

Abstract
The retinoblastoma tumor suppressor gene (RB) product has been implicated in epigenetic control of gene expression owing to its ability to physically bind to many chromatin modifiers. However, the biological and clinical significance of this activity was not well elucidated. To address this, we performed genetic and epigenetic analyses in an Rb-deficient mouse thyroid C cell tumor model. Here we report that the genetic interaction of Rb and ATM regulates DNMT1 protein stability and hence controls the DNA methylation status in the promoters of at least the Ink4a, Shc2, FoxO6, and Noggin genes. Furthermore, we demonstrate that inactivation of pRB promotes Tip60 (acetyltransferase)-dependent ATM activation; allows activated ATM to physically bind to DNMT1, forming a complex with Tip60 and UHRF1 (E3 ligase); and consequently accelerates DNMT1 ubiquitination driven by Tip60-dependent acetylation. Our results indicate that inactivation of the pRB pathway in coordination with aberration in the DNA damage response deregulates DNMT1 stability, leading to an abnormal DNA methylation pattern and malignant progression.
INTRODUCTION
The retinoblastoma tumor suppressor gene (RB) is prevalently mutated at the initiation of retinoblastoma, osteosarcoma, and small-cell lung cancer. However, in the majority of human cancers, retinoblastoma protein (pRB) inactivation is frequently detected during tumor progression and is implicated in various facets of malignant behaviors (1). This suggests that pRB may possess more functions beyond its well-known roles in the control of the cell cycle and differentiation (2, 3). Previous studies demonstrated the direct interaction of pRB with a number of epigenetic modifiers carrying the LXCXE motif, including DNA methyltransferase 1 (DNMT1) (4, 5). pRB has been suggested to play a certain role in nuclear reprogramming in embryonic stem (ES) cells, induced pluripotent stem (iPS) cells, mouse embryonic fibroblasts (MEFs), as well as tumor cells (6–10). Furthermore, RB loss was recently shown to contribute to the progression of human retinoblastoma via epigenetic modifications (11). However, the precise mechanism and target genes of the pRB epigenetic function are still largely unknown.
pRB inactivation induces DNA double-strand breaks (DSBs) via multiple pathways (12–17). The MRN (Mre11, Rad50, and NBS1) sensor complex detects DSBs, rapidly activates ataxia telangiectasia mutated (ATM), and recruits ATM to the site of DSBs. ATM then activates a number of transducers, such as MDC1, 53BP1, and BRCA1, and effector kinases, such as Chk2. These proteins subsequently activate molecules required for DSB repair, including the histone variant H2AX (18–20). Our previous study demonstrated that some components of the DNA damage response (DDR) are activated upon Rb loss in vivo and in vitro (15). We therefore speculated that ATM might mediate pRB functions to regulate the DDR pathway.
C cell (calcitonin-producing cell) tumors developed in Rb+/− mice are precancerous and display many features of the DDR and cellular senescence. Early-stage Rb-deficient C cell tumors exhibited many features of the DDR, including activated ATM, but few features of cellular senescence (15). In contrast, cellular senescence markers were predominant in late-stage Rb-deficient C cell tumors, and simultaneous deletion of senescence-inducing genes, such as Ink4a, Arf, or Suv39h1, allowed Rb-deficient C cells to directly develop into highly malignant tumors (15). Therefore, we thought further genetic analysis of Rb+/−-based composite knockout mice (15, 21–23) might enable us to elucidate the mechanism that links DDR signals to cellular senescence in vivo. To address this, we initiated the current study by analyzing mice lacking both Rb and ATM. We then discovered that pRB and ATM coordinately regulate DNA methylation by affecting DNMT1 protein stability.
MATERIALS AND METHODS
Mouse strains and genotyping.
Rb+/− mice (a gift from T. Jacks) (21) that had been backcrossed to the C57BL/6J strain for 5 to 7 generations and inbred were crossed with Ink4a−/− (a gift from N. Sharpless) (24) or ATM+/− (a gift from J. McKinnon) (25) mice similarly backcrossed to the C57BL/6J strain for at least 5 generations. The resultant F1 progenies were intercrossed to generate F2 mice. Progenies obtained by intercrossing F2 mice were inbred and used in this study. Mouse genotyping was performed as described previously (21, 24, 25). Animals were handled in accordance with the guidelines of Kanazawa University.
Retroviruses and plasmids.
pCDNA3-YFP-Flag-ATM and -YFP-Flag-ATMS1981A were gifts from David Chen (26). MaRXIVfhygro-hDNMT1 was a gift from E. Hara. pEGFPcl-Tip60 and pOZN-Tip60-Flag-HA (27) were gifts from T. Ikura. pLXSB-RB was described previously (15). pRK5-HA-Ubiquitin-WT (28) was purchased from Addgene (Boston, MA). For retrovirus production, EcoPak2 (Clontech) cells were used. Transfection of MEFs or the C cell tumor cell line was performed by using FuGene6 (Roche). MEFs or AC232 cells infected with pBabe-puro or pLXSB vectors were selected with 4 μg/ml puromycin or 8 μg/ml blasticidin S, respectively, for approximately 36 to 48 h.
Cell culture.
Primary C cell adenocarcinoma cell lines were derived as described previously (15). MEFs were prepared from embryonic day 10.0 (E10.0) or E12.5 embryos derived by intercrossing Rb+/−; ATM+/− or Rb+/−; Ink4a−/− mice. The 3T3 protocol (3-day transfer, inoculum of 3 × 105 cells) assay was performed as described previously (15).
Antibodies.
For immunohistochemistry, immunofluorescence (IF), immunoblotting, or chromatin immunoprecipitation (ChIP) assays, mouse or rabbit antibodies specific to the following proteins were used: proliferating cell nuclear antigen (PCNA) (catalog no. SC-7907; Santa Cruz), Ki67 (catalog no. AB15580; Abcam), γH2AX (catalog no. 05-636; Upstate), ATM phosphorylated at serine 1981 (ATMpS1981) (catalog no. 200-301-500; Rockland) (for immunohistochemistry), p53pS15 (catalog no. PC386; Calbiochem), histone H3 trimethylated at lysine 9 (H3K9me3) (catalog no. 07-442; Upstate), heterochromatin protein 1γ (HP1γ) (catalog no. MAB3450; Chemicon), p16INK4a (catalog no. SC-1661), calcitonin (catalog no. SC-20725), carcinoembryonic antigen (CEA) (catalog no. AB33562), ATMpS1981 (catalog no. 200-301-400; Rockland), Suv39H1 (catalog no. AB12405; Abcam), ARF (catalog no. AB80), pRBL2 (catalog no. SC-56178), Chk2pT68 (catalog no. AB38461), p21 (catalog no. 556430; BD), α-tubulin (catalog no. CP06; Calbiochem), pRB (catalog no. 554136 [BD], MK-15-3 [MBL], and SC-50), DNMT1 (catalog no. 70-203 [Bioacademia Inc.] and AB13537), ubiquitin (catalog no. 3933, Cell Signaling), acetylated lysine (catalog no. 9441; Cell Signaling), HDAC1 (catalog no. SC-7872), UHRF1 (catalog no. AB57083), Tip60 (catalog no. AB79373), DNMT3b (catalog no. SC-81252), ataxia telangiectasia Rad-3-related protein kinase (ATR) (catalog no. SC-1666328), ATM (catalog no. AB78), hemagglutinin (HA) (catalog no. 3F10; Roche), anti-FLAG (catalog no. F4042; Sigma), histone H3K4me2 (catalog no. AB32356), and histone H3K9me3 (catalog no. AB8898). Staining of genetically unmodified (wild-type) thyroid tissues was performed in our previous work (15) for most of the proteins examined in the current study.
Tagged proteins.
HA-tagged DNMT1 was constructed by inserting a PCR-amplified fragment of MaRXIVfhygro-hDNMT1 into pBabe-puro. pEF-BOS-3HA-ATM, -3HA-ATM-CD (D2870A, kinase dead), -3HA-ATM-BD, -3HA-ATM-ES, -3HA-ATM-KS, -3HA-ATM-PM, and -3HA-ATM-KD were constructed by inserting PCR-amplified or restriction-digested fragments of human ATM (hATM) into pEF-BOS (29). pEF-BOS-FLAG-DNMT1-MS, -FLAG-DNMT1-KS, -3HA-DNMT1-D2, -3HA-DNMT1-D3, -3HA-DNMT1-D4, and -3HA-DNMT1-D5 were constructed by inserting a PCR-amplified fragment of hDNMT1 cDNA into pEF-BOS. Structures of these tagged ATM and DNMT1 fragments are shown in Fig. S8 in the supplemental material.
Immunohistochemistry.
