amplified by PCR, separated in a 3% agarose gel, and visualized by ethidium bromide staining.
Insulin-stimulated induction of Egr1 in adipocytes is mediated, at least partially, by mTORC1 as rapamycin and PP242 inhibit this effect (Fig. 5A). In parallel, both inhibitors block the negative effect of insulin on ATGL expression (Fig. 5A) and lipolysis (Fig. 5B) in insulin-treated adipocytes.
Fig 5.

mTORC1 controls ATGL expression and lipolysis in 3T3-L1 adipocytes via Egr1. (A) 3T3-L1 adipocytes were treated with 100 nM insulin in the presence (+) or absence (−) of 100 nM rapamycin (Rapa) or PP242 (10 nM) for 16 h. Total cell lysates were analyzed by Western blotting. The dotted line indicates that irrelevant lanes have been spliced out. GAPDH served as loading control. (B) 3T3-L1 adipocytes were treated with 100 nM insulin in the presence (+) or absence (−) of 100 nM rapamycin (Rapa) or PP242 (10 nM) for 16 h. After that, cells were transferred to phenol red-free DMEM with 2% fatty acid free BSA without (white bars) or with (black bars) 10 μM isoproterenol (Iso) for 2 h. Glycerol was measured in medium aliquots in triplicate and normalized based on the protein concentration in whole-cell lysates. Data are expressed as means ± the SD. (C) 3T3-L1 adipocytes were incubated either in nutrient-free Krebs-Ringer-HEPES buffer (KRH) or in nutrient-enriched DMEM in the presence or absence of 100 nM insulin for 3 h. Levels of Egr1 mRNA were determined in triplicate by quantitative PCR and normalized by 36B4 mRNA. (D) 3T3-L1 adipocytes were incubated in nutrient-free Krebs-Ringer-HEPES buffer (KRH) for 3 h in the presence or absence of 5 mM leucine, 5 mM glucose, and 100 nM insulin as indicated. Total cell lysates were analyzed by Western blotting for phospho-4E-BP. Actin served as a loading control. The top panel shows the levels of Egr1 mRNA determined in triplicate by quantitative PCR and normalized based on the 36B4 mRNA. *, P < 0.05; **, P < 0.01 (all panels).
In order to confirm the connection between mTORC1 and Egr1, we determined the effect of nutrients on the expression of Egr1 mRNA in the absence and in the presence of insulin. We found that withdrawal of nutrients blocked insulin-stimulated induction of Egr1 mRNA (Fig. 5C). On the contrary, treatment of cultured 3T3-L1 adipocytes with leucine alone activated the mTORC1 pathway and stimulated Egr1 expression (Fig. 5D).
In the next experiment, we analyzed protein expression in mice that received a high-fat diet for 14 weeks. As reported earlier (52), the activity of mTORC1 in the fat tissues of these animals is increased (Fig. 6A). According to our model, activation of mTORC1 should decrease expression of ATGL, and ATGL levels are indeed decreased in mice fed high-fat diet (Fig. 6A). This observation, although supportive of the previous findings (41, 53), may seem to contradict a well-known fact that plasma FFA are commonly increased in obesity (10). This, however, may reflect the overall increase in adipose tissue, whereas lipolysis normalized per kg of fat mass is actually decreased in obesity (reviewed in reference 54), a finding consistent with results shown in Fig. 6A. Finally, we have determined, by qPCR, that a high-fat diet increases the expression of Egr1 (Fig. 6B).
Fig 6.
