

This manuscript describes an improved set of alignments of Drosophilid miRNA hairpins, including nearly 150 corrections to ortholog calls and nucleotide sequences in the available global 12-way Drosophilid alignments. Analysis of these reveals the previously unappreciated impact of biogenesis strategy and genomic location on the evolutionary dynamics of miRNAs, and affirms that miRNAs do not evolve as a unitary class.
Keywords: microRNA evolution, structure evolution, microRNA clusters, mirtrons, Drosophila
Abstract
The molecular evolutionary signatures of miRNAs inform our understanding of their emergence, biogenesis, and function. The known signatures of miRNA evolution have derived mostly from the analysis of deeply conserved, canonical loci. In this study, we examine the impact of age, biogenesis pathway, and genomic arrangement on the evolutionary properties of Drosophila miRNAs. Crucial to the accuracy of our results was our curation of high-quality miRNA alignments, which included nearly 150 corrections to ortholog calls and nucleotide sequences of the global 12-way Drosophilid alignments currently available. Using these data, we studied primary sequence conservation, normalized free-energy values, and types of structure-preserving substitutions. We expand upon common miRNA evolutionary patterns that reflect fundamental features of miRNAs that are under functional selection. We observe that melanogaster-subgroup-specific miRNAs, although recently emerged and rapidly evolving, nonetheless exhibit evolutionary signatures that are similar to well-conserved miRNAs and distinct from other structured noncoding RNAs and bulk conserved non-miRNA hairpins. This provides evidence that even young miRNAs may be selected for regulatory activities. More strikingly, we observe that mirtrons and clustered miRNAs both exhibit distinct evolutionary properties relative to solo, well-conserved miRNAs, even after controlling for sequence depth. These studies highlight the previously unappreciated impact of biogenesis strategy and genomic location on the evolutionary dynamics of miRNAs, and affirm that miRNAs do not evolve as a unitary class.
INTRODUCTION
MicroRNAs (miRNAs) are ∼22-nucleotide (nt) RNAs that play broad roles in post-transcriptional gene regulation in most eukaryotes (Flynt and Lai 2008; Axtell et al. 2011). In animals, canonical primary miRNA (pri-miRNA) transcripts are first cropped by the RNase III enzyme Drosha to produce precursor miRNA (pre-miRNA) hairpins. These are further processed in the cytoplasm by a Dicer-class RNase III enzyme to yield small RNA duplexes. One duplex strand, termed the mature miRNA, is preferentially retained in an Argonaute complex and guides it to target mRNA transcripts, while its complementary (miRNA*, or “star”) strand is preferentially degraded. Beyond this basic framework, other aspects of miRNA processing have emerged over the years. For example, star strands are not simply passive passengers, since a substantial proportion of star strands are subject to strict nucleotide constraint and contribute to endogenous regulatory networks (Okamura et al. 2008; Yang et al. 2011). Large-scale analysis of hairpin variants has defined other features that promote mammalian pri-miRNA cropping (Auyeung et al. 2013). Finally, a variety of noncanonical miRNA biogenesis pathways have emerged in the past few years, including both Drosha-independent and Dicer-independent mechanisms (Yang and Lai 2011).
Comparative genomics permits large-scale assessment of the evolutionary features, and thus presumably functional importance, of sequence elements across the genome. This has enabled the identification of novel protein-coding genes (Siepel et al. 2007), transcription factor–binding sites, and miRNA-binding sites (Nobrega et al. 2003; Friedman et al. 2009). Comparative approaches also permitted identification of novel conserved miRNAs (Lai et al. 2003; Lim et al. 2003a,b; Bentwich et al. 2005) and provided insights into miRNA evolution (Lai et al. 2003; Lim et al. 2003a). Early findings based on limited species comparisons described a common signature in which functional mature miRNA sequences exhibit higher constraint than star sequences, and lower constraint of terminal loops than the remainder of pre-miRNA hairpins (Lai et al. 2003). The characteristic “saddle” shape of hairpin evolution remains the most informative signature of miRNA sequence evolution (Lai et al. 2003; Altuvia et al. 2005).