Paraffin-embedded mouse C cell tumors, human medullary thyroid carcinomas (MTCs), and human esophageal cancers were immunostained as described previously (15).
Immunofluorescence.
Texas Red-conjugated goat anti-mouse or Alexa Fluor 488-conjugated goat anti-rabbit antibody (catalog no. T862 and A11034, respectively; Molecular Probes) was used as the secondary antibody. DNA was visualized by staining with 4′,6-diamino-2-phenylindole (DAPI).
Immunoblotting.
Whole-cell lysates were prepared in MLB buffer or in acid extraction buffer as described previously (15).
Cell fractionation.
Collected cells were lysed in solution A (10 mM HEPES [pH 7.9], 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose, 10% glycerol, 1 mM dithiothreitol, protease and phosphatase inhibitors, and 0.1% Triton X-100). The cytoplasmic fraction (C) was harvested from supernatants after centrifugation at 1,300 rpm for 5 min. The nuclear pellet was washed once with solution A and relysed in solution B (3 mM EDTA, 0.2 mM EGTA, 1 mM dithiothreitol, and protease and phosphatase inhibitors). The soluble nuclear fraction (N) was obtained from supernatants after centrifugation at 1,700 rpm for 5 min. The chromatin pellet (Ch) was washed twice in solution B, collected by centrifugation at 10,000 rpm for 3 min, and resolved in a loading buffer for SDS-PAGE.
Immunoprecipitation.
Cell lysates were immunoreacted with specific antibodies and analyzed as described previously (15).
Colony formation assays.
A colony formation assay was performed as described previously (15).
RNA interference.
Mission TRC short hairpin RNA (shRNA) target sets for ATM (TRCN0000012643 and TRCN0000012647), CDKN2A (TRCN0000077815 and TRCN0000077816), DNMT1 (TRCN0000225698 and TRCN0000225700), and Tip60 (TRCN0000039302 and TRCN0000039303) and the TurboGFP shRNA control vector (SHC004) were purchased from Sigma-Aldrich.
DNA methylation analysis.
Genomic DNA sequences retrieved from the Mouse Genome Informatics (MGI) database were searched for CpG islands and specific primers by using the Methyl sequence software provided by Applied Bioscience. Bisulfite modification of DNA was performed by using a Methyl Easy Xceed Rapid DNA bisulfite modification kit (catalog no. ME002; Human Genetic Signature) and amplified by PCR using the bisulfite sequencing-specific primers listed in Table S4 in the supplemental material. The amplified DNA fragment was subcloned into T-Vector pMD20 (catalog no. 3270; TaKaRa Bio). Clones were screened by colony PCR, and plasmid DNA was sequenced by using the KB-3130 sequencer (Applied Biosystems). Bisulfite sequencing data were analyzed by using the Quantification Tool for Methylation Analysis (QUMA) provided by RIKEN (http://quma.cdb.riken.jp/). Statistical analysis was performed by using the same program.
ChIP assay.
A ChIP assay was performed as described previously (15). Briefly, cells were cross-linked with 1% formaldehyde for 10 min at room temperature, and the reaction was terminated with 0.125 M glycine. The cells were then lysed in lysis buffer (catalog no. SC-45000), resuspended in high-salt lysis buffer (catalog no. SC-45001), and sonicated for 5 cycles of 30 s. Five hundred micrograms of protein was then immunoprecipitated with anti-H3K4me2 (catalog no. AB32356), anti-H3K9me3 (catalog no. AB8898), or anti-DNMT1 (catalog no. AB13537) antibodies. The DNA was phenol extracted, and the promoters of genes were amplified by PCR (ChIP). Quantitative PCR (quantitative ChIP) was performed by using Fast SYBR green master mix (Applied Biosystems) with a Light Cycler 480 II instrument (Roche), according to the protocols provided by the manufacturers. The sequences of primers used for these amplifications are indicated in Table S5 in the supplemental material, and their locations on the Ink4a promoter region are shown in Fig. S6B in the supplemental material.
DNMT1 activity assay.
DNMT1 activity was measured by using the DNMT1 direct activity assay kit (catalog no. 52050L; BPS Bioscience), according to the manufacturer's protocol. Briefly, protein lysates were incubated with the reaction mixture for 1 h with rocking at 4°C. The reaction mixture was placed onto a substrate-coated plate, incubated with anti-5-methylcytosine antibody, and then incubated with horseradish peroxidase (HRP)-labeled secondary antibody and HRP chemiluminescent substrate. Chemiluminescence of the samples was immediately measured by using a Fluoroscan Ascent FL instrument (Thermo Electric Corporation).
Microarray.
Rb−/−; ATM−/− MEFs were transfected using nucleofection (nucleofected) with 0.5 μg pBabe-puro plus 5.0 μg pCDNA3-YFP-Flag-ATM or an empty vector or infected with lentiviruses expressing shRNAs that target DNMT1 and selected with 4 μg/ml puromycin or 8 μg/ml blasticidin S for 36 to 48 h. Selected cells were grown for a further 48 h in normal culture medium supplemented with 10% fetal bovine serum (FBS) and harvested on ice, and RNA was extracted by using the RNeasy kit (Qiagen Inc.). The quality of the RNA samples was assessed by using the RNA 6000 Nano LabChip kit (Bioanalyzer 2100; Agilent Technologies). One-color microarray analysis was performed by using an Agilent mouse whole-genome 4X44k Oligo Microarray (Agilent Technologies), as previously described (30).
Human tumors.
Tumors from patients with MTCs were surgically removed at the Department of Endocrine Surgery; were diagnosed at the Department of Pathology, Tokyo Women's Medical University; and were analyzed according to the guidelines of Tokyo Women's Medical University. Esophageal squamous cell carcinoma (SCC) tumors were surgically removed at the Department of Gastroenterological Surgery; were diagnosed at the Department of Pathology, Graduate school of Medicine, Osaka University; and were analyzed according to the guidelines of Osaka University. Patients did not receive chemotherapy or radiation preoperatively.
Comet assay.
A Comet assay reagent kit was purchased from Trevigen (catalog no. 4250-050K), and assays were performed as previously described (15).
BrdU incorporation.
Bromodeoxyuridine (BrdU) incorporation was measured as described previously (15).
Bisulfite PCR.
Methylation of the Ink4a promoter was evaluated by nested PCR with unmethylation- and methylation-specific primers by using the 5′ untranslated region of Ink4a (Gene bank accession no. U47018), as previously described (24, 31). Bisulfite modification of DNA was performed by using a MethylEasy DNA bisulfite modification kit (catalog no. ME001; Human Genetic Signature). The sequences of the unmethylation- or methylation-specific PCR primers are listed in Table S2 in the supplemental material.
RT-PCR.
Total RNA was extracted by using the RNeasy minikit from Qiagen (catalog no. 74104), and reverse transcription-PCR (RT-PCR) was performed by using the TaKaRa RNA PCR kit (catalog no. RR019A). The sequences of specific primers are shown in Table S3 in the supplemental material.
Microarray data accession number.
The microarray data described in this study have been deposited in the NCBI Gene Expression Omnibus database under accession number GSE47501 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE47501).
RESULTS
ATM status affects p16Ink4a in Rb-deficient tumors.
Rb+/− mice develop several types of “Rb-deficient” tumors as a consequence of normal allele inactivation in somatic cells (3). We analyzed primary thyroid C cell tumors developed in Rb+/−; ATM+/+ (average age at examination [AE] ± standard error of the mean [SEM] = 12.5 ± 1.5 months), Rb+/−; ATM+/− (AE = 7.1 ± 1.3 months), and Rb+/−; ATM−/− (AE = 5.4 ± 1.3 months) mice. C cell tumors were not detected in Rb+/+; ATM−/− (AE = 9.4 ± 1.4 months) mice (Fig. 1A). Carcinoembryonic antigen (CEA) and Ki67 were frequently expressed in both Rb-deficient ATM+/− and Rb-deficient ATM−/− tumors but were infrequently expressed in Rb-deficient ATM+/+ tumors (Fig. 1A; see also Table S1 in the supplemental material). Rb+/−; ATM−/− mice exhibited significantly poorer survival than did Rb+/−; ATM+/− mice (Fig. 1C). As we previously reported (15), primary Rb-deficient ATM+/+ tumors exhibited frequent expression of DDR markers, including ATM phosphorylated at serine 1981 (ATMpS1981), histone variant H2AX phosphorylated at serine 139 (γH2AX), and p53 phosphorylated at serine 15 (p53pS15) (see Fig. S1 in the supplemental material). Ataxia telangiectasia Rad-3-related protein kinase (ATR) was frequently expressed in Rb-deficient, ATM-deficient tumors, and Rb-deficient ATM−/− tumors showed significant accumulation of γH2AX. ATM heterozygosity reduced the frequency of ATMpS1981 to <10% (see Fig. S1 in the supplemental material); this may explain the haploid insufficiency of the ATM locus in Rb-deficient C cell tumors. Consistent with our previous report, Rb-deficient tumors frequently expressed senescence markers, including histone H3 trimethylated at lysine 9 (H3K9me3), heterochromatin protein 1γ (HP1γ), and p16Ink4a (Fig. 1B). In contrast to the previously studied Rb-deficient N-ras−/− tumors (15), Rb-deficient ATM+/− or Rb-deficient ATM−/− tumors retained frequent expression of H3K9me3, HP1γ, and p19Arf. However, p16Ink4a expression was very infrequent in these tumors, in sharp contrast to the frequent DNMT1 expression (Fig. 1B). Because promoter methylation is a common mechanism for Ink4a gene inactivation, we suspected that ATM activity might influence the Ink4a expression level via regulation of DNMT1.
