

Abstract
Leucine zipper tumor suppressor 2 (LZTS2) is implicated in several cancers; however, its biological mechanisms in non-small cell lung cancer (NSCLC) are not yet understood. We found that low levels of LZTS2 in NSCLC were correlated with tumor and nodal status. LZTS2 could inhibit cell proliferation and cell cycle transition at the G1/S phase and was implicated in the regulation of proteins associated with the canonical Wnt pathway, including GSK3β and β-catenin through inactivating the Akt pathway. These results provide novel mechanistic insight into the biological roles of LZTS2 in lung cancer cells.
Keywords: LZTS2, NSCLC, β-catenin, Akt, GSK3β
Introduction
There have been numerous studies on the loss of heterozygosity (LOH) and mutation analyses in different tumor tissues, indicating that multiple tumor suppressor genes (TSG) are present within the human chromosome 8p21-22 and 10q23-24 regions (Carter et al. 1990; Gray et al. 1995; Kagan et al. 1995; Bova et al. 1996; Ittmann 1996; Cairns et al. 1997; Kim et al. 1998; Petersen et al. 1998; Whang et al. 1998; Ishii et al. 1999). It has been reported that leucine zipper tumor suppressor 1 (LZTS1), a putative TSG located on 8p22, may function as a cell growth modulator (Ishii et al. 1999; Cabeza-Arvelaiz et al. 2001a). An LZTS1 related gene, LZTS2 (synonym, LAPSER1), has been found to map to a sub-region of human chromosome 10q24.3, which is deleted in various cancers, along with its neighboring PTEN locus (Cabeza-Arvelaiz et al. 2001b). Overexpression of LZTS2 cDNA strongly inhibits cell growth and the colony forming efficiencies of most cancer cells, including LNCaP, TRSUPr1, PC3, U2OS, HEK-293T, AT6.2, and Rat-1 cells; this suggests that the LZTS2 gene is also involved in the regulation of cell growth and that loss of LZTS2 function may contribute to cancer development (Cabeza-Arvelaiz et al. 2001a).
β-catenin is a multifunctional protein (Polakis, Hart, and Rubinfeld 1999; Barker, Morin, and Clevers 2000), having an essential role in cell-cell adhesion by interacting with the intracellular tail of E-cadherin and linking to the actin filament network via α-catenin. β-catenin is also a key component in the Wnt/Wingless signaling pathway, which has a critical role in cell development, proliferation, and differentiation (Wodarz and Nusse 1998). In the absence of a Wnt/Wingless signal, β-catenin locates primarily at adherens junctions, and any free β-catenin interacts with high-molecular-mass complexes, including adenomatous polyposis coli (APC) (Rubinfeld et al. 1993; Su, Vogelstein, and Kinzler 1993), GSK3β (Rubinfeld et al. 1996), and Axin (Behrens et al. 1998; Hart et al. 1998). Nuclear localization of β-catenin, and its binding to the T-cell factor/lymphoid enhancing factor (TCF/LEF) family, may enhance the expression of several Wnt-targeted genes, such as Tcf4, c-Myc, and CyclinD1 (Zhang, Gaspard, and Chung 2001; Easwaran et al. 2003; Boon et al. 2004; Kaler et al. 2009).
GSK3β inhibition has also been shown to have a central role in the Wnt/Wingless signaling pathway by binding to and stabilizing β-catenin and, thereby, modulating Lef/Tcf-dependent transcription (Polakis, Hart, and Rubinfeld 1999; Barker, Morin, and Clevers 2000). Phosphorylation of β-catenin by GSK3β (Ikeda et al. 1998) targets β-catenin for degradation via the ubiquitin-proteasome system (Aberle et al. 1997). Activated non-phospho-GSK3β, a known inhibitor of β-catenin, also primes β-catenin for ubiquitination and proteasomal degradation via sequential phosphorylation of the β-catenin N-terminal region (Fuchs et al. 2005). Deactivation of GSK3β due to reduced expression or via Akt phosphorylation may facilitate stabilization of β-catenin in the cytoplasm, or translocation into the nucleus, where it has a role in transcriptional regulation of several genes (Jiang and Liu 2009).
Thyssen et al. proposed that LZTS2, which acts as a β-catenin-interacting protein, suppresses β-catenin-regulated transcription by mediating regulation of β-catenin nuclear export protein (Thyssen et al. 2006). Their study was based on colorectal cancer cell lines in which β-catenin is mainly located in the cytoplasm; however, the role of LZTS2 in lung cancer cell lines, where β-catenin is mainly expressed on the membrane, is still unclear.
In the present study, we found that low levels of LZTS2 in non-small cell lung cancer (NSCLC) were correlated with tumor and nodal status. We then examined the relationship between LZTS2 and β-catenin-regulated pathways, and specifically analyzed the effect of LZTS2 on the Akt/GSK3β pathway in A549 and H1299 cells. Having determined that Lef/Tcf-dependent transcription was inhibited by LZTS2 in these cell lines, we sought to define the signaling pathways used by LZTS2 to deactivate Lef/Tcf-dependent transcription in lung cancer.
