

Abstract
Small-molecule inhibition of hypoxia-inducible factor prolyl 4-hydroxylases (HIF-P4Hs) is being explored for the treatment of anemia. Previous studies have suggested that HIF-P4H-2 inhibition may also protect the heart from an ischemic insult. Hif-p4h-2gt/gt mice, which have 76 to 93% knockdown of Hif-p4h-2 mRNA in endothelial cells, fibroblasts, and cardiomyocytes and normoxic stabilization of Hif-α, were subjected to ligation of the left anterior descending coronary artery (LAD). Hif-p4h-2 deficiency resulted in increased survival, better-preserved left ventricle (LV) systolic function, and a smaller infarct size. Surprisingly, a significantly larger area of the LV remained perfused during LAD ligation in Hif-p4h-2gt/gt hearts than in wild-type hearts. However, no difference was observed in collateral vessels, while the size of capillaries, but not their number, was significantly greater in Hif-p4h-2gt/gt hearts than in wild-type hearts. Hif-p4h-2gt/gt mice showed increased cardiac expression of endothelial Hif target genes for Tie-2, apelin, APJ, and endothelial nitric oxide (NO) synthase (eNOS) and increased serum NO concentrations. Remarkably, blockage of Tie-2 signaling was sufficient to normalize cardiac apelin and APJ expression and resulted in reversal of the enlarged-capillary phenotype and ischemic cardioprotection in Hif-p4h-2gt/gt hearts. Activation of the hypoxia response by HIF-P4H-2 inhibition in endothelial cells appears to be a major determinant of ischemic cardioprotection and justifies the exploration of systemic small-molecule HIF-P4H-2 inhibitors for ischemic heart disease.
INTRODUCTION
Hypoxia-inducible factor (HIF) prolyl 4-hydroxylase 2 (HIF-P4H-2, also known as PHD2 and EglN1) is the main regulator of the oxygen-dependent proteasomal degradation of the HIF-α subunit (1–3). Inhibition of HIF-P4H-2 stabilizes HIF-α, which initiates the cellular hypoxia response involving the upregulation of several hundred genes, such as those for erythropoietin and vascular endothelial growth factor (1–3). Hif-p4h-2/EglN1 deletion is embryonic lethal, and universal conditional inactivation of Hif-p4h-2/EglN1 leads to severe erythrocytosis, hyperactive angiogenesis, and congestive heart failure (CHF) (4, 5). CHF does not appear to be a major consequence of Hif-p4h-2/EglN1 inactivation in the heart but rather a cause of the severe polycythemia observed in these mice (6). Mice with cardiomyocyte-specific Hif-p4h-2/EglN1 deletion develop mild cardiac dysfunction after 8 months of age (6, 7) but show a reduced infarct size and improved mechanical recovery following ligation of the left anterior descending coronary artery (LAD) (7).
By using the gene trap (gt) strategy, we have generated a mouse line with tissue-specific Hif-p4h-2/EglN1 inactivation (Hif-p4h-2gt/gt) (8). These mice have a 92% reduction of cardiac Hif-p4h-2 mRNA levels and normoxic stabilization of Hif-1α and Hif-2α in the heart (8). Hif-p4h-2gt/gt mice show 85 to 15% reductions in Hif-p4h-2 mRNA levels in other tissues, but unlike mice with broad-spectrum conditional inactivation of Hif-p4h-2, they do not have polycythemia (8). Hif-p4h-2/EglN1 deficiency in the hearts of Hif-p4h-2gt/gt mice leads to the induction of several Hif target genes and improved recovery of the isolated hearts from ex vivo ischemia-reperfusion (I-R) injury (8).
Small-molecule inhibitors that target HIF-P4Hs and stabilize HIF-α in normoxia are currently in clinical trials for the treatment of anemia (9). Inhibition of Hif-p4h-2 in Hif-p4h-2gt/gt mice is not restricted to one cell type of the heart, which makes them a more suitable model of the in vivo effects of therapeutic HIF-P4H inhibitors than mice with cardiomyocyte-only Hif-p4h-2 deletion. In this study, Hif-p4h-2gt/gt mice were subjected to acute myocardial infarction (AMI) and cardiac I-R injury. We found that Hif-p4h-2 deficiency enhanced survival and resulted in better-preserved left ventricle (LV) systolic function following ischemia. The hearts of Hif-p4h-2gt/gt mice had a smaller area at risk (AR), increased capillary size, enhanced nitric oxide (NO) production, and increased Tie-2 receptor expression. Finally, we show that Tie-2 is the major effector of stabilized HIF in the endothelium, since blockade of Tie-2 signaling was sufficient to reverse the enlarged-capillary phenotype and prevent ischemic cardioprotection.
MATERIALS AND METHODS
Hif-p4h-2gt/gt mice.
Generation of the Hif-p4h-2gt/gt mouse line has been described earlier (8). Shortly, a gt targeting vector was introduced into intron 1 of the Hif-p4h-2/EglN1 gene in the embryonic stem cells used for the generation of Hif-p4h-2gt/gt mutant mice in the C57BL/6 background. Surprisingly, the targeted alleles yield, in addition to the truncated trapped Hif-p4h-2 mRNA, some wild-type Hif-p4h-2 mRNA because of partial skipping of the targeting vector. The level of wild-type Hif-p4h-2 mRNA varied between tissues, being the lowest in the heart. Heterozygous matings of Hif-p4h-2wt/gt mice resulted in live pups with two targeted alleles but in a reduced Mendelian ratio. However, the Hif-p4h-2gt/gt mice born appeared to have a normal life span.
Fractioning of cardiac cells.
Adult ventricular myocytes were isolated from 5-month-old mice as described previously (10). Briefly, each mouse was deeply anesthetized with isoflurane and the heart was excised rapidly, cannulated through the aorta, and retrogradely perfused with HEPES-buffered Tyrode's solution supplemented with 0.1% collagenase type II (Worthington) and 2,3-butandione-monoxime. The ventricular tissue was homogenized, and myocytes were collected by low-speed centrifugation (18 × g for 2 min). After calcium reintroduction, the myocytes were plated onto laminin-coated plates in minimum essential medium–5% fetal bovine serum (FBS)–insulin-transferrin-selenin (Invitrogen)–10 mM 2,3-butandione-monoxime–2 mM l-glutamine–penicillin-streptomycin (Sigma). After myocyte attachment, the plates were washed with Tyrode's solution to remove nonadherent cells and frozen at −70°C. After low-speed centrifugation to pellet the myocytes, the supernatant was collected and centrifuged at 300 × g for 5 min to pellet the fibroblasts and endothelial cells. These cells were resuspended in phosphate-buffered saline (PBS)–2 mM EDTA–2% FBS and filtered through 40-μm nylon mesh (BD Falcon). The endothelial cells were isolated by immunomagnetic cell separation with rat anti-mouse CD31 antibody (BD Biosciences) and anti-rat IgG microbeads (Miltenyi Biotec) with MS columns and a VarioMACS separator (Miltenyi Biotec). The CD31-positive endothelial fraction and the CD31-negative fibroblast fraction were frozen at −70°C.
