

Abstract
The essential yeast kinases Mec1 and Rad53, or human ATR and Chk1, are crucial for checkpoint responses to exogenous genotoxic agents, but why they are also required for DNA replication in unperturbed cells remains poorly understood. Here we report that even in the absence of DNA-damaging agents, the rad53-4AQ mutant, lacking the N-terminal Mec1 phosphorylation site cluster, is synthetic lethal with a deletion of the RAD9 DNA damage checkpoint adaptor. This phenotype is caused by an inability of rad53-4AQ to activate the downstream kinase Dun1, which then leads to reduced basal deoxynucleoside triphosphate (dNTP) levels, spontaneous replication fork stalling, and constitutive activation of and dependence on S phase DNA damage checkpoints. Surprisingly, the kinase-deficient rad53-K227A mutant does not share these phenotypes but is rendered inviable by additional phosphosite mutations that prevent its binding to Dun1. The results demonstrate that ultralow Rad53 catalytic activity is sufficient for normal replication of undamaged chromosomes as long as it is targeted toward activation of the effector kinase Dun1. Our findings indicate that the essential S phase function of Rad53 is comprised by the combination of its role in regulating basal dNTP levels and its compensatory kinase function if dNTP levels are perturbed.
INTRODUCTION
DNA replication stress, in the form of transiently stalled or irreversibly collapsed DNA replication forks, is an important source of genome instability and contributes to cancer onset and progression (1, 2). Processivity of the replication machinery may be hindered if cellular levels of deoxyribonucleoside triphosphates (dNTPs) are too low, if the DNA template is damaged or distorted, or if protein complexes remain bound to the DNA (3). In response to excessive replication fork stalling, the budding yeast checkpoint kinases Mec1 (ATR) and Rad53 (Chk2) are known to inhibit the firing of additional replication origins while damage persists (4) and to prevent the irreversible collapse of stalled replication forks (5, 6).
Mec1 kinase can phosphorylate and activate Rad53 via two parallel pathways, the Mrc1-dependent replication checkpoint (7, 8) or the Rad9-dependent DNA damage checkpoint (9, 10) (Fig. 1A). Once phosphorylated by Mec1, Rad53 in turn activates the downstream kinase Dun1 via phosphorylation of T380 in the Dun1 activation loop (11). In order to bind Dun1, Rad53 has to be phosphorylated on two adjacent Mec1 target motifs in the N-terminal Rad53 SQ/TQ-cluster domain 1 (SCD1), which then serve as a docking site for the Dun1-FHA domain (12). Active Dun1 increases ribonucleotide reductase (RNR) protein levels and activity in response to DNA damage and therefore promotes a substantial rise in cellular dNTP levels, which are required for efficient DNA repair (13, 14). This occurs via the phosphorylation and inactivation of the transcriptional repressor Crt1 (15), the nuclear trafficking protein Dif1 (16), and the RNR inhibitor Sml1 (17) (Fig. 1A).
Fig 1.

Impaired SCD1 phosphorylation leads to spontaneous checkpoint activation. (A) Schematic representation of yeast S phase checkpoint responses. (B) Tetrad dissection of a diploid strain heterozygous for rad53-4AQ, rad9Δ, and sml1Δ. Sibling spores are displayed in a column. Circles and squares denote indicated genotypes. (C) Western blot comparing Rad9, Rad53, and γH2A phosphorylation in WT and rad53-4AQ cells released into the cell cycle from α-factor-induced G1 arrest. The positive control is the WT treated with 0.05% MMS for 60 min. (D) Rad53 IP kinase activity in WT and rad53-4AQ cells at the indicated time points after α-factor release (mean ± SEM; n = 10 for the WT; n = 2 for the rad53-4AQ strain). (E) Tetrad dissection of a diploid strain heterozygous for rad53-4AQ, mrc1Δ, and sml1Δ, similar to panel B. (F) Western blot comparing Rad53 activation and H2A phosphorylation in asynchronous diploid strains of the indicated genotypes.
In addition to their critical functions in response to exogenous genotoxins, the essential MEC1 and RAD53 genes also have poorly understood functions in normal S phase, since their absence leads to catastrophic fork collapse even during the replication of undamaged DNA (18, 19). The lethality of a mec1Δ or rad53Δ null allele can be suppressed by genetically engineered RNR hyperactivity, for example, via deletion of SML1 (20) or RNR3 overexpression (21), which indicates that provision of adequate dNTP pools for DNA replication is a central component of the essential function of these upstream checkpoint kinases. However, despite its direct role in checkpoint-dependent regulation of RNR activity in normal S phase (17, 22) and 30 to 50% lower dNTP levels in dun1Δ cells (23), DUN1 itself is not required for cell viability, suggesting that the essential function of the checkpoint pathway may involve additional functions beyond dNTP regulation. Furthermore, the severely checkpoint-deficient rad53-K227A Rad53 catalytic domain mutant is viable without the need for extragenic suppressors, which creates a conundrum as to how Dun1 is activated during unperturbed DNA replication.
Here, we show that minimal intrinsic Rad53 catalytic activity is sufficient to support its essential S phase checkpoint function as long as it is targeted toward Dun1 activation by the same phospho-SCD1/FHA scaffold mechanism that is operational in response to exogenous DNA damage. However, if basal dNTP pool regulation is impaired due to Dun1 activation defects, increased Rad53 kinase activity becomes essential to execute a compensatory S phase checkpoint response even in the absence of exogenous DNA-damaging agents. These data support a unifying model where the essential function of RAD53 comprises the regulation of basal dNTP levels via Dun1 and that this is complemented by its conventional checkpoint function to stabilize spontaneously stalled replication forks upon nucleotide starvation.
MATERIALS AND METHODS
Yeast strains and cultures.
All yeast strains are derived from W303-1A with corrected RAD5 (20): MATa ade2-1 can1-100 his3-11,15 leu2-3,112 trp1-1 ura3-1 RAD5. All deletion strains lack the entire open reading frame. rad53 point mutation alleles were generated as previously described (24, 25). All cultures were grown using YPD (1% yeast extract, 2% peptone, and 2% glucose), except for sporulations in 1% potassium acetate. Zymolyase 20T (Saikagaku)-digested tetrads were dissected on YPD plates, photographed after 3 to 4 days, and genotyped using auxotrophic markers or PCR and restriction analysis for point mutations. All DNA damage samples were treated with 0.05% methyl methanesulfonate (MMS) for 60 min. For Hug1 induction, cells were treated with 100 mM hydroxyurea for 2 h as described elsewhere (26). Cells were synchronized with 20 μg/ml α-factor for up to 2 h, washed extensively, and released into prewarmed YPD.
General procedures.
For Western blots, lysates were prepared using glass beads and urea buffer for 7% or 15% SDS-PAGE gel electrophoresis (25). Anti-Rad53 antibodies were rabbit polyclonal (27) for gel shifts and mouse monoclonal F9A1 (28) for activated Rad53; rabbit anti-Dun1 was raised and affinity purified using a recombinant Dun1-FHA domain (29), anti-Rad9 was kindly provided by N. Lowndes (National University of Ireland), anti-Sml1 was kindly provided by R. Rothstein (Columbia University), anti-Hug1 was kindly provided by Munira Basrai (National Cancer Institute), anti-γH2A was from Abcam (ab15083), and antiactin was from Millipore (MAB1501).