Materials and Methods
Patient Recruitment and Collection of Tumor Specimens
This study was conducted with the approval of the Institutional Review Board at China Medical University. Written consent was given by the participants for their personal details to be stored in the hospital database and for their specimens to be used in this study. All clinical investigations were conducted according to the principles expressed in the Declaration of Helsinki. Primary tumor specimens were obtained from 89 patients (46 males and 43 females) diagnosed with lung squamous cell carcinoma (SCC) and adenocarcinoma and who had undergone complete resection in The First Affiliated Hospital of China Medical University between 2001 and 2004. Large cell carcinoma, adenosquamous carcinoma, or other NSCLC subtypes were not included in this study. Follow-up information was obtained by reviewing the patients’ medical records. None of the patients had received radiotherapy or chemotherapy before surgical resection, and all patients were treated with routine chemotherapy after the operation. The histological diagnosis and grade of differentiation were evaluated using hematoxylin and eosin-stained sections according to the World Health Organization guidelines of classification. All 89 specimens were re-evaluated with respect to histological subtype, differentiation stage, and tumor stage. SCC was identified in 40 cases, and adenocarcinoma in 49 cases. Lymph node metastases were identified in 35 out of 89 patients. The p-TNM cancer staging system of the International Union Against Cancer (7th edition) was used to classify specimens. We collected 6 pairs of fresh lung cancer tissues (4 cases of adenocarcinoma and 2 cases of squamous cell carcinoma) for western blot.
Cell Culture
A549 and H1299 cell lines were obtained from the American Type Culture Collection (Manassas, VA). Cells were cultured in RPMI-1640 (Invitrogen; Carlsbad, CA) containing 10% fetal calf serum, 2 mM glutamine, 100 UI/ml penicillin, and 100 mg/ml streptomycin at 37C, 5% CO2 in a humidified atmosphere. Cells were grown on sterile tissue culture dishes and were maintained in logarithmic growth phase by using 0.25% trypsin (Invitrogen) every 2 days. The PI3K/Akt inhibitor, LY294002 (10 µM; 4 hr) (Tang, Zhou, and Zhou 2009), was purchased from Beyotime Institute of Biotechnology, China, and the GSK3β inhibitor, SB216763 (10 µM; 4 hr) (Coghlan et al. 2000), was purchased from Selleck Chemicals (Houston, TX).
Immunohistochemistry
Surgically excised tumor samples were fixed in 10% neutral formalin and embedded in paraffin, and 4-μm sections were prepared. Immunostaining was performed using the avidin–biotin–peroxidase complex method (Ultra Sensitive TM kit; Maixin, Fuzhou, China). The sections were deparaffinized in xylene, rehydrated through a graded alcohol series, and boiled in 0.01 M citrate buffer (pH 6.0) for 2 min in an autoclave. Endogenous peroxidase activity was blocked with hydrogen peroxide (0.3%), followed by incubation with normal goat serum to reduce nonspecific binding. Tissue sections were incubated with LZTS2 (1:150; Sigma, Mundelein, IL) and p-Akt (1:100; Cell Signaling Technology, Boston, MA) antibodies. Rabbit immunoglobulin (at the same concentration as the antigen-specific antibody; Maixin, China) was used as a negative control. Staining for primary antibody was performed at room temperature for 2 hr. Biotinylated goat anti-rabbit serum IgG (ready-to-use; Maixin) was used as the secondary antibody. After rinsing in PBS, the sections were incubated with horseradish peroxidase (HRP)-conjugated streptavidin–biotin, followed by 3,3’-diaminobenzidine tetrahydrochloride to develop the peroxidase reaction. The sections were counterstained with hematoxylin and dehydrated in ethanol before mounting. All tumor slides were randomly examined by two independent investigators, with five views per slide, and 100 cells examined per view at 400× magnification. We replaced the LZTS2 antibody with phosphate-buffered saline (PBS) in the negative control (Fig. 1A). Immunostaining of LZTS2 was scored following a semi-quantitative scale by evaluating the intensity and percentage of cells showing higher immunostaining in representative tumor areas compared with control cells. Nuclear staining of the tumor cells was considered as positive immunostaining. The intensity of LZTS2 nuclear staining was scored as 0 (no staining), 1 (weak), or 2 (marked). Percentage scores were assigned as follows: 1 (1–25%), 2 (26–50%), 3 (51–75%), and 4 (76–100%). The scores for each tumor sample were multiplied to give a final score between 0 and 8, and the total expression of LZTS2 was designated as either negative or low expression ([–]; score < 4) or positive or high expression ([+]; score ≥ 4), respectively.
Figure 1.

(A) Phosphate-buffered saline (PBS) was used in place of leucine zipper tumor suppressor (LZTS)-2 antibody as the negative control. The expression of LZTS2 by immunohistochemistry in (B) alveolar and bronchial epithelia; (C, D) squamous cell carcinoma; and (E, F) adenocarcinoma. (H) The expression of LZTS2 in normal lung tissues and non-small cell lung cancer (NSCLC) by western blot analysis. (G) The level of LZTS2 in cancer tissue is much lower than that in normal lung tissue in this sample. *p<0.05. Bars A-F = 100 µm; G = 200 µm.