Fig 1.

Analysis of Rb-deficient C cell tumors with various ATM genotypes. (A) Thyroid tumors developed in mice of the indicated genotypes at the indicated months of age were photographed, and the tissues were then analyzed by hematoxylin-eosin (HE) staining and immunostaining with antibodies to the indicated proteins. The frequency of immunopositive cells per 200 normal thyroid or tumor cells was quantified. Columns show means and SEM. Scale bars, 1 mm in macroscopic and 100 μm in microscopic pictures. (B) Serial sections of the same tumors were immunostained with antibodies to the indicated proteins and analyzed as described above for panel A. (C) Survival rate in mice of the indicated genotypes. Data are expressed as percent survival.
Human sporadic medullary thyroid carcinomas (MTCs) can be classified into two groups: one with high-level pRB expression and the other with low-level pRB expression (15). A total of 200 cells were randomly selected and evaluated for immunopositive nuclear staining in all tumors, and tumors were classified according to the frequency of immunopositive cells into two groups, with cutoff values of ≥50% for high-level expression and <50% for low-level expression. In the current study, we analyzed 5 cases with high-level and 5 cases with low-level pRB expression. Five out of five cases with low pRB levels showed low expression levels of ATMpS1981. Four out of the five cases with low pRB levels showed high DNMT1 expression levels; all four of these cases showed low p16INK4a expression levels (see Fig. S2A to D in the supplemental material). One case with low pRB and exceptionally low DNMT1 levels showed very high p16INK4a expression levels. Because pRB inactivation is frequently detected in esophageal cancers, we also tested this correlation in human esophageal squamous cell carcinomas (SCCs). We then detected 8 out of 10 cases with low pRB expression levels. Six out of the eight cases with low pRB levels showed high ATMpS1981, low DNMT1, and high p16INK4a expression levels (see Fig. S2E to G in the supplemental material). Although the ATM activation status varied in these two types of human cancers, the strict correlation of ATMpS1981, DNMT1, and p16INK4a in low-level pRB-expressing MTCs and esophageal SCCs supports our hypothesis that ATM status might determine p16INK4a expression via regulation of DNMT1 in the absence of pRB.
ATM status affects p16Ink4a in Rb-deficient mouse embryonic fibroblasts.
Biallelic loss of ATM shortened the life span of Rb−/− embryos (average life span, ∼E13.5) by approximately 3 days. Therefore, we prepared MEFs of varied Rb and ATM genotypes from E10.0 embryos. Compared to cells derived from Rb-deficient ATM−/− C cell tumors, Rb−/−; ATM−/− MEFs adapted easily to adhesive culture conditions. Thus, we extensively analyzed MEFs. Upon a 3T3 protocol assay, Rb−/−; ATM−/− and Rb−/−; ATM+/− MEFs achieved infinite proliferation significantly earlier than did Rb−/− or ATM−/− MEFs (Fig. 2A). In addition, Rb−/−; ATM+/− and Rb−/−; ATM−/− MEFs but not Rb−/− or ATM−/− MEFs could form colonies when plated at a low cell density (Fig. 2B, left). These signs of immortalization were more evident in Rb−/−; ATM−/− than in Rb−/−; ATM+/− MEFs (Fig. 2A and B). Consistently, immunoblotting showed a tight correlation between DNMT1 and p16Ink4a levels in Rb-negative MEFs depending on the ATM status (Fig. 2B).
Fig 2.

Analysis of Rb−/− MEFs of various ATM genotypes. (A) 3T3 protocol assay of MEFs with the indicated genotypes. MEFs were freshly prepared from embryos of timed pregnant inbred mice and cultivated for 3 passages before the start of the 3T3 assay. The cumulative multiplicity in the representative cultures at the indicated passage number is illustrated in the graph. WT, wild type. (B) Representative results from colony formation assays with MEFs of the indicated genotypes plated at 1 × 103 cells per 60-mm dish and cultured for 14 days. (C) Immunoblotting analysis of MEFs of the indicated genotypes with antibodies to the indicated proteins. (D) Rb−/− MEFs infected with retroviruses expressing the indicated proteins were selected and analyzed by immunoblotting. (E) Rb−/−; ATM−/− MEFs infected with lentiviruses expressing shRNAs that target the indicated genes were analyzed by a colony formation assay and immunoblotting with antibodies to the indicated proteins. #1 and #2 indicate independently designed shRNAs targeting DNMT1. (F) Rb+/+ or Rb−/− MEFs infected with lentiviruses expressing the shRNAs that target the indicated genes were selected and analyzed by immunoblotting. GFP, green fluorescent protein. (G) Rb−/−; ATM−/− MEFs transduced with vectors expressing the indicated proteins were selected and analyzed by immunoblotting.
Both the abundance and activity of ATM (ATMpS1981) were markedly increased, and the DNMT1 level was downregulated in Rb−/− MEFs compared to Rb+/+ cells (Fig. 2C). A previous report, however, indicated that RB loss upregulates DNMT1 transcription by severalfold (32). These findings suggested that the genetic interaction between Rb and ATM might regulate DNMT1 at the posttranscriptional level.
Rb deletion upregulated p16Ink4a, p21Waf1/Cip1, and Suv39h1 with concomitant increases in the levels of H3K9me3 and HP1γ. Simultaneous deletion of ATM remarkably decreased the level of p16Ink4a and reciprocally upregulated DNMT1 and proliferating cell nuclear antigen (PCNA) (Fig. 2C). Heterozygous deletion of ATM also upregulated DNMT1 to a level slightly lower than that with the homozygous deletion (Fig. 2B, right). Furthermore, Rb deletion induced Chk2pT68 and p53pS15, which was significantly attenuated by the simultaneous deletion of ATM. In contrast, Rb−/−; ATM−/− MEFs showed marked increases in the levels of γH2AX, ATR, and Chk1pS345 (Fig. 2C). Introduction of pRB into Rb−/− MEFs decreased ATMpS1981 and increased DNMT1 levels (Fig. 2D). Depletion of DNMT1 significantly increased p16Ink4a levels in Rb−/−; ATM−/− MEFs and eliminated their ability to form colonies when plated at a low density (Fig. 2E). We previously demonstrated that an additional Ink4a deletion immortalized Rb−/− MEFs at early passage (15). All these observations suggest that suppression of the Ink4a gene by DNMT1 might be the major mechanism underlying the immortal growth of Rb−/−; ATM−/− MEFs.
To assess the role of induced and activated ATM in Rb−/− MEFs, we depleted ATM from Rb+/+ and Rb−/− MEFs. ATM depletion in Rb−/− MEFs reduced Chk2pT68 and p53pS15 levels and induced γH2AX, consistent with the findings for Rb−/−; ATM−/− MEFs. However, Chk1pS345 was also attenuated by ATM depletion, contrary to our observations of Rb−/−; ATM−/− MEFs. Importantly, ATM depletion in Rb−/− MEFs upregulated DNMT1 to a level similar to that seen in Rb−/−; ATM−/− MEFs, with reciprocal downregulation of p16Ink4a. Interestingly, ATM depletion from Rb+/+ cells rather slightly increased both DNMT1 and p16Ink4a levels (Fig. 2F). These findings strongly indicate that the Rb status is a critical determinant of the regulatory interaction of ATM, DNMT1, and p16Ink4a. Immunofluorescence (IF) analysis and Comet assays of ATM-depleted cells showed results consistent with those obtained by immunoblotting (see Fig. S3A in the supplemental material). In addition, ATM depletion allowed Rb−/− but not Rb+/+ MEFs to form colonies under low-density plating conditions (see Fig. S3D in the supplemental material). Interestingly, the simultaneous depletion of both ATM and p16Ink4a was sufficient to immortalize Rb+/+ MEFs (see Fig. S3E in the supplemental material). These findings suggest that inactivation of the CDKIs-pRB pathway in combination with a deficient DDR is sufficient to immortalize MEFs.