Real-time quantitative PCR (qPCR).
Hearts were quickly dissected at sacrifice and snap-frozen in liquid nitrogen. Total RNA was isolated with the TriPure isolation reagent (Roche Applied Science), further purified with an EZNA total RNA kit (OMEGA Bio-tek), and subjected to reverse transcription with an iScript cDNA synthesis kit (Bio-Rad). The baseline samples were collected and processed in the same way. The EZNA total RNA kit (OMEGA Bio-tek) and the iScript cDNA synthesis kit (Bio-Rad) were used to isolate RNA and synthesize cDNA from the fractionated cardiomyocytes, fibroblasts, and endothelial cells. qPCR was performed with iTaq SYBR green Supermix and ROX (Bio-Rad) in a Stratagene MX3005 thermocycler or by TaqMan chemistry in an ABI Prism 7700 sequence detection system (Applied Biosystems). For the sequences of the primers and probes used for qPCR, see Table S1 in the supplemental material.
In vivo cardiac surgery.
The surgical procedures were performed by two experienced surgeons (E.G. and L.V.) as previously described (11). All animals in a given experiment were operated on by the same surgeon at the same time. In summary, mice were anesthetized with 2% isoflurane inhalation but not ventilated. A small skin cut (1.2 cm) was made over the left chest, and a purse string suture was made. The pectoral major and minor muscles were dissected and retracted, and the fourth intercostal space was exposed to make a small hole in it with a mosquito clamp to open the pleural membrane and pericardium. With the clamp slightly open, the heart was gently popped out through the hole. The LAD was located and ligated at a site 2 to 3 mm from its origin with a 6-0 silk suture. The ligation was deemed successful when the anterior wall of the LV turned pale. After ligation, the heart was immediately placed back into the intrathoracic space; this was followed by manual evacuation of air and closure of the muscle and skin by means of the previously placed purse string suture. The mouse was then allowed to breathe room air. The average procedure time was about 1.2 min. The animals were monitored for 3 or 24 h after ischemia. In the case of I-R, the internal needle end of a slipknot suture was cut as short as possible and the other end of the suture, about 0.8 cm long, remained outside the chest. After 30 min of ischemia, the slipknot was released by pulling the long end of the slipknot suture smoothly and gently until a feeling of release was sensed, at which time the myocardium began being reperfused. The permanent LAD ligations were performed with female mice 8 to 9 months old, and the I-R studies were performed with male mice 3 to 8 months old. All animal experiments were performed according to protocols approved by the Provincial State Office of Southern Finland.
Echocardiography.
The mice were anesthetized with isoflurane and analyzed for cardiac dimensions and function with a Visualsonics Vevo 2100 high-resolution ultrasound imaging system. LV end-systolic and end-diastolic dimensions and the thicknesses of the interventricular septum and posterior wall (PW) were measured. LV fractional shortening and the ejection fraction were calculated from the M mode.
Determination of AR and infarct sizes.
Infarct size and the size of the AR were determined as previously described (11). At the end of the 24-h reperfusion period, the mice were reanesthetized and the ligature around the LAD was retied through the previous ligation site. Following this, 2% Evans blue dye was injected and allowed to circulate uniformly in the areas of the heart perfused by the open coronary arteries. In the case of AMI, the ligature was present and no retying was performed. The heart was then quickly excised and cut into five sections from the apex to the base. These sections were then incubated in a 1% triphenyltetrazolium chloride (TTC) (Sigma) solution and photographed with a Canon EOS 450D digital camera with a 100-mm macro lens. The area not at risk (ANAR; Evans blue-stained area) and the AR (including both the TTC staining-positive [noninfarct, red] and TTC staining-negative [infarct, white] areas) were measured with the Nikon NIS-Elements BR 2.30 program.
Serum analyses.
The serum fraction was collected from the terminal blood of the mice at sacrifice, and serum cardiac troponin-I (s-cTNI) was measured in the 24-h post-I-R samples with a high-sensitivity mouse cTNI enzyme-linked immunosorbent assay kit (Life Diagnostics Inc.). Serum NO concentrations in the samples collected at 24 h after I-R were measured with a Parameter total NO/nitrite/nitrate immunoassay (R&D Systems).
Histological analyses.
The dissected hearts were fixed in 10% formalin overnight at room temperature, washed with 0.15 M NaCl in 0.02 M phosphate (pH 7.4), and transferred to 70% ethanol. They were then dehydrated and embedded in paraffin, and 5-μm sections were cut. For the cryosections, paraformaldehyde-fixed specimens were transferred to 15% sucrose at 4°C for 1 h, followed by overnight incubation in 30% sucrose at 4°C and embedding in O.C.T. compound (Sakura). Five-micrometer sections were cut. In order to detect apoptotic cells, the sections were stained with a fluorescein in situ cell death detection kit (Roche). The stained heart sections were viewed and photographed with an Olympus BX51 microscope equipped with an Olympus XC50 camera. The number of terminal deoxynucleotidyltransferase-mediated dUTP-biotin nick end labeling (TUNEL)-positive cells per section was determined from the photographs by the Dot Count program.
The heart sections were stained with Pecam-1 (sc-1506 [Santa Cruz Biotechnology, CA] or MEC13.3 [BD Pharmingen]) to identify the endothelial cells by using a Nikon Eclipse 50i microscope and DS-5 M-L2 camera, a Leica DM LB2 microscope and DFC 320 camera, or an Olympus BX51 microscope and DP50 camera to view and photograph the specimens. The numbers of capillaries per field and capillary lumen area were measured in five representative high-power fields (40× objective) by the Nikon NIS-Elements BR 2.30 program. Five corresponding fields were chosen from the LV of each sample for comparison. Masson's trichrome staining of the consecutive heart sections was performed to visualize cardiomyocytes. The slices were viewed and photographed with a Nikon Eclipse 50i microscope and DS-5 M-L2 camera, and the area of the cardiomyocytes was measured by the Nikon NIS-Elements BR 2.30 program. The capillary area/cardiomyocyte area ratio was calculated.
To quantify the number of collateral vessels, α-smooth muscle cell actin (α-SMA) antibody (Ab5694; Abcam) and biotinylated isolectin B4 (B-1205, Vector Laboratories) costaining of slices from three distinct levels of the heart were performed and viewed and photographed with a Nikon Eclipse 80i microscope and DS-Qi1Mc camera. Masson's trichrome or periodic acid-Schiff (PAS) staining was used in addition to detect collateral vessels in the smooth muscle cell layer of the heart slices. The collateral lumen area was calculated from 18 to 29 representative high-power field (40× objective) pictures per heart taken from Masson's trichrome- or PAS-stained heart sections with a 40× objective on a Nikon Eclipse 80i microscope and DS-Qi1Mc camera and quantified by the ImageJ program. These data were justified to the heart area.
Resin casting of coronary arteries and optical projection tomography (OPT).