Nuclear and Cytosolic Fractionation and Western Blot Analysis
Fresh lung cancer tissues and the cultured cells were trypsinized and washed with cold PBS, and the cell pellets were resuspended in ice-cold lysis buffer (210 mM mannitol, 70 mM sucrose, 5 mM Tris, pH 7.5, 1 mM EDTA) supplemented with protease inhibitors. After being incubated on ice for 15 min, the cells were homogenized. The nuclei were separated by centrifugation (10 min, 12000round/min, 4C), and the supernatant containing the cytosolic fraction was boiled in sample buffer. The pellet containing the nuclei was washed with PBS and then resuspended in RIPA buffer (Santa Cruz Biotechnology; Santa Cruz, CA) for 5 min on ice. The pellet was then centrifuged again, and the supernatant (nuclear fraction) was boiled in sample buffer. Tissues and nuclear and cytosolic proteins were loaded on 10% SDS polyacrylamide gels, electrotransferred to polyvinylidene fluoride (PVDF) membranes (Millipore; Billerica, MA), and then incubated overnight at 4C with the following antibodies: LZTS2 (1:1000; Sigma); β-actin (1:500); β-catenin (1:1000; BD Biosciences, San Jose, CA), cyclin D1 (1:1000); c-Myc (1:400); phospho-Akt (1:500); total Akt (1:500); phospho-GSK3β (Ser9) (1:500; Cell Signaling Technology); and phospho-β-catenin (1:500; Santa Cruz Biotechnology). After incubation with HRP-conjugated anti-mouse IgG (Santa Cruz Biotechnology) at 37C for 2 hr, bound proteins were visualized by enhanced chemoluminescence (ECL; Thermo Fisher Scientific, Waltham, MA) and detected using a bio-imaging system (UVP; Upland, CA). The relative protein levels were calculated using β-actin as a loading control.
Plasmid Transfection and Small Interfering RNA Treatment
The pCMV6-LZTS2 plasmid was purchased from Origene (Rockville, MD), and pCMV6 empty vector was used as a negative control. Small interfering RNA (siRNA) for LZTS2 (M-012409-01) and non-targeting siRNA #1 (D-001810-01) were purchased from Genepharma (Shanghai, China). The plasmid and siRNA were transfected into cells using Attractene Transfection Reagent (Qiagen; Hilden, Germany). Cells were seeded in a 6-well plate 24 hr before transfection, and protein levels were assessed 48 hr posttransfection.
Cell Proliferation Assay and Colony Formation Assay
The cell proliferation assay was performed using Cell Counting Kit-8 solution (Dojindo; Gaithersburg, MD) according to the manufacturer’s protocol. Briefly, cells were seeded at a concentration of 5 × 103 cells/100 μl/well in 96-well culture plates and treated with 10 μl/well of Cell Counting Kit-8 solution during the last 4 hr of culture. Optical density was measured at 450 nm using a microplate reader.
For the colony formation assay, cells were plated into three 6-cm cell culture dishes (1000 cells per dish) and incubated at 37C, 5% CO2 in a humidified atmosphere for 12 days. Plates were washed with PBS and stained using Giemsa stain. Colonies with more than 50 cells were counted.
Luciferase Reporter Assay
For the luciferase activity assay, cells at 80% confluence in 24-well plates were co-transfected with 0.2 μg firefly luciferase reporter vector along with 0.02 μg Renilla luciferase reporter vector (Promega; Madison, WI) for 12 hr using Attractene Transfection Reagent (Qiagen), according to the manufacturers’ protocols, and Topflash reporter plasmids (Biotime Biotechnology; Alameda, CA). The luciferase activity was measured in the cellular extracts using a dual luciferase reporter gene assay kit (Promega). Relative reporter gene activity was calculated by dividing the firefly luciferase signal by the Renilla luciferase signal.
Cell Cycle Analysis
Cells (5 × 105) were seeded into 6-cm tissue culture dishes. After 12 hr, the cells were subjected to serum starvation for 24 hr and then transfected with plasmid or siRNA, as described above. Cells were harvested at the times specified above, fixed in 1% paraformaldehyde, washed with PBS, and stained using 5 mg/ml propidium iodide in PBS supplemented with RNase A (Roche; Indianapolis, IN) for 30 min at room temperature. Data were collected using the BD analysis system, with debris and cell aggregates excluded. Cell cycle was divided into three parts: G1 phase, S phase, and G2/M phase.
Immunoprecipitation
Cells were washed twice with 5 ml of PBS followed by incubation on ice for 5 min with lysis buffer containing 0.5% NP-40, 50 mM Tris, 150 mM NaCl, 1 mM phenylmethylsulfonyl fluoride, 5 mg/ml leupeptin, 2 mg/ml aprotinin, 1 mM sodium orthovanadate, and 1 mM EDTA. Cells were harvested from the plates and transferred to a 1.5-ml tube. The lysate was centrifuged at 16,000 xg for 5 min at 4C and the supernatant was transferred to a new tube. Lysates were quantified by Bradford assay and equal amounts of total protein were used for immunoprecipitation with the anti-β-catenin mAb overnight. The immunocomplexes were then subjected to SDS-PAGE.
Statistical Analysis
SPSS v16.0 for Windows was used for all statistical analyses (SPSS Inc., Chicago, IL). The Chi-square test was used to examine possible correlations between LZTS2 expression and clinicopathological factors. The Student’s t-test was used to compare all other data. The p values were based on two-sided statistical analysis; p<0.05, p<0.01, and p<0.001 were considered statistically significant. Experiments were performed in triplicate, and the results were reported as mean ± standard deviation. The p values presented in figures, tables, and text were not corrected for multiplicity of tests.