For flow cytometry, cells were fixed in 70% ethanol, treated with RNase A and 10 μg/ml propidium iodide, sonicated, and analyzed using a BD FACSCalibur flow cytometer. Pulsed-field gel electrophoresis was performed as described previously (30). For two-dimensional (2D) agarose gels, genomic DNA from 50- to 100-ml cultures was extracted in agarose plugs as described previously (31), digested overnight with 250 U NcoI, and electrophoresed using an Amersham Gene Navigator pulsed-field gel electrophoresis (PFGE) system, essentially as described previously (32). DNA was transferred to nylon membranes for Southern blot analysis using 32P-labeled MAT (chromosome III; PFGE), AFI1 (chromosome XV; PFGE), or EMC1 (chromosome III; 2D gels) probes and exposed to PhosphorImager screens (Molecular Dynamics) for 1 to 10 days. Signals were quantified using ImageQuant software (GE Healthcare).
dNTP pool measurements.
dNTP pools were measured in asynchronous cultures as described previously (33) with minor changes. Briefly, cells were harvested by filtration at a density of 0.4 × 107 to 0.5 × 107 cells/ml, and nucleoside triphosphates (NTPs) and dNTPs were extracted in trichloroacetic acid and MgCl2 followed by a Freon-trioctylamine mix. dNTPs were separated using boronate columns (Affigel 601; Bio-Rad) and analyzed by high-performance liquid chromatography (HPLC) on a LaChrom Elite UV detector (Hitachi) with a Partisphere SAX column (Hichrom, United Kingdom).
Rad53 IP kinase assay.
Native protein lysates were prepared in phosphate-buffered saline (PBS) containing 0.5% Tween 20, 5 mM EDTA, 50 mM NaF, 50 mM β-glycerophosphate, 5 mM sodium pyrophosphate, and 1× protease inhibitor cocktail (Sigma), and Rad53 was immunoprecipitated using 1 μg EL7E1 monoclonal antibody (28) and protein G Sepharose (GE Healthcare). After extensive washing of the beads with lysis buffer and PBS, kinase assays were performed for 15 min at 30°C in 20 mM Tris (pH 7.4), 10 mM Mg-acetate, 0.5 mM dithiothreitol (DTT), 0.05% Tween 20, 250 μM ATP, 2.5 μg/μl substrate peptide (LKKLTRRASFSGQ), and up to 15 μCi [γ32P]ATP. Reaction mixtures were spotted onto phosphocellulose paper and washed with 75 mM phosphoric acid, and signals were quantified using a TriCarb 2900TR (PerkinElmer) scintillation counter. Specific activities were calculated based on sample counts above the background level relative to input radioactive phosphate in the reaction mix and total amount of protein in the lysate, quantified by bicinchoninic acid (BCA) assay (Pierce/Thermo Scientific), and expressed as picomoles of transferred phosphate groups/min/mg of total protein.
Mass spectrometry methods.
The peptide nanoflow liquid chromatography-tandem mass spectrometry (LC-MS/MS) experiments were carried out using an LTQ-Orbitrap hybrid mass spectrometer (Thermo) equipped with a nanoelectrospray ion source (New Objective) and an Agilent binary high-performance liquid chromatography pump (Agilent Technologies) with an autosampler (Thermo). The scan cycle was initiated with a full-scan survey MS (m/z 320 to 1,600) experiment in the Orbitrap instrument, followed by data-dependent MS/MS experiments in the linear ion trap on the 12 most abundant ions detected in the full-MS scan. The raw files were analyzed by the software program MaxQuant (1.3.0.5) (34) for protein identification in the Saccharomyces cerevisiae protein database.
RESULTS
Impaired Rad53-SCD1 phosphorylation leads to spontaneous DNA damage checkpoint activation during normal S phase.
During attempts to combine the previously described rad53-4AQ SCD1 phosphosite mutation (12) with a deletion of the critical DNA damage checkpoint mediator Rad9, we could never recover viable rad53-4AQ rad9Δ double mutant haploid colonies in >100 tetrad dissections of a RAD53/rad53-4AQ RAD9/rad9Δ SML1/sml1Δ heterozygous parental strain (Fig. 1B, circles), indicating that the rad53-4AQ strain depends on RAD9 for viability. In contrast, morphologically normal triple mutant colonies containing rad53-4AQ rad9Δ in combination with sml1Δ were readily obtained in the same dissections (Fig. 1B, squares), suggesting that the lethality of rad53-4AQ rad9Δ SML1 cells may be related to impaired DNA replication.
To corroborate these findings, we tested if in rad53-4AQ mutants the Rad9 pathway was spontaneously activated during S phase, which can be detected by mobility shifts on Western blots. In wild-type (WT) control cells, Rad9 exhibited modest, heterogeneous mobility shifts relative to G1 samples between 30 and 70 min after release from α-factor arrest, consistent with its cell cycle-dependent phosphorylation by cyclin-dependent kinase (35, 36) (Fig. 1C). In addition to these cell cycle-dependent shifts, rad53-4AQ SML1 cells also contained pronounced supershifted Rad9 bands from 50 to 90 min after α-factor release that were similar to the mobility of activated Rad9 in the DNA-damaged (MMS) control (Fig. 1C). Since Rad9 activation depends in part on binding of its BRCT domain to phospho-Ser129 of the general DNA damage marker phospho-histone H2A (γH2A) (37), we also monitored γH2A levels in these samples. Similar to previous reports (38, 39), WT cells contained modest γH2A levels during normal S phase, but S phase γH2A levels were markedly increased in rad53-4AQ cells, again comparable to results for the MMS-treated control (Fig. 1C). These elevated γH2A levels are likely to be functionally important, since rad53-4AQ double mutants with either the BRCT domain-inactivating rad9-K1088M mutation (see Fig. S1A in the supplemental material) or the H2A phosphorylation site-deficient hta-S129* allele (see Fig. S1B) exhibited strong colony growth defects that could again be suppressed by sml1Δ.
In these Western blot analyses, we also noted that Rad53 itself was partially shifted in the rad53-4AQ mutant but not the WT from relatively early S phase time points (30 min after release) even before increased γH2A and Rad9 supershifts were detectable (Fig. 1C). To determine if these Rad53 mobility shifts reflect its catalytic activation, we developed a novel quantitative Rad53 immunoprecipitation (IP) kinase assay that utilizes the monoclonal antibody EL7E1 (28) to efficiently precipitate endogenous Rad53 and a synthetic substrate peptide (LKKLTRRAS*FSGQ) that contains a preferential Rad53 SF phosphorylation site motif (40). In these assays, basal Rad53 kinase activity was ∼10-fold higher in undamaged WT cells than in the kinase-deficient rad53-K227A control cells (see Fig. S1E in the supplemental material) but was not detectably increased in S phase compared to that for G1 samples (Fig. 1D). In contrast, Rad53 kinase activity was ∼5-fold higher than WT levels in rad53-4AQ SML1 cells at 50 to 70 min post-α-factor release (Fig. 1D), which approximately coincides with the peak of elevated H2A phosphorylation and Rad9 supershifts in this strain (Fig. 1C). Thus, an absence of SCD1 phosphorylation sites appears to result in ectopic catalytic activation of Rad53 during normal S phase.
The notion that the Rad53 mobility shifts preceded the Rad9 shifts during S phase in rad53-4AQ cells (Fig. 1C) suggested they may reflect Rad53 phosphorylation via the Mrc1-dependent replication checkpoint pathway (Fig. 1A). To test this, we crossed the rad53-4AQ mutant with an mrc1Δ strain or a strain carrying the replication checkpoint-deficient mrc1-AQ allele. Interestingly, although double mutants were viable, both rad53-4AQ mrc1Δ and rad53-4AQ mrc1-AQ resulted in a strong synthetic sickness phenotype that could again be suppressed by sml1Δ (Fig. 1E; see also Fig. S1C in the supplemental material), and as expected, the early S phase mobility shifts of Rad534AQ were suppressed by mrc1-AQ (see Fig. S1D). Finally, to assess if increased checkpoint activation resulting from the rad53-4AQ allele was due to the loss of a function normally performed by SCD1 versus the gain of a pathological function, we analyzed its effect on spontaneous checkpoint activation in diploid cells that were homozygous for SML1. Similar to results for haploid strains, increased basal γH2A levels and Rad53 shifts were observed in a rad53-4AQ homozygous strain but not in two independently generated RAD53/rad53-4AQ heterozygotes (Fig. 1F), demonstrating that this phenotype is recessive and is therefore most likely due to the loss of a physiological SCD1 function.
Altogether, these data indicate that defective Rad53-SCD1 phosphorylation leads to the loss of a normal S phase function of Rad53 and subsequent compensatory activation of and dependence on both the Mrc1-dependent DNA replication checkpoint and the Rad9-dependent DNA damage checkpoint even in the absence of exogenous DNA-damaging agents.