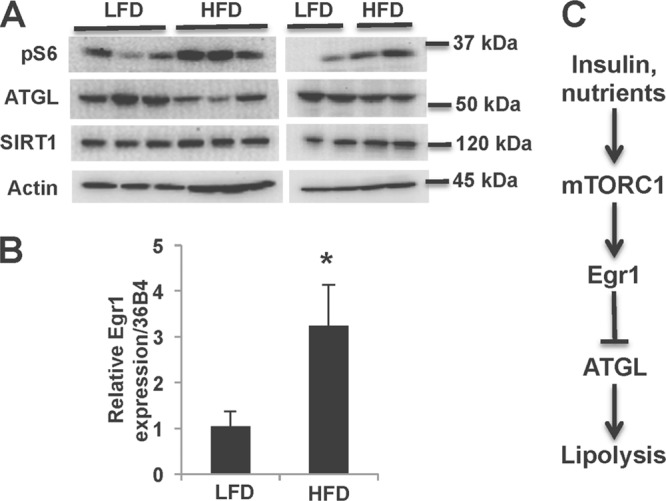
A high-fat diet activates mTORC1, increases the levels of Egr1, and decreases ATGL expression in epididymal white adipose tissue. (A) Male C57/BL6 mice were fed a low-fat or high-fat diet for 14 weeks; epididymal fat pads were dissected and frozen. Total lysates were analyzed by Western blotting. The panel shows two independent experiments with, respectively, 6 and 4 mice. (B) The levels of Egr1 mRNA were determined in triplicate by quantitative PCR and normalized based on the 36B4 mRNA levels. (C) Insulin and nutrients inhibit ATGL expression and lipolysis via the mTORC1-Egr1 regulatory axis.
Thus, our experiments identify the evolutionarily conserved mTORC1-Egr1-ATGL regulatory axis (Fig. 6C) as a novel mechanism of the antilipolytic effect of insulin. We want to emphasize here that our results do not negate the previously established mechanism of short-term insulin action on lipolysis by inhibition of cAMP-mediated signaling to HSL and perilipin (9, 23, 24). Rather, these findings demonstrate a new level of such regulation that is essential for better understanding and restraining the metabolic disease. For example, the downregulation of ATGL via this pathway may represent an essential compensatory mechanism that may be needed for maintaining physiological concentrations of circulating FFA in obesity.
ACKNOWLEDGMENTS
We thank Michael Sherman (Boston University Medical School [BUSM]), Cynthia Smas (Toledo College of Medicine), Andy Greenberg (Tufts Medical School), and Erhard Hofer (Medical University of Vienna, Vienna, Austria) for their generous gifts of reagents, Matthew Layne (BUSM) and Olga Kandror (Harvard Medical School) for helpful and insightful discussions, and Taylor English and Linda Hong for help with several experiments.
This study was supported by research grants DK52057 and AG039612 from the National Institutes of Health, research grant 7-11-BS-76 from the American Diabetes Association, and a research award from the Allen Foundation to K.V.K.
Footnotes
Published ahead of print 15 July 2013
REFERENCES
- 1.Guilherme A, Virbasius JV, Puri V, Czech MP. 2008. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat. Rev. Mol. Cell. Biol. 9:367–377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Savage DB, Petersen KF, Shulman GI. 2007. Disordered lipid metabolism and the pathogenesis of insulin resistance. Physiol. Rev. 87:507–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stumvoll M, Goldstein BJ, van Haeften TW. 2008. Type 2 diabetes: pathogenesis and treatment. Lancet 371:2153–2156 [DOI] [PubMed] [Google Scholar]
- 4.Qatanani M, Lazar MA. 2007. Mechanisms of obesity-associated insulin resistance: many choices on the menu. Genes Dev. 21:1443–1455 [DOI] [PubMed] [Google Scholar]
- 5.Coen PM, Goodpaster BH. 2012. Role of intramyocellular lipids in human health. Trends Endocrinol. Metab. 23:391–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.McGarry JD. 1992. What if Minkowski had been ageusic? An alternative angle on diabetes. Science 258:766–770 [DOI] [PubMed] [Google Scholar]
- 7.McGarry JD. 2002. Banting lecture 2001: dysregulation of fatty acid metabolism in the etiology of type 2 diabetes. Diabetes 51:7–18 [DOI] [PubMed] [Google Scholar]
- 8.Zechner R, Kienesberger PC, Haemmerle G, Zimmermann R, Lass A. 2009. Adipose triglyceride lipase and the lipolytic catabolism of cellular fat stores. J. Lipid Res. 50:3–21 [DOI] [PubMed] [Google Scholar]
- 9.Duncan RE, Ahmadian M, Jaworski K, Sarkadi-Nagy E, Sul HS. 2007. Regulation of lipolysis in adipocytes. Annu. Rev. Nutr. 27:79–101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lafontan M, Langin D. 2009. Lipolysis and lipid mobilization in human adipose tissue. Prog. Lipid Res. 48:275–297 [DOI] [PubMed] [Google Scholar]