Results
Low Expression of LZTS2 in NSCLC and Association with Clinicopathological Parameters
First, we examined LZTS2 expression levels in a panel of 89 NSCLCs and 30 corresponding normal lung tissue samples by immunohistochemistry. LZTS2 displayed a strong expression and was primarily localized in the nuclear compartment in 28 out of 30 (93.33%) normal lung tissue samples (Fig. 1B). LZTS2 was positive in 35 out of 89 (39.33%) NSCLC samples (Figs. 1C-1F), showing that the positive rate of LZTS2 in NSCLC samples was much lower than that in normal lung tissues (Fig. 1G). We compared LZTS2 expression among six fresh lung cancer samples and their paired normal lung tissues by western blot and found that lung cancer tissues showed a much lower level of LZTS2 than their paired normal lung tissues (Fig. 1H). When we analyzed the relationship between LZTS2 expression and clinicopathological parameters (Table 1), we identified significant associations between LZTS2 expression and histology (p=0.022), tumor status (p=0.010), and nodal status (p=0.023), independent of patient age, gender, or differentiation, indicating that LZTS2 may play an important role as a tumor suppressor in the lung cancer developmental process.
Table 1.
Distribution of Leucine Zipper Tumor Suppressor 2 (LZTS2) Status in Non-small Cell Lung Cancer (NSCLC) According to Clinicopathological Characteristics .
| Characteristic | No. of Patients | LZTS2 Negative, No. (%) | LZTS2 Positive, No. (%) | p Value |
|---|---|---|---|---|
| Age | ||||
| <60 | 43 | 28 (65.12) | 15 (34.88) | 0.407 |
| ≥60 | 46 | 26 (56.52) | 20 (43.48) | |
| Gender | ||||
| Male | 46 | 30 (65.22) | 16 (34.78) | 0.364 |
| Female | 43 | 24 (55.81) | 19 (44.19 ) | |
| Histology | ||||
| Adenocarcinoma | 49 | 35 (71.43) | 14 (28.57) | 0.022 |
| Squamous cell carcinoma | 40 | 19 (47.50) | 21 (52.50) | |
| Differentiation | ||||
| Well | 28 | 14 (50.00) | 14 (50.00) | 0.163 |
| Moderate–poor | 61 | 40 (65.57) | 21 (34.43) | |
| TNM stage | ||||
| I | 40 | 19 (47.50) | 21 (52.50) | 0.022 |
| II and III | 49 | 35 (71.43) | 14 (28.57) | |
| Tumor status | ||||
| T1 | 29 | 12 (41.38) | 17 (58.62) | 0.010 |
| T2 and T3 | 60 | 42 (70.00) | 18 (30.00) | |
| Nodal status | ||||
| N0 | 54 | 27 (50.00) | 27 (50.00) | 0.023 |
| N1, N2, and N3 | 35 | 27 (77.14) | 8 (22.86) | |
TNM, tumor, nodal and metastases.
ecti LZTS2 Inhibits Cell Proliferation and G1/S Cell Cycle Transition
We observed LZTS2 protein expression in six lung cancer cell lines (A549, LTEP-a-2, SPC, H1299, H157, and H460), human bronchial epithelial (HBE) cell line, and human embryonic kidney (HEK) 293T cell line (Fig. 2A). We selected A549 and H1299 lung cancer cell lines to examine the role of LZTS2 in lung cancer cells (Fig. 2B).
Figure 2.

(A) The expression of LZTS2 in six lung cancer cell lines, normal human bronchial epithelial (HBE) cells, and transformed human embryonic kidney (HEK) 293T cells. (B) Overexpression of LZTS2 in H1299 cells and silenced LZTS2 in A549 cells.
MTT and colony formation assays were performed to analyze the effect of LZTS2 on cell proliferation. Overexpression of LZTS2 in H1299 cells resulted in cell growth inhibition (Vector vs. LZTS2: 1.11 ± 0.06 vs. 0.70 ± 0.08, p<0.05) (Fig. 3A) and a marked reduction in colony numbers compared with the empty vector (Vector vs. LZTS2: 452 ± 13 vs. 153 ± 9, p<0.001) (Fig. 3B). In LZTS2-transfected cells, cell cycle analysis by fluorescence activated cell sorting flow cytometry showed an increase in the percentage of cells in the G1 phase (Vector vs. LZTS2: 51.62 ± 0.78% vs. 61.57 ± 1.46%, p<0.05) and a decrease in S phase cells (Vector vs. LZTS2: 39.76 ± 0.59% vs. 31.50 ± 1.58%, p<0.05), showing that LZTS2 could inhibit G1 to S phase transition (Fig. 3C).
Figure 3.

The effect of LZTS2 on cell proliferation by (A) MTT assay, (B) colony formation, and (C) cell cycle analysis by fluorescence activated cell sorting in two lung cancer cell lines, H1299 and A549 cells. Abbreviations: V, vector; LZ, LZTS2, NC, negative control; Si, SiRNA for LZTS2.