SCD1 phosphorylation is required for normal S phase progression.
To evaluate the functional significance of the constitutive S phase checkpoint activation, we monitored if S phase progression itself was impaired in rad53-4AQ cells. Fluorescence-activated cell sorting (FACS) analysis of bulk DNA content showed that rad53-4AQ cells entered S phase with kinetics similar to those of the WT but were considerably delayed at 35 to 40 min postrelease and seemed to take longer than the WT to achieve 2n DNA content (Fig. 2A). These observations were confirmed by pulsed-field gel electrophoresis using conditions where only intact chromosomes can enter the gel but replicating or damaged chromosomes remain in the well (see Fig. S2A in the supplemental material). Based on Southern blot quantification of two separate chromosomes, rad53-4AQ and WT cells initiated DNA replication with similar kinetics, but in both cases the peak of complete replication was delayed by 20 min in the mutant (Fig. 2B; see also Fig. S2B), and completion of chromosome replication was considerably more asynchronous in the rad53-AQ population, possibly as a consequence of spontaneous checkpoint activation (Fig. 1C and D).
Fig 2.

SCD1 phosphorylation is required for normal DNA replication. (A) FACS analysis of cell cycle progression in WT and rad53-4AQ cells at the indicated time points after release from α-factor G1 arrest. (B) Southern blot quantification of chromosomal bands separated by pulsed-field gel electrophoresis, normalized to signal intensity at G1, in WT and rad53-4AQ cells at the indicated time points after release from α-factor arrest. (C) Schematic representation of the genomic locus analyzed in panel D. “N” denotes relevant NcoI sites, gray boxes are genes, the black box is the replication origin, and the thick black line depicts the position of the probe used for Southern blots. Arrows identify the transcription start sites for the highly transcribed genes PDI1 and GLK1. (D) Southern blots of neutral-neutral 2D agarose gels of replication intermediates at the locus shown in panel C. WT and rad53-4AQ cells were released from α-factor arrest into YPD, and samples were taken at the indicated time points. (E) Schematic diagram of relevant replication intermediates and their relative migration in 2D agarose gels. (F) Quantification of replication structures from samples in panel D, relative to total signals within each sample. (G) 2D gel analysis of replication intermediates at the locus shown in panel C. Indicated strains were arrested in G1, and samples were taken at 40 min postrelease.
Since the rad53-4AQ mutation leads to both delayed gross chromosomal DNA replication (Fig. 2A and B) and increased S phase γH2A formation (Fig. 1C), we monitored replication fork progression using neutral-neutral 2D gel electrophoresis at a locus that has been associated with very slow fork progression rates (19) and highly elevated basal γH2A levels (39). This locus is very close to the highly efficient, early-firing origin ARS305, so that it is replicated in early S phase, before checkpoint activation is detected in rad53-4AQ cells (Fig. 1D). Replication forks originating from ARS305 replicate this region from left to right (Fig. 2C) and appear on 2D gels as single Y molecules after NcoI digestion (Fig. 2D; diagram of replication intermediate mobility, Fig. 2E). As expected, in WT cells, there was a characteristic increase in large Y intermediates relative to small Y's at 40 to 50 min after α-factor release, indicative of slower fork progression in the distal part of this NcoI fragment (Fig. 2D and F). In rad53-4AQ cells, the relative proportion of large Y's was increased by a further 2- to 3-fold, consistent with exacerbated fork stalling (Fig. 2D and F). Interestingly, X-shaped molecules, which typically include restart intermediates of stalled or collapsed forks, were also increased by ∼3-fold in the rad53-4AQ mutant compared to results for the WT (Fig. 2D and F). Similar to the other phenotypes of rad53-4AQ (Fig. 1), both the increased fork stalling (large Y's) and the increased formation of X-shaped molecules were completely suppressed by sml1Δ (Fig. 2G). In addition, rad53-4AQ rad52Δ double mutants exhibited an SML1-dependent severe synthetic sickness phenotype (see Fig. S2C in the supplemental material), supporting the notion that S phase completion in rad53-4AQ cells involves recombination-dependent fork restart mechanisms. Thus, increased replication fork slowing and delayed S phase progression indicate that Rad53 SCD1 phosphorylation sites are essential for normal replication of undamaged chromosomes, particularly in difficult-to-replicate regions.
Impaired Rad53 SCD1 phosphorylation causes more severe spontaneous phenotypes than Rad53 kinase deficiency or loss of the effector kinase Dun1.
Since Rad53-SCD1 phosphorylation is involved in both Rad53 and Dun1 kinase activation (11, 12, 41, 42), we tested if defective signaling by one of these kinases during normal S phase could explain the rad53-4AQ phenotypes. However, neither the kinase-impaired rad53-K227A allele nor dun1Δ was synthetic lethal with rad9Δ (Fig. 3A and B) or synthetic sick in combination with mrc1Δ (data not shown). Likewise, although S phase γH2A levels were modestly elevated in rad53-K227A and dun1Δ cells compared to results for the WT, these were substantially lower than those in the corresponding rad53-4AQ samples (Fig. 3C). Interestingly, while there was a clear Rad53 mobility shift in dun1Δ cells at 30 min after α-factor release, this was much more transient than results for rad53-4AQ cells (Fig. 3C). Thus, the much stronger phenotypes in rad53-4AQ cells than in rad53-K227A and dun1Δ cells demonstrate that these defects cannot simply be due to an isolated activation defect of Rad53 or Dun1 during normal S phase.
Fig 3.
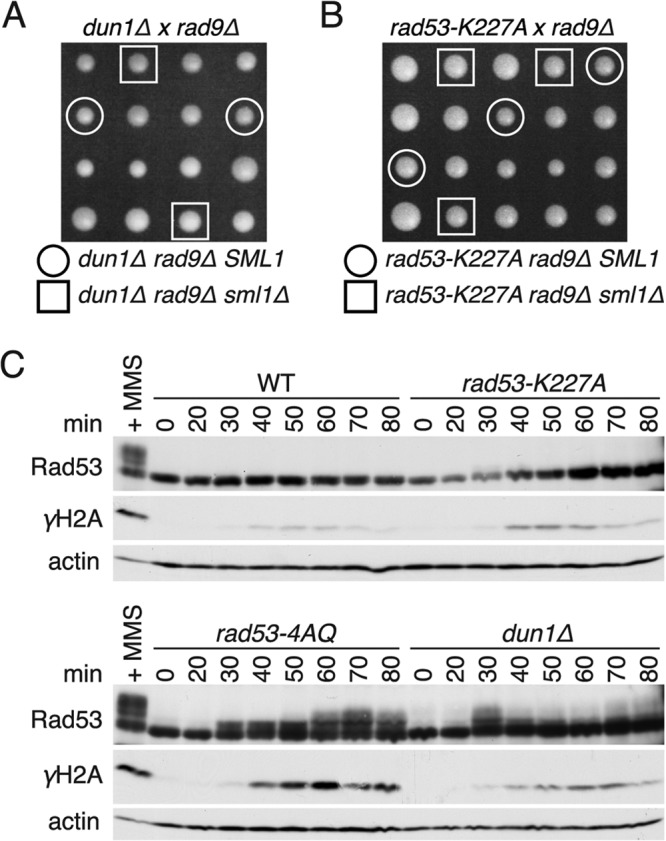
Comparison of rad53-4AQ cells with rad53-K227A and dun1Δ cells. (A and B) Tetrad dissection of diploid strains heterozygous for rad9Δ, sml1Δ, and either dun1Δ or rad53-K227A. Colonies originating from the same tetrad are in columns. Indicated genotypes are marked with circles or squares. (C) Western blot analysis comparing WT, rad53-4AQ, rad53-K227A, and dun1Δ cells at the indicated time points after release from G1 arrest. All four strains were processed at the same time, and the same WT + MMS reference sample was used for both gels.
