

Trm5 enzymes catalyze methyl transfer from S-adenosyl methionine (AdoMet) to the N1(G37) critical modification, preventing ribosome frameshift errors. Trm5 is specific to eukaryotes and archaea, but most information on Trm5 derives from the archaeal enzyme. It is shown that the human Trm5 has retained key features of catalytic properties of the archaeal enzyme. It is further shown that a single substitution in the human enzyme improves the resistance to proteolysis.
Keywords: burst kinetics, S-adenosyl methionine, m1G37-tRNA, pH-activity profile, structure-guided mutagenesis
Abstract
Enzymes of the Trm5 family catalyze methyl transfer from S-adenosyl methionine (AdoMet) to the N1 of G37 to synthesize m1G37-tRNA as a critical determinant to prevent ribosome frameshift errors. Trm5 is specific to eukaryotes and archaea, and it is unrelated in evolution from the bacterial counterpart TrmD, which is a leading anti-bacterial target. The successful targeting of TrmD requires detailed information on Trm5 to avoid cross-species inhibition. However, most information on Trm5 is derived from studies of the archaeal enzyme Methanococcus jannaschii (MjTrm5), whereas little information is available for eukaryotic enzymes. Here we use human Trm5 (Homo sapiens; HsTrm5) as an example of eukaryotic enzymes and demonstrate that it has retained key features of catalytic properties of the archaeal MjTrm5, including the involvement of a general base to mediate one proton transfer. We also address the protease sensitivity of the human enzyme upon expression in bacteria. Using the tRNA-bound crystal structure of the archaeal enzyme as a model, we have identified a single substitution in the human enzyme that improves resistance to proteolysis. These results establish conservation in both the catalytic mechanism and overall structure of Trm5 between evolutionarily distant eukaryotic and archaeal species and validate the crystal structure of the archaeal enzyme as a useful model for studies of the human enzyme.
INTRODUCTION
The extensive base and backbone modifications present in natural tRNAs serve to promote translational fidelity and efficiency (Bjork et al. 1999; Motorin and Helm 2010; Yi and Pan 2011). Of the more than 100 modifications identified to date, the m1G37 base modification is strictly conserved in evolution for tRNAs specific for leucine (CUN codons, N being one of the four natural nucleotides), proline (CCN codons), and one of the arginine isoacceptors (the CGG codon) (Bjork et al. 2001). This modification is also present in mitochondria and chloroplasts and even in Mycoplasma spp., the latter of which has one of the smallest sequenced genomes known to date. The m1G37 modification replaces the imino proton of N1 of guanosine with a methyl group, thus preventing base-pairing on the 3′ side of the anticodon in +1 frameshifts (Fig. 1A,B). Indeed, m1G37 is essential for cell viability; its elimination leads to accumulation of frameshifts and delayed entry of charged tRNA to the ribosome (Bjork et al. 1989; Hagervall et al. 1993; Li and Bjork 1995). In yeast, elimination of m1G37 also promotes misacylation of tRNAAsp with arginine (Putz et al. 1994). For eukaryotic tRNAPhe specifically, m1G37 is the substrate for conversion to the hypermodified wybutosine via four successive reactions (Noma et al. 2006). In archaea, m1G37 is important for synthesis of cysteinyl-tRNACys required as a substrate for decoding cysteine codons (Hauenstein et al. 2008; Zhang et al. 2008).
FIGURE 1.

Synthesis of m1G37-tRNA. (A) Trm5 catalyzes the conversion of guanosine to 1-methyl-guanosine (m1G37) in tRNA, using AdoMet as the methyl donor. (B) The sequence and cloverleaf secondary structure of human tRNACys, which was used as the substrate for HsTrm5 in this study. The target G37 is circled.
While m1G37 is conserved in tRNAs, the enzymes that catalyze its synthesis are unrelated in evolution. Eukaryotes and archaea employ the Trm5 enzyme (Bjork et al. 2001; Christian et al. 2004), whereas bacteria use the TrmD enzyme (Bystrom and Bjork 1982a,b). While both enzymes use AdoMet as the methyl donor to convert G37-tRNA to m1G37-tRNA, they are fundamentally distinct in structure (Ahn et al. 2003; Christian et al. 2004; Goto-Ito et al. 2008, 2009), in kinetics (Christian et al. 2010b), and in substrate recognition (Christian and Hou 2007; Lahoud et al. 2011; Sakaguchi et al. 2012). The lack of similarity between TrmD and Trm5 has led to the suggestion that specific targeting of TrmD, without affecting the Homo sapiens Trm5 (HsTrm5), would be a highly attractive strategy in developing the next generation of antibiotics (White and Kell 2004).
The potential of TrmD as a leading anti-microbial target emphasizes the need to better understand HsTrm5 for two reasons. First, when an inhibitor is identified for TrmD, it must be critically evaluated against HsTrm5 to eliminate cross-inhibition in order to ensure species-specific targeting. However, key aspects of the evaluation remain completely void for HsTrm5, including kinetic parameters of methyl transfer necessary for analysis of cross-inhibition and a structural model of the enzyme for modeling and identification of potential cross-reacting chemical groups of inhibitors. This lack of information is hindering the development of inhibitors into drugs. Second, given the importance of Trm5 in maintaining tRNA translational fidelity, it is essential that fundamental knowledge of the enzyme in human cells be made readily available. However, in the only report on HsTrm5 (Brule et al. 2004), the enzyme was studied using nonhuman tRNA substrates. In general, eukaryotic Trm5 enzymes are poorly understood. The only other eukaryotic Trm5 that has been studied is the enzyme of Saccharomyces cerevisiae (Lee et al. 2007), which was reported to recognize mitochondrial initiator tRNAMet.