In contrast, when LZTS2 was silenced in A549 cells, the proliferation rate (negative control vs. SiLZTS2: 0.75 ± 0.08 vs. 1.30 ± 0.09, p<0.05) and colony formation ability (negative control vs. SiLZTS2: 115 ± 8 vs. 342 ± 15, p<0.001) were increased (Figs. 3A and 3B). Cell cycle analysis showed that the percentage of cells at G1 phase was decreased (negative control vs. SiLZTS2: 62.86 ± 0.55% vs. 53.83 ± 0.71%, p<0.05), and the percentage of cells at S phase was increased (negative control vs. SiLZTS2: 23.07 ± 1.55% vs. 32.64 ± 1.01%, p<0.05) (Fig. 3C).
LZTS2 Inhibits Lef/Tcf-dependent Transcription Activity through Downregulation of β-Catenin
We found that overexpression of LZTS2 in H1299 cells downregulated β-catenin, cyclin D1, and c-Myc and suppressed Topflash (Tcf4 reporter luciferase activity), whereas RNA interference of LZTS2 in A549 cells upregulated these proteins and promoted Topflash (Fig. 4A). Unlike the study of Thyssen and colleagues (2006), LZTS2 tends to influence the degradation of β-catenin, for the level of phospho-β-catenin was obviously changed. Moreover, β-catenin levels in nuclear and cytoplasmic extracts were downregulated by LZTS2 (Fig. 4B).
Figure 4.

The influence of LZTS2 on the expression of (A) β-catenin, cyclin D1, c-Myc, and Topflash (Tcf4 reporter luciferase activity); and (B) cytoplasmic and nuclear β-catenin using western blot in two lung cancer cell lines, H1299 and A549 cells.
We also found that LZTS2 was primarily located in the nucleus, while β-catenin was mainly located on the membrane in these cell lines (Fig. 5A). When we immunoprecipitated β-catenin or LZTS2 from the cell lysates and detected the complexes using different antibodies as indicated, we found no interaction between β-catenin and LZTS2 in H1299 and A549 lung cancer cells, but LZTS2 bound to β-catenin in SW480 cells (Fig. 5B). There were no associations between β-catenin and LZTS2 in nuclear extracts (Supplemental Fig. 2). This is in accordance with the observation that β-catenin and LZTS2 are localized to different regions in lung cancer cells.
Figure 5.

(A) The location of LZTS2 and β-catenin in H1299 and A549 lung cancer cell lines. (B) Whole cell lysates of H1299, A549, and SW480 were immunoprecipitated (IP) with normal rabbit IgG, β-catenin or LZTS2 and then analyzed by western blotting with different antibodies as indicated. Bars = 10 µm.
LZTS2 Promotes β-Catenin Degradation through GSK3β/Akt Pathway
We then examined whether LZTS2 had a functional effect on GSK3β, a known β-catenin inhibitor and a component of the degradation complex. Our findings showed that overexpression of LZTS2 in H1299 cells downregulated phospho-GSK3β (Ser9) but did not alter the level of total GSK3β. Meanwhile, phospho-GSK3β was upregulated in A549 cells upon silencing LZTS2 (Fig. 6A). Furthermore, when H1299 cells transfected with the LZTS2 plasmid were treated with SB216367, a GSK3β inhibitor, LZTS2 inhibition of Tcf4 reporter luciferase activity was reversed (Fig. 6B).
Figure 6.
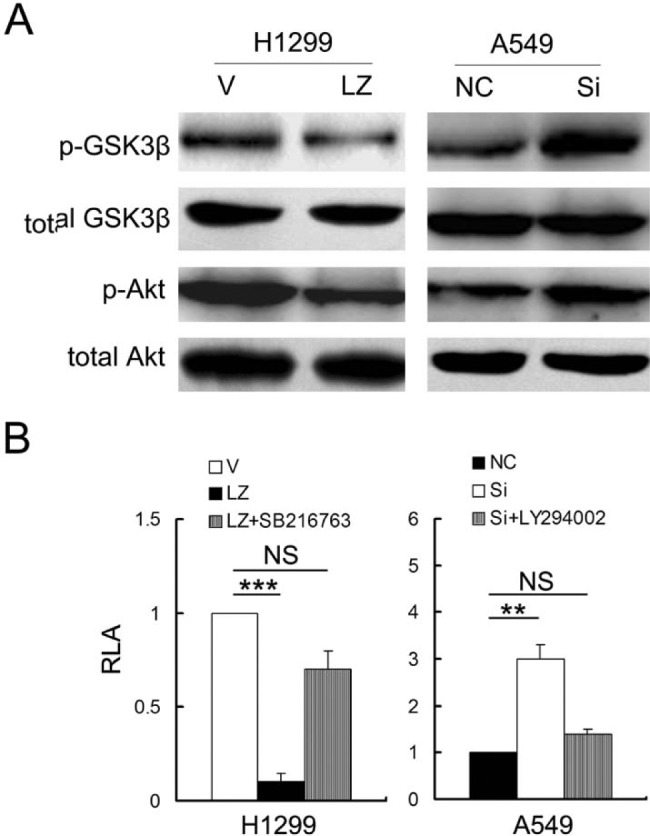
(A) The effect of LZTS2 on p-Akt and p-GSK3β (Ser9) in H1299 and A549 lung cancer cells. (B) LY294002 (an Akt/PI3K pathway inhibitor) and SB216763 (a GSK3β inhibitor) were used to check LZTS2’s role in the Akt/GSK3β pathway in H1299 and A549 cells.