Rad53 SCD1 phosphorylation sites but not intact kinase activity are required for regulation of basal dNTP pools and S phase-specific Sml1 degradation.
The finding that all defects of rad53-4AQ cells were consistently suppressed by sml1Δ (Fig. 1 and 2) prompted us to test if altered dNTP levels could contribute to these phenotypes. As previously reported (17, 23, 43), deletion of Dun1, which regulates S phase RNR activity via Sml1 degradation, led to a reduction of dNTP pools to ∼50% of WT levels (Fig. 4A), and a similar reduction of dNTP pools was also observed in rad53-4AQ cells (Fig. 4A). Strikingly, in contrast to rad53-4AQ cells, kinase-impaired rad53-K227A cells maintained WT dNTP levels. To corroborate these findings, we also monitored Sml1 levels in these strains. Again, G1 Sml1 levels were similar in WT and rad53-K227A cells, and Sml1 degradation was only modestly attenuated in rad53-K227A cells compared to that in WT cells (Fig. 4B). In contrast, both rad53-4AQ and dun1Δ cells had considerably elevated basal Sml1 levels during G1 phase and impaired Sml1 degradation in early S phase (Fig. 4B). Thus, although Dun1 and the SCD1 are clearly required for Sml1 degradation and dNTP regulation during normal S phase, Rad53 kinase activity seems to be largely dispensable in this context.
Fig 4.
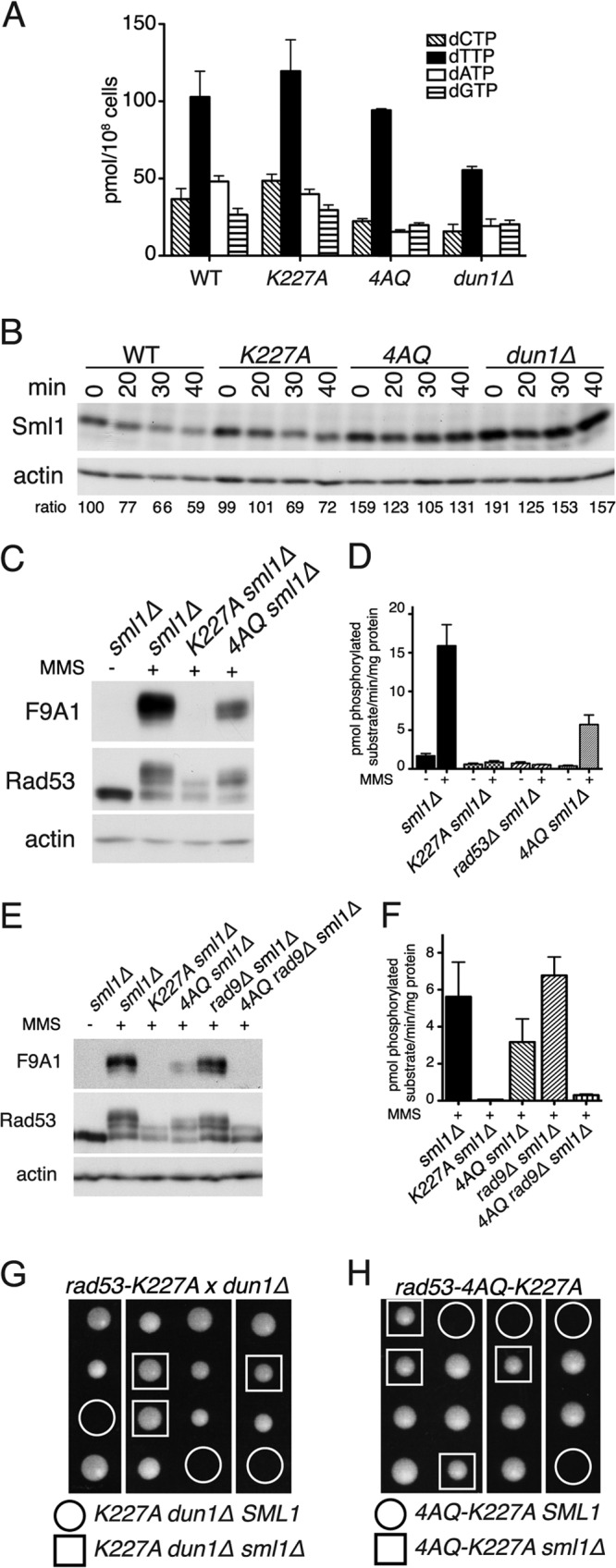
The rad53-4AQ phenotype reflects a combination of defective nucleotide regulation and impaired Rad53 kinase activation. (A) Measurement of endogenous dNTP pools in asynchronous cultures of the indicated strains (mean ± SEM; n = 2). (B) Western blot analysis of Sml1 levels in cells of the indicated genotypes at indicated times (min) after release from G1 arrest into YPD. Numbers below each lane are Sml1/actin ratios relative to those for WT G1, which was assigned an arbitrary value of 100. (C) Western blot analysis of the indicated strains after treatment with 0.05% MMS for 60 min (+) and untreated control. The F9A1 antibody is specific for activated Rad53 (28). (D) Rad53 IP kinase assay in indicated strains after treatment with 0.05% MMS for 60 min and untreated controls (mean ± SEM; n = 5 for WT−, WT+, K227A+, and 4AQ+; n = 2 for others). (E) Similar to panel C; Western blot analysis of the indicated strains after treatment with 0.05% MMS for 60 min. (F) Rad53 IP kinase assay of the indicated strains after treatment with 0.05% MMS for 60 min (mean ± SEM; n = 2). (G) Tetrad dissection of a diploid strain heterozygous for rad53-K227A, dun1Δ, and sml1Δ. (H) Tetrad dissection of a RAD53/rad53-4AQ-K227A SML1/sml1Δ diploid.
Rad53 kinase activity compensates for insufficient basal dNTP pools.
Reduced dNTP levels can explain the DNA replication defects (Fig. 2) and spontaneous compensatory checkpoint activation (Fig. 1C and D) in rad53-4AQ cells. However, low dNTP pools alone cannot be the reason for the severe rad53-4AQ S phase phenotypes, as these are not shared by the similarly dNTP-deficient dun1Δ cells (Fig. 3A and C; Fig. 4A and B). Likewise, it was striking that rad53-4AQ phenotypes were generally more severe than those of the catalytically impaired rad53-K227A allele, which prompted us to analyze Rad534AQ catalytic activity in more detail. To avoid interference from impaired dNTP pools, we used sml1Δ strains for these analyses. In mobility shift Western blots following MMS treatment as a crude measure of Rad53 activation (27, 44), Rad534AQ shifted less than the WT Rad53 protein but contained considerably more supershifted forms than kinase-deficient Rad53K227A, phosphorylation of which is presumably limited to Mec1/Tel1-dependent sites (Fig. 4C, middle panel). Rad534AQ but not Rad53K227A was also detected by the activation-state-specific Rad53 monoclonal antibody F9A1 (28), with ∼30% of the intensity of the WT Rad53 control (Fig. 4C, top panel). Likewise, Rad534AQ retained ∼30 to 50% of WT kinase activity after MMS treatment in our IP kinase assay, where any residual activity of Rad53K227A was indistinguishable from the background level detected in rad53Δ cells devoid of the Rad53 protein (Fig. 4D and F). Thus, using three independent Rad53 kinase activity measures, we show that although modestly impaired, the Rad534AQ protein retains substantial catalytic activity, particularly in comparison to Rad53K227A.
Since this modest impairment of Rad534AQ kinase activity, even if coupled with reduced dNTP pools (Fig. 4A and B), was sufficient for viability of rad53-4AQ SML1 cells in the presence of Rad9 (Fig. 1B), we next tested if loss of Rad9 compounds the Rad534AQ activation defect. Strikingly, whereas MMS-induced Rad53 mobility shifts, detection of its active conformation by the F9A1 antibody, and IP kinase activity were grossly normal in rad9Δ sml1Δ cells and again were only partially impaired in rad53-4AQ sml1Δ cells, all three assays demonstrated dramatically reduced Rad53 catalytic activity in rad53-4AQ rad9Δ sml1Δ mutants, almost to the level of that of the kinase-impaired rad53-K227A sml1Δ strain (Fig. 4E and F). Thus, these experiments indicate that rad53-4AQ rad9Δ double mutants may be inviable because they suffer both from dNTP starvation, due to the SCD1 mutation alone (Fig. 4A and B), and from an almost complete failure to activate Rad53 as a synergistic defect of both mutations (Fig. 4E and F).