The availability of 12 sequenced Drosophila genomes allowed additional features to be assessed (Drosophila 12 Genomes Consortium et al. 2007; Ruby et al. 2007b; Stark et al. 2007a; Nozawa et al. 2010). These include stronger constraint of paired than unpaired bases, high conservation of internal bulges within the mature region, and stronger conservation at the termini of the mature:star duplex relative to central sites. These evolutionary characteristics have shed light on our understanding of miRNA biology as well. For example, the higher constraint near the termini of the mature:star duplex reflects its precise enzymatic excision from the precursor hairpin, as well as appropriate strand selection, and internal bulge sites proved to control appropriate sorting of miRNA duplex strands into different Argonaute effector complexes (Czech and Hannon 2010).
All of these features were discerned from a collection of relatively ancient miRNAs. They may not be representative of all miRNAs, especially of recently evolved transcripts that potentially have more heterogeneous characteristics that have not yet been honed by evolutionary pressures. Indeed, miRNA annotations continue to be updated irrespective of conservation, by taking advantage of the power of deep sequencing of small RNAs. In this way, miRNA catalogs in vertebrates (Chiang et al. 2010; Friedlander et al. 2012; Ladewig et al. 2012) and invertebrates (Flynt et al. 2010; Berezikov et al. 2011; Chung et al. 2011) have been extended, comprising loci of diverse ages, genomic arrangements, and biogenesis pathways (Yang and Lai 2011). For example, a substantial class of alternative miRNA substrate derives from short hairpin introns termed mirtrons, in which splicing bypasses the normal requirement for Drosha cleavage to generate the pre-miRNA hairpin (Okamura et al. 2007; Ruby et al. 2007a). Altogether, these recently annotated genes provide opportunities to investigate properties of miRNA evolution more broadly.
We focus our present study on the well-annotated miRNAs of Drosophila melanogaster. As a foundation of this study, we created a high-quality resource of Drosophila miRNA orthologs that resurrects many loci erroneously missing in available genome-wide Drosophilid alignments and corrects many fly orthologs containing sequence errors. We use these to examine conservation features across finer partitions of the miRNA hairpins than previously assessed. Next, by segregating miRNAs according to evolutionary age, while focusing on loci with strict evidence for specific processing, we identify shared and novel themes in their sequence and structure characteristics. Finally, we investigate the impact of biogenesis pathway and genomic arrangement on miRNA evolution. We show that both splicing-derived miRNAs and genomically clustered miRNAs exhibit distinct features of evolutionary divergence relative to canonical miRNAs that were not cotranscribed with other miRNAs (i.e., “solo” canonical miRNAs). Therefore, although miRNA genes share some fundamental features of evolutionary selection, the appropriate grouping of miRNA loci reveals many subtype-specific selection pressures. These findings emphasize the diversity of miRNA genes and the evolutionary dynamics of their emergence and disappearance.
RESULTS
Extensive corrections to miRNA orthologs in the 12 Drosophila global alignments
Comparative genomic studies are predicated on accurate whole-genome sequences and multiple species alignments. Although the 12 Drosophila whole-genome multiple alignment provides a tremendous resource for comparative genomics (Stark et al. 2007b), we recently noted that some alignment errors exist among miRNA loci (Berezikov et al. 2010). We therefore considered it essential to conduct a thorough reassessment to generate Drosophila miRNA alignments of the highest-possible quality, on which we would base our subsequent analysis. Starting from the original MULTIZ whole-genome alignment, we extracted alignment blocks for all miRNAs and implemented extensive changes of two types: (1) replacement of entire orthologs, and (2) correction of genomic bases (Table 1; Supplemental Tables S3–S8).
TABLE 1.