The major question in the study of HsTrm5, and more broadly eukaryotic enzymes, is the relationship with their archaeal counterparts. While eukaryotic and archaeal Trm5 enzymes are grouped as one family, the correlation between the two divisions in sequence, structure, and catalytic mechanism is not known. At present, the archaeal MjTrm5 (Methanococcus jannaschii) is the best-characterized enzyme, having an extensive set of kinetic data (Christian et al. 2010b) and well-defined crystal structures, including a binary structure with sinefungin (an inactive AdoMet analog) and a ternary structure with AdoMet and tRNA (Goto-Ito et al. 2008, 2009). However, whether MjTrm5 is a valid model to study HsTrm5 or other eukaryotic enzymes remains an open question and cannot be predicted based on sequence alone. Indeed, a sequence alignment by T-coffee (Notredame et al. 2000) revealed only moderate homology between HsTrm5 and MjTrm5, showing 29% identity and 53% similarity, in contrast to 80% identity and 95% similarity among TrmDs. Also, there exist species-specific differences between archaeal and eukaryotic enzymes (Christian et al. 2010a), whose meaning is unknown. Furthermore, purification yields of HsTrm5 in bacteria have been noted as low (Brule et al. 2004), in contrast to high yields of MjTrm5 (Christian et al. 2004), indicating at least in part the possibility of protease sensitivity of the human enzyme relative to the archaeal enzyme.
Here we address the correlation between HsTrm5 and MjTrm5 in two parts, with the first using a kinetic approach to evaluate the conservation of catalytic properties of the two enzymes and the second using the tRNA-bound structure of the archaeal enzyme as a model to evaluate the overall similarity with the human enzyme. We show that, despite their modest sequence homology, the two enzymes are fundamentally similar in both the catalytic mechanism and overall structure, suggesting that the available structures of MjTrm5 can provide a useful model to study HsTrm5. This study provides direct information for HsTrm5 and a framework necessary and most relevant to human health in the anti-microbial targeting of TrmD.
RESULTS AND DISCUSSION
Protease sensitivity of HsTrm5 in Escherichia coli
HsTrm5 was expressed in Escherichia coli with a C-terminal His tag, based on the Kazusa sequence (Nagase et al. 2000). The Kazusa sequence differs from the annotated genomic sequence by lacking the N-terminal peptide V2LWILWRP9 of the genomic sequence, and by containing a K394 residue instead of glutamate (numbering based on the Kazusa sequence) (Supplemental Fig. S1) at a nonconserved position. The lack of the N-terminal peptide was to facilitate cloning, while the E394K substitution was in the Kazusa sequence and not an artifact. While the expression level of the recombinant HsTrm5 was high upon induction (MW: ∼60 kDa), only a small fraction was soluble, and the purity after elution from a metal resin was poor (15%–20%) (Supplemental Fig. S2A), consistent with a previous report (Brule et al. 2004). Further purification of the affinity-purified enzyme on monoS, while reaching 90% homogeneity, resulted in significant losses of protein quantity. Because the affinity-purified enzyme was as active as the monoS-purified enzyme on the transcript of human tRNACys as a substrate (Fig. 1B; Supplemental Fig. S2B), we used the affinity-purified enzyme throughout this work.
Among the impurities in the affinity-purified enzyme, a 27-kDa protein was most persistent, which was also observed upon expression of MjTrm5 in E. coli but to a lesser extent (Christian and Hou 2007). An LC/MS/MS analysis of the smaller protein showed that it was a degradation product, representing the C-terminal half of the protein. Detailed MS/MS analysis showed that the cleavage sites occurred at the N terminus to M252, V254, M261, and T263 (Supplemental Fig. S3). The persistent appearance of the 27-kDa protein suggested that the soluble fraction of HsTrm5 when expressed in E. coli was sensitive to proteolysis, which was one of the reasons for the poor yield of the enzyme relative to MjTrm5.
Kinetic analysis of HsTrm5
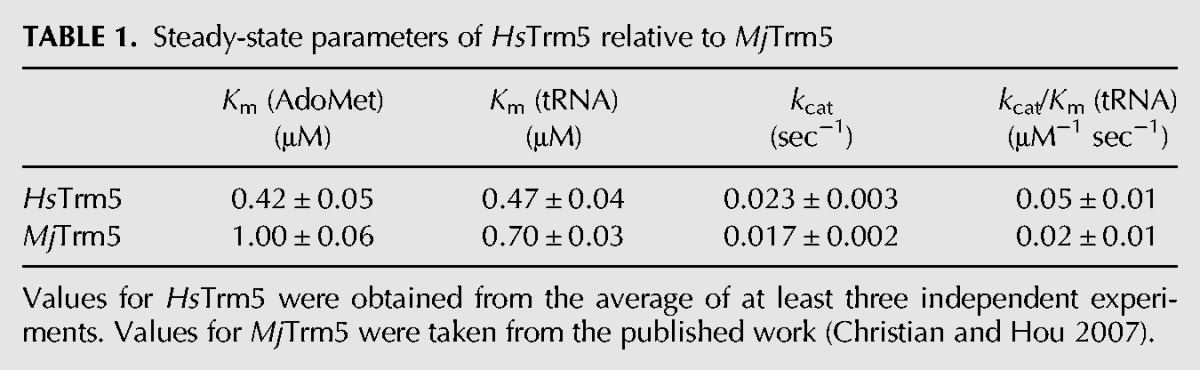
The affinity-purified HsTrm5 was examined for its steady-state activity on the transcript of human tRNACys, similar to the previous analysis of MjTrm5 with a transcript of the archaeal tRNACys (Christian et al. 2004, 2010b; Sakaguchi et al. 2012). The transcript of human tRNACys had a capacity of methylation to ∼70% levels in extended time. The methylation reaction monitored methyl transfer from [3H-methyl]-AdoMet to G37-tRNA, producing 3H-m1G37-tRNA and releasing S-adenosyl homocysteine (AdoHcys). Steady-state activity as a function of AdoMet concentration with saturating tRNA revealed Km (AdoMet) of 0.42 ± 0.08 μM and kcat of 0.023 ± 0.003 sec−1, closely similar to values of MjTrm5 [Km (AdoMet) of 1.0 ± 0.1 μM and kcat of 0.017 ± 0.002 sec−1] (Table 1; Christian et al. 2006). Separately, steady-state activity as a function of tRNA in the presence of saturating AdoMet revealed Km (tRNA) of 0.47 ± 0.04 μM and kcat/Km (tRNA) of 0.05 ± 0.01 μM−1 sec−1, also closely similar to values for MjTrm5 [Km (tRNA) of 0.70 ± 0.05 μM and kcat/Km (tRNA) of 0.024 ± 0.003 μM−1 sec−1) (Table 1; Christian et al. 2006).