If the combined inability to provide sufficient dNTPs and to activate Rad53 causes cell death, the rad53-K227A mutation should start to depend on sml1Δ if it is combined with a dNTP regulation defect. Consistently, combination of rad53-K227A (kinase impaired) and dun1Δ (dNTP deficient) resulted in lethality that could be rescued by sml1Δ (Fig. 4G). Furthermore, a “double-defective” rad53 allele harboring both the 4AQ and K227A mutations was also inviable but sustained morphologically normal colony growth indistinguishable from that of WT cells when SML1 was deleted (Fig. 4H). Thus, these results indicate that checkpoint-proficient Rad53 kinase activity only becomes necessary for the replication of undamaged DNA as a compensatory mechanism when physiological dNTP pool regulation fails.
Differential functions of individual threonines within the Rad53-SCD1.
Rad53-SCD1 monophosphorylation on any one of its four threonines has been proposed to suffice for Rad53 kinase activation (41). We thus tested if preservation of any TQ motif in the SCD1 could prevent synthetic lethality with rad9Δ using a complete set of SCD1 “single-TQ” alleles (Fig. 5A). Surprisingly, only T8 as the sole TQ motif in the SCD1 was able to fully prevent synthetic genetic interactions with rad9Δ in the presence of SML1 (Fig. 5B). In contrast, T5, T12, or T15 as a sole SCD1-TQ still resulted in high levels of synthetic lethality or synthetic growth defects in combination with rad9Δ (Fig. 5B) or mrc1Δ (see Fig. S3A in the supplemental material), again suppressible by sml1Δ. Likewise, compared to results for unsynchronized rad53-4AQ SML1 cultures, increased basal γH2A levels and elevated Rad9 shifts were restored to the WT level only in rad53-T8-3AQ cultures but remained partially elevated in rad53-T5-3AQ, rad53-T12-3AQ, and rad53-T15-3AQ cultures unless SML1 was deleted (Fig. 5C).
Fig 5.

SCD1 diphosphorylation is required for S phase dNTP regulation. (A) Diagram of relevant rad53 SCD1 and catalytic domain mutations. (B) Tetrad dissections of compound heterozygous diploids for the indicated rad53 SCD1-mutated alleles, rad9Δ and sml1Δ. (C) Western blot analysis of Rad53, Rad9, and H2A phosphorylation in asynchronous cultures of indicated SCD1 mutant strains that are also SML1 (+) or sml1Δ (Δ). (D) Rad53 IP kinase activity in indicated strains after treatment with 0.05% MMS for 60 min (mean ± SEM; n = 2). (E) dNTP pool measurement in asynchronous cultures of the indicated strains (mean ± SEM; n = 2). (F) Tetrad dissections of RAD53/rad53-T8-3AQ-K227A or rad53-T5T8-2AQ-K227A SML1/sml1Δ diploids.
We next tested if this T8 add-back effect reflects restored Rad53 activation or improved dNTP pools. Remarkably, maintaining T8 as the sole TQ motif in SCD1 restored MMS-induced Rad53 kinase activation in the absence of Rad9 to ∼50% of WT or rad9Δ levels, whereas any of the other three individual threonines only poorly improved Rad53 kinase activity compared to that for rad53-4AQ rad9Δ sml1Δ (Fig. 5D; see also Fig. S3B in the supplemental material). Conversely, neither rad53-T8-3AQ nor the other single-TQ add-back alleles had a beneficial effect on dNTP levels (Fig. 5E), further supporting the notion that improved checkpoint signaling compared to that with rad53-4AQ rad9Δ is able to support viability of these dNTP-starved cells.
SCD1 diphosphorylation is required for viability of kinase-impaired rad53-K227A cells.
Dun1 activation in response to exogenous DNA damage depends on simultaneous phosphorylation of two adjacent threonines within the Rad53-SCD1 (12). Consistently, compared to the single TQ alleles, the “double-TQ add-back” rad53-T5T8-2AQ mutant contained markedly improved basal dNTP pools at near-WT levels (Fig. 5E), suggesting that Dun1-dependent dNTP regulation during normal S phase and that after exogenous DNA damage utilize similar mechanisms. We thus tested if two adjacent TQ motifs in the SCD1 are also necessary and sufficient for viability of the kinase-impaired rad53-K227A allele, and we generated RAD53/rad53-T8-3AQ-K227A SML1/sml1Δ and RAD53/rad53-T5T8-2AQ-K227A SML1/sml1Δ diploids for tetrad dissections. As predicted, rad53-T5T8-2AQ-K227A haploid cells were able to form colonies even in the presence of WT SML1, whereas rad53-T8-3AQ-K227A cells, containing the “preferred” single-TQ motif for kinase activation (Fig. 5D), were viable only when this was in combination with sml1Δ (Fig. 5F). These results indicate that diphospho-SCD1-dependent, Dun1-mediated dNTP pool regulation is essential for viability of severely Rad53 kinase-impaired cells.
We also tested if T5-T8 is the only di-TQ-SCD1 constellation that supports activation of the Dun1 branch by generating a rad53-T12-T15-2AQ allele. Importantly, the availability of a second TQ motif in the SCD1 in rad53-T12-T15-2AQ cells completely suppressed the synthetic lethality/sickness phenotype with rad9Δ (see Fig. S4A in the supplemental material), as well as the spontaneous Rad53 and Rad9 shifts (see Fig. S4B), compared to results for the respective rad53-T12-3AQ and rad53-T15-3AQ single-T mutants. Moreover, rad53-T12-T15-2AQ was as efficient as rad53-T5-T8-2AQ in promoting induction of Hug1, as a reliable in vivo reporter for Rad53-dependent Dun1 activation (45), in response to hydroxyurea treatment (see Fig. S4C).
Rad53K227A retains minimal catalytic activity sufficient for residual Dun1 kinase activation.
Since Dun1 kinase activity is required for basal Sml1 degradation during unperturbed S phase (17), these results suggested that Rad53K227A might retain some catalytic activity for S phase-specific Dun1 regulation. To test this, we used mass spectrometry to monitor Dun1 phosphorylation on its T-loop activation site, T380, and the autophosphorylation sites S10, S139, and S142 (11). While phosphorylation on any of these sites was too low during unperturbed S phase to be accurately quantified, in response to MMS treatment, considerably higher Dun1 autophosphorylation levels were preserved in rad53-K227A sml1Δ cells than in rad53-4AQ sml1Δ cells (Fig. 6A and C). To investigate if this was indeed due to minute residual Rad53 kinase activity in rad53-K227A cells, we generated another Rad53 kinase-deficient allele, rad53-D339A, that in contrast to rad53-K227A was previously reported to be lethal in plasmid shuffle experiments (46). As expected, the rad53-D339A strain was inviable in the absence of the sml1Δ suppressor (Fig. 6E). Interestingly, although Rad53 mobility supershifts and detection by the active state-specific F9A1 antibody were similarly impaired in rad53-D339A and rad53-K227A cells (Fig. 6D), in contrast to rad53-K227A cells, Dun1 autophosphorylation was almost completely abolished in rad53-D339A cells (Fig. 6A and C). Importantly, while very low levels of T380 phosphorylation were detectable in rad53-K227A cells (∼1% of WT control), only unrelated contaminating ions were detected in the same mass range in rad53-D339A cells (Fig. 6B and C). Thus, these data indicate that Rad53K227A is not entirely kinase-dead but retains minute intrinsic catalytic activity sufficient to partially activate Dun1 in vivo.
Fig 6.