Introduction of ATM into Rb−/−; ATM−/− MEFs recovered the DDR, downregulated DNMT1 but not DNMT3b, and rescued p16Ink4a (Fig. 2G). ATM reconstitution significantly suppressed colony formation by Rb−/−; ATM−/− MEFs when cultured at a low density (see Fig. S4A in the supplemental material). On the other hand, the effect of gene transduction on cell proliferation became visible only after day 14 (see Fig. S4B and C in the supplemental material). All our assessments of cellular proteins were done at days 10 to 12 after gene transduction. This suggests that most of the above-mentioned changes preceded the effects on the cell cycle possibly given by ATM. ATM introduced into Rb−/−; ATM−/− MEFs exhibited a high level of phosphorylation and induced DDR activation, which was significantly antagonized by pRB (see Fig. S4D in the supplemental material). A mutant form of ATM in which serine 1981 was changed to alanine (ATMS1981A) significantly failed to achieve these functions (see Fig. S4 in the supplemental material). Introduction of ATM or ATMS1981A into an Rb-deficient ATM−/− C cell adenocarcinoma cell line showed results highly similar to those obtained with Rb−/−; ATM−/− MEFs (see Fig. S5 in the supplemental material). Taken together, these findings suggest that Rb and ATM cooperatively regulate MEF immortalization by modulating the p16Ink4a level via control of DNMT1 abundance.
ATM regulates the DNA methylation status of the Ink4a promoter.
To assess the significance of the reciprocal correlation of DNMT1 and p16Ink4a, we examined the methylation status of CpG islands residing in the Ink4a promoter by bisulfite sequencing. We observed that Rb loss significantly decreased DNA methylation in the Ink4a promoter region (Fig. 3A and C). Surprisingly, simultaneous ATM deletion induced hypermethylation in the same region of the Ink4a promoter (Fig. 3B and C). Introduction of ATM but not ATMS1981A into Rb−/−; ATM−/− MEFs carrying Ink4a promoter hypermethylation and following cultivation under normal conditions for 10 days remarkably reduced the number of methylated CpG nucleotides in the Ink4a promoter (Fig. 3B and C; see also Fig. S6A in the supplemental material). Bisulfite PCR with either unmethylation- or methylation-specific primers showed similar results (see Fig. S7A in the supplemental material). Both treatment with the ATM-specific kinase inhibitor KU-55933 and, more importantly, the introduction of pRB antagonized the ATM function to induce the hypomethylation status of the Ink4a promoter (Fig. 3B and C; see also Fig. S6A in the supplemental material). Reintroduction of only pRB into Rb−/−; ATM−/− MEFs caused an insignificant change in the Ink4a promoter methylation status (Fig. 3B and C; see also Fig. S6A in the supplemental material). Treatment of Rb−/−; ATM−/− MEFs expressing exogenous ATM and pRB with KU-55933 caused an insignificant increase in the Ink4a promoter methylation level compared with the marked increase in the Ink4a promoter methylation level induced by ATM kinase inhibition in Rb-deficient MEFs (Fig. 3B and C; see also Fig. S6A in the supplemental material). We thought that this variability of ATM function reflected different levels of ATM activity because exogenously introduced pRB inhibited ATM activation (ATMpS1981 and p53pS15), increased DNMT1 abundance, and suppressed p16Ink4a to levels comparable to those observed in Rb-deficient MEFs treated with KU-55933. Therefore, further treatment of Rb−/−; ATM−/− MEFs expressing exogenous ATM and pRB with KU-55933 did not cause significant changes in either ATM activation, DNMT1 abundance, or the p16Ink4a protein level (Fig. 3D). Interestingly, DNMT1 suppression by specific shRNAs or methyltransferase inhibitors significantly reduced DNA methylation, particularly in the CpG nucleotides closer to the transcription start site of the Ink4a gene (Fig. 3C; see also Fig. S6A in the supplemental material).
Fig 3.

DNA methylation status of the Ink4a promoter region in MEFs of various Rb and ATM genotypes. (A) Results of bisulfite sequencing of the Ink4a promoter region in Rb+/+ or Rb−/− MEFs. (B) Rb−/−; ATM−/− MEFs were transfected with the vectors expressing the indicated proteins, selected, cultured for an additional 10 days, and thereafter treated with the indicated reagent (KU-55933 at 10 μM for 7 days) and analyzed. (C) Statistical analysis of the results of bisulfite sequencing of the Ink4a promoter region in MEFs of the indicated genotypes that were transfected with the expression vectors or infected with lentiviruses expressing shRNAs that target the indicated genes, selected for 72 h, and treated with the indicated reagents (trichostatin A [TSA] at 2 μM for 3 days, KU-55933 at 10 μM for 5 days, and 5-AzadC at 2 μM for 3 days). *, P < 0.01 by Mann-Whitney test. (D) Immunoblotting analysis of MEFs of the indicated genotypes with antibodies to the indicated proteins. MEFs were transfected with the vectors expressing the indicated proteins, selected, cultured for an additional 10 days, and thereafter treated with the indicated reagent (KU-55933 at 10 μM for 5 days) and analyzed. (E) ChIP assay of the Ink4a promoter region in MEFs of the indicated genotypes. Rb−/−; ATM−/− MEFs were transduced with vectors expressing the indicated proteins, selected, and analyzed for binding of the indicated proteins by PCR with specific primers (see Fig. S6B and Table S5 in the supplemental material). Proximal, middle, and distal regions are relative to the transcription start site (see Fig. S6B in the supplemental material). (F) Quantitative ChIP assay of the Ink4a promoter region (positions ∼−44 to −149) in MEFs of the indicated genotypes. Rb−/−; ATM−/− MEFs were transduced with vectors expressing the indicated proteins, selected, and analyzed for DNMT1 binding by qPCR with specific primers (see Fig. S6B and Table S5 in the supplemental material).
ATM inhibition in the Rb-negative context suppressed the Ink4a mRNA level (see Fig. S7B in the supplemental material). Chromatin immunoprecipitation (ChIP) indicated that simultaneous deletion of Rb and ATM significantly enhanced binding of DNMT1 and the bivalent histone mark to the Ink4a promoter. However, ATM reconstitution in Rb−/−; ATM−/− MEFs diminished DNMT1 binding, with insignificant changes in the histone mark, particularly in the proximal promoter region (Fig. 3E). The effect of Rb and ATM statuses on DNMT1 binding to the proximal region of the Ink4a promoter was confirmed by quantitative ChIP analysis (Fig. 3F). However, the transcription level of DNMT1 was insignificantly affected by the ATM status in an Rb-negative context (see Fig. S7C in the supplemental material). Taken together, these observations imply that activated ATM influences the Ink4a promoter methylation status by regulating DNMT1 primarily at the posttranscriptional level, and pRb antagonizes these ATM functions by attenuating its activation.
Active ATM dictates ubiquitination of DNMT1 in the absence of Rb.
To further explore the functional relationship between ATM and DNMT1, we introduced ATM or ATMS1981A into Rb−/−; ATM−/− MEFs and observed the fate of endogenous DNMT1 by IF. Compared to the vector control, ATM but not ATMS1981A significantly reduced the intensity of the DNMT1 signal in the nucleus. In addition, we detected a certain degree of colocalization of exogenous ATMpS1891 and endogenous DNMT1 in the nucleus. However, the ATMS1981A and DNMT1 signals were mutually exclusive in the majority of examined cells (Fig. 4A).
Fig 4.