TABLE 1.
Steady-state parameters of HsTrm5 relative to MjTrm5
Pre-steady-state assays were also performed, with the enzyme in molar excess of AdoMet and with tRNA saturating to permit only one methyl transfer on the enzyme. The rate constant of each time course represented a composite term for all reaction steps from enzyme-substrates binding and up to and including the formation of m1G37-tRNA (Johnson 1998). All time courses were well fit to a single exponential equation (Supplemental Fig. S4A), indicating rapid equilibrium binding. Control experiments confirmed that the mixing order did not affect the rate of methyl transfer. Fitting the data of kobs as a function of enzyme concentration to a hyperbola (Supplemental Fig. S4B) revealed the dissociation constant Kd (AdoMet) (1.1 ± 0.1 μM), which is closely similar to the value of MjTrm5 (Christian et al. 2010b). Similarly, fitting the data of kobs as a function of enzyme concentration in excess of tRNA and with saturating AdoMet revealed Kd (tRNA) of 1.8 ± 0.1 μM (Supplemental Fig. S4C,D), also closely similar to the Kd (tRNA) of 1.4 ± 0.1 μM of MjTrm5 (Table 2; Christian et al. 2010b). The maximum rate constant kchem (0.09 ± 0.01 sec−1), which included both the pre-chemistry isomerization of the enzyme and the chemistry of methyl transfer, was again similar to that of MjTrm5 (Table 2).
TABLE 2.
Pre-steady-state parameters of HsTrm5 relative to MjTrm5
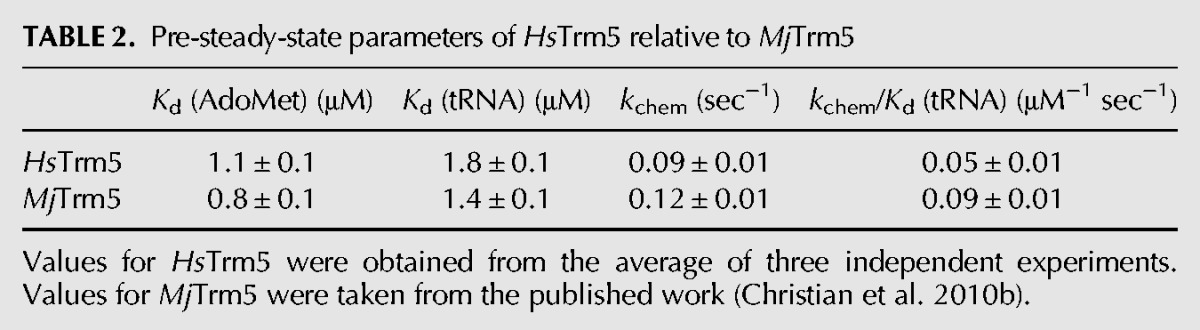
The Kd values of pre-steady-state assays were interpreted as the equilibrium dissociation constant, due to the rapid equilibrium binding conditions (Johnson 1998). This interpretation has been confirmed for MjTrm5, using the intrinsic enzyme tryptophan fluorescence as a probe for equilibrium binding of tRNA in the presence of saturating sinefungin (Sakaguchi et al. 2012). The addition of tRNA to the enzyme resulted in quenching of the intrinsic fluorescence and the measurement of the quench as a function of tRNA revealed a Kd (tRNA) of 1.3 ± 0.6 μM, closely similar to the kinetic Kd (tRNA) determined for MjTrm5 (1.4 ± 0.1 μM) and for HsTrm5 (1.8 ± 0.1 μM). Based on the kinetic Kd of each enzyme as the equilibrium binding constant, the catalytic efficiency of methyl kchem/Kd (tRNA) was closely similar for the two enzymes (0.05 ± 0.01 μM−1 sec−1 for HsTrm5 and 0.09 ± 0.01 μM−1 sec−1 for MjTrm5).
The rate-determining step
MjTrm5 previously showed a rapid burst of product synthesis in a pre-steady-state assay, followed by a slower and linear synthesis over time (Christian et al. 2010b). The burst suggested that product synthesis in the first turnover was fast, but that product release from the enzyme was slow, thus limiting the rate of subsequent turnovers. In contrast, TrmD showed a linear synthesis of product over time, suggesting that the slow step occurred before or during methyl transfer (Christian et al. 2010b). Similar to MjTrm5, HsTrm5 also showed a rapid burst of product synthesis followed by a steady-state rate (Fig. 2). Fitting the burst data yields kchem of 0.13 ± 0.02 sec−1 and kcat of 0.020 ± 0.002 sec−1, similar to values determined by separate steady-state and pre-steady-state analysis (Table 1). Extrapolation of the burst plot to the ordinate yielded an estimate for the active enzyme fraction at 15%–20%, which corresponds closely to the purity of the affinity-isolated enzyme. This active-site fraction was used to correct the enzyme concentration as was done for MjTrm5 (Christian et al. 2010b).
FIGURE 2.

A rapid burst of product synthesis catalyzed by HsTrm5. Time courses of m1G37-tRNA synthesis under pre-steady-state conditions, showing a rapid burst of product synthesis in the first enzyme turnover, followed by a slower steady-state rate. Error bars represent standard errors of three independent experiments. The rate constant of the first enzyme turnover is shown as kchem, while that of subsequence enzyme turnovers is shown as kcat.
The pH-activity profile
The high pKa (∼9.5) of the N1 imino proton of G37 suggested that enzyme-catalyzed deprotonation would be necessary for methyl transfer. Indeed the pH-activity profile of MjTrm5 showed that catalysis was dependent on one proton transfer (Christian et al. 2010a), most likely the transfer of the N1 proton of G37 to a general base on the enzyme. A pH-activity profile of HsTrm5 also revealed a steep increase of kobs as the proton concentration was lowered, up to an asymptote at pH 8.0 (Fig. 3A). We measured kobs at each pH, rather than Kd and kchem, due to the inability to produce large quantities of HsTrm5. The logarithmic plot of kobs (log[kobs]) versus pH revealed a slope of 0.5 (Fig. 3B), most consistent with one proton transfer upon deprotonation of the N1 of G37. The value of 0.5 suggests the possibility that the kobs of the human enzyme might be a function of both kchem and Kd (tRNA) and that only one of the two (probably kchem) exhibited a dependence on pH. This is a notable difference from MjTrm5, which displayed a value close to 1.0 (Christian et al. 2010a). For the human enzyme, fitting the data of kobs as a function of pH to a one-proton transfer equation revealed a pKa of 7.1 ± 0.1 (Fig. 3A), higher by 0.6 units than the pKa of MjTrm5 (6.5 ± 0.2) (Christian et al. 2010a).