Hearts of deeply isoflurane-anesthetized mice were dissected, and the aorta was cannulated to initiate Langendorff perfusion. The hearts were fixed with 3% paraformaldehyde in PBS at a speed of 2 ml/min, followed by resin casting with PU4ii resin (VasQtec) at the same speed. The casts were allowed to harden overnight at room temperature. The hearts were placed in 100% glycerol for 1 h prior to OPT to induce optical clearing. OPT scanning of the heart was then performed in glycerol with a Bioptonics 3001 device in the Texas Red channel (560/40-nm exciter, 610-nm long-pass emitter) exploiting the autofluorescence of the cast material.
Transmission electron microscopy (TEM) analysis.
Heart samples were fixed in 1% glutaraldehyde and 4% formaldehyde in 0.1 M phosphate buffer, postfixed in 1% osmium tetroxide, dehydrated in acetone, and embedded in Epon LX 112 (Ladd Research Industries). Thin sections were cut with a Leica Ultracut UCT ultramicrotome, stained in uranyl acetate and lead citrate, and examined in a Philips CM100 transmission electron microscope. The images were captured with a Morada charge-coupled device camera (Olympus Soft Imaging Solutions).
Western blotting (WB).
Cardiac homogenates were prepared in 150 mM NaCl–50 mM Tris-HCl (pH 7.5)–0.5% NP-40. The antibodies used were against HIF-1α (NB100-479; Novus Biologicals), HIF-2α (NB100-122; Novus Biologicals), Tie-2 (05-584 [Millipore] and sc-324 [Santa Cruz]), P-Tie-2 (P-Tyr ab 4G10; Millipore), vascular endothelium cadherin (VE-cadherin) (V1514; Sigma-Aldrich), and tubulin (B-6199; Sigma-Aldrich). Bound antibodies were detected with horseradish peroxidase-conjugated secondary antibodies (Dako) and enhanced-chemiluminescence detection reagents (Thermo Scientific). Agarose-conjugated sc-324 beads (Santa Cruz) were used for immunoprecipitation.
Analysis of endogenous Tie-2 signaling in human umbilical vein endothelial cells (HUVECs).
HUVECs were stimulated for 15 min with cartilage oligomeric protein (COMP)–angiopoietin-1 (Ang-1), which contains the Tie-2 receptor binding part of Ang-1 but in which the coiled-coil domains of Ang-1 are replaced with COMP (1.5 μg/ml), the mouse Tie-2 extracellular domain (ECD; 1.5 μg/ml), or a combination of the two. The cells were lysed in buffer containing 0.5% Triton X-100 and 0.5% NP-40 supplemented with protease inhibitors (Complete; Roche). Immunoprecipitation was performed with a goat anti-human Tie-2 antibody (R&D Systems) and WB with mouse anti-P-Tyr antibody (Millipore). Band densitometry was done with ImageJ software.
Blocking of Tie-2 signaling in vivo.
Adeno-associated virus 9 (AAV9) that expresses Tie-2 ECD-Flag and an enhanced green fluorescent protein (EGFP) control was generated as described earlier (12). Virus particles (1011) were injected into 4-week-old isoflurane-anesthetized mice via the vena jugularis.
Statistical analyses.
Statistical analyses were performed with Student's two-tailed t test. The data are shown as means ± the standard error of the means. Pearson's correlation coefficient was calculated to compare linear dependences between two variables. P values of <0.05 were considered statistically significant (symbols in figures: *, P < 0.05; **, P < 0.01; ***, P < 0.005).
RESULTS
Hif-p4h-2 deficiency is significant in all resident cell types in the Hif-p4h-2gt/gt mouse heart.
We first fractionated from wild-type and Hif-p4h-2gt/gt hearts the resident cardiac cell types, i.e., cardiomyocytes, fibroblasts, and endothelial cells (Fig. 1A), all of which can contribute to recovery following ischemic injury, and analyzed their Hif-p4h-2 expression levels. qPCR analysis showed that Hif-p4h-2 mRNA was reduced by 93% in the cardiomyocytes, 77% in the fibroblasts, and 76% in the endothelial cells extracted from Hif-p4h-2gt/gt hearts (Fig. 1B). Thus, the reduction of Hif-p4h-2 in Hif-p4h-2gt/gt mutant hearts is not restricted to the cardiomyocytes as in earlier studies (6, 7).
Fig 1.
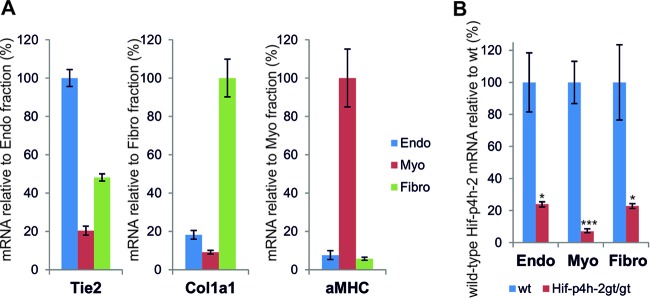
Expression of Hif-p4h-2 mRNA is significantly lower in all resident cardiac cell types in Hif-p4h-2gt/gt mice. (A) qPCR analysis of the expression of Tie-2, Col1a1, and α-MHC mRNAs in pools of fractionated cardiac cells (wild-type hearts, n = 3; Hif-p4h-2gt/gt hearts, n = 3). Tie-2, Col1a1, and α-MHC (aMHC) were used as markers for endothelial cells, fibroblasts, and cardiomyocytes, respectively. (B) Expression of wild-type (wt) Hif-p4h-2 mRNA in endothelial cells (Endo), cardiomyocytes (Myo), and fibroblasts (Fibro) in the hearts of Hif-p4h-2gt/gt mice relative to that in the wild type, studied by qPCR (wild-type hearts, n = 3; Hif-p4h-2gt/gt hearts, n = 3).
Hif-p4h-2gt/gt mice show increased survival of cardiac ischemia.
Echocardiography analyses of 4-week- and 4-month-old Hif-p4h-2gt/gt mutant mice showed no difference from the wild type in LV function or structure at the baseline (see Table S2 in the supplemental material). A mild increase in LV PW thickness was observed in the hearts of 10- to 11-month-old Hif-p4h-2gt/gt mice relative to that in the wild type, but in contrast to earlier published data obtained with cardiomyocyte-specific Hif-p4h-2-deficient hearts (6), no significant differences between the genotypes in LV systolic or diastolic function were found (see Table S3). We then subjected age- and sex-matched Hif-p4h-2gt/gt mutant and wild-type mice to ligation of the LAD. While 26.5% of the wild-type mice died during the first 30 min of ischemia, 100% of the Hif-p4h-2gt/gt mice survived.
Cardiac Hif-p4h-2 deficiency results in improved recovery following AMI.