The rad53-K227A mutant retains residual catalytic activity. (A and B) For mass spectrometry, samples were prepared from the indicated rad53 strains grown for 45 min after release from α-factor into YPD plus 0.05% MMS (“light” samples L1 to L3) and mixed with a “heavy” (H) stable isotope labeling with amino acids in cell culture (SILAC) reference sample of an unsynchronized isogenic strain (CAN1 arg4::KAN lys2::NAT) grown in minimal medium supplemented with l-lysine-13C6,15N2 and l-arginine-13C6,15N4 (Isotec) and treated with 0.05% MMS for 2 h (see Fig. S6 in the supplemental material for a schematic outline of the mass spectrometry procedure). Panel A shows precursor mass spectra of an unphosphorylated input control and spectra for the three phosphorylated Dun1 peptides indicated above. Note the similarity of peak heights of the three rad53 input controls (L1 to L3) to those of the heavy (H) reference sample but varying peak heights for the phosphopeptides in these rad53 strains compared to those of the same reference sample. Panel B shows precursor mass spectra of Dun1 T380 phosphorylation in the rad53-K227A or rad53-D339A sample (light) compared to those in the WT sample (heavy). The right panels are magnifications of the light samples in the middle panel; m/z values for correct Dun1 pT380-containing tryptic peptide ions are underlined. (C) Quantification of two independent MS/MS analyses of MMS-induced Dun1 phosphorylation in the indicated rad53 mutant strains. (D) Western blot analysis of Rad53 in the indicated strains with or without treatment with 0.05% MMS for 60 min. (E) Tetrad dissection of a RAD53/rad53-D339A SML1/sml1Δ diploid.
DISCUSSION
Here we have shown that the absence of SCD1 phosphorylation sites in the rad53-4AQ mutant leads to considerably lower dNTP pools (Fig. 4A) and increased replication fork slowing and conversion to X-shaped intermediates at early-replicating loci during normal, unperturbed S phase (Fig. 2D and F). This most likely generates spontaneous DNA damage-like signals that elicit increased γH2A and Rad9 phosphorylation (Fig. 1C and 3C) and compensatory Rad53 kinase activation (Fig. 1D), presumably to stabilize stalled forks by the same DNA damage checkpoint pathways that are also active after exogenous DNA damage. Interestingly, Rad53 exhibits partial mobility shifts in rad53-4AQ cells but not WT cells during early S phase prior to increased H2A and Rad9 phosphorylation and Rad53 kinase activation (Fig. 1C and D). These early shifts are absent in rad53-4AQ mrc1-AQ cells (see Fig. S1D in the supplemental material), indicating that the Mec1/Mrc1 pathway may respond to the increased replication fork stalling in rad53-4AQ cells but is unable to activate Rad53 kinase in the absence of SCD1 phosphorylation sites. These data suggest that under normal conditions, the Mec1/Mrc1 pathway phosphorylates Rad53-SCD1 very early during S phase, which may then provide a docking platform for partial Dun1 activation, followed by limited Sml1 degradation to complete replication of undamaged chromosomes (Fig. 7B, left panel). However, if SCD1 phosphorylation fails, increased fork stalling and collapse lead to catalytic activation of Rad53 via the Mec1/γH2A/Rad9 S phase checkpoint pathway to complete DNA replication under dNTP-starved conditions (Fig. 7B, right panel).
Fig 7.

Essential functions of Rad53 during normal S phase. (A) Summary of dNTP pools, kinase activity and viability of the indicated yeast mutants. “*” denotes phenotypes that were deduced from other experimental evidence or implied from other mutant strains. For example, low dNTP pools for the rad53-D339A strain were inferred from its inability to activate Dun1 (see Fig. 6), and very low Rad53 kinase activity in the rad53-4AQ-K227A strain was implied by rad53-K227A data (see Fig. 4C to F and Fig. 6). (B) Schematic diagram of Rad53 activation during unperturbed S phase in WT and rad53-4AQ cells. Numbers indicate the order of events, and dashed lines indicate defects in the mutant strain.
The central DNA damage checkpoint kinases Mec1 and Rad53 are essential for normal DNA replication (18, 19), but the molecular basis of their essential function has remained unresolved. Suppression of the lethality of mec1Δ and rad53Δ null alleles by RNR3 overexpression or deletion of SML1 indicated that S phase-specific dNTP pool regulation was a central part of their essential function (20, 21), but it has remained unresolved whether atypically high dNTP levels are prophylactic under these conditions in a manner that obscures other essential functions of these kinases (21, 47). Likewise, it has remained unresolved why the two entirely checkpoint-deficient Rad53 catalytic domain mutation alleles rad53-K227A (viable) and rad53-D339A (lethal without extragenic suppressors) differ in their ability to support the essential functions of RAD53 (46). While it has been speculated that the rad53-K227A mutant may retain some residual kinase activity above that of the rad53-D339A mutant (46), convincing experimental evidence for this assumption has been lacking. Here we have resolved both of these conundrums by directly showing that Rad53K227A but not Rad53D339A indeed retains low-level catalytic activity for DNA damage-induced transphosphorylation of the Dun1 activation site T380 in vivo (Fig. 6) and that this minute intrinsic kinase activity (≤1% of WT activity) is sufficient for the essential S phase function of RAD53 as long as it is directly targeted to the activation of Dun1 (Fig. 4 to 6). The reason why only very low Rad53 kinase activity is required for its essential function is most likely that only a relatively modest proportion of Sml1 needs to be degraded for physiological dNTP supply during unperturbed DNA replication (∼40%) (Fig. 4B), in contrast to almost complete Sml1 degradation in response to exogenous genotoxic stress (17, 43).
These data thus indicate that the primary function of RAD53 during normal S phase is its Dun1-mediated regulation of basal dNTP pools and that this is complemented by its conventional checkpoint function when the physiological dNTP supply is impaired for other reasons (e.g., the absence of Dun1 or Dun1-FHA docking sites in Rad53-SCD1). Thus, the mec1 and rad53 mutants and other mutants that are defective for both dNTP supply and checkpoint-proficient Rad53 kinase activity (e.g., the rad53-4AQ rad9Δ, dun1Δ rad53-K227A, rad53-4AQ-K227A, and rad53-D339A mutants) are inviable without extragenic suppressors, whereas mutation combinations that preserve at least one of these functions (e.g., rad53-K227A, dun1Δ, rad53-T8-3AQ rad9Δ, and rad53-T5-T8-K227A) remain viable (Fig. 7A). It should be noted that in contrast to almost complete loss of Rad53 kinase activity in the rad53-4AQ rad9Δ mutant (Fig. 4), loss of the Mrc1 pathway has only a modest effect of residual Rad534AQ kinase activity (our unpublished data), which explains why rad53-4AQ is lethal with rad9Δ but not with mrc1Δ or mrc1-AQ (Fig. 1; see also Fig. S1 in the supplemental material). Along these lines, our findings may also explain the previously reported synthetic lethality of dun1Δ with the viable but checkpoint-deficient hypomorphic alleles rad53-1, rad53-11, rad53-17, and rad53-H622Y (17, 48, 49). (Note that in contrast to these severely kinase-deficient alleles, rad53-4AQ, with relatively modest kinase-deficiency, remains viable with dun1Δ [see Fig. S5 in the supplemental material].)
It is interesting to note that Rad53 kinase activity becomes essential in cells where nucleotide levels are genetically reduced to ∼60% of WT levels (Fig. 5), suggesting that an ∼40% reduction in dNTP pools already leads to replication stress and checkpoint activation. Taken together with the observations that spontaneous replication fork stalling and histone H2A phosphorylation are detectable in WT cells (Fig. 1C and 3C) (38, 50) and that fork progression is increased in sml1Δ cells compared to that in WT cells (51), this indicates that yeast cells replicate their DNA at dNTP levels that are at the lower end of their replicative needs and thus may occasionally drop below the threshold required to completely prevent fork stalling during normal S phases. This spontaneous fork slowing may in fact prompt the limited Mec1 activation required for Rad53-SCD1 phosphorylation during normal S phase, which in turn promotes Dun1-dependent Sml1 degradation and a rise in cellular dNTP pools to assist replication fork progression.