We then tested whether LZTS2 had an effect on the Akt pathway, which is known to inhibit GSK3β activation. We noticed that downregulation of LZTS2 in A549 cells caused an upregulation in phospho-Akt, whereas overexpression of LZTS2 in H1299 inactivated the Akt pathway (Fig. 6A). We then treated LZTS2-deleted A549 cells with LY294002, a PI3K/Akt pathway inhibitor, and found that LY294002 could reverse the promoting effect of silencing LZTS2 on Tcf4 reporter luciferase activity (Fig. 6B).
Next, we explored the p-Akt levels in 89 NSCLC samples and studied the relationship between LZTS2 and p-Akt. In accordance with the result in cell lines, we found that the overexpression of LZTS2 was related to the low levels of p-Akt (Fig. 7 and Table 2).
Figure 7.
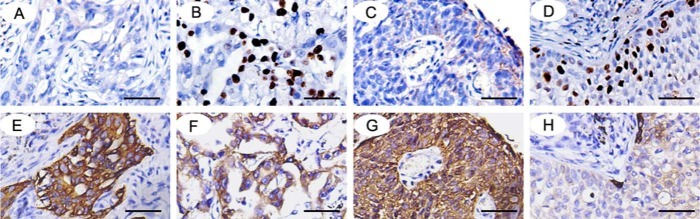
The expression of LZTS2 in (A, B) adenocarcinoma and (C, D) squamous cell carcinoma. The expressionof p-Akt in the same section (E-H). Bars = 50 µm.
Table 2.
LZTS2’s Association with p-Akt in 89 NSCLC Samples.
| Characteristic | No. of Patients | LZTS2 Negative, No. (%) | LZTS2 Positive, No. (%) | p Value |
|---|---|---|---|---|
| p-Akt negative | 39 | 18 (46.15) | 21 (53.85) | 0.013 |
| p-Akt positive | 50 | 36 (72.00) | 14 (28.00) |
Discussion
Previous reports on the biological functions of LZTS2 in prostate cancer, colorectal cancer, and glioma have shown that overexpression of LZTS2 inhibits cell proliferation and tumor growth, suggesting that LZTS2 may act as a tumor suppressor (Cabeza-Arvelaiz et al. 2001a).
In this study, we investigated LZTS2 expression levels in 89 lung cancer tissues. Immunohistochemistry demonstrated a much lower positive rate of LZTS2 in NSCLC samples than that in normal lung tissues. We also found that LZTS2 overexpression was correlated with histological type, tumor status, and nodal status in NSCLC tissue samples. Furthermore, western blotting in six paired lung cancer tissues showed that LZTS2 expression was lower in NSCLC compared with normal lung tumor tissues. These findings support the premise that LZTS2 may be a tumor suppressor in lung cancer.
We examined the expression levels of LZTS2 in several lung cancer cells and chose H1299 and A549 cell lines for the gain- and loss-of-function analyses. We found that LZTS2 could inhibit cell proliferation, colony formation, and G1 to S phase transition during cell cycle progression. In order to study the possible mechanisms by which LZTS2 works in lung cancer cells, we examined the expression levels of several Wnt-related proteins and focused on LZTS2’s effects on β-catenin, GSK3β, and Akt function. We found that LZTS2 enhanced the ability of GSK3β to activate the destruction of β-catenin by inactivating the Akt pathway. In addition, we also found that LZTS2 correlated with p-Akt negatively in the 89 NSCLC tissues.
Thyssen et al. (2006) demonstrated that LZTS2 was localized in the cytoplasm and interacted with cytoplasmic β-catenin to regulate the cellular localization of β-catenin in colorectal cancers. However, we did not detect the a similar interaction between LZTS2 and β-catenin in lung cancer, which seems contradictory to Thyssen’s data.
We observed that LZTS2 was primarily located in the nucleus, whereas β-catenin was mainly located on the membrane in most lung cancer tissues and cell lines. The different localization of LZTS2 and β-catenin in lung cancer cells likely explains the reason why there was little association between LZTS2 and β-catenin in co-immunoprecipitation assays.
It has been reported that more than 80% of colorectal cancers possess inactive mutants of the APC protein, which correlates with increased levels of free β-catenin (Miyoshi et al. 1992; Powell et al. 1992). Most of these mutations result in truncated proteins, which are unable to bind to Axin and other regulatory proteins, and lead to β-catenin accumulation (Polakis 2000; Fodde, Smits, and Clevers 2001). However, in lung cancer, APC is integrated in the destruction complex and maintains β-catenin at a relatively low level in the nuclear compartment. As shown in Supplemental Fig. 1, the Topflash activity, which reflects the nuclear level of β-catenin in H1299 and A549, is much lower than that in SW480. Therefore, the failure of detecting LZTS2’s interaction with β-catenin might be due to the little amount of β-catenin in the nuclear parts of H1299 and A549 cells.
Interestingly, we found that LZTS2 was also involved in the regulation of Dishevelled (Dvl)-1 but not Dvl-2 or Dvl-3, and co-immunoprecipitation indicated that only Dvl-1 associated with LZTS2 (data not shown). Given that Dvl proteins are located primarily in the nucleus in most lung cancer cells and participate in regulating Lef/Tcf-dependent transcription activity (Behrens et al. 1996; Molenaar et al. 1996), along with LZTS2, we hypothesize that LZTS2 may also interact with Dvl-1.