Summary of miRNA alignment correction
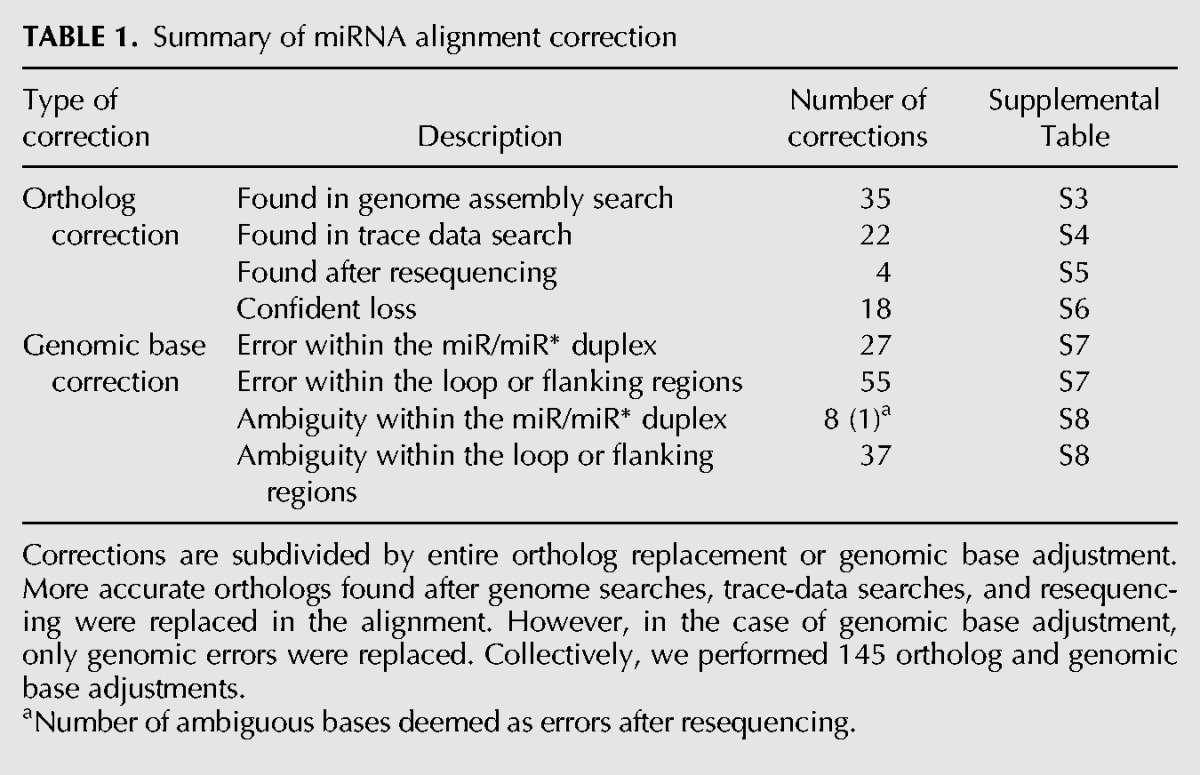
This analysis yielded 61 updated ortholog calls within 37 miRNA loci. In 35 cases, we were able to identify better orthologs than those in the global Drosophila alignment by searching the genome assembly. For example, the originally aligned sequence corresponding to the dwi-mir-318 (Drosophila willistoni) ortholog contains several substitutions in its mature miRNA sequence. Moreover, the MULTIZ alignment indicates a genomic rearrangement between the dwi-mir-318 and dwi-mir-994 loci, which are clustered and adjacent in the other Drosophila genomes. After correction, both miRNAs were ordered correctly on the same scaffold, and the updated dwi-mir-318 ortholog proved to be ultraconserved with respect to the other flies (Fig. 1A).
FIGURE 1.

Examples of miRNA alignment corrections. Original and corrected orthologs identified in genome assembly search; (A) dwi-mir-318, and in trace-data search; (B) dpe-mir-285. (C) Four miRNA orthologs missing from trace data, but recovered after targeted PCR and sequencing of genomic gaps. (D) Validation of genuine substitutions based on trace evidence within the mature and star regions of mir-316, an otherwise well-conserved miRNA. (E) Genomic base corrections within the dsi-mir-4916, dsi-mir-985, and der-mir-985 orthologs based on trace-data evidence.
More strikingly, we identified 22 miRNA orthologs that were only present within the trace data and thus were never incorporated within their respective genome assemblies. For example, the Drosophila persimilis ortholog of mir-285 is missing within both the MULTIZ alignment and the D. persimilis genome assembly, but was recovered in several Sanger trace reads (Fig. 1B). Fifteen of these cases occurred in D. simulans and six in D. persimilis, correlating with the lower sequencing coverage of these species. It remains unclear why the genome assembler used, ARACHNE (Batzoglou et al. 2002), failed to incorporate these clearly orthologous sequences. However, we note that many resided toward the ends of longer shotgun reads (>600 nt).