Mechanical recovery of cardiac function was analyzed by echocardiography 24 h after LAD ligation. We observed a significantly better-preserved LV ejection fraction (%EF) in Hif-p4h-2gt/gt than in wild-type hearts following AMI (Fig. 2A; Table 1). The cTNI concentration in serum collected from terminal blood taken at sacrifice 24 h after LAD ligation was significantly lower in Hif-p4h-2gt/gt mice (Fig. 2B). Significant reductions of 55 and 35%, respectively, were observed in the size-of-infarct/LV (I/LV) and I/AR ratios in Hif-p4h-2gt/gt hearts relative to the wild type determined by Evans blue and TTC staining (Fig. 2C). Interestingly, the AR/LV ratio was reduced by 30% in Hif-p4h-2gt/gt hearts (Fig. 2C), suggesting better perfusion of the heart following LAD ligation in mice lacking Hif-p4h-2 than in wild-type mice.
Fig 2.
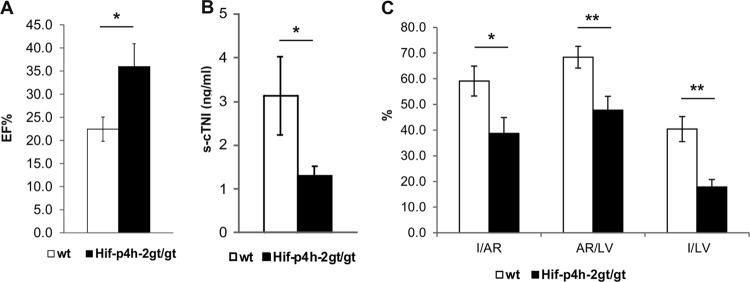
Hif-p4h-2gt/gt mice have better-preserved LV systolic function, a smaller infarct size, and a reduced AR following AMI. (A) %EF at 24 h after AMI (n = 9 for wild-type [wt] mice; n = 8 for Hif-p4h-2gt/gt mice). (B) s-cTNI concentrations at 24 h after AMI (wild-type serum samples, n = 9; Hif-p4h-2gt/gt serum samples, n = 6). (C) Data for Evans blue- and TTC-stained hearts 24 h after AMI. I/AR, AR/LV area (wild-type hearts, n = 9; Hif-p4h-2gt/gt hearts, n = 6), and I/LV (wild-type heart n = 9, Hif-p4h-2gt/gt heart n = 9) ratios were compared.
Table 1.
Hif-p4h-2gt/gt mutant mice show preserved cardiac function 24 h after AMIa
| Genotype (no.) of mice | LVID, dc (mm) | LVID, sd (mm) | PW, de (mm) | %FSf | %EF | IVRTg |
|---|---|---|---|---|---|---|
| Wild type (7) | 4.46 ± 0.12 | 4.01 ± 0.14 | 0.59 ± 0.04 | 10.3 ± 1.3 | 22.4 ± 2.6 | 17.7 ± 1.7 |
| Hif-p4h-2gt/gt (6) | 4.39 ± 0.25 | 3.64 ± 0.28 | 0.62 ± 0.06 | 17.3 ± 2.6b | 36.0 ± 5.0b | 17.4 ± 1.8 |
Mice were subjected to AMI by ligation of the LAD. They were then monitored for 24 h and analyzed for cardiac function with a Visualsonics Vevo 2100 high-resolution ultrasound imaging system at 24 h after AMI. Values are averages and standard errors of means.
P < 0.05 versus the wild type.
LVID, d, LV end-diastolic diameter.
LVID, s, LV end-systolic diameter.
PW, d, PW thickness, diastole.
%FS, percent fractional shortening.
IVRT, isovolumetric relaxation time.
Lack of Hif-p4h-2 improves cardiac function and reduces apoptosis following I-R injury.
We next subjected the mice to 30 min of ischemia by LAD ligation, followed by 3 or 24 h of reperfusion. The Hif-p4h-2gt/gt mice had a significantly better-preserved %EF 24 h after I-R than the wild-type mice did (Fig. 3A and B, Table 2). The number of TUNEL-positive cells at 3 h after I-R was significantly lower in the hearts of Hif-p4h-2gt/gt mice than in the hearts of wild-type mice (Fig. 3C). s-cTNI collected 24 h after I-R showed a tendency toward a lower level in Hif-p4h-2gt/gt than in wild-type mice (Fig. 3D). In order to determine AR and infarct sizes with Evans blue and TTC staining, the LAD was retied under terminal anesthesia 24 h after I-R (Fig. 3E). There was a trend toward a lower I/LV ratio in Hif-p4h-2gt/gt hearts than in wild-type hearts and a significant negative correlation between I/LV and %EF; i.e., a smaller infarct size correlated with a higher ejection fraction (r = −0.66, P < 0.05) but there was no significant difference in I/AR between the genotypes (Fig. 3F). Interestingly, and similar to that in the AMI model (Fig. 2C), AR/LV ratio was significantly lower in Hif-p4h-2gt/gt hearts than in wild-type hearts (Fig. 3F).
Fig 3.

Hif-p4h-2gt/gt hearts are protected against cardiac I-R injury. (A) %EF at 24 h after I-R (wild-type [wt] hearts, n = 9; Hif-p4h-2gt/gt hearts, n = 5). (B) Representative M-mode images of wild-type and Hif-p4h-2gt/gt hearts at 24 h following I-R. The systolic function in wild-type mice is notably impaired compared with that in Hif-p4h-2gt/gt mice. The two genotypes show similar cardiac dimensions. (C) Average number of TUNEL-positive cells in LV sections of hearts at 3 h after I-R (wild-type hearts, n = 12; Hif-p4h-2gt/gt hearts, n = 6). (D) s-cTNI concentrations in mice at 24 h after I-R (wild-type serum samples, n = 8; Hif-p4h-2gt/gt serum samples, n = 4). (E) Representative samples of Evans blue- and TTC-stained hearts. The blue-stained area became perfused during LAD ligation, while the red-stained area is noninfarcted tissue and the white area is infarcted tissue. (F) Quantification of the data for TTC-stained hearts 24 h after I-R. I/AR, AR/LV (wild-type hearts, n = 8; Hif-p4h-2gt/gt hearts, n = 4), and I/LV (wild-type hearts, n = 9; Hif-p4h-2gt/gt hearts, n = 4) ratios are shown for wild-type and Hif-p4h-2gt/gt hearts.
Table 2.
Hif-p4h-2gt/gt mutant mice show preserved cardiac function 24 h after I-R injurya
| Genotype (no.) of mice | LVID, dc (mm) | LVID, sd (mm) | PW, de (mm) | %FSf | %EF |
|---|---|---|---|---|---|
| Wild type (9) | 4.38 ± 0.11 | 3.64 ± 0.16 | 0.98 ± 0.05 | 17.2 ± 2.0 | 35.6 ± 3.9 |
| Hif-p4h-2gt/gt (5) | 4.43 ± 0.13 | 3.40 ± 0.14 | 0.92 ± 0.09 | 23.4 ± 1.3b | 46.9 ± 2.2b |
Mice were subjected to I-R injury by ligation of the LAD and release of the slipknot after 30 min to allow reperfusion of the myocardium. They were then monitored for 24 h and analyzed for cardiac function 24 h after I-R with a Visualsonics Vevo 2100 high-resolution ultrasound imaging system. Values are averages and standard errors of means.
P < 0.05 versus the wild type.
LVID, d, LV end-diastolic diameter.
LVID, s, LV end-systolic diameter.