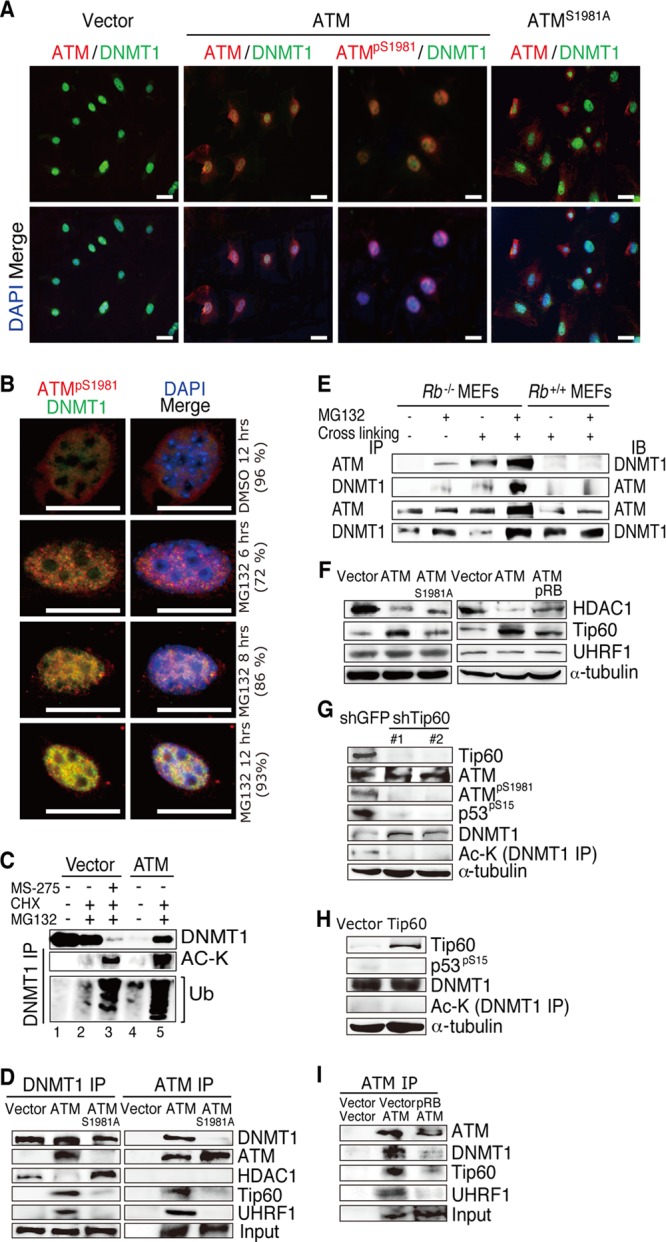
Effects of pRB and ATM on the DNMT1 degradation complex. (A) IF analysis of the indicated proteins in Rb−/−; ATM−/− MEFs transduced with vectors expressing the indicated proteins. (B) IF analysis of Rb−/−; ATM−/− MEFs transduced with vectors expressing the indicated proteins and treated with vehicle (DMSO) or 5 μM MG132 for the indicated periods of time. Numbers in parentheses indicate percentages of the representative phenotype that appeared in all analyzed cells. (C) Lysates from Rb−/−; ATM−/− MEFs transduced with vectors expressing the indicated proteins and treated with the indicated reagents were immunoprecipitated with DNMT1-specific antibody and analyzed by immunoblotting. Concentrations and durations of treatments are as follows: MS275 at 5 μM for 48 h, cycloheximide (CHX) at 2.5 μg/ml for 12 h, and MG132 at 5 μM for 12 h. Ub, ubiquitin (D) Lysates from Rb−/−; ATM−/− MEFs transduced with vectors expressing the indicated proteins were treated with 5 μM MG132 for 12 h, immunoprecipitated with antibodies to DNMT1 or ATM, and analyzed by immunoblotting. (E) Lysates from MEFs of the indicated genotypes arrested in S phase and treated with the indicated reagents were immunoprecipitated (IP) with antibodies to DNMT1 or ATM and analyzed by immunoblotting (IB). Cells were arrested in S phase by a double-thymidine block at 2 mM for 18 h (first block), released for 9 h, and blocked for another 17 h (second block); MG132 was used at 5 μM for 12 h; and cross-linking was performed with a 1% formaldehyde solution for 10 min at room temperature and terminated with 0.125 M glycine. (F) Rb−/−; ATM−/− MEFs were transduced with vectors expressing the indicated proteins, selected with 4 μg/ml puromycin and 8 μg/ml blasticidin S for 5 days, and analyzed by immunoblotting using antibodies specific to the indicated proteins. (G) Rb−/−; ATM−/− MEFs transduced with vectors expressing ATM were infected with lentiviruses expressing shRNAs that target the indicated genes, selected, and analyzed by immunoblotting. (H) Rb−/−; ATM−/− MEFs transduced with vectors expressing the indicated proteins were analyzed by immunoblotting. Ac-K, acetylated lysine. (I) Lysates from Rb−/−; ATM−/− MEFs transduced with vectors expressing the indicated proteins were treated with 5 μM MG132 for 12 h, immunoprecipitated with ATM-specific antibody, and analyzed by immunoblotting.
DNMT1 is regulated by ubiquitination-dependent degradation at the proteasome (33, 34). Treatment of the ATM-reconstituted Rb−/−; ATM−/− MEFs with a proteasome inhibitor, MG132, increased the DNMT1 level in the nucleus and enhanced its colocalization with ATMpS1891 in a time-dependent manner (Fig. 4B). In addition, MG132 treatment of vector-transduced Rb−/−; ATM−/− MEFs did not affect the DNMT1 protein level; however, it did dramatically increase the DNMT1 level in ATM-reconstituted cells. Furthermore, MG132 treatment induced accumulation of acetylated and polyubiquitinated DNMT1 in ATM-reconstituted Rb−/−; ATM−/− MEFs (Fig. 4C, lane 5), indicating that ATM accelerates DNMT1 ubiquitination in the absence of Rb. Treatment of Rb−/−; ATM−/− MEFs simultaneously with MS275 (HDAC1 inhibitor) and MG132 decreased DNMT1 abundance and enhanced accumulation of the acetylated and polyubiquitinated forms of DNMT1 (Fig. 4C, lane 3), suggesting that increased acetylation of DNMT1 is sufficient to trigger its ubiquitination. Taken together, these findings suggest that ATM promotes acetylation-driven ubiquitination following degradation of DNMT1 in Rb-deficient cells.
Next, we asked whether activated ATM regulates the functions of enzymes involved in regulation of DNMT1 acetylation. The immunoprecipitation assay of lysates from MG132-treated cells with DNMT1-specific antibody indicated that DNMT1 is bound to HDAC1 in vector-transduced Rb−/−; ATM−/− MEFs. Introduction of ATM precluded HDAC1 from the DNMT1 complex and instead recruited Tip60 acetyltransferase and UHRF1 E3 ligase (Fig. 4D). However, the introduced ATMS1981A largely failed to exhibit this function (Fig. 4D). The immunoprecipitation assay with an ATM-specific antibody provided results consistent with those obtained in the experiments described above (Fig. 4D). In addition, the immunoprecipitation assay of lysates from Rb−/− MEFs with ATM or DNMT1 antibodies indicated that endogenous ATM is bound to DNMT1, and treatment with MG132 or a formaldehyde cross-linker enhanced their binding. Although the ATM level is lower than that in Rb−/− MEFs, we so far did not detect endogenous ATM-DNMT1 binding in Rb+/+ MEFs even when treated with MG132 or by formaldehyde cross-linking (Fig. 4E). In addition, the introduction of ATM into Rb−/−; ATM−/− MEFs upregulated Tip60 and suppressed the HDAC1 protein level; however, pRB antagonized this effect of ATM (Fig. 4F), suggesting that activated ATM controls DNMT1 acetylation even by regulating the abundances of HDAC1 and Tip60. Tip60 was recently shown to destabilize DNMT1 via an acetylation-driven ubiquitination mechanism (35); however, the current study proposes that activated ATM plays a critical role in the assembly of the DNMT1 degradation complex. Tip60 depletion from ATM-reconstituted Rb−/−; ATM−/− MEFs induced a significant downregulation of ATMpS1981 and rescued DNMT1 via suppression of its acetylation (Fig. 4G). In contrast, without ATM, overexpressed Tip60 did not affect either the abundance or acetylation of DNMT1 (Fig. 4H), implying that ATM essentially mediates Tip60-induced DNMT1 acetylation. Furthermore, introduction of pRB into ATM-reconstituted Rb−/−; ATM−/− MEFs disrupted the ATM-DNMT1-Tip60-UHRF1 (DNMT1 degradation) complex (Fig. 4I). Taken together with our findings that pRb regulates ATM activation (Fig. 2D) and that ATM-mediated Tip60 functions to acetylate DNMT1 (Fig. 4F to H), these observations indicated a key role for pRB in regulating the DNMT1 degradation complex.
We next assessed the direct effects of cell lysates from MEFs with various Rb and ATM statuses on the activity of DNMT1 in vitro. Cell lysates from Rb−/−; ATM−/− MEFs increased the DNMT1 activity in a dose-dependent manner, whereas cell lysates from Rb−/−; ATM+/+ MEFs significantly attenuated DNMT1 activity. Lysates from wild-type MEFs showed no significant effects on DNMT1 activity (Fig. 5A), whereas lysates from ATM-reconstituted Rb−/−; ATM−/− MEFs significantly decreased DNMT1 activity. Finally, lysates from Rb−/−; ATM−/− MEFs transduced with ATMS1981A slightly attenuated DNMT1 activity (Fig. 5B). These findings support the notion that pRb stabilizes DNMT1 by antagonizing ATM activation.