Finally, several species appeared to lack orthologs of miRNAs present in closely related Drosophilids, based on their absence from both genome assembly and from exhaustive searches of trace data. Eighteen of these cases were deemed confidently lost (Supplemental Table S6), as supported by common loss events across adjacent miRNAs or clades. Surprisingly, however, by amplifying and sequencing genomic segments from gaps that we suspected might contain miRNAs, we were able to resurrect four miRNA orthologs that were absent from all available genomic resources (Fig. 1C; Supplemental Fig. S1; Supplemental Table S5). The recovery of miRNA loci absent even from raw genomic traces underscores the challenges in comprehensive ortholog identification even among well-studied genomes. We earlier identified scores of “missing” chicken miRNA loci by analyzing chicken small RNA reads (Yang et al. 2011), and similar observations were recently made regarding miRNA loci that are erroneously “missing” from the rat genome (Uva et al. 2013).
We also analyzed evidence of nucleotide accuracy within miRNA orthologs, using genome trace data and available small RNA reads (Berezikov et al. 2010). We evaluated all cases but focused in particular on conserved miRNA nucleotides that diverged in individual species, because it seemed plausible that these might be artifactual. In the case of mir-316, all species maintain precisely the same mature and star products as in D. melanogaster, except for Drosophila mojavensis, which actually has mutations on both mature and star arms. Inspection of trace reads confirmed that, despite the ultraconserved nature of the mir-316 locus, the D. mojavensis ortholog bears a diverged nucleotide on miR-316 and two divergences on miR-316* (Fig. 1D). Moreover, we documented extensive cases in which apparently diverged nucleotides present in the global Drosophilid genome alignments were not supported by trace data. For instance, the D. simulans ortholog of mir-4916 contained three contiguous, erroneous bases within its seed region (Fig. 1E). Another salient example was mir-985, for which different nucleotides were called as diverged in both D. simulans and Drosophila erecta; neither of these was supported by the dominant sequences in their respective trace data.
In total, we corrected 84 genomic errors spanning 53 miRNAs (Supplemental Tables S7, S8). Importantly, 28 replacements were made within the mature or star regions. These were particularly important to distinguish from other cases of genuinely diverged miRNA sequences. Consistent with our ortholog replacements, 92% of these were within D. simulans. We could not confidently assess from trace evidence the validity of 8 bases within the miR or miR* region of five simulans miRNA orthologs. Upon resequencing these loci, only 1 base was deemed an error, which supports the putative validity of most genomic bases flagged as ambiguous and our avoidance of modifying these within the alignment (Supplemental Fig. S1). Altogether, we performed nearly 150 changes, comprising substantial improvements to Drosophila miRNA alignments. This revised collection of miRNA alignments and the associated changes are available online and can be downloaded in FASTA format. This well-curated set of improved alignments provides a strong foundation for our study of miRNA evolution in Drosophila.
Classification of miRNAs according to evolutionary age
Canonical miRNA primary sequences exhibit distinct conservation signatures within various partitions of their hairpin secondary structure. These patterns are driven by the processing activity of Drosha and Dicer, the sorting and loading of the mature miRNAs into effector Argonaute complexes, and base-identity constraint imposed by mRNA targets. Earlier analysis of Drosophila miRNA evolution focused primarily on well-conserved miRNAs (Drosophila 12 Genomes Consortium et al. 2007) or on a limited set of recently evolved miRNAs (Aravin et al. 2003; Lai et al. 2003; Nozawa et al. 2010). The current collection includes a sizable number (101) of melanogaster-subgroup miRNAs and mirtrons (Ruby et al. 2007b; Berezikov et al. 2011; Chung et al. 2011), offering the opportunity to evaluate whether patterns of miRNA evolution differ with age. In addition, we were able to incorporate data on dominant miRNA/star duplexes defined from a billion small RNA reads (Berezikov et al. 2011), providing a functionally based partitioning of miRNA hairpins.