These data suggest that LZTS2 may act through the Akt pathway to inhibit cell proliferation in lung cancer.
Supplementary Material
Footnotes
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: The National Natural Science Foundation of China (NSFC No. 81071905, 81272606).
Supplementary material for this article is available on the Journal of Histochemistry & Cytochemistry Web site at http://jhc.sagepub.com/supplemental.
References
- Aberle H, Bauer A, Stappert J, Kispert A, Kemler R. 1997. Beta-catenin is a target for the ubiquitin-proteasome pathway. EMBO J. 16:3797–3804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barker N, Morin PJ, Clevers H. 2000. The yin-yang of TCF/beta-catenin signaling. Adv Cancer Res. 77:1–24 [DOI] [PubMed] [Google Scholar]
- Behrens J, Jerchow BA, Wurtele M, Grimm J, Asbrand C, Wirtz R, Kuhl M, Wedlich D, Birchmeier W. 1998. Functional interaction of an axin homolog, conductin, with beta-catenin, APC, and GSK3beta. Science. 280:596–599 [DOI] [PubMed] [Google Scholar]
- Behrens J, von Kries JP, Kuhl M, Bruhn L, Wedlich D, Grosschedl R, Birchmeier W. 1996. Functional interaction of beta-catenin with the transcription factor LEF-1. Nature. 382:638–642 [DOI] [PubMed] [Google Scholar]
- Boon EM, Keller JJ, Wormhoudt TA, Giardiello FM, Offerhaus GJ, van der Neut R, Pals ST. 2004. Sulindac targets nuclear beta-catenin accumulation and Wnt signalling in adenomas of patients with familial adenomatous polyposis and in human colorectal cancer cell lines. Br J Cancer. 90:224–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bova GS, MacGrogan D, Levy A, Pin SS, Bookstein R, Isaacs WB. 1996. Physical mapping of chromosome 8p22 markers and their homozygous deletion in a metastatic prostate cancer. Genomics. 35:46–54 [DOI] [PubMed] [Google Scholar]
- Cabeza-Arvelaiz Y, Sepulveda JL, Lebovitz RM, Thompson TC, Chinault AC. 2001a. Functional identification of LZTS1 as a candidate prostate tumor suppressor gene on human chromosome 8p22. Oncogene. 20:4169–4179 [DOI] [PubMed] [Google Scholar]
- Cabeza-Arvelaiz Y, Thompson TC, Sepulveda JL, Chinault AC. 2001b. LAPSER1: a novel candidate tumor suppressor gene from 10q24.3. Oncogene. 20:6707–6717 [DOI] [PubMed] [Google Scholar]
- Cairns P, Okami K, King P, Bonacum J, Ahrendt S, Wu L, Mao L, Jen J, Sidransky D. 1997. Genomic organization and mutation analysis of Hel-N1 in lung cancers with chromosome 9p21 deletions. Cancer Res. 57:5356–5359 [PubMed] [Google Scholar]
- Carter BJ, de Vroom E, Long EC, van der Marel GA, van Boom JH, Hecht SM. 1990. Site-specific cleavage of RNA by Fe(II).bleomycin. Proc Natl Acad Sci U S A. 87:9373–9377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coghlan MP, Culbert AA, Cross DA, Corcoran SL, Yates JW, Pearce NJ, Rausch OL, Murphy GJ, Carter PS, Roxbee Cox L, Mills D, Brown MJ, Haigh D, Ward RW, Smith DG, Murray KJ, Reith AD, Holder JC. 2000. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol. 7:793–803 [DOI] [PubMed] [Google Scholar]
- Easwaran V, Lee SH, Inge L, Guo L, Goldbeck C, Garrett E, Wiesmann M, Garcia PD, Fuller JH, Chan V, Randazzo F, Gundel R, Warren RS, Escobedo J, Aukerman SL, Taylor RN, Fantl WJ. 2003. Beta-catenin regulates vascular endothelial growth factor expression in colon cancer. Cancer Res. 63:3145–3153 [PubMed] [Google Scholar]
- Fodde R, Smits R, Clevers H. 2001. APC, signal transduction and genetic instability in colorectal cancer. Nat Rev Cancer. 1:55–67 [DOI] [PubMed] [Google Scholar]
- Fuchs SY, Ougolkov AV, Spiegelman VS, Minamoto T. 2005. Oncogenic beta-catenin signaling networks in colorectal cancer. Cell Cycle. 4:1522–1539 [DOI] [PubMed] [Google Scholar]
- Gray IC, Phillips SM, Lee SJ, Neoptolemos JP, Weissenbach J, Spurr NK. 1995. Loss of the chromosomal region 10q23-25 in prostate cancer. Cancer Res. 55:4800–4803 [PubMed] [Google Scholar]
- Hart MJ, de los Santos R, Albert IN, Rubinfeld B, Polakis P. 1998. Downregulation of beta-catenin by human Axin and its association with the APC tumor suppressor, beta-catenin and GSK3 beta. Curr Biol. 8:573–581 [DOI] [PubMed] [Google Scholar]