PW, d, PW thickness, diastole.
%FS, percent fractional shortening.
Capillary size is increased in Hif-p4h-2gt/gt hearts.
Analysis of cardiac vasculature showed no gross alterations in the branching of the LAD or the right coronary artery (RCA) between the genotypes (Fig. 4A; see Videos S1 and S2 in the supplemental material). Quantification of the number and size of collateral vessels by α-SMA and isolectin costaining revealed no difference in the anatomy of arteries between wild-type and Hif-p4h-2gt/gt hearts (Fig. 4B and C). The number of capillaries in Pecam-1-stained sections of Hif-p4h-2gt/gt hearts was comparable to that in wild-type hearts. However, the capillary size was markedly increased in Hif-p4h-2gt/gt hearts (6.1 ± 0.3 μm2 in Hif-p4h-2gt/gt hearts versus 4.5 ± 0.2 μm2 in wild-type hearts, P = 0.01) (Fig. 4D and E; see Fig. S1 in the supplemental material). Importantly, the difference in capillary size also persisted following I-R injury (see Fig. S2 in the supplemental material). The average number of nuclei in a capillary was also significantly greater in Hif-p4h-2gt/gt hearts than in wild-type hearts (Fig. 4G). TEM analysis showed enlarged capillaries in Hif-p4h-2gt/gt hearts, often with a wavy endothelial layer (Fig. 4F).
Fig 4.

Hif-p4h-2gt/gt hearts have larger capillaries. (A) Resin casts of heart vasculature. The LAD and RCA are indicated by dotted lines. (B and C) Baseline α-SMA (red) and isolectin (green) costaining of hearts and quantification of the number (wild-type [wt] hearts, n = 5; Hif-p4h-2gt/gt hearts, n = 4) and size (wild-type hearts, n = 6; Hif-p4h-2gt/gt hearts, n = 6) of collateral vessels. (D and E) Baseline Pecam-1-stained hearts and quantification of the number and size of capillaries (wild-type hearts, n = 3; Hif-p4h-2gt/gt hearts, n = 3). (F) TEM of wild-type and Hif-p4h-2gt/gt hearts. Large capillaries (c) are visible in the latter. (G) Quantification of the number of nuclei per capillary on the basis of TEM analysis (wild-type hearts, n = 3; Hif-p4h-2gt/gt hearts, n = 3).
Lack of Hif-p4h-2 modulates the expression of endothelial Hif target genes in the heart.
Analysis of endothelium-specific Hif targets and known angiogenic genes showed significantly higher endothelial angiopoietin receptor tyrosine kinase Tie-2, apelin, apelin receptor APJ, and Ang-2 mRNA levels in Hif-p4h-2gt/gt hearts than in wild-type hearts (Fig. 5A). However, no significant changes in Tie-1, Ang-1, Vegfa, Vefgb, Vegfc, Vegf-R1, Vegf-R2, Nrp1, eNOS, or inducible NO synthase mRNA expression were found (Fig. 5A). The increased Tie-2-encoding gene expression correlated with higher Tie-2 protein levels in Hif-p4h-2gt/gt hearts than in wild-type hearts, whereas equal amounts of VE-cadherin, another endothelial protein, were detected (Fig. 5B). Importantly, the Tie-2 protein in Hif-p4h-2gt/gt hearts was phosphorylated, suggesting increased signaling through Tie-2 in the endothelial cells of these hearts (Fig. 5B). Ang-2 is regarded as an antagonist of Tie-2, while Ang-1 is the main agonist (13), although in effect it is the Ang-1/Ang-2 ratio that is an essential determinant of Tie-2 activation. The Ang-1/Ang-2 ratio was 10 in Hif-p4h-2gt/gt hearts and 12.5 in wild-type hearts, suggesting that agonistic signaling prevails in the former hearts.
Fig 5.

Increased expression of endothelial genes in Hif-p4h-2gt/gt hearts. (A) qPCR analysis of baseline endothelial and vascular gene mRNA levels in hearts (wild-type hearts, n = 5; Hif-p4h-2gt/gt hearts, n = 5). (B) WB for baseline Tie-2, phosphorylated Tie-2 (P-Tie-2), and VE-cadherin levels in wild-type (wt) and Hif-p4h-2gt/gt hearts. Hif-1α and Hif-2α are shown as controls, and tubulin is a loading control. Where indicated, Tie-2 was immunoprecipitated (IP) from heart homogenate with anti-Tie-2 (sc-324) beads and detected with P-Tyr and anti-Tie-2 antibody (Ab33). On the basis of densitometry, the expression level of Tie-2/tubulin in Hif-p4h-2gt/gt hearts was 134% ± 24% (n = 3) compared to 100% ± 23% (n = 3) in the wild type. (C and D) qPCR analysis of eNOS mRNA from endothelial cells at the baseline (wild-type cells, n = 6; Hif-p4h-2gt/gt cells, n = 5) and whole hearts 3 h after I-R (wild-type hearts, n = 5; Hif-p4h-2gt/gt hearts, n = 4). (E) NO concentrations in the serum of mice at 3 h after I-R (wild-type serum samples, n = 11; Hif-p4h-2gt/gt serum samples, n = 4). iNOS, induced NO synthase.
The major source of NO in the myocardium under physiological conditions is eNOS (14). There was no difference in the baseline eNOS mRNA expression level between Hif-p4h-2gt/gt and wild-type total hearts, but it was significantly induced in the endothelial cell fraction isolated from Hif-p4h-2gt/gt hearts (Fig. 5C). Following I-R injury, significantly higher eNOS mRNA levels were found in Hif-p4h-2gt/gt whole-heart extracts than in wild-type whole-heart extracts (Fig. 5D), and they were accompanied by increased serum NO concentrations in Hif-p4h-2gt/gt mice (Fig. 5E).
Increased capillary size in Hif-p4h-2gt/gt hearts can be reversed by blocking Tie-2 signaling.
Overexpression of the main Tie-2 agonist, Ang-1, has been reported to result in an increased vessel diameter (15). To reveal whether increased Tie-2 expression was responsible for the enlarged capillaries in Hif-p4h-2gt/gt hearts, we blocked Tie-2-mediated signaling in 4-week-old mice by AAV9-mediated overexpression of the Tie-2 ECD, which binds angiopoietins but does not transduce the signal inside the cell, thereby blocking endogenous Tie-2 signaling, as shown in cultured HUVECs (see Fig. S3 in the supplemental material). Four weeks after AAV9-mediated gene transfer, equal levels of Tie-2 ECD were observed in the hearts of both genotypes (Fig. 6A) and echocardiography analysis showed no difference in their LV function (not shown). Tie-2 blockage had normalized the apelin and APJ mRNA levels in Hif-p4h-2gt/gt hearts (Fig. 6B), and notably, no difference in capillary size existed between the genotypes (compare Fig. 6C and D to 4D and E).
Fig 6.