From 238 D. melanogaster miRNAs in miRBase Release 19, we categorized loci as pan-Drosophilid, Sophophoran, or melanogaster-subgroup specific, based on their phylogenetic distribution of confident orthologs (see Materials and Methods) (Fig. 2). The classification of pan-Drosophilid miRNAs is largely straightforward and has been done previously (Ruby et al. 2007b; Stark et al. 2007a); our main addition to their analysis was to incorporate our many revised sequence calls and the identification of genuine orthologs not present in the global Drosophilid alignments. We grouped a limited set of 10 Sophophoran miRNAs and mirtrons together and analyzed their conservation features separately, due to their limited numbers.
FIGURE 2.
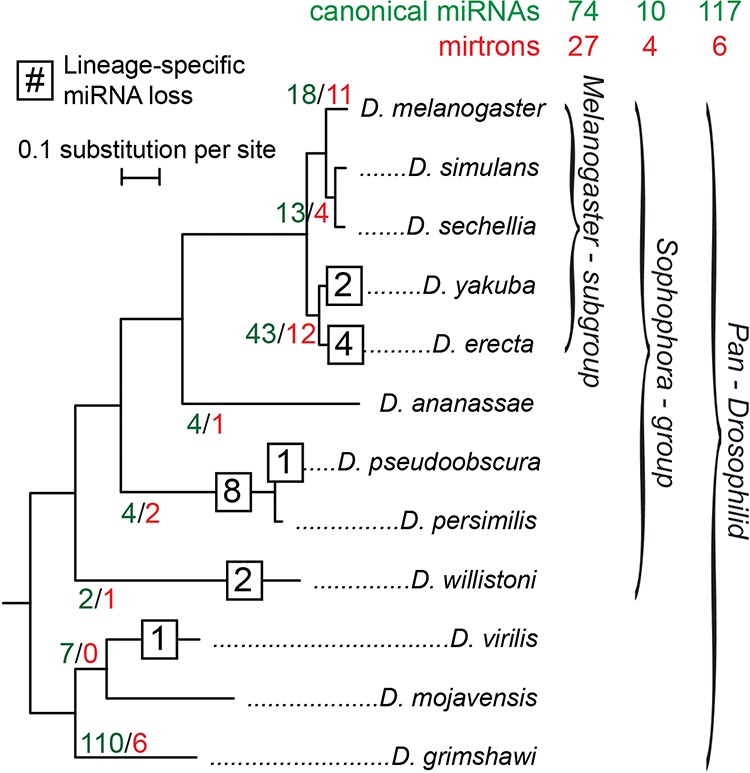
Age distribution of 238 D. melanogaster miRNAs and mirtrons within 12 Drosophila species. miRNAs are further classified into three presence–depth groups: pan-Drosophilid, Sophophoran, and melanogaster subgroup. The number of D. melanogaster canonical miRNAs (green) and mirtrons (red) with putative functional orthologs up to the indicated branch are labeled within the tree. The numbers of miRNAs with confident lineage- or species-specific miRNA death events are shown in squares at their respective branch. The total numbers of canonical miRNAs and mirtrons for each of the three presence–depth groups are shown above.
More challenging was the assignment of miRNA orthologs newly emerging among melanogaster-subgroup species. For these, it was less clear that aligned sequences were subject to endogenous processing, as can be reasonably assumed when alignments are deeply conserved. To avoid dilution of potential conservation signals by the inclusion of non-miRNA sequences, we focused on melanogaster-subgroup-specific miRNA loci with clear hairpin orthologs within D. erecta and/or Drosophila yakuba; i.e., miRNAs that were present in at least four species (Fig. 2). We segregated 17 loci with only Dmel/Dsim/Dsec orthologs (13 canonical miRNAs and four mirtrons) (Fig. 2) and analyzed them separately, since we were less confident that their aligned sequences were retained for miRNA-generating potential. Finally, we set aside 29 D. melanogaster loci that lacked confident orthologs in other species, since no further evolutionary analysis was possible for them.
Altogether, we segregated 55 canonical miRNA and mirtron loci with confident hairpin alignments within the five melanogaster-subgroup species (Fig. 2). Along with the expanded collection of pan-Drosophilid canonical miRNAs (117) and mirtrons (6) and a limited number of Sophophoran loci available in the current annotation, we were able to conduct a much broader survey of miRNA sequence and structure evolution across presence–depth groups than previously possible.