FIGURE 3.

A pH-activity profile of HsTrm5. (A) A plot of the observed rate constant kobs against pH. (B) A logarithmic plot of kobs as a function of pH from 6.0 to 9.5. Error bars represent standard errors of at least three independent experiments.
Kinetic isotope effect
While the pH-activity analysis demonstrated the importance of one-proton transfer for catalysis, the structure of MjTrm5 did not identify an appropriately positioned general base for proton abstraction of the N1 of G37. Instead, the general base for MjTrm5 was proposed to be E185, which is strictly conserved as glutamate or aspartate but is located >5 Å away from the N1 of G37 (Christian et al. 2010a). The location of E185 far away from the N1 of G37 suggested that proton abstraction had occurred before the active-site structure was captured in the crystal, most likely during the induced-fit rearrangement of the enzyme with G37 of tRNA. In this model, the kobs represented the induced-fit process rather than the chemical step of proton abstraction and thus would not be sensitive to the isotope effect at the N1 of G37. This hypothesis was confirmed for both MjTrm5 and HsTrm5 by evaluating the kinetic isotope effect of the methyl transfer reaction, where the N1 proton of G37 had been readily exchanged to deuterium in the presence of deuterium water (McConnell et al. 1983; Snoussi and Leroy 2001). Pre-steady-state kinetic assays showed that kobs for MjTrm5 (0.11 sec−1) and kobs for HsTrm5 (0.012 sec−1) remained unaffected in deuterium water relative to hydrogen water (Fig. 4A,B), indicating that the chemical step of proton abstraction was not the rate-determining step for these enzymes. The lack of an effect for both enzymes supports the notion that they share a common kinetic mechanism involving proton abstraction during a slower and rate-determining process of induced fit. This induced-fit and proton abstraction would require the action of the general base and a conserved arginine side chain to facilitate the rearrangement of electrons (Supplemental Fig. S5).
FIGURE 4.

Analysis of kinetic isotope effect. (A) Kinetics of m1G37-tRNA synthesis catalyzed by MjTrm5, showing a virtually identical kobs of 0.11 sec−1 in H2O and in D2O, with standard errors of 0.02. (B) Kinetics of m1G37-tRNA synthesis catalyzed by HsTrm5, showing a virtually identical kobs of 0.012 sec−1 with standard errors of 0.001. Data for H2O are shown in closed triangles; data for D2O are shown in closed circles.
Evaluation of the active-site structure
The conservation of kinetic features between HsTrm5 and MjTrm5 suggests the conservation of the active-site structure. We tested this hypothesis by evaluating mutations in HsTrm5 (Table 3), based on the ternary structure of MjTrm5 (Goto-Ito et al. 2009). Due to the limitation in enzyme quantities, we again performed assays for HsTrm5 variants at a single enzyme concentration in excess to tRNA in the presence of saturating AdoMet. To verify the proposed mechanism, we showed that the E288A mutation of HsTrm5, equivalent to the E185A mutation of MjTrm5 at the general base position, reduced kobs by ∼330-fold, similar to the reduction observed for MjTrm5 (Christian et al. 2010a). Also, a highly conserved aspartate residue (D172 in MjTrm5 and D275 in HsTrm5) in the flexible loop preceding the proposed general base (Fig. 5A,B) may be involved in the induced-fit movement of the catalytic process. We showed that the D275A mutation in HsTrm5 decreased kobs by 33-fold in a magnitude similar to the 24-fold decrease by the D172A mutation of the archaeal enzyme, supporting the notion that both enzymes engage this aspartate likely in the movement of the general base to the proximity of the N1 of G37.
TABLE 3.
Pre-steady-state parameters with respect to tRNA for wild-type (WT) and mutants of HsTrm5
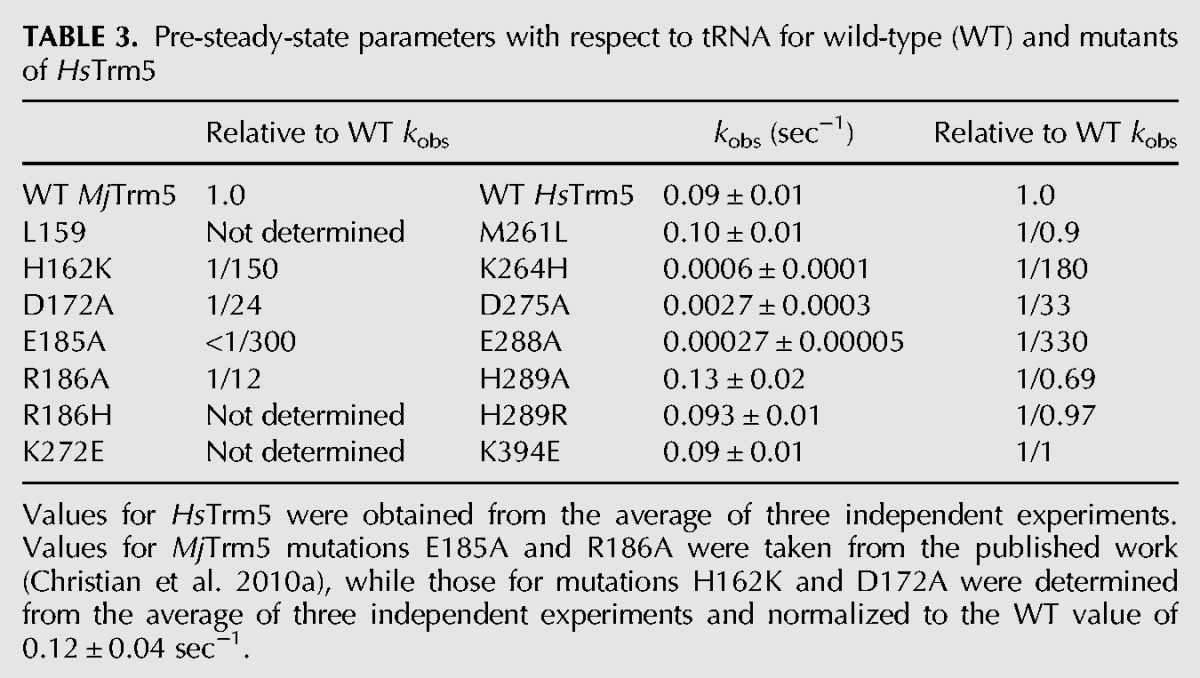
FIGURE 5.