- 11.Coleman RA, Mashek DG. 2011. Mammalian triacylglycerol metabolism: synthesis, lipolysis, and signaling. Chem. Rev. 111:6359–6386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zimmermann R, Strauss JG, Haemmerle G, Schoiswohl G, Birner-Gruenberger R, Riederer M, Lass A, Neuberger G, Eisenhaber F, Hermetter A, Zechner R. 2004. Fat mobilization in adipose tissue is promoted by adipose triglyceride lipase. Science 306:1383–1386 [DOI] [PubMed] [Google Scholar]
- 13.Villena JA, Roy S, Sarkadi-Nagy E, Kim KH, Sul HS. 2004. Desnutrin, an adipocyte gene encoding a novel patatin domain-containing protein, is induced by fasting and glucocorticoids: ectopic expression of desnutrin increases triglyceride hydrolysis. J. Biol. Chem. 279:47066–47075 [DOI] [PubMed] [Google Scholar]
- 14.Jenkins CM, Mancuso DJ, Yan W, Sims HF, Gibson B, Gross RW. 2004. Identification, cloning, expression, and purification of three novel human calcium-independent phospholipase A2 family members possessing triacylglycerol lipase and acylglycerol transacylase activities. J. Biol. Chem. 279:48968–48975 [DOI] [PubMed] [Google Scholar]
- 15.Smirnova E, Goldberg EB, Makarova KS, Lin L, Brown WJ, Jackson CL. 2006. ATGL has a key role in lipid droplet/adiposome degradation in mammalian cells. EMBO Rep. 7:106–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haemmerle G, Lass A, Zimmermann R, Gorkiewicz G, Meyer C, Rozman J, Heldmaier G, Maier R, Theussl C, Eder S, Kratky D, Wagner EF, Klingenspor M, Hoefler G, Zechner R. 2006. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science 312:734–737 [DOI] [PubMed] [Google Scholar]
- 17.Bezaire V, Mairal A, Ribet C, Lefort C, Girousse A, Jocken J, Laurencikiene J, Anesia R, Rodriguez AM, Ryden M, Stenson BM, Dani C, Ailhaud G, Arner P, Langin D. 2009. Contribution of adipose triglyceride lipase and hormone-sensitive lipase to lipolysis in hMADS adipocytes. J. Biol. Chem. 284:18282–18291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miyoshi H, Perfield JW, 2nd, Obin MS, Greenberg AS. 2008. Adipose triglyceride lipase regulates basal lipolysis and lipid droplet size in adipocytes. J. Cell. Biochem. 105:1430–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kershaw EE, Hamm JK, Verhagen LA, Peroni O, Katic M, Flier JS. 2006. Adipose triglyceride lipase: function, regulation by insulin, and comparison with adiponutrin. Diabetes 55:148–157 [PMC free article] [PubMed] [Google Scholar]
- 20.Gronke S, Mildner A, Fellert S, Tennagels N, Petry S, Muller G, Jackle H, Kuhnlein RP. 2005. Brummer lipase is an evolutionary conserved fat storage regulator in Drosophila. Cell Metab. 1:323–330 [DOI] [PubMed] [Google Scholar]
- 21.Kurat CF, Natter K, Petschnigg J, Wolinski H, Scheuringer K, Scholz H, Zimmermann R, Leber R, Zechner R, Kohlwein SD. 2006. Obese yeast: triglyceride lipolysis is functionally conserved from mammals to yeast. J. Biol. Chem. 281:491–500 [DOI] [PubMed] [Google Scholar]
- 22.Chakrabarti P, English T, Shi J, Smas CM, Kandror KV. 2010. The mTOR complex 1 suppresses lipolysis, stimulates lipogenesis and promotes fat storage. Diabetes 59:775–781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kitamura T, Kitamura Y, Kuroda S, Hino Y, Ando M, Kotani K, Konishi H, Matsuzaki H, Kikkawa U, Ogawa W, Kasuga M. 1999. Insulin-induced phosphorylation and activation of cyclic nucleotide phosphodiesterase 3B by the serine-threonine kinase Akt. Mol. Cell. Biol. 19:6286–6296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Choi SM, Tucker DF, Gross DN, Easton RM, DiPilato LM, Dean AS, Monks BR, Birnbaum MJ. 2010. Insulin regulates adipocyte lipolysis via an Akt-independent signaling pathway. Mol. Cell. Biol. 30:5009–5020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim JY, Tillison K, Lee JH, Rearick DA, Smas CM. 2006. The adipose tissue triglyceride lipase ATGL/PNPLA2 is downregulated by insulin and TNF-alpha in 3T3-L1 adipocytes and is a target for transactivation by PPARγ. Am. J. Physiol. Endocrinol. Metab. 291:E115–E127 [DOI] [PubMed] [Google Scholar]