Conservation properties of pan-Drosophilid and newly emerged miRNAs
To evaluate the utility of our improved miRNA alignments and expanded miRNA annotations, we analyzed hairpin partitions earlier analyzed in the 12 Drosophila genomes project (Supplemental Fig. S2, results and hairpin-partitioning diagram; Drosophila 12 Genomes Consortium et al. 2007). We computed conservation scores using the statistical phylogenetic method phyloP (Pollard et al. 2010; Hubisz et al. 2011). While score differences between the original and corrected alignments were modest (mean phyloP score = 0.028), two partitions exhibited significant differences. First, both paired and unpaired regions of the mature strand showed small but significant differences (Mann–Whitney Test [MWT], P = 0.02; mature-unpaired, P < 10−3; duplex-paired). Second, although it was previously concluded that unpaired sites of mature miRNAs evolve more slowly than paired ones (Drosophila 12 Genomes Consortium et al. 2007), our analyses showed the opposite to be the case. We attribute this to the larger number of conserved loci available for study, as well as to the extensive corrections we made to the miRNA alignments.
Next, we investigated conservation properties across an extended, finer partitioning scheme that including the lower-stem region of the pri-miRNA hairpin, replacing the “mature-complementary” region (Drosophila 12 Genomes Consortium et al. 2007) with the explicit star sequence, and subdivides the mature and star regions into seed, mid, and distal segments (Fig. 3A). We reaffirm established concepts of the evolution of well-conserved miRNAs (Fig. 3B), namely, that (1) the lowest mean conservation score occurs in the terminal loop, (2) mature miRNA sequences are generally better conserved than their partner star sequences, (3) paired sites are generally better conserved than unpaired sites throughout the hairpin stem, and (4) the seed and distal paired subpartitions of both mature and star strands are better conserved than central duplex regions (see Supplemental Fig. S5 for higher resolution).
FIGURE 3.

Primary sequence and secondary structure evolutionary characteristics for miRNAs and other structured RNA classes. (A) Diagram of an extended hairpin partitioning scheme for which phyloP conservation scores were computed. (B,C) Mean phyloP conservation scores computed within extended partitions for (B) 116 pan-Drosophilid miRNAs and (C) 37 melanogaster-subgroup miRNAs. Mirtrons, melanogaster-only, and “3-species” canonical miRNAs and CDS miRNAs were excluded. Error bars indicate the standard error of the mean. Horizontal dashed lines portray the mean phyloP conservation score for other reference genomic classes. Blue values specify the number of miRNAs represented within the partition, and lack of a number indicates that all miRNAs are represented. (D,E) Properties of structure evolution for miRNAs, non-miRNA conserved hairpin structures, and other structured RNAs (snRNAs, tRNAs). Numbers on the x-axis indicate the number of loci represented in each class. Melanogaster-subgroup miRNAs have (D) proportions of free-energy difference and (E) consistent-to-compensatory substitution ratios (CCSRs), which are similar to pan-Drosophilid miRNAs but distinct from other structured RNAs.
Having demonstrated that we could recapitulate and extend known patterns of evolution of well-conserved miRNAs, we turned our attention to the 37 recently evolved, melanogaster-subgroup-specific miRNAs (Fig. 3C). As expected, these miRNAs exhibited less sequence constraint than pan-Drosophilid miRNAs in relation to the protein-coding sequence (CDS) and transfer RNA (tRNA) reference classes. In part, this reduction could reflect the reduced depth of the melanogaster-specific alignments, but we continued to observe reduced conservation scores when we considered scores for pan-Drosophilid miRNAs based on only the melanogaster-subgroup clade and a similar sample size (Supplemental Fig. S4). Nevertheless, we observed similarities in the pattern of evolutionary divergence of the “old” and “young” miRNAs. All partitions, including the loop, showed higher phyloP scores than those observed in neutral or intergenic regions. Moreover, both sets showed elevated constraint on paired residues across the hairpin stem, indicating selection to maintain secondary structure (MWT; mean P-value = 0.02 × 10−6) (Fig. 3C). In addition, there was evidence of constraint on the pairing in the lower- and upper-stem regions, consistent with selection to maintain the structural requirements for Drosha and Dicer cleavage, respectively. This contrasted with “3-species” miRNAs, in that the former lacked specific patterns of nucleotide and structural constraint across the hairpin (Supplemental Fig. S8). As mentioned above, it is not clear that the absence of constraint on these very newly emerged miRNAs reflects a lack of endogenous function or whether the signal for conservation in this set is diluted by the inclusion of some putative orthologs that are not actually subject to miRNA biogenesis. Analysis of nine Sophophoran miRNAs showed a similar reduction in constraint and near-identical patterns to that of the melanogaster-subgroup miRNAs (Supplemental Fig. S9A). However, we avoided merging the two sets in order to provide unbiased estimates of evolutionary rate.