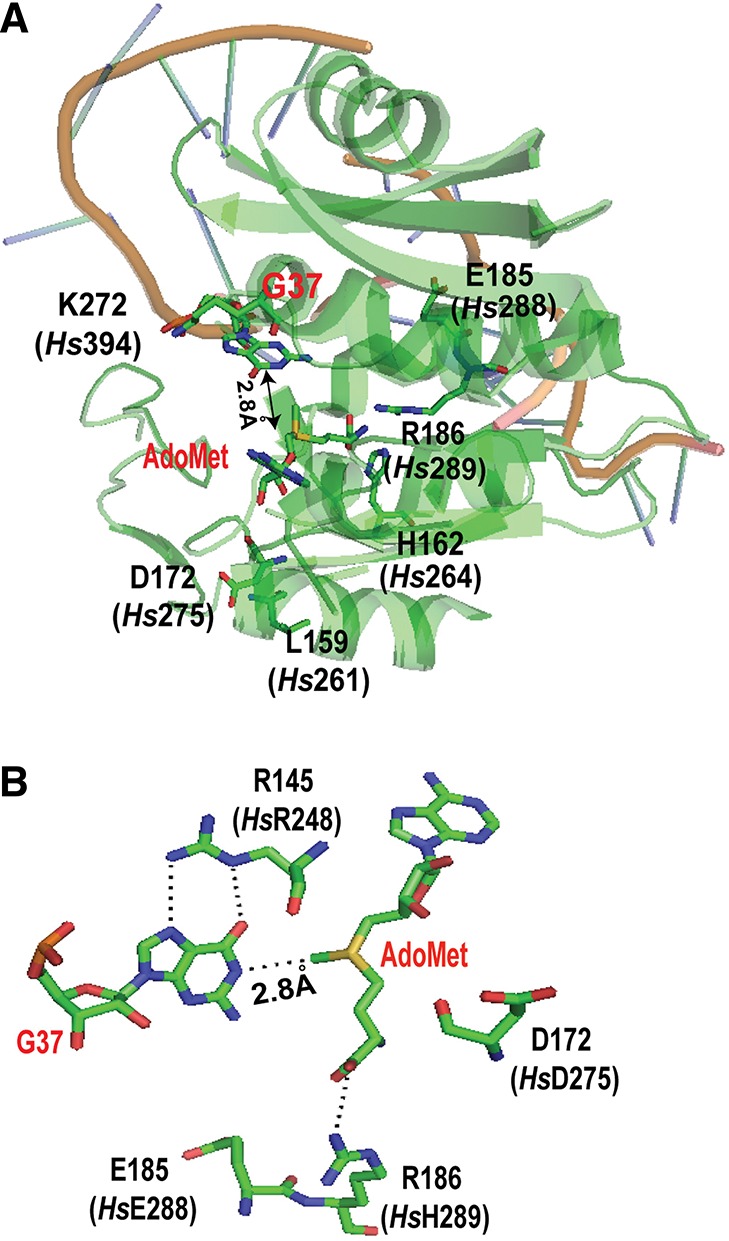
A structure-guided mutational analysis of HsTrm5. (A) An overall view of the active site in the crystal structure of MjTrm5 in complex with tRNA and AdoMet (PDB 2ZZN). All of the residues that have been tested in MjTrm5 and the corresponding residues in HsTrm5 (shown in parentheses) are highlighted. The G37 in the tRNA is flipped out from stacking with the anticodon nucleotides, and its N1 is directly opposite from the methyl group of the bound AdoMet by 2.8 Å. (B) A close-up view of the stereochemistry of the active site in MjTrm5, showing the enzyme stabilization of the O6-carbonyl of G37 by R145 (HsR248) and stabilization of the carboxyl group of AdoMet by R186 (HsH289), allowing the placement of the methyl group directly opposite from the N1-proton of G37 by 2.8 Å. The catalytically important E185 (HsE288), which likely acts as a general base, and the highly conserved D172 (HsD275), which may participate in the movement of the general base, are both shown.
Within the conserved active-site structure of MjTrm5 and HsTrm5, we tested archaea- and eukaryote-specific residues. Of interest was the residue immediately C-terminal to the proposed general base, where the R186A mutation in the archaeal enzyme decreased kobs by 12-fold, whereas the corresponding H289A mutation in the human enzyme had no effect. The lack of an effect in HsTrm5 was further confirmed by the H289R mutation, representing a switch from the eukaryote-specific histidine to the archaea-specific arginine. Thus, while this position in the archaeal enzyme appears important, stabilizing AdoMet binding with a charged hydrogen bond to the carboxylate of the methyl donor (Fig. 5B), it is not important for HsTrm5. However, at other positions where archaeal versus eukaryotic sequences differed, we found that sequence differences do not lead to a different effect on catalysis. For example, at a position adjacent to the amino group of AdoMet (Fig. 5A), while H162 in MjTrm5 is largely conserved as histidine (∼75%) or tyrosine (∼25%) among archaeal enzymes (Christian et al. 2010a), the corresponding K264 of HsTrm5 has a wider diversity (e.g., lysine, serine, glutamate, valine, and isoleucine). However, we found that the H162K mutation in the archaeal enzyme, which introduced the human lysine, and the reciprocal K264H mutation of the human enzyme, decreased kobs similarly by 150-fold to 180-fold, supporting the notion that the active site of the two enzymes is generally well conserved. Also, we showed that the K394E substitution in HsTrm5, which reproduced the genomic sequence, had no effect on kobs, indicating that this substitution is not deleterious for catalysis and that the enzyme produced by the Kazusa sequence is essentially identical to that encoded by the genomic sequence.