In support of our model, Schizosaccharomyces pombe double mutants defective in both dNTP regulation and checkpoint activation are also inviable, even though these functions are regulated by two separate pathways (52). Similarly, hydroxyurea-induced depletion of mammalian dNTP pools also leads to replication fork stalling and a reliance on fork stabilizing checkpoint functions (53, 54). Thus, despite potential differences in the respective signaling pathways, the synthetic interactions between nucleotide regulation pathways and checkpoint kinases are likely to be conserved in higher eukaryotes. In this context, it is important to mention that the mammalian RNR subunits p53R2 and RNR2 (55, 56) are both regulated in a checkpoint kinase-dependent manner, indicating that ATR and Chk1 could be essential due to a similar combination of functions in dNTP regulation and fork stability. Since the checkpoint response is a well-documented tumor suppressor pathway (2) and oncogene-induced replication stress caused by dNTP depletion has been proposed as a major contributor to the genomic instability that drives cancer onset and progression (57), further studies of the relationship between checkpoint responses and dNTP regulation in mammalian cells may lead to the discovery of novel cellular targets for cancer therapy.
Supplementary Material
ACKNOWLEDGMENTS
We thank Rodney Rothstein, Hannah Klein, Munira Basrai, and Noel Lowndes for reagents.
This work was supported by grants and fellowships from the National Health and Medical Research Council of Australia (NHMRC) and in part by the Victorian Government's Operational Infrastructure Support Program (to J.H.), grant EX95-9508NI from the National Health Research Institute (NHRI), an Academia Sinica Investigator Award (to M.-D.T.), AIRC (IG grant no. 10343, to A.P.), and the Swedish Foundation for Strategic Research, the Swedish Research Council, and the Swedish Cancer Society (to A.C.).
N.C.H. performed most of the experiments; E.S.-W.C., S.-C.W., and M.-D.T. provided mass spectrometry results; R.B. and A.C. provided dNTP measurements; A.F. helped with tetrad dissections; A.P. provided reagents; N.C.H., E.S.-W.C., R.B., A.H., A.P., A.C., M.-D.T., and J.H. analyzed, interpreted, and discussed data; and N.C.H. and J.H. wrote the article.
Alessandro Fazio was on exchange from the Department of Systems Biology, Technical University of Denmark, Kongens Lyngby, Denmark.
We declare that we have no conflict of interest.
Footnotes
Published ahead of print 10 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.00474-13.
REFERENCES
- 1. Branzei D, Foiani M. 2010. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 11: 208– 219 [DOI] [PubMed] [Google Scholar]
- 2. Halazonetis TD, Gorgoulis VG, Bartek J. 2008. An oncogene-induced DNA damage model for cancer development. Science 319: 1352– 1355 [DOI] [PubMed] [Google Scholar]
- 3. Mirkin EV, Mirkin SM. 2007. Replication fork stalling at natural impediments. Microbiol. Mol. Biol. Rev. 71: 13– 35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Santocanale C, Diffley JF. 1998. A Mec1- and Rad53-dependent checkpoint controls late-firing origins of DNA replication. Nature 395: 615– 618 [DOI] [PubMed] [Google Scholar]
- 5. Lopes M, Cotta-Ramusino C, Pellicioli A, Liberi G, Plevani P, Muzi-Falconi M, Newlon CS, Foiani M. 2001. The DNA replication checkpoint response stabilizes stalled replication forks. Nature 412: 557– 561 [DOI] [PubMed] [Google Scholar]
- 6. Segurado M, Diffley JF. 2008. Separate roles for the DNA damage checkpoint protein kinases in stabilizing DNA replication forks. Genes Dev. 22: 1816– 1827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJ, Bousset K, Furuya K, Diffley JF, Carr AM, Elledge SJ. 2001. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat. Cell Biol. 3: 958– 965 [DOI] [PubMed] [Google Scholar]
- 8. Osborn AJ, Elledge SJ. 2003. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev. 17: 1755– 1767 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sun Z, Hsiao J, Fay DS, Stern DF. 1998. Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science 281: 272– 274 [DOI] [PubMed] [Google Scholar]
- 10. Vialard JE, Gilbert CS, Green CM, Lowndes NF. 1998. The budding yeast Rad9 checkpoint protein is subjected to Mec1/Tel1-dependent hyperphosphorylation and interacts with Rad53 after DNA damage. EMBO J. 17: 5679– 5688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen SH, Smolka MB, Zhou H. 2007. Mechanism of Dun1 activation by Rad53 phosphorylation in Saccharomyces cerevisiae. J. Biol. Chem. 282: 986– 995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lee H, Yuan C, Hammet A, Mahajan A, Chen ES, Wu MR, Su MI, Heierhorst J, Tsai MD. 2008. Diphosphothreonine-specific interaction between an SQ/TQ cluster and an FHA domain in the Rad53-Dun1 kinase cascade. Mol. Cell 30: 767– 778 [DOI] [PubMed] [Google Scholar]
- 13. Chabes A, Georgieva B, Domkin V, Zhao X, Rothstein R, Thelander L. 2003. Survival of DNA damage in yeast directly depends on increased dNTP levels allowed by relaxed feedback inhibition of ribonucleotide reductase. Cell 112: 391– 401 [DOI] [PubMed] [Google Scholar]
- 14. Sabouri N, Viberg J, Goyal DK, Johansson E, Chabes A. 2008. Evidence for lesion bypass by yeast replicative DNA polymerases during DNA damage. Nucleic Acids Res. 36: 5660– 5667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Huang M, Zhou Z, Elledge SJ. 1998. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell 94: 595– 605 [DOI] [PubMed] [Google Scholar]
- 16. Lee YD, Wang J, Stubbe J, Elledge SJ. 2008. Dif1 is a DNA-damage-regulated facilitator of nuclear import for ribonucleotide reductase. Mol. Cell 32: 70– 80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Zhao X, Rothstein R. 2002. The Dun1 checkpoint kinase phosphorylates and regulates the ribonucleotide reductase inhibitor Sml1. Proc. Natl. Acad. Sci. U. S. A. 99: 3746– 3751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zheng P, Fay DS, Burton J, Xiao H, Pinkham JL, Stern DF. 1993. SPK1 is an essential S-phase-specific gene of Saccharomyces cerevisiae that encodes a nuclear serine/threonine/tyrosine kinase. Mol. Cell. Biol. 13: 5829– 5842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cha RS, Kleckner N. 2002. ATR homolog Mec1 promotes fork progression, thus averting breaks in replication slow zones. Science 297: 602– 606 [DOI] [PubMed] [Google Scholar]
- 20. Zhao X, Muller EG, Rothstein R. 1998. A suppressor of two essential checkpoint genes identifies a novel protein that negatively affects dNTP pools. Mol. Cell 2: 329– 340 [DOI] [PubMed] [Google Scholar]
- 21. Desany BA, Alcasabas AA, Bachant JB, Elledge SJ. 1998. Recovery from DNA replicational stress is the essential function of the S-phase checkpoint pathway. Genes Dev. 12: 2956– 2970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhou Z, Elledge SJ. 1993. DUN1 encodes a protein kinase that controls the DNA damage response in yeast. Cell 75: 1119– 1127 [DOI] [PubMed] [Google Scholar]
- 23. Fasullo M, Tsaponina O, Sun M, Chabes A. 2010. Elevated dNTP levels suppress hyper-recombination in Saccharomyces cerevisiae S-phase checkpoint mutants. Nucleic Acids Res. 38: 1195– 1203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Erdeniz N, Mortensen UH, Rothstein R. 1997. Cloning-free PCR-based allele replacement methods. Genome Res. 7: 1174– 1183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pike BL, Hammet A, Heierhorst J. 2001. Role of the N-terminal forkhead-associated domain in the cell cycle checkpoint function of the Rad53 kinase. J. Biol. Chem. 276: 14019– 14026 [DOI] [PubMed] [Google Scholar]