Overall, we take the similarity of divergence patterns of “old” and “young” miRNAs to reflect that newly emerged Drosophilid miRNAs are under detectable selection for functionality as regulatory species. This is particularly significant since conservation analysis of seed-complementary regions, the usual strategy for computational inference of miRNA functionality, does not provide sufficient signal to identify targets of recently emerged miRNAs.
Evolution of melanogaster-subgroup miRNA hairpin structures
To further investigate the hypothesis that recently emerged Drosophilid miRNAs evolve in a manner that is characteristic of ancient miRNAs, we examined the structural evolutionary properties of these new miRNAs and contrasted them with other classes of structured noncoding RNAs, such as tRNAs and small nuclear RNAs (snRNAs), and conserved non-miRNA hairpin loci (Materials and Methods) (see Supplemental Fig. S6 for illustration). In particular, we considered normalized minimum-free-energy scores for extant miRNA sequences and consistent-to-compensatory substitution ratios (CCSRs). Briefly, consistent substitutions were defined as a single substitution preserving the pairing relationship at a single position of the hairpin structure, whereas compensatory substitutions were defined as two substitutions on both arms at a single hairpin position (see Materials and Methods).
By both measures, melanogaster-subgroup miRNAs showed structural evolutionary patterns that were more similar to pan-Drosophilid miRNAs than to other structured loci. First, melanogaster-subgroup miRNAs and pan-Drosophilid miRNAs showed similar normalized minimum-free-energy values (Kolmogorov-Smirnov [KS] test [KST], P ≅ 0.02), yet scores significantly lower than the scores of conserved, non-miRNA hairpin loci (KST, P < 10−10) (Fig. 3D). This result indicates that recently evolved miRNAs are as thermodynamically stable as older miRNA genes, despite the faster divergence rates of their primary sequences.
Nei and colleagues reported that well-conserved miRNAs were structurally stable after measuring an excess of paired-to-paired and unpaired-to-unpaired substitutions over structure-breaking substitutions (Nozawa et al. 2010). We examined paired-to-paired substitutions further by studying consistent-to-compensatory substitution ratios (CCSRs; see Materials and Methods), in order to understand the behavior of recently evolved miRNAs. As previously demonstrated, pan-Drosophilid miRNAs exhibited predominantly higher CCSRs, whereas other structured functional RNAs showed lower ratios (Stark et al. 2007a; Srivastava et al. 2011). We found that melanogaster-subgroup miRNAs also showed elevated CCSRs, but not as elevated as those for pan-Drosophilid miRNAs (Fig. 3E) (both differences significant by a Fisher’s exact test). These results indicate a high level of structural stability in all of the analyzed miRNAs, with slightly higher stability in the older, pan-Drosophilid group than in the younger, melanogaster-specific group. We note that for this analysis we filtered nucleotides near gaps to avoid alignment artifacts, but the results held without filtering as well (Supplemental Fig. S7).
Collectively, these results indicate that older and younger miRNA genes share characteristic evolutionary themes in both primary sequence and secondary structure, which distinguish them from other structured noncoding RNAs and even from bulk conserved hairpins that lack small RNA-generating potential. We take this to suggest that, despite their comparatively rapid evolution, newly emerged miRNAs have been detectably incorporated into beneficial regulatory networks.
Distinct evolutionary rates of canonical miRNA and mirtron loci
We recently proposed that mirtrons generally evolve faster than canonical miRNAs (Berezikov et al. 2010), based on the small number of well-conserved mirtrons and their comparatively greater representation among recently evolved Drosophila miRNA loci. Here, we performed detailed comparisons of the primary sequence evolutionary properties of Drosophila mirtrons and canonical miRNA loci. We restricted these analyses to pre-miRNA regions, since mirtrons lack the lower-stem region that is bound by the Drosha/Pasha complex. For reasons that will become evident later, we focused our comparison of mirtrons with solo canonical miRNAs.