Evaluation of the overall structure
The conservation of catalytic features between HsTrm5 and MjTrm5, however, does not mean the conservation of their overall structure. To probe the similarity of their overall structure, we addressed the challenge of the protease sensitivity of the human enzyme relative to the archaeal enzyme. If the two enzymes share a similar structure, then a structure-guided site-specific substitution in the human enzyme to model the archaeal enzyme should confer an archaeal-like stability to the human enzyme. Because the protease-sensitive sites of the human enzymes M252, V254, M261, and T263 (Supplemental Fig. S3) are nonconserved and outside of the active site, this made the modeling more challenging. Modeling of these residues to the tRNA-bound structure of MjTrm5 showed that, while M252 and V254 are localized in a loop that stabilizes G37, M261 and T263 are in a helix more distant from G37 (Fig. 5A). We found that, indeed, the single M261L substitution that recapitulated the archaeal residue minimized the 27-kDa protease product upon enzyme expression in E. coli, indicating improved stability. In contrast, the T263I substitution had no effect, whereas the double substitutions (M261L and T261I) had the same effect as the single M261L substitution (Supplemental Fig. S6). Importantly, the single M261L substitution retained the full enzyme activity (Table 3), indicating that the substitution is not involved in catalysis but in structural stability, at least for expression in E. coli. The successful remodeling of the human enzyme for expression in E. coli, based on modeling with the archaeal structure, demonstrates the overall structural similarity of the two enzymes and provides validation that the archaeal structure is a reasonable working model for studies of the human enzyme.
CONCLUSION
The interest in Trm5 structure and mechanism has been high because of its fundamental distinction from the bacterial TrmD, which is a leading target in developing the next generation of antibiotics (White and Kell 2004). However, the information on HsTrm5, in particular, has been limited, thus hampering the progress of drug development against TrmD. For HsTrm5, its moderate sequence similarity with MjTrm5 is not sufficient to support structural and mechanistic similarity. We show here that, despite the moderate sequence similarity, HsTrm5 is fundamentally similar to MjTrm5 in catalysis (Figs. 2–4; Tables 1, 2), including the occurrence of one-proton transfer in an induced-fit process of methyl transfer. We also show that HsTrm5 is similar to MjTrm5 not only in the active site (as inferred from kinetic data) but also in broader regions of the overall structure that control stability (Table 3). However, this study also identifies two features where HsTrm5 differs from MjTrm5. In one, the value of 0.5-proton transfer of HsTrm5 versus one-proton transfer of MjTrm5 (Fig. 3) suggests a more complex meaning of kobs for the human enzyme. In the other, while the residue C-terminal to the general base plays an important role for the archaeal enzyme by stabilizing the carboxyl end of AdoMet (R186), it is not important for the human enzyme (H289). Further studies are necessary to illuminate the structural basis of these differences.
An important contribution of this study is the validation of the MjTrm5 ternary structure as a useful model for HsTrm5. The successful utilization of the structure to remodel HsTrm5 for protease resistance, even at nonconserved positions outside of the active site, is a strong testament for the validation. This validation of the archaeal structure is important, given the difficulty to obtain large quantities of HsTrm5 for structural analysis. However, caution should be exercised during modeling of the archaeal structure. For example, yeast Trm5 is localized in the nucleus, where it catalyzes m1G37 synthesis after tRNAPhe is modified with Cm32 and Gm34 (Nm = 2′-O-methylation in nucleotide N) in the cytoplasm and sent back to the nucleus (Murthi et al. 2010). The Trm5 product m1G37-tRNA is then re-exported to the cytoplasm for conversion of m1G37 to yW37 (Ohira and Suzuki 2011). Such a complex tRNA trafficking mechanism does not exist in archaea, suggesting the possibility that eukaryotic Trm5 may act on Cm32- and Gm34-modified tRNAPhe in a distinct G37-recognition mechanism relative to MjTrm5. This possibility must be addressed with specific analysis of tRNAPhe and with high-resolution structures of HsTrm5 in complex with tRNAPhe and an analog of AdoMet. Because of the importance of HsTrm5, efforts to obtain its structures should be given priority in order to produce insight into how eukaryotic Trm5 is diverged from its archaeal counterpart and how it is separated from the bacterial TrmD.
MATERIALS AND METHODS
Cloning and expression of HsTrm5
The cDNA sequence for HsTrm5 was amplified from the KIAA 1393 plasmid of Kazusa DNA Research Institute (Kisarazu, Chiba, Japan). This cDNA encodes F3 to T500 (Supplemental Fig. S1) and was cloned into the NdeI and XhoI restriction sites of pET22b. The protein sequence as expressed starts with M1 encoded by the NdeI site, followed by F2SGRF6 to T500, and ends with a His-tag. The coding sequence differs from the genomic sequence (http://www.uniprot.org/uniprot/Q32P41) by having K394 instead of E394. The recombinant enzyme was expressed in E. coli BL21(DE3)-RIL and purified by affinity binding to the His-Link resin (Promega [V882A] in a sonication buffer [20 mM HEPES at pH 7.5, 250 mM NaCl2, 10 mM β-mercaptoethanol, and 0.2 mM PMSF]). Mutations of the enzyme were created using the Quik-Change protocol and confirmed by DNA sequencing analysis. Enzyme mutants were assayed in the same way as the wild-type enzyme. Transcripts of HstRNACys were synthesized by T7 RNA polymerase based on a DNA template created from two overlapping oligonucleotides (Zhang et al. 2008) and were purified by a denaturing 12% PAGE/7 M urea gel.
Mass spectrometry analysis
The analysis, performed as a service at the proteomic center of Ohio State University, is described in the legend to Supplemental Figure S3.