- 26.Lake AC, Sun Y, Li JL, Kim JE, Johnson JW, Li D, Revett T, Shih HH, Liu W, Paulsen JE, Gimeno RE. 2005. Expression, regulation, and triglyceride hydrolase activity of adiponutrin family members. J. Lipid Res. 46:2477–2487 [DOI] [PubMed] [Google Scholar]
- 27.Fortier M, Wang SP, Mauriege P, Semache M, Mfuma L, Li H, Levy E, Richard D, Mitchell GA. 2004. Hormone-sensitive lipase-independent adipocyte lipolysis during beta-adrenergic stimulation, fasting, and dietary fat loading. Am. J. Physiol. Endocrinol. Metab. 287:E282–E288 [DOI] [PubMed] [Google Scholar]
- 28.Nielsen TS, Vendelbo MH, Jessen N, Pedersen SB, Jorgensen JO, Lund S, Moller N. 2011. Fasting, but not exercise, increases adipose triglyceride lipase (ATGL) protein and reduces G0/G1 switch gene 2 (G0S2) protein and mRNA content in human adipose tissue. J. Clin. Endocrinol. Metab. 96:E1293–E1297 [DOI] [PubMed] [Google Scholar]
- 29.Alsted TJ, Nybo L, Schweiger M, Fledelius C, Jacobsen P, Zimmermann R, Zechner R, Kiens B. 2009. Adipose triglyceride lipase in human skeletal muscle is upregulated by exercise training. Am. J. Physiol. Endocrinol. Metab. 296:E445–E453 [DOI] [PubMed] [Google Scholar]
- 30.Chakrabarti P, Kandror KV. 2009. FoxO1 controls insulin-dependent ATGL expression and lipolysis in adipocytes. J. Biol. Chem. 284:13296–13300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chakrabarti P, English T, Karki S, Qiang L, Tao R, Kim J, Luo Z, Farmer SR, Kandror KV. 2011. SIRT1 controls lipolysis in adipocytes via FOXO1-mediated expression of ATGL. J. Lipid Res. 52:1693–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luong N, Davies CR, Wessells RJ, Graham SM, King MT, Veech R, Bodmer R, Oldham SM. 2006. Activated FOXO-mediated insulin resistance is blocked by reduction of TOR activity. Cell Metab. 4:133–142 [DOI] [PubMed] [Google Scholar]
- 33.Birse RT, Choi J, Reardon K, Rodriguez J, Graham S, Diop S, Ocorr K, Bodmer R, Oldham S. 2010. High-fat-diet-induced obesity and heart dysfunction are regulated by the TOR pathway in Drosophila. Cell Metab. 12:533–544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu Z, Kandror KV. 2002. Translocation of small preformed vesicles is responsible for the insulin activation of glucose transport in adipose cells: evidence from the in vitro reconstitution assay. J. Biol. Chem. 277:47972–47975 [DOI] [PubMed] [Google Scholar]
- 35.Kumbrink J, Kirsch KH, Johnson JP. 2010. EGR1, EGR2, and EGR3 activate the expression of their coregulator NAB2 establishing a negative-feedback loop in cells of neuroectodermal and epithelial origin. J. Cell. Biochem. 111:207–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lucerna M, Pomyje J, Mechtcheriakova D, Kadl A, Gruber F, Bilban M, Sobanov Y, Schabbauer G, Breuss J, Wagner O, Bischoff M, Clauss M, Binder BR, Hofer E. 2006. Sustained expression of early growth response protein-1 blocks angiogenesis and tumor growth. Cancer Res. 66:6708–6713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang J, Manning BD. 2008. The TSC1-TSC2 complex: a molecular switchboard controlling cell growth. Biochem. J. 412:179–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Festuccia WT, Laplante M, Berthiaume M, Gelinas Y, Deshaies Y. 2006. PPARγ agonism increases rat adipose tissue lipolysis, expression of glyceride lipases, and the response of lipolysis to hormonal control. Diabetologia 49:2427–2436 [DOI] [PubMed] [Google Scholar]