Fig 5.
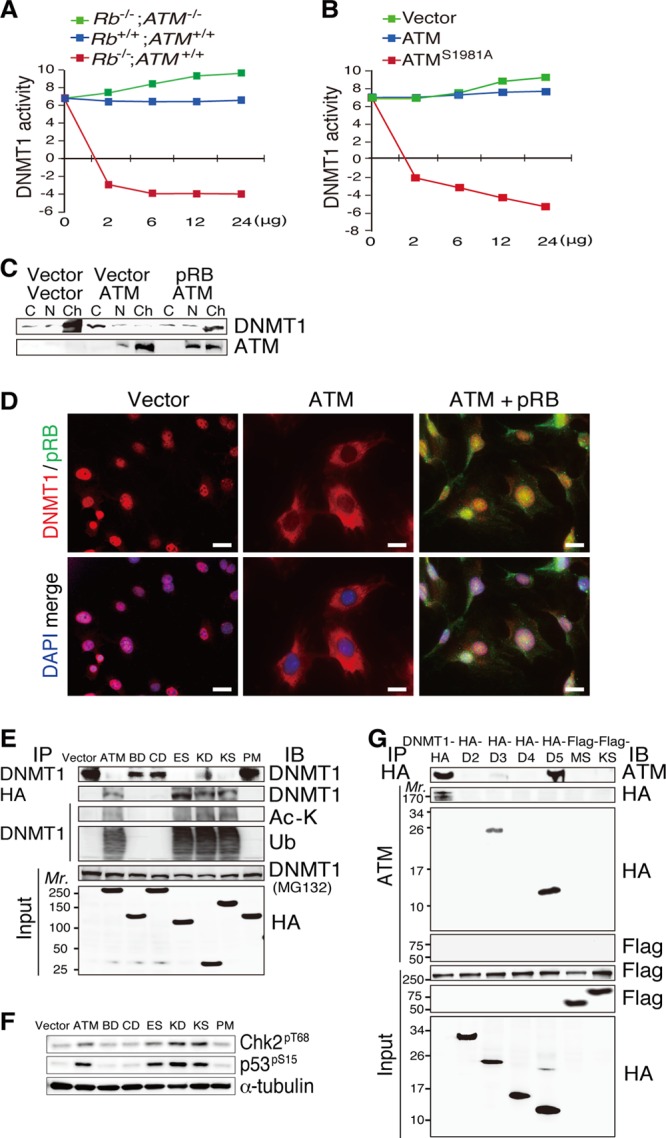
Physical interaction between ATM and DNMT1. (A) Measurement of the dose-dependent effects of the lysates from MEFs of the indicated genotypes on DNMT1 enzyme activity. (B) Measurement of the dose-dependent effects of the lysates from Rb−/−; ATM−/− MEFs transduced with vectors expressing the indicated proteins on DNMT1 enzyme activity. (C) Cytosolic (C), nuclear (N), or chromatin (Ch) protein fractions from Rb−/−; ATM−/− MEFs transduced with vectors expressing the indicated proteins and treated with 5 μM MG132 for 12 h were analyzed by immunoblotting. (D) IF analysis with antibodies specific to the indicated proteins in Rb−/−; ATM−/− MEFs transduced with vectors expressing the indicated proteins. Scale bars, 20 μm. (E) Lysates from Rb−/−; ATM−/− MEFs transduced with vectors expressing the indicated proteins (see Fig. S8A in the supplemental material) were untreated or treated with 5 μM MG132 for 12 h (indicated in parentheses), immunoprecipitated, and analyzed by immunoblotting. (F) Lysates from cells prepared as described above for panel E were analyzed by immunoblotting with antibodies to the indicated proteins. (G) Lysates from FLAG-ATM-reconstituted Rb−/−; ATM−/− MEFs transduced with vectors expressing the indicated proteins (see Fig. S8B in the supplemental material) were treated with 5 μM MG132 for 12 h, immunoprecipitated with antibodies specific to the indicated proteins, and analyzed by immunoblotting.
Subcellular fractionation and IF analysis of MG132-treated Rb−/−; ATM−/− MEFs indicated that endogenous DNMT1 is localized predominantly in the chromatin fraction in vector-transduced Rb−/−; ATM−/− MEFs, while introduction of ATM significantly reduced the abundance of DNMT1 in the chromatin fraction and increased it in the cytosolic fraction, and this was antagonized by the simultaneous introduction of pRB (Fig. 5C and D). In the same setting, pRB reduced the level of exogenous ATM in the chromatin fraction. These findings suggest that the level of insoluble DNMT1 (chromatin fraction) is affected by the status of ATM and pRB, and the localization of ATM to chromatin is dynamically controlled by pRB.
The ATM kinase domain interacts with the DNMT1 catalytic domain.
To further characterize the physical interaction between ATM and DNMT1, we generated various HA-tagged ATM fragments (see Fig. S8A in the supplemental material) and HA- or FLAG-tagged DNMT1 fragments (see Fig. S8B) and examined their interactions when introduced into Rb−/−; ATM−/− MEFs. We found that ATM fragments containing the kinase domain were able to bind to and trigger acetylation and degradation of endogenous DNMT1 when introduced into Rb−/−; ATM−/− MEFs (Fig. 5E). Consistent with this, these ATM fragments were able to activate downstream targets, including Chk2 and p53, when overexpressed (Fig. 5F). Furthermore, DNMT1 fragments containing the catalytic domain were able to bind to exogenous ATM when both were introduced into Rb−/−; ATM−/− MEFs (Fig. 5G). In summary, these findings indicate that the region in the ATM kinase domain (positions 2740 to 3056) and the region in the DNMT1 catalytic domain (positions 1504 to 1632) are necessary for ATM-DNMT1 binding.
The pRB-ATM-DNMT1 pathway regulates DNA methylation status.
We next surveyed for genes possibly regulated by the pRB-ATM-DNMT1 pathway. We performed microarray analysis of Rb−/−; ATM−/− MEFs to identify genes that simultaneously fit two criteria, i.e., (i) being repressed by endogenous DNMT1 and (ii) being upregulated by exogenous ATM, conceivably through DNMT1 inactivation. We identified 318 genes commonly upregulated in 2 combinations of experiments (cutoff point = 2.0×), namely, Rb−/−; ATM−/− MEFs depleted for DNMT1 versus the control and those reconstituted with ATM versus the control (Fig. 6A; see also Table S6 in the supplemental material). Of these genes, we focused on 3 cancer-related genes, Shc2, FoxO6, and Noggin. The frequency of methylated CpG nucleotides in the promoter regions of these genes was invariably high in Rb−/−; ATM−/− MEFs (Fig. 6B to G). Importantly, reintroduction of ATM into Rb−/−; ATM−/− MEFs markedly reduced the frequency of methylated CpGs in the promoter regions of these genes after 10 days of cultivation under normal conditions. Furthermore, additional introduction of pRB antagonized the ATM-induced suppression of CpG island methylation of the promoter regions of these genes (Fig. 6B to G; see also Fig. S9 in the supplemental material). Taken together, these findings suggest that in the Rb-negative context, the ATM-DNMT1 interaction regulates the DNA methylation status of gene promoters.
Fig 6.

Genome-wide survey for genes epigenetically regulated by the pRB-ATM-DNMT1 nexus. (A) Venn diagram of microarray analysis. Rb−/−; ATM−/− MEFs were transduced with vectors expressing the indicated proteins or infected with lentiviruses expressing the shRNAs that target the indicated genes, selected with 4 μg/ml puromycin or 8 μg/ml blasticidin S for 72 h, cultured for an additional 4 days under normal culture conditions, and analyzed by microarray analysis. Commonly upregulated genes in the 2 combinations of experiments are shown in red. A list of genes and their ontology are indicated in Table S6 in the supplemental material. (B and C) Results of bisulfite sequencing of the Shc2 promoter region in MEFs of the indicated genotypes transduced with vectors expressing the indicated proteins and their statistical analysis. (D and E) Results of bisulfite sequencing of the FoxO6 promoter region in MEFs of the indicated genotypes transduced with vectors expressing the indicated proteins and their statistical analysis. (F and G) Results of bisulfite sequencing of the Noggin promoter region in Rb−/−; ATM−/− MEFs transduced with vectors expressing the indicated proteins and their statistical analysis. A detailed statistical analysis of the effects of pRB and ATM on the frequency and location of methylated CpG (Me-CpG) nucleotides in the promoters of these three representative genes is shown in Fig. S9 in the supplemental material.