Kinetic assays
Steady-state kinetic assays of HsTrm5 were performed at 37°C in the same buffer as for MjTrm5 (Christian et al. 2004). Assays for tRNA parameters were performed with 25 μM [3H-methyl]-AdoMet (80 Ci/mmol) (PerkinElmer NET 155H), 0.25–6 μM of HstRNACys transcript, and 50 nM of the enzyme, while those for AdoMet parameters were measured with tRNA saturated at 6 μM and 0.25–5 μM of AdoMet. Aliquots were removed at time points and precipitated in 5% TCA (trichloroacetic acid), and counts were measured and corrected for backgrounds and a quenching efficiency of ∼55%. Parameters were determined from fitting data as a function of concentration to the Michaelis–Menten equation. Each value was determined from the average of at least three independent analyses, showing standard errors, which were obtained by dividing the standard deviation of each value by the root square of the sample size.
Pre-steady-state kinetics was monitored on an RQF3 KinTek instrument (Christian et al. 2010b). One syringe contained enzyme (0.5–10 μM) with tRNA (0.5 μM) and the other syringe contained [3H-methyl]-AdoMet (25 μM). Data were fit to the single exponential equation:
 |
where y0 is the y intercept, A is the scaling constant, kapp is the apparent or observed rate constant, and t is the time in seconds to determine kapp. The analysis of kapp versus enzyme concentration was fit to the hyperbolic equation:
where kchem is the rate constant for the chemistry step, and E0 is the enzyme concentration. Measurement of Kd (AdoMet) was as described (Christian et al. 2010b); one syringe contained enzyme (0.5–10 μM) with [3H-methyl]-AdoMet (0.3 μM), and the other syringe contained tRNA (6 μM). Data were fit to the single exponential equation as above.
Burst analysis was monitored as described (Christian et al. 2010b), upon rapid mixing of HsTrm5 (1 μM) in one syringe with tRNA (10 μM) and 3H-AdoMet (25 μM) in the second syringe. The data were fit to the burst equation:
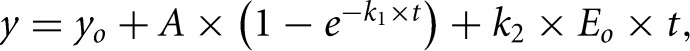 |
where y0 is the y intercept, A is the amplitude of the initial exponential phase, k1 is the apparent rate constant of the initial exponential regression, k2 is the apparent rate constant of the steady-state phase, and t is the time in seconds.
The pH-activity profile
The experiments were performed as described (Christian et al. 2010a). Buffers used for different pH values were as follows: sodium cacodylate (pH 5.9, 6.1); MES (pH 6.1, 6.3, 6.4, 6.6); MOPS (pH 6.6, 6.8, 7.0, 7.1); glycyl glycine (pH 7.1, 7.3, 7.6, 8.1); and glycine (pH 9.1). If the pH was lower than the normal value, drops of 5 M KOH were added to the 5× solution such that the 1× solution became properly adjusted. Reactions at pH lower than 7.3 were monitored by hand sampling, while those at higher pH values were done on the RQF3 instrument. No differences in rate were observed for reactions run at pH values where two different buffers were used. HsTrm5 enzyme was used at 3 μM, 37°C, in the reaction buffer containing a final concentration of 0.1 M buffer, 0.1 M KCl, 6 mM MgCl2, 0.1 mM EDTA, 4 mM DTT, and 0.024 mg/mL BSA. Upon rapid mixing with 0.5 μM tRNA and 25 μM AdoMet in the same buffer, the time course of m1G37 synthesis was monitored. Substrate saturation at pH 6.0, 8.1, and 9.8 was demonstrated by showing that the kobs remained unchanged upon reducing the enzyme and tRNA concentrations by twofold. The consistency in the measured kobs across all three pH values established that both tRNA and AdoMet were stable at all relevant pH values. The data were fit to the equation:
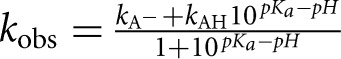 |
where kobs is the observed reaction rate at a specific pH, kAH is the activity of the protonated form of G37 (kAH = 0), kA− is the activity of the deionized form of G37, and Ka is the equilibrium constant for the dissociation of the proton.
Kinetic isotope effect
The solvent-exchangeable N1 imino proton of G37 in tRNA was replaced with deuterium by incubating the tRNA (previously dried down) in D2O (Cambridge Isotope Laboratories) for a few minutes in one syringe of RQF3. The enzyme and AdoMet were incubated in the water-based buffer in the other syringe. Upon rapid mixing, the time course of methyl transfer was monitored by taking aliquots and measuring acid-precipitable counts. The kinetics of MjTrm5 was measured at the near-saturating 6 μM of the enzyme, while that of HsTrm5 was measured at 2 μM, due to the difficulty in obtaining large quantities of the latter. The average value of kobs from two independent measurements was reported.
Molecular modeling
Figures were drawn with PyMol (PDB 2ZZN), corresponding to the ternary complex of MjTrm5-tRNACys–AdoMet complex.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
This work is supported by NIH grant GM081601 to Y.M.H. We thank Takao Igarashi for construction of the HsTrm5 expression clone, and Albert J. Chang, Cuiping Liu, Reiko Sakaguchi for assistance with figures.