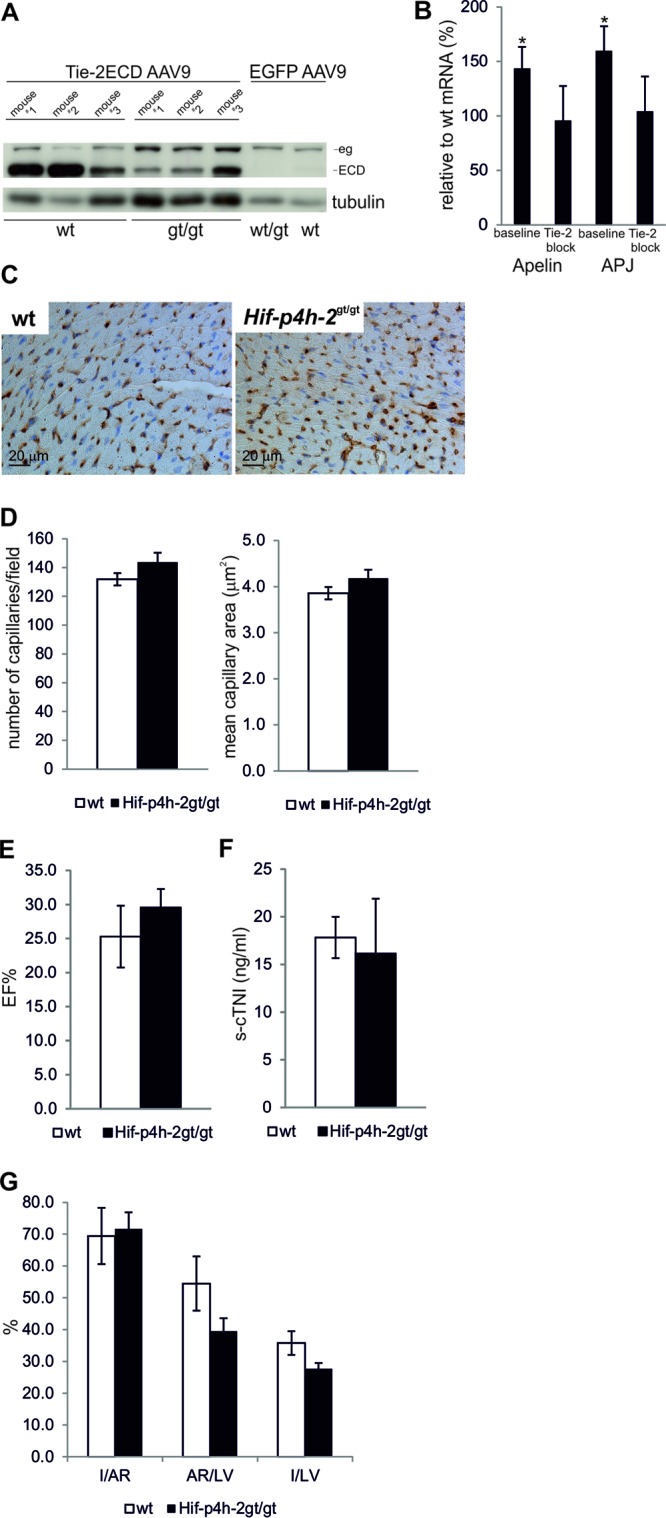
Changes in endothelial phenotype and ischemic cardioprotection in Hif-p4h-2gt/gt hearts are normalized by blockage of Tie-2 signaling. (A) Expression of the Tie-2 ECD transgene in the hearts of wild-type (wt) and Hif-p4h-2gt/gt mice analyzed by WB 4 weeks after AAV9 injection. Migration of endogenous Tie-2 (eg) and Tie-2 ECD (ECD) is indicated by arrows. EGFP AAV9 was used as a negative control. On the basis of densitometry, the expression level of endogenous Tie-2/tubulin in Hif-p4h-2gt/gt hearts was 129% ± 17% (n = 3) compared to 100% ± 7% (n = 3) in the wild type. (B) qPCR analysis of apelin and APJ mRNA expression in the hearts of Hif-p4h-2gt/gt mice 4 weeks after Tie-2 block relative to that in the wild type and to the baseline. (C and D) Pecam-1 staining and quantification of the number and size of capillaries of hearts 4 weeks after a Tie-2 block. In panels A, C, and D, n = 6 for wild-type and Hif-p4h-2gt/gt mice. (E) %EF at 24 h after AMI (n = 4 for wild type; n = 5 for Hif-p4h-2gt/gt). (F) s-cTNI concentrations at 24 h after AMI (wild-type serum samples, n = 4; Hif-p4h-2gt/gt serum samples, n = 5). (G) Data for Evans blue- and TTC-stained hearts 24 h after AMI (wild-type hearts, n = 4; Hif-p4h-2gt/gt hearts, n = 5).
Blocking of Tie-2 signaling reverses ischemic cardioprotection in Hif-p4h-2gt/gt hearts.
To study whether the Tie-2 induction-mediated increased capillary area was responsible for the observed ischemic cardioprotection seen, we performed AMI 4 weeks after in vivo Tie-2 blockage on both wild-type and Hif-p4h-2gt/gt mice. There was no longer any observed difference between the genotypes in the LV ejection fraction 24 h after LAD ligation (Fig. 6E) (P = 0.45). In accordance there was no significant difference in the concentration of s-cTNI 24 h after LAD ligation between the Hif-p4h-2gt/gt and wild-type mice (Fig. 6F) (P = 0.80); however, the s-cTNI concentration was more than 5-fold higher in both genotypes than that after AMI in wild-type mice with no Tie-2 blockage (Fig. 2B). No significant differences were observed between Hif-p4h-2gt/gt and wild-type hearts in the I/LV and I/AR ratios determined by Evans blue and TTC staining (Fig. 6G). However, the difference in AR/LV between wild-type and Hif-p4h-2gt/gt hearts was not fully reversed following Tie-2 blockage (Fig. 6G).
DISCUSSION
Recent studies have suggested that modulation of the hypoxia response pathway can lead to an improved outcome following cardiac ischemia (16–19). However, this has not been elucidated with genetic loss-of-function models involving substantial HIF-P4H inhibition in more than one cell type. We show here that Hif-p4h-2 deficiency results in better-preserved LV function and enhanced survival following AMI and I-R injury. Evans blue staining showed increased perfusion of Hif-p4h-2gt/gt hearts following LAD ligation, and microscopy analysis of cardiac tissue sections revealed enlarged capillaries in the hearts of Hif-p4h-2gt/gt mice. Coronary flow reserve (CFR), defined as the maximum increase in blood flow through the coronary arteries above the normal resting volume, is inversely related to infarct size (20) and is a strong predictor of LV function recovery after AMI (21, 22). During conditions of increased oxygen demand in the myocardium, there is a reduction in the resistance of the coronary arteries, facilitating increased blood flow. However, during maximal dilation of the coronary vasculature, capillary resistance becomes the main determinant of coronary blood flow. Under normal conditions, only a fraction of the capillaries are recruited, whereas under conditions of increased oxygen demand, there is a marked increase in the recruitment of capillaries. Since the capillaries are placed in parallel, their volume, determined by capillary density and area, is the main determinant of resistance and a change in their diameter (area) affects CFR more than a change in capillary density does (23, 24). Thus, enlarged capillaries in Hif-p4h-2gt/gt hearts are consistent with the observed AR reduction and improved outcome following LAD ligation.