- Ikeda S, Kishida S, Yamamoto H, Murai H, Koyama S, Kikuchi A. 1998. Axin, a negative regulator of the Wnt signaling pathway, forms a complex with GSK-3beta and beta-catenin and promotes GSK-3beta-dependent phosphorylation of beta-catenin. EMBO J. 17:1371–1384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii H, Baffa R, Numata SI, Murakumo Y, Rattan S, Inoue H, Mori M, Fidanza V, Alder H, Croce CM. 1999. The FEZ1 gene at chromosome 8p22 encodes a leucine-zipper protein, and its expression is altered in multiple human tumors. Proc Natl Acad Sci U S A. 96:3928–3933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ittmann MM. 1996. Loss of heterozygosity on chromosomes 10 and 17 in clinically localized prostate carcinoma. Prostate. 28:275–281 [DOI] [PubMed] [Google Scholar]
- Jiang BH, Liu LZ. 2009. PI3K/PTEN signaling in angiogenesis and tumorigenesis. Adv Cancer Res. 102:19–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kagan J, Stein J, Babaian RJ, Joe YS, Pisters LL, Glassman AB, von Eschenbach AC, Troncoso P. 1995. Homozygous deletions at 8p22 and 8p21 in prostate cancer implicate these regions as the sites for candidate tumor suppressor genes. Oncogene. 11:2121–2126 [PubMed] [Google Scholar]
- Kaler P, Godasi BN, Augenlicht L, Klampfer L. 2009. The NF-kappaB/AKT-dependent induction of Wnt signaling in colon cancer cells by macrophages and IL-1beta. Cancer Microenviron. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SK, Ro JY, Kemp BL, Lee JS, Kwon TJ, Hong WK, Mao L. 1998. Identification of two distinct tumor-suppressor loci on the long arm of chromosome 10 in small cell lung cancer. Oncogene. 17:1749–1753 [DOI] [PubMed] [Google Scholar]
- Miyoshi Y, Nagase H, Ando H, Horii A, Ichii S, Nakatsuru S, Aoki T, Miki Y, Mori T, Nakamura Y. 1992. Somatic mutations of the APC gene in colorectal tumors: mutation cluster region in the APC gene. Hum Mol Genet. 1:229–233 [DOI] [PubMed] [Google Scholar]
- Molenaar M, van de Wetering M, Oosterwegel M, Peterson-Maduro J, Godsave S, Korinek V, Roose J, Destree O, Clevers H. 1996. XTcf-3 transcription factor mediates beta-catenin-induced axis formation in Xenopus embryos. Cell. 86:391–399 [DOI] [PubMed] [Google Scholar]
- Petersen S, Rudolf J, Bockmuhl U, Gellert K, Wolf G, Dietel M, Petersen I. 1998. Distinct regions of allelic imbalance on chromosome 10q22-q26 in squamous cell carcinomas of the lung. Oncogene. 17:449–454 [DOI] [PubMed] [Google Scholar]
- Polakis P. 2000. Wnt signaling and cancer. Genes Dev. 14:1837–1851 [PubMed] [Google Scholar]
- Polakis P, Hart M, Rubinfeld B. 1999. Defects in the regulation of beta-catenin in colorectal cancer. Adv Exp Med Biol. 470:23–32 [DOI] [PubMed] [Google Scholar]
- Powell SM, Zilz N, Beazer-Barclay Y, Bryan TM, Hamilton SR, Thibodeau SN, Vogelstein B, Kinzler KW. 1992. APC mutations occur early during colorectal tumorigenesis. Nature. 359:235–237 [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. 1996. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science. 272:1023–1026 [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Souza B, Albert I, Muller O, Chamberlain SH, Masiarz FR, Munemitsu S, Polakis P. 1993. Association of the APC gene product with beta-catenin. Science. 262:1731–1734 [DOI] [PubMed] [Google Scholar]
- Su LK, Vogelstein B, Kinzler KW. 1993. Association of the APC tumor suppressor protein with catenins. Science. 262:1734–1737 [DOI] [PubMed] [Google Scholar]
- Tang XD, Zhou X, Zhou KY. 2009. Dauricine inhibits insulin-like growth factor-I-induced hypoxia inducible factor 1alpha protein accumulation and vascular endothelial growth factor expression in human breast cancer cells. Acta Pharmacol Sin. 30:605–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyssen G, Li TH, Lehmann L, Zhuo M, Sharma M, Sun Z. 2006. LZTS2 is a novel beta-catenin-interacting protein and regulates the nuclear export of beta-catenin. Mol Cell Biol. 26:8857–8867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whang YE, Wu X, Suzuki H, Reiter RE, Tran C, Vessella RL, Said JW, Isaacs WB, Sawyers CL. 1998. Inactivation of the tumor suppressor PTEN/MMAC1 in advanced human prostate cancer through loss of expression. Proc Natl Acad Sci U S A. 95:5246–5250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wodarz A, Nusse R. 1998. Mechanisms of Wnt signaling in development. Annu Rev Cell Dev Biol. 14:59–88 [DOI] [PubMed] [Google Scholar]
- Zhang X, Gaspard JP, Chung DC. 2001. Regulation of vascular endothelial growth factor by the Wnt and K-ras pathways in colonic neoplasia. Cancer Res. 61:6050–6054 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.