DISCUSSION
The current study revealed at least a part of the mechanism that links the DDR to cellular senescence in the Rb-deficient context and also a previously unexpected role of the genetic interaction between RB and ATM in controlling DNMT1 protein stability (Fig. 7). pRB has been linked to the ATM activation status via DSB formation (13). Our current study proposed additional mechanisms including control of Tip60-dependent ATM autophosphorylation and recruitment of ATM to chromatin. We further demonstrated that activated ATM physically binds to DNMT1 in a complex with Tip60 and UHRF1 and promotes the acetylation-driven ubiquitination of DNMT1. Recently, Du et al. reported a similar regulation of DNMT1 ubiquitination by Tip60 (35). We demonstrated here that Tip60 requires the presence of ATM to initiate DNMT1 acetylation and following ubiquitination. In addition, Tip60 is also known to acetylate ATMK3016 that further induces ATM autophosphorylation at multiple sites, including ATMS1981, which is required for DDR activation (35, 36) and DNMT1 degradation (observed in this study). Therefore, there is a possibility that Tip60 regulates both ATM and DNMT1 in the same complex (Fig. 7). Our data also indicate that the unavailability of pRB disables HDAC-mediated protection and promotes the Tip60-mediated inhibition of DNMT1 via ATM modifications (Fig. 7).
Fig 7.

Model for the functional interaction of ATM and DNMT1 in the absence of pRB. The molecular species participating in the DNMT1 protein stability regulation complex are dynamically altered depending on the status of pRB and ATM, and these changes actively modulate the DNA methylation status of target gene promoters. DKO, double knockout; AC, acetylation.
The current study indicated that the high level of DNMT1 in cells lacking both Rb and ATM is a rate-limiting step in maintaining the high level of DNA methylation in several cancer-related gene promoters, including Ink4a. A previous report, using a somatic knockout of DNMT1 in HCT116 cell clones, showed an insignificant difference in the INK4a promoter methylation status between DNMT1+/+ and DNMT1−/− cells (37). Previous work by the same group indicated that DNMT3B, which was unaffected by pRb or ATM in our Rb−/−; ATM−/− cells (Fig. 2G), is functionally redundant with DNMT1 in maintaining the methylation status of the INK4a promoter (38). Fournel et al. also demonstrated with another cell line that DNMT1 downregulation was sufficient to reduce the DNA methylation of the INK4a promoter (39). In addition, Robert et al. demonstrated that DNMT1 inhibition is a major mechanism by which 5-aza-2’-deoxycytidine (5-AzadC) reactivates the INK4a promoter (40). Taken together, these findings indicate that the role of DNMT1 can be rate limiting in the regulation of at least the Ink4a promoter methylation status and that this might depend on the cellular context and functional redundancy with other DNA methyltransferases.
DDR activation has been demonstrated to contribute to oncogene-induced senescence (OIS) (41); however, the mechanism linking the DDR to cellular senescence was not known. Most oncogenic signals merge upon the transcriptional upregulation of D-type cyclins, which, in cooperation with CDK4/6, inactivate pRB by initiating its phosphorylation. Thus, we suspect that pRB inactivation and the consequent ATM activation might be a common mechanism of various types of OIS.
Furthermore, our data indicated a possibility that both pRB and ATM are simultaneously inactivated with reciprocal upregulation of DNMT1 in half of sporadic human MTC cases. A previous study demonstrated simultaneous mutations of RB and ATM in lung adenocarcinomas (42). These observations suggest that the pRB-ATM-DNMT1 nexus might be frequently deregulated in human cancers. Even without mutations in RB and ATM, a variety of oncogenic signals can affect the activation statuses of both pRB and ATM. Furthermore, tumor cells typically exhibit a certain level of DSBs, as depicted, for instance, by positive γH2AX staining observed in their nucleus; this might lead to varied levels of ATM activation. Therefore, there is a possibility that a significant portion of cellular information is translated onto epigenetic modifications via the pRB-ATM-DNMT1 nexus. If so, this scenario may provide at least a part of the mechanism of the abnormal DNA methylation pattern prevalently found in human cancers. This expectation may then endow us with an interesting insight into the future rationale for epigenetic therapy of cancers. Finally, it is noteworthy that Rb deficiency in mouse embryos showed abnormal stem cell activity in many lineages (3, 10). There is a possibility that pRB controls the epigenetic status during developmental processes through the pRB-ATM-DNMT1 nexus.
Supplementary Material
ACKNOWLEDGMENTS
We thank J. McKinnon, A. Hirao, T. Jacks, N. Sharpless, D. Chen, E. Hara, and T. Ikura for providing materials; M. Asano and E. Kamimura for support in animal experiments; M. Ewen, D. Peeper, and M. Monden for encouragement; Y. Kido for technical and N. Nagatani for secretarial assistance; and all the colleagues in the Takahashi laboratory for valuable discussion.
This work was supported by the Funding Program for Next Generation World-Leading Researchers (NEXT) to C.T., a grant-in-aid for scientific research (MEXT) to C.T. and A.S., the Astellas Foundation for Research on Metabolic Disorders, the Takeda Science Foundation, the Naito Foundation, the Daiichi-Sankyo Foundation for Life Science, the NOVARTIS Foundation (Japan) for the Promotion of Science, and the Hokkoku Foundation for Cancer Research to C.T.
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 10 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/MCB.01597-12.
REFERENCES
- 1.Burkhart DL, Sage J. 2008. Cellular mechanisms of tumour suppression by the retinoblastoma gene. Nat. Rev. Cancer 8:671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Viatour P, Sage J. 2011. Newly identified aspects of tumor suppression by RB. Dis. Model. Mech. 4:581–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Takahashi C, Sasaki N, Kitajima S. 2012. Twists in views on RB functions in cellular signaling, metabolism and stem cells. Cancer Sci. 103:1182–1188 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robertson KD, Ait-Si-Ali S, Yokochi T, Wade PA, Jones PL, Wolffe AP. 2000. DNMT1 forms a complex with Rb, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nat. Genet. 25:338–342 [DOI] [PubMed] [Google Scholar]
- 5.Dick FA. 2007. Structure-function analysis of the retinoblastoma tumor suppressor protein—is the whole a sum of its parts? Cell Div. 2:26. 10.1186/1747-1028-2-26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lin W, Cao J, Liu J, Beshiri ML, Fujiwara Y, Francis J, Cherniack AD, Geisen C, Blair LP, Zou MR, Shen X, Kawamori D, Liu Z, Grisanzio C, Watanabe H, Minamishima YA, Zhang Q, Kulkarni RN, Signoretti S, Rodig SJ, Bronson RT, Orkin SH, Tuck DP, Benevolenskaya EV, Meyerson M, Kaelin WG, Jr, Yan Q. 2011. Loss of the retinoblastoma binding protein 2 (RBP2) histone demethylase suppresses tumorigenesis in mice lacking Rb1 or Men1. Proc. Natl. Acad. Sci. U. S. A. 108:13379–13386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lopez-Bigas N, Kisiel AT, DeWaal DC, Holmes KB, Volkert TL, Love J, Murray HL, Young RA, Benevolenskaya EV. 2008. Genome-wide analysis of the H3K4 histone demethylase RBP2 reveals a transcriptional program controlling differentiation. Mol. Cell 31:520–530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li F, He Z, Shen J, Huang Q, Li W, Lui X, He Y, Wolf F, Li CY. 2010. Apoptotic caspases regulate induction of iPSCs from human fibroblasts. Cell Stem Cell 7:508–520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Liu Y, Clem B, Zuba-Surma EK, El-Naggar S, Telang S, Jenson AB, Wang Y, Shao H, Ratajczak MZ, Chesney J, Dean DC. 2009. Mouse fibroblasts lacking RB1 function form spheres and undergo reprogramming to a cancer stem cell phenotype. Cell Stem Cell 4:336–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sage J. 2012. The retinoblastoma tumor suppressor and stem cell biology. Genes Dev. 26:1409–1420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang J, Benavente CA, McEvoy J, Flores-Otero J, Ding L, Chen X, Ulyanov A, Wu G, Wilson M, Wang J, Brennan R, Rusch M, Manning AL, Ma J, Easton J, Shurtleff S, Mullighan C, Pounds S, Mukatira S, Gupta P, Neale G, Zhao D, Lu C, Fulton RS, Fulton LL, Hong X, Dooling DJ, Ochoa K, Naeve C, Dyson NJ, Mardis ER, Bahrami A, Ellison D, Wilson RK, Downing JR, Dyer MA. 2012. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature 481:329–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duensing S, Munger K. 2002. The human papillomavirus type 16 E6 and E7 oncoproteins independently induce numerical and structural chromosome instability. Cancer Res. 62:7075–7082 [PubMed] [Google Scholar]