- 26. Hammet A, Pike BL, Heierhorst J. 2002. Posttranscriptional regulation of the RAD5 DNA repair gene by the Dun1 kinase and the Pan2-Pan3 poly(A)-nuclease complex contributes to survival of replication blocks. J. Biol. Chem. 277: 22469– 22474 [DOI] [PubMed] [Google Scholar]
- 27. Pike BL, Yongkiettrakul S, Tsai MD, Heierhorst J. 2003. Diverse but overlapping functions of the two forkhead-associated (FHA) domains in Rad53 checkpoint kinase activation. J. Biol. Chem. 278: 30421– 30424 [DOI] [PubMed] [Google Scholar]
- 28. Fiorani S, Mimun G, Caleca L, Piccini D, Pellicioli A. 2008. Characterization of the activation domain of the Rad53 checkpoint kinase. Cell Cycle 7: 493– 499 [DOI] [PubMed] [Google Scholar]
- 29. Hammet A, Pike BL, Mitchelhill KI, Teh T, Kobe B, House CM, Kemp BE, Heierhorst J. 2000. FHA domain boundaries of the dun1p and rad53p cell cycle checkpoint kinases. FEBS Lett. 471: 141– 146 [DOI] [PubMed] [Google Scholar]
- 30. Pike BL, Heierhorst J. 2007. Mdt1 facilitates efficient repair of blocked DNA double-strand breaks and recombinational maintenance of telomeres. Mol. Cell. Biol. 27: 6532– 6545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Ide S, Kobayashi T. 2010. Analysis of DNA replication in Saccharomyces cerevisiae by two-dimensional and pulsed-field gel electrophoresis. Curr. Protoc. Cell Biol. 49: 22.14.1–22.14.12. 10.1002/0471143030.cb2214s49 [DOI] [PubMed] [Google Scholar]
- 32. Brewer BJ, Fangman WL. 1987. The localization of replication origins on ARS plasmids in S. cerevisiae. Cell 51:463– 471 [DOI] [PubMed] [Google Scholar]
- 33. Kumar D, Abdulovic AL, Viberg J, Nilsson AK, Kunkel TA, Chabes A. 2011. Mechanisms of mutagenesis in vivo due to imbalanced dNTP pools. Nucleic Acids Res. 39: 1360– 1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Cox J, Mann M. 2008. MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat. Biotechnol. 26: 1367– 1372 [DOI] [PubMed] [Google Scholar]
- 35. Bonilla CY, Melo JA, Toczyski DP. 2008. Colocalization of sensors is sufficient to activate the DNA damage checkpoint in the absence of damage. Mol. Cell 30: 267– 276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Granata M, Lazzaro F, Novarina D, Panigada D, Puddu F, Abreu CM, Kumar R, Grenon M, Lowndes NF, Plevani P, Muzi-Falconi M. 2010. Dynamics of Rad9 chromatin binding and checkpoint function are mediated by its dimerization and are cell cycle-regulated by CDK1 activity. PLoS Genet. 6: e1001047. 10.1371/journal.pgen.1001047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hammet A, Magill C, Heierhorst J, Jackson SP. 2007. Rad9 BRCT domain interaction with phosphorylated H2AX regulates the G1 checkpoint in budding yeast. EMBO Rep. 8:851– 857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Szilard RK, Jacques PE, Laramee L, Cheng B, Galicia S, Bataille AR, Yeung M, Mendez M, Bergeron M, Robert F, Durocher D. 2010. Systematic identification of fragile sites via genome-wide location analysis of gamma-H2AX. Nat. Struct. Mol. Biol. 17: 299– 305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kitada T, Schleker T, Sperling AS, Xie W, Gasser SM, Grunstein M. 2011. GammaH2A is a component of yeast heterochromatin required for telomere elongation. Cell Cycle 10: 293– 300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Smolka MB, Albuquerque CP, Chen SH, Zhou H. 2007. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc. Natl. Acad. Sci. U. S. A. 104: 10364– 10369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lee SJ, Schwartz MF, Duong JK, Stern DF. 2003. Rad53 phosphorylation site clusters are important for Rad53 regulation and signaling. Mol. Cell. Biol. 23: 6300– 6314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Bashkirov VI, Bashkirova EV, Haghnazari E, Heyer WD. 2003. Direct kinase-to-kinase signaling mediated by the FHA phosphoprotein recognition domain of the Dun1 DNA damage checkpoint kinase. Mol. Cell. Biol. 23: 1441– 1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhao X, Chabes A, Domkin V, Thelander L, Rothstein R. 2001. The ribonucleotide reductase inhibitor Sml1 is a new target of the Mec1/Rad53 kinase cascade during growth and in response to DNA damage. EMBO J. 20: 3544– 3553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pellicioli A, Lucca C, Liberi G, Marini F, Lopes M, Plevani P, Romano A, Di Fiore PP, Foiani M. 1999. Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J. 18: 6561– 6572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Basrai MA, Velculescu VE, Kinzler KW, Hieter P. 1999. NORF5/HUG1 is a component of the MEC1-mediated checkpoint response to DNA damage and replication arrest in Saccharomyces cerevisiae. Mol. Cell. Biol. 19: 7041– 7049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Fay DS, Sun Z, Stern DF. 1997. Mutations in SPK1/RAD53 that specifically abolish checkpoint but not growth-related functions. Curr. Genet. 31: 97– 105 [DOI] [PubMed] [Google Scholar]
- 47. Zegerman P, Diffley JF. 2009. DNA replication as a target of the DNA damage checkpoint. DNA Repair (Amst.) 8: 1077– 1088 [DOI] [PubMed] [Google Scholar]
- 48. Tsaponina O, Barsoum E, Astrom SU, Chabes A. 2011. Ixr1 is required for the expression of the ribonucleotide reductase Rnr1 and maintenance of dNTP pools. PLoS Genet. 7: e1002061. 10.1371/journal.pgen.1002061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Gardner R, Putnam CW, Weinert T. 1999. RAD53, DUN1 and PDS1 define two parallel G2/M checkpoint pathways in budding yeast. EMBO J. 18:3173– 3185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Deshpande AM, Newlon CS. 1996. DNA replication fork pause sites dependent on transcription. Science 272: 1030– 1033 [DOI] [PubMed] [Google Scholar]
- 51. Poli J, Tsaponina O, Crabbe L, Keszthelyi A, Pantesco V, Chabes A, Lengronne A, Pasero P. 2012. dNTP pools determine fork progression and origin usage under replication stress. EMBO J. 31: 883– 894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Holmberg C, Fleck O, Hansen HA, Liu C, Slaaby R, Carr AM, Nielsen O. 2005. Ddb1 controls genome stability and meiosis in fission yeast. Genes Dev. 19: 853– 862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Cliby WA, Roberts CJ, Cimprich KA, Stringer CM, Lamb JR, Schreiber SL, Friend SH. 1998. Overexpression of a kinase-inactive ATR protein causes sensitivity to DNA-damaging agents and defects in cell cycle checkpoints. EMBO J. 17: 159– 169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Snyder RD. 1984. The role of deoxynucleoside triphosphate pools in the inhibition of DNA-excision repair and replication in human cells by hydroxyurea. Mutat. Res. 131: 163– 172 [DOI] [PubMed] [Google Scholar]
- 55. D'Angiolella V, Donato V, Forrester FM, Jeong YT, Pellacani C, Kudo Y, Saraf A, Florens L, Washburn MP, Pagano M. 2012. Cyclin F-mediated degradation of ribonucleotide reductase M2 controls genome integrity and DNA repair. Cell 149: 1023– 1034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Tanaka H, Arakawa H, Yamaguchi T, Shiraishi K, Fukuda S, Matsui K, Takei Y, Nakamura Y. 2000. A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage. Nature 404: 42– 49 [DOI] [PubMed] [Google Scholar]
- 57. Bester AC, Roniger M, Oren YS, Im MM, Sarni D, Chaoat M, Bensimon A, Zamir G, Shewach DS, Kerem B. 2011. Nucleotide deficiency promotes genomic instability in early stages of cancer development. Cell 145: 435– 446 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.