To investigate the underlying mechanism, we analyzed the expression of genes known to affect vessel structure and formation and found increased expression of the gene for endothelial angiopoietin receptor Tie-2, a HIF target gene (25), in Hif-p4h-2gt/gt hearts. Ang-1, the main agonist for Tie-2, is essential in the mouse vasculature in response to injury (26), and overexpression of Ang-1 has been reported to increase vessel diameter (15). Hif-p4h-2 deficiency also increased the cardiac expression of apelin and its receptor APJ, which have been shown to be involved in the regulation of blood vessel diameter during angiogenesis (27). We then blocked Tie-2 signaling by AAV9-mediated overexpression of Tie-2 ECD, which was sufficient to normalize cardiac apelin and APJ mRNA levels and to reverse the capillary enlargement in Hif-p4h-2gt/gt hearts, confirming the key role of this pathway regulating the capillary phenotype. Tie-2 blockage reversed the better-preserved LV function and smaller infarct size in Hif-p4h-2gt/gt hearts than in the wild type following AMI, suggesting that Hif-p4h-2 deficiency-mediated Tie-2 induction is at least partly responsible for the improved ischemia tolerance of Hif-p4h-2gt/gt hearts. The high induction of s-cTNI following AMI in wild-type and Hif-p4h-2gt/gt mice after Tie-2 blockage further suggests that Tie-2 signaling offers protection from myocardial ischemia. Our data do not, however, exclude the possibility that some paracrine factors from cardiomyocytes and/or fibroblasts in the Hif-p4h-2-deficient heart induced Tie-2 expression in endothelial cells or stimulated the proliferation of Tie-2-expressing endothelial cells.
It was recently reported that mice haplodeficient in Hif-p4h-2 have normal capillary density in the heart but increased collateral artery growth that is due to activation of the NF-κB pathway in Hif-p4h-2+/− macrophages (28). Hif-p4h-2gt/gt mice, however, have a more complete cardiac inhibition of HIF-P4H-2, which may be necessary for development of the observed endothelial phenotype.
As another determinant of the crucial role of the endothelial cells in ischemic cardioprotection, we found increased induction of cardiac eNOS mRNA, increased serum NO levels, and reduced cardiac apoptosis in Hif-p4h-2gt/gt mice following ischemic injury. NO is known to mediate several beneficial effects promoting cardiac ischemia survival (29). Regulation of eNOS mRNA has been shown to be under the control of multiple regulators, including the Foxo transcription factors that take part in postnatal vessel formation and vessel maturation (30). A recent study showed, interestingly, that pharmacological induction of HIF-1α in endothelial cells promotes the protection of cardiomyocytes from hypoxia-and-reoxygenation injury, this action being NO dependent and associated with inhibition of the opening of the mitochondrial permeability transition pore (31). Interestingly, even though Tie-2 blockage resulted in similar LV function levels and infarct sizes in Hif-p4h-2gt/gt and wild-type hearts following AMI, a trend toward a difference in AR/LV remained. This suggests that some difference in capillary/vascular phenotype still exists following Tie-2 blockage or that additional factors, such as increased NO, contribute to improved perfusion even in the presence of Tie-2 blockage.
We show here that Hif-p4h-2 deficiency improves survival during ischemia and enhances LV systolic function following I-R and AMI. Our data suggest that inhibition of HIF-P4H-2 in endothelial cells plays a crucial role in protection from cardiac ischemic injury. Hif-p4h-2 deficiency enhances NO production and Tie-2 signaling, with the latter resulting in larger capillaries in the heart and improved perfusion of the myocardium during ischemia. While further studies are needed, systemic inhibition of HIF-P4H-2 may be a valid approach to the treatment of patients susceptible to cardiac ischemia.
Supplementary Material
ACKNOWLEDGMENTS
We thank T. Aatsinki, M. Arbelius, E. Lehtimäki, and K. Salo for their excellent technical assistance and L. Eklund for helpful discussions.
We thank the Core Facilities for Imaging and Physiological Analysis of Transgenic Animals at Biocenter Oulu, University of Oulu (cofunded by Biocenter Finland). This study was supported by Academy of Finland grants 120156, 140765, and 218129 (P.K.), 218044 and 263731 (R.K.), 200471, and 202469, by Center of Excellence 2012-2017 grant 251314 (J.M.), by the S. Jusélius Foundation (P.K., R.K., and J.M.), by the Finnish Foundation for Cardiovascular Research (R.K.), and by FibroGen, Inc. (J.M.).
Footnotes
Published ahead of print 17 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00432-13.
REFERENCES
- 1.Kaelin WG, Jr, Ratcliffe PJ. 2008. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell 30:393–402 [DOI] [PubMed] [Google Scholar]
- 2.Semenza GL. 2011. Oxygen sensing, homeostasis, and disease. N. Engl. J. Med. 365:537–547 [DOI] [PubMed] [Google Scholar]
- 3.Fraisl P, Aragones J, Carmeliet P. 2009. Inhibition of oxygen sensors as a therapeutic strategy for ischaemic and inflammatory disease. Nat. Rev. Drug Discov. 8:139–152 [DOI] [PubMed] [Google Scholar]
- 4.Minamishima YA, Moslehi J, Bardeesy N, Cullen D, Bronson RT, Kaelin WG., Jr 2008. Somatic inactivation of the PHD2 prolyl hydroxylase causes polycythemia and congestive heart failure. Blood 111:3236–3244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Takeda K, Cowan A, Fong GH. 2007. Essential role for prolyl hydroxylase domain protein 2 in oxygen homeostasis of the adult vascular system. Circulation 116:774–781 [DOI] [PubMed] [Google Scholar]
- 6.Moslehi J, Minamishima YA, Shi J, Neuberg D, Charytan DM, Padera RF, Signoretti S, Liao R, Kaelin WG., Jr 2010. Loss of hypoxia-inducible factor prolyl hydroxylase activity in cardiomyocytes phenocopies ischemic cardiomyopathy. Circulation 122:1004–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hölscher M, Silter M, Krull S, von Ahlen M, Hesse A, Schwartz P, Wielockx B, Breier G, Katschinski DM, Zieseniss A. 2011. Cardiomyocyte-specific prolyl-4-hydroxylase domain 2 knock out protects from acute myocardial ischemic injury. J. Biol. Chem. 286:11185–11194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hyvärinen J, Hassinen IE, Sormunen R, Maki JM, Kivirikko KI, Koivunen P, Myllyharju J. 2010. Hearts of hypoxia-inducible factor prolyl 4-hydroxylase-2 hypomorphic mice show protection against acute ischemia-reperfusion injury. J. Biol. Chem. 285:13646–13657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Myllyharju J. 2009. HIF prolyl 4-hydroxylases and their potential as drug targets. Curr. Pharm. Des. 15:3878–3885 [DOI] [PubMed] [Google Scholar]