- 13.Pickering MT, Kowalik TF. 2006. Rb inactivation leads to E2F1-mediated DNA double-strand break accumulation. Oncogene 25:746–755 [DOI] [PubMed] [Google Scholar]
- 14.Frame FM, Rogoff HA, Pickering MT, Cress WD, Kowalik TF. 2006. E2F1 induces MRN foci formation and a cell cycle checkpoint response in human fibroblasts. Oncogene 25:3258–3266 [DOI] [PubMed] [Google Scholar]
- 15.Shamma A, Takegami Y, Miki T, Kitajima S, Noda M, Obara T, Okamoto T, Takahashi C. 2009. Rb regulates DNA damage response and cellular senescence through E2F-dependent suppression of N-ras isoprenylation. Cancer Cell 15:255–269 [DOI] [PubMed] [Google Scholar]
- 16.van Harn T, Foijer F, van Vugt M, Banerjee R, Yang F, Oostra A, Joenje H, te Riele H. 2010. Loss of Rb proteins causes genomic instability in the absence of mitogenic signaling. Genes Dev. 24:1377–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sage J, Straight AF. 2010. RB's original CIN? Genes Dev. 24:1329–1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Polo SE, Jackson SP. 2011. Dynamics of DNA damage response proteins at DNA breaks: a focus on protein modifications. Genes Dev. 25:409–433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bensimon A, Aebersold R, Shiloh Y. 2011. Beyond ATM: the protein kinase landscape of the DNA damage response. FEBS Lett. 585:1625–1639 [DOI] [PubMed] [Google Scholar]
- 20.Lukas J, Bartek J. 2009. DNA repair: new tales of an old tail. Nature 458:581–583 [DOI] [PubMed] [Google Scholar]
- 21.Takahashi C, Bronson RT, Socolovsky M, Contreras B, Lee KY, Jacks T, Noda M, Kucherlapati R, Ewen ME. 2003. Rb and N-ras function together to control differentiation in the mouse. Mol. Cell. Biol. 23:5256–5268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takahashi C, Contreras B, Bronson RT, Loda M, Ewen ME. 2004. Genetic interaction between Rb and K-ras in the control of differentiation and tumor suppression. Mol. Cell. Biol. 24:10406–10415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Takahashi C, Contreras B, Iwanaga T, Takegami Y, Bakker A, Bronson RT, Noda M, Loda M, Hunt JL, Ewen ME. 2006. Nras loss induces metastatic conversion of Rb1-deficient neuroendocrine thyroid tumor. Nat. Genet. 38:118–123 [DOI] [PubMed] [Google Scholar]
- 24.Sharpless NE, Bardeesy N, Lee KH, Carrasco D, Castrillon DH, Aguirre AJ, Wu EA, Horner JW, DePinho RA. 2001. Loss of p16Ink4a with retention of p19Arf predisposes mice to tumorigenesis. Nature 413:86–91 [DOI] [PubMed] [Google Scholar]
- 25.Herzog KH, Chong MJ, Kapsetaki M, Morgan JI, McKinnon PJ. 1998. Requirement for Atm in ionizing radiation-induced cell death in the developing central nervous system. Science 280:1089–1091 [DOI] [PubMed] [Google Scholar]
- 26.So S, Davis AJ, Chen DJ. 2009. Autophosphorylation at serine 1981 stabilizes ATM at DNA damage sites. J. Cell Biol. 187:977–990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ikura T, Ogryzko VV, Grigoriev M, Groisman R, Wang J, Horikoshi M, Scully R, Qin J, Nakatani Y. 2000. Involvement of the TIP60 histone acetylase complex in DNA repair and apoptosis. Cell 102:463–473 [DOI] [PubMed] [Google Scholar]
- 28.Lim KL, Chew KC, Tan JM, Wang C, Chung KK, Zhang Y, Tanaka Y, Smith W, Engelender S, Ross CA, Dawson VL, Dawson TM. 2005. Parkin mediates nonclassical, proteasomal-independent ubiquitination of synphilin-1: implications for Lewy body formation. J. Neurosci. 25:2002–2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kobayashi M, Ono H, Mihara K, Tauchi H, Komatsu K, Shibata T, Shimizu H, Uchida K, Yamamoto K. 2006. ATM activation by a sulfhydryl-reactive inflammatory cyclopentenone prostaglandin. Genes Cells 11:779–789 [DOI] [PubMed] [Google Scholar]
- 30.Abe K, Naruse C, Kato T, Nishiuchi T, Saitou M, Asano M. 2011. Loss of heterochromatin protein 1 gamma reduces the number of primordial germ cells via impaired cell cycle progression in mice. Biol. Reprod. 85:1013–1024 [DOI] [PubMed] [Google Scholar]
- 31.Patel AC, Anna CH, Foley JF, Stockton PS, Tyson FL, Barrett JC, Devereux TR. 2000. Hypermethylation of the p16 (Ink4a) promoter in B6C3F1 mouse primary lung adenocarcinomas and mouse lung cell lines. Carcinogenesis 21:1691–1700 [DOI] [PubMed] [Google Scholar]
- 32.McCabe MT, Davis JN, Day ML. 2005. Regulation of DNA methyltransferase 1 by the pRb/E2F1 pathway. Cancer Res. 65:3624–3632 [DOI] [PubMed] [Google Scholar]
- 33.Qin W, Leonhardt H, Pichler G. 2011. Regulation of DNA methyltransferase 1 by interactions and modifications. Nucleus 2:392–402 [DOI] [PubMed] [Google Scholar]
- 34.Bronner C. 2011. Control of DNMT1 abundance in epigenetic inheritance by acetylation, ubiquitylation, and the histone code. Sci. Signal. 4:pe3. 10.1126/scisignal.2001764 [DOI] [PubMed] [Google Scholar]
- 35.Du Z, Song J, Wang Y, Zhao Y, Guda K, Yang S, Kao HY, Xu Y, Willis J, Markowitz SD, Sedwick D, Ewing RM, Wang Z. 2010. DNMT1 stability is regulated by proteins coordinating deubiquitination and acetylation-driven ubiquitination. Sci. Signal. 3:ra80. 10.1126/scisignal.2001462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun Y, Jiang X, Price BD. 2010. Tip60: connecting chromatin to DNA damage signaling. Cell Cycle 9:930–936 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rhee I, Jair KW, Yen RW, Lengauer C, Herman JG, Kinzler KW, Vogelstein B, Baylin SB, Schuebel KE. 2000. CpG methylation is maintained in human cancer cells lacking DNMT1. Nature 404:1003–1007 [DOI] [PubMed] [Google Scholar]
- 38.Rhee I, Bachman KE, Park BH, Jair KW, Yen RW, Schuebel KE, Cui H, Feinberg AP, Lengauer C, Kinzler KW, Baylin SB, Vogelstein B. 2002. DNMT1 and DNMT3b cooperate to silence genes in human cancer cells. Nature 416:552–556 [DOI] [PubMed] [Google Scholar]
- 39.Fournel M, Sapieha P, Beaulieu N, Besterman JM, MacLeod AR. 1999. Down-regulation of human DNA-(cytosine-5) methyltransferase induces cell cycle regulators p16ink4A and p21WAF/Cip1 by distinct mechanisms. J. Biol. Chem. 274:24250–24256 [DOI] [PubMed] [Google Scholar]
- 40.Robert MF, Morin S, Beaulieu N, Gauthier F, Chute IC, Barsalou A, MacLeod AR. 2003. DNMT1 is required to maintain CpG methylation and aberrant gene silencing in human cancer cells. Nat. Genet. 33:61–65 [DOI] [PubMed] [Google Scholar]
- 41.Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre' M, Nuciforo PG, Bensimon A, Maestro R, Pelicci PG, d'Adda di Fagagna F. 2006. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 444:638–642 [DOI] [PubMed] [Google Scholar]
- 42.Ding L, Getz G, Wheeler DA, Mardis ER, McLellan MD, Cibulskis K, Sougnez C, Greulich H, Muzny DM, Morgan MB, Fulton L, Fulton RS, Zhang Q, Wendl MC, Lawrence MS, Larson DE, Chen K, Dooling DJ, Sabo A, Hawes AC, Shen H, Jhangiani SN, Lewis LR, Hall O, Zhu Y, Mathew T, Ren Y, Yao J, Scherer SE, Clerc K, Metcalf GA, Ng B, Milosavljevic A, Gonzalez-Garay ML, Osborne JR, Meyer R, Shi X, Tang Y, Koboldt DC, Lin L, Abbott R, Miner TL, Pohl C, Fewell G, Haipek C, Schmidt H, Dunford-Shore BH, Kraja A, Crosby SD, Sawyer CS, et al. 2008. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 455:1069–1075 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.