REFERENCES
- Ahn HJ, Kim HW, Yoon HJ, Lee BI, Suh SW, Yang JK 2003. Crystal structure of tRNA(m1G37)methyltransferase: Insights into tRNA recognition. EMBO J 22: 2593–2603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjork GR, Wikstrom PM, Bystrom AS 1989. Prevention of translational frameshifting by the modified nucleoside 1-methylguanosine. Science 244: 986–989 [DOI] [PubMed] [Google Scholar]
- Bjork GR, Durand JM, Hagervall TG, Leipuviene R, Lundgren HK, Nilsson K, Chen P, Qian Q, Urbonavicius J 1999. Transfer RNA modification: Influence on translational frameshifting and metabolism. FEBS Lett 452: 47–51 [DOI] [PubMed] [Google Scholar]
- Bjork GR, Jacobsson K, Nilsson K, Johansson MJ, Bystrom AS, Persson OP 2001. A primordial tRNA modification required for the evolution of life? EMBO J 20: 231–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brule H, Elliott M, Redlak M, Zehner ZE, Holmes WM 2004. Isolation and characterization of the human tRNA-(N1G37) methyltransferase (TRM5) and comparison to the Escherichia coli TrmD protein. Biochemistry 43: 9243–9255 [DOI] [PubMed] [Google Scholar]
- Bystrom AS, Bjork GR 1982a. Chromosomal location and cloning of the gene (trmD) responsible for the synthesis of tRNA (m1G) methyltransferase in Escherichia coli K-12. Mol Gen Genet 188: 440–446 [DOI] [PubMed] [Google Scholar]
- Bystrom AS, Bjork GR 1982b. The structural gene (trmD) for the tRNA(m1G)methyltransferase is part of a four polypeptide operon in Escherichia coli K-12. Mol Gen Genet 188: 447–454 [DOI] [PubMed] [Google Scholar]
- Christian T, Hou YM 2007. Distinct determinants of tRNA recognition by the TrmD and Trm5 methyl transferases. J Mol Biol 373: 623–632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Evilia C, Williams S, Hou YM 2004. Distinct origins of tRNA(m1G37) methyltransferase. J Mol Biol 339: 707–719 [DOI] [PubMed] [Google Scholar]
- Christian T, Evilia C, Hou YM 2006. Catalysis by the second class of tRNA(m1G37) methyl transferase requires a conserved proline. Biochemistry 45: 7463–7473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Lahoud G, Liu C, Hoffmann K, Perona JJ, Hou YM 2010a. Mechanism of N-methylation by the tRNA m1G37 methyltransferase Trm5. RNA 16: 2484–2492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian T, Lahoud G, Liu C, Hou YM 2010b. Control of catalytic cycle by a pair of analogous tRNA modification enzymes. J Mol Biol 400: 204–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goto-Ito S, Ito T, Ishii R, Muto Y, Bessho Y, Yokoyama S 2008. Crystal structure of archaeal tRNA(m1G37)methyltransferase aTrm5. Proteins 72: 1274–1289 [DOI] [PubMed] [Google Scholar]
- Goto-Ito S, Ito T, Kuratani M, Bessho Y, Yokoyama S 2009. Tertiary structure checkpoint at anticodon loop modification in tRNA functional maturation. Nat Struct Mol Biol 16: 1109–1115 [DOI] [PubMed] [Google Scholar]
- Hagervall TG, Tuohy TM, Atkins JF, Bjork GR 1993. Deficiency of 1-methylguanosine in tRNA from Salmonella typhimurium induces frameshifting by quadruplet translocation. J Mol Biol 232: 756–765 [DOI] [PubMed] [Google Scholar]
- Hauenstein SI, Hou YM, Perona JJ 2008. The homotetrameric phosphoseryl-tRNA synthetase from Methanosarcina mazei exhibits half-of-the-sites activity. J Biol Chem 283: 21997–22006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson KA 1998. Advances in transient-state kinetics. Curr Opin Biotechnol 9: 87–89 [DOI] [PubMed] [Google Scholar]
- Lahoud G, Goto-Ito S, Yoshida K, Ito T, Yokoyama S, Hou YM 2011. Differentiating analogous tRNA methyltransferases by fragments of the methyl donor. RNA 17: 1236–1246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Kramer G, Graham DE, Appling DR 2007. Yeast mitochondrial initiator tRNA is methylated at guanosine 37 by the Trm5-encoded tRNA (guanine-N1-)-methyltransferase. J Biol Chem 282: 27744–27753 [DOI] [PubMed] [Google Scholar]
- Li JN, Bjork GR 1995. 1-Methylguanosine deficiency of tRNA influences cognate codon interaction and metabolism in Salmonella typhimurium. J Bacteriol 177: 6593–6600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell B, Rice DJ, Uchima FD 1983. Exceptional characteristics of amino proton exchange in guanosine compounds. Biochemistry 22: 3033–3037 [DOI] [PubMed] [Google Scholar]
- Motorin Y, Helm M 2010. tRNA stabilization by modified nucleotides. Biochemistry 49: 4934–4944 [DOI] [PubMed] [Google Scholar]
- Murthi A, Shaheen HH, Huang HY, Preston MA, Lai TP, Phizicky EM, Hopper AK 2010. Regulation of tRNA bidirectional nuclear–cytoplasmic trafficking in Saccharomyces cerevisiae. Mol Biol Cell 21: 639–649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagase T, Kikuno R, Ishikawa KI, Hirosawa M, Ohara O 2000. Prediction of the coding sequences of unidentified human genes. XVI. The complete sequences of 150 new cDNA clones from brain which code for large proteins in vitro. DNA Res 7: 65–73 [DOI] [PubMed] [Google Scholar]
- Noma A, Kirino Y, Ikeuchi Y, Suzuki T 2006. Biosynthesis of wybutosine, a hyper-modified nucleoside in eukaryotic phenylalanine tRNA. EMBO J 25: 2142–2154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Notredame C, Higgins DG, Heringa J 2000. T-Coffee: A novel method for fast and accurate multiple sequence alignment. J Mol Biol 302: 205–217 [DOI] [PubMed] [Google Scholar]
- Ohira T, Suzuki T 2011. Retrograde nuclear import of tRNA precursors is required for modified base biogenesis in yeast. Proc Natl Acad Sci 108: 10502–10507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Putz J, Florentz C, Benseler F, Giege R 1994. A single methyl group prevents the mischarging of a tRNA. Nat Struct Biol 1: 580–582 [DOI] [PubMed] [Google Scholar]
- Sakaguchi R, Giessing A, Dai Q, Lahoud G, Liutkeviciute Z, Klimasauskas S, Piccirilli J, Kirpekar F, Hou YM 2012. Recognition of guanosine by dissimilar tRNA methyltransferases. RNA 18: 1687–1701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snoussi K, Leroy JL 2001. Imino proton exchange and base-pair kinetics in RNA duplexes. Biochemistry 40: 8898–8904 [DOI] [PubMed] [Google Scholar]
- White TA, Kell DB 2004. Comparative genomic assessment of novel broad-spectrum targets for antibacterial drugs. Comp Funct Genomics 5: 304–327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi C, Pan T 2011. Cellular dynamics of RNA modification. Acc Chem Res 44: 1380–1388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang CM, Liu C, Slater S, Hou YM 2008. Aminoacylation of tRNA with phosphoserine for synthesis of cysteinyl-tRNACys. Nat Struct Mol Biol 15: 507–514 [DOI] [PMC free article] [PubMed] [Google Scholar]