- 39.Kershaw EE, Schupp M, Guan HP, Gardner NP, Lazar MA, Flier JS. 2007. PPARγ regulates adipose triglyceride lipase in adipocytes in vitro and in vivo. Am. J. Physiol. Endocrinol. Metab. 293:E1736–E1745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nielsen R, Pedersen TA, Hagenbeek D, Moulos P, Siersbaek R, Megens E, Denissov S, Borgesen M, Francoijs KJ, Mandrup S, Stunnenberg HG. 2008. Genome-wide profiling of PPARγ:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis. Genes Dev. 22:2953–2967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Eguchi J, Wang X, Yu S, Kershaw EE, Chiu PC, Dushay J, Estall JL, Klein U, Maratos-Flier E, Rosen ED. 2011. Transcriptional control of adipose lipid handling by IRF4. Cell Metab. 13:249–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Estruch F, Carlson M. 1993. Two homologous zinc finger genes identified by multicopy suppression in a SNF1 protein kinase mutant of Saccharomyces cerevisiae. Mol. Cell. Biol. 13:3872–3881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boyle KB, Hadaschik D, Virtue S, Cawthorn WP, Ridley SH, O'Rahilly S, Siddle K. 2009. The transcription factors Egr1 and Egr2 have opposing influences on adipocyte differentiation. Cell Death Differ. 16:782–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang W, Huang L, Huang Y, Yin JW, Berk AJ, Friedman JM, Wang G. 2009. Mediator MED23 links insulin signaling to the adipogenesis transcription cascade. Dev. Cell 16:764–771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carnevalli LS, Masuda K, Frigerio F, Le Bacquer O, Um SH, Gandin V, Topisirovic I, Sonenberg N, Thomas G, Kozma SC. 2010. S6K1 plays a critical role in early adipocyte differentiation. Dev. Cell 18:763–774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Shen N, Yu X, Pan FY, Gao X, Xue B, Li CJ. 2011. An early response transcription factor, egr-1, enhances insulin resistance in type 2 diabetes with chronic hyperinsulinism. J. Biol. Chem. 286:14508–14515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gokey NG, Lopez-Anido C, Gillian-Daniel AL, Svaren J. 2011. Early growth response 1 (egr1) regulates cholesterol biosynthetic gene expression. J. Biol. Chem. 286:29501–29510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brand E, Herrmann SM, Nicaud V, Evans A, Ruidavets JB, Arveiler D, Luc G, Cambien F, Soubrier F. 2000. Identification of two polymorphisms in the early growth response protein-1 gene: possible association with lipid variables. J. Mol. Med. (Berlin) 78:81–86 [DOI] [PubMed] [Google Scholar]
- 49.Thiel G, Cibelli G. 2002. Regulation of life and death by the zinc finger transcription factor Egr-1. J. Cell Physiol. 193:287–292 [DOI] [PubMed] [Google Scholar]
- 50.Sartipy P, Loskutoff DJ. 2003. Expression profiling identifies genes that continue to respond to insulin in adipocytes made insulin-resistant by treatment with tumor necrosis factor-alpha. J. Biol. Chem. 278:52298–52306 [DOI] [PubMed] [Google Scholar]
- 51.Kralisch S, Klein J, Lossner U, Bluher M, Paschke R, Stumvoll M, Fasshauer M. 2005. Isoproterenol, TNFα, and insulin downregulate adipose triglyceride lipase in 3T3-L1 adipocytes. Mol. Cell. Endocrinol. 240:43–49 [DOI] [PubMed] [Google Scholar]
- 52.Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G. 2004. Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431:200–205 [DOI] [PubMed] [Google Scholar]
- 53.Jocken JW, Langin D, Smit E, Saris WH, Valle C, Hul GB, Holm C, Arner P, Blaak EE. 2007. Adipose triglyceride lipase and hormone-sensitive lipase protein expression is decreased in the obese insulin-resistant state. J. Clin. Endocrinol. Metab. 92:2292–2299 [DOI] [PubMed] [Google Scholar]
- 54.Karpe F, Dickmann JR, Frayn KN. 2011. Fatty acids, obesity, and insulin resistance: time for a reevaluation. Diabetes 60:2441–2449 [DOI] [PMC free article] [PubMed] [Google Scholar]