- 10.Martini JS, Raake P, Vinge LE, DeGeorge BR, Jr, Chuprun JK, Harris DM, Gao E, Eckhart AD, Pitcher JA, Koch WJ. 2008. Uncovering G protein-coupled receptor kinase-5 as a histone deacetylase kinase in the nucleus of cardiomyocytes. Proc. Natl. Acad. Sci. U. S. A. 105:12457–12462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao E, Lei YH, Shang X, Huang ZM, Zuo L, Boucher M, Fan Q, Chuprun JK, Ma XL, Koch WJ. 2010. A novel and efficient model of coronary artery ligation and myocardial infarction in the mouse. Circ. Res. 107:1445–1453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Holopainen T, Saharinen P, D'Amico G, Lampinen A, Eklund L, Sormunen R, Anisimov A, Zarkada G, Lohela M, Helotera H, Tammela T, Benjamin LE, Yla-Herttuala S, Leow CC, Koh GY, Alitalo K. 2012. Effects of angiopoietin-2-blocking antibody on endothelial cell-cell junctions and lung metastasis. J. Natl. Cancer Inst. 104:461–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Augustin HG, Koh GY, Thurston G, Alitalo K. 2009. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat. Rev. Mol. Cell Biol. 10:165–177 [DOI] [PubMed] [Google Scholar]
- 14.Ignarro LJ, Napoli C, Loscalzo J. 2002. Nitric oxide donors and cardiovascular agents modulating the bioactivity of nitric oxide: an overview. Circ. Res. 90:21–28 [DOI] [PubMed] [Google Scholar]
- 15.Thurston G, Suri C, Smith K, McClain J, Sato TN, Yancopoulos GD, McDonald DM. 1999. Leakage-resistant blood vessels in mice transgenically overexpressing angiopoietin-1. Science 286:2511–2514 [DOI] [PubMed] [Google Scholar]
- 16.Bao W, Qin P, Needle S, Erickson-Miller CL, Duffy KJ, Ariazi JL, Zhao S, Olzinski AR, Behm DJ, Pipes GC, Jucker BM, Hu E, Lepore JJ, Willette RN. 2010. Chronic inhibition of hypoxia-inducible factor prolyl 4-hydroxylase improves ventricular performance, remodeling, and vascularity after myocardial infarction in the rat. J. Cardiovasc. Pharmacol. 56:147–155 [DOI] [PubMed] [Google Scholar]
- 17.Cai Z, Zhong H, Bosch-Marce M, Fox-Talbot K, Wang L, Wei C, Trush MA, Semenza GL. 2008. Complete loss of ischaemic preconditioning-induced cardioprotection in mice with partial deficiency of HIF-1 alpha. Cardiovasc. Res. 77:463–470 [DOI] [PubMed] [Google Scholar]
- 18.Eckle T, Kohler D, Lehmann R, El Kasmi K, Eltzschig HK. 2008. Hypoxia-inducible factor-1 is central to cardioprotection: a new paradigm for ischemic preconditioning. Circulation 118:166–175 [DOI] [PubMed] [Google Scholar]
- 19.Huang M, Chan DA, Jia F, Xie X, Li Z, Hoyt G, Robbins RC, Chen X, Giaccia AJ, Wu JC. 2008. Short hairpin RNA interference therapy for ischemic heart disease. Circulation 118:S226–S233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Van Herck PL, Paelinck BP, Haine SE, Claeys MJ, Miljoen H, Bosmans JM, Parizel PM, Vrints CJ. 2013 Impaired coronary flow reserve after a recent myocardial infarction: correlation with infarct size and extent of microvascular obstruction. Int. J. Cardiol. 167:351–356 [DOI] [PubMed] [Google Scholar]
- 21.Bax M, de Winter RJ, Schotborgh CE, Koch KT, Meuwissen M, Voskuil M, Adams R, Mulder KJ, Tijssen JG, Piek JJ. 2004. Short- and long-term recovery of left ventricular function predicted at the time of primary percutaneous coronary intervention in anterior myocardial infarction. J. Am. Coll. Cardiol. 43:534–541 [DOI] [PubMed] [Google Scholar]
- 22.Suryapranata H, Zijlstra F, MacLeod DC, van den Brand M, de Feyter PJ, Serruys PW. 1994. Predictive value of reactive hyperemic response on reperfusion on recovery of regional myocardial function after coronary angioplasty in acute myocardial infarction. Circulation 89:1109–1117 [DOI] [PubMed] [Google Scholar]
- 23.Jayaweera AR, Wei K, Coggins M, Bin JP, Goodman C, Kaul S. 1999. Role of capillaries in determining CBF reserve: new insights using myocardial contrast echocardiography. Am. J. Physiol. 277:H2363–H2372 [DOI] [PubMed] [Google Scholar]
- 24.Kaul S, Jayaweera AR. 2008. Myocardial capillaries and coronary flow reserve. J. Am. Coll. Cardiol. 52:1399–1401 [DOI] [PubMed] [Google Scholar]
- 25.Tian H, McKnight SL, Russel DW. 1997. Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev. 11:72–82 [DOI] [PubMed] [Google Scholar]
- 26.Jeansson M, Gawlik A, Anderson G, Li C, Kerjaschki D, Henkelman M, Quaggin SE. 2011. Angiopoietin-1 is essential in mouse vasculature during development and in response to injury. J. Clin. Invest. 121:2278–2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kidoya H, Ueno M, Yamada Y, Mochizuki N, Nakata M, Yano T, Fujii R, Takakura N. 2008. Spatial and temporal role of the apelin/APJ system in the caliber size regulation of blood vessels during angiogenesis. EMBO J. 27:522–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Takeda Y, Costa S, Delamarre E, Roncal C, Leite de Oliveira R, Squadrito ML, Finisguerra V, Deschoemaeker S, Bruyere F, Wenes M, Hamm A, Serneels J, Magat J, Bhattacharyya T, Anisimov A, Jordan BF, Alitalo K, Maxwell P, Gallez B, Zhuang ZW, Saito Y, Simons M, De Palma M, Mazzone M. 2011. Macrophage skewing by Phd2 haplodeficiency prevents ischaemia by inducing arteriogenesis. Nature 479:122–126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dezfulian C, Raat N, Shhiva S, Gladwin MT. 2007. Role of the anion nitrite in ischemia-reperfusion cytoprotection and therapeutics. Cardiovasc. Res. 75:327–338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Potente M, Urbich C, Sasaki K, Hofmann WK, Heeschen C, Aicher A, Kollipara R, DePinho RA, Zeiher AM, Dimmeler S. 2005. Involvement of Foxo transcription factors in angiogenesis and postnatal neovascularization. J. Clin. Invest. 115:2382–2392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Leucker TM, Bienengraeber M, Muravyeva M, Baotic I, Weihrauch D, Brzezinska AK, Warltier DC, Kersten JR, Pratt PF., Jr 2011. Endothelial-cardiomyocyte crosstalk enhances pharmacological cardioprotection. J. Mol. Cell. Cardiol. 51:803–811 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.