Only six mirtrons are conserved across the Drosophilid phylogeny, but their evolutionary characteristics are notable given that they otherwise appear to have identical biochemical activity as canonical miRNAs (Okamura et al. 2007; Ruby et al. 2007a). Of the six deeply conserved mirtrons, four (mir-1003, mir-1007, mir-1011, and mir-1017) harbor substitutions within their mature arms (Fig. 4B). In contrast, the mature arms of pan-Drosophilid canonical miRNAs are highly stable. In aggregate, the mature miRNA sequences of deeply conserved mirtrons were less conserved than those of solo canonical miRNAs, and the same was true of the associated star sequences (Fig. 5A). Nevertheless, the patterns of divergence in deeply conserved mirtrons are similar to those in canonical miRNAs in that they display the lowest levels of conservation within their terminal loops, higher conservation of paired compared with unpaired sites, and still higher conservation in mature-unpaired seed and distal subpartitions than the mid region (Fig. 5A). In essence, deeply conserved mirtrons experience an overall reduction in purifying selection compared with canonical miRNAs.
FIGURE 4.

Characteristics of mirtrons and clustered canonical miRNAs. (A) Classification of canonical miRNAs and mirtrons by mature and star arm conservation patterns. Pan-Drosophilid miRNA genes with both mature and star arm divergences are generally either clustered canonical miRNAs or mirtrons, whereas melanogaster-subgroup genes show homogeneity in arm divergences. (B) Alignment for dme-mir-1011, a pan-Drosophilid mirtron with divergent mature and star arms, and seed mutations. These patterns are discordant from solo canonical miRNAs. (C) Proportions of solo to clustered miRNAs at differing presence–depth groups. Melanogaster-subgroup miRNAs are rarely clustered, unlike older miRNAs. (D,E) Similar to mirtrons, clustered miRNAs also show dual arm divergences like dme-mir-309, a pan-Drosophilid miRNA (D), and mir-2498, a melanogaster-subgroup miRNA (E). The functional 7- to 8-nt “seed” regions within pan-Drosophilid clustered miRNAs are ultraconserved, unlike recently evolved clustered miRNAs. Not all clustered miRNAs evolve similarly, however; mir-309 is the only miRNA to have divergent mature and star sequence (yellow and blue stars), unlike other members of its cluster. (F) Illustration of dynamic miRNA turnover with miRNA clusters. In the mir-959 → 964 cluster, several members have emerged and died across Drosophilid evolution. The absence of mir-959 and mir-961 from ancestral outgroup insect species suggests that these miRNAs were born during Drosophilid radiation.
FIGURE 5.

PhyloP conservation score profile and variance for three miRNA classes with different biogenesis and emergence properties. miRNA classes include solo and clustered canonical miRNAs, and mirtrons. (A,B) Partition-specific conservation scores for (A) pan-Drosophilid and (B) melanogaster-subgroup miRNAs and mirtrons. Pan-Drosophilid mirtrons show partition-specific patterns similar to canonical miRNAs, such as higher conservation of paired than unpaired duplex sites. (C) Variance of conservation scores per miRNA class. At all presence–depths, clustered miRNAs have greater variance of phyloP scores than other miRNA classes. Numbers within each panel (A,B) or on the x-axis (C) indicate the number of miRNAs present in each class.
The proportion of mirtrons among recently evolved miRNAs is much higher than among deeply conserved miRNAs, and this trend has continued as many new miRNAs have been annotated in D. melanogaster (Fig. 2; Berezikov et al. 2011; Chung et al. 2011). This supports the notion that the emergence of mirtron substrates is more facile than canonical miRNA substrates (Axtell et al. 2011). Curiously, the 11 melanogaster-subgroup-specific mirtrons (i.e., those that had hairpin orthologs in each of the five melanogaster-subgroup species) showed levels of constraint roughly equivalent to those of the 29 solo canonical miRNAs of similar age (Fig. 5B). This observation also applied to the four Sophophoran mirtrons when compared with six solo canonical miRNAs of the same evolutionary age (Supplemental Fig. S9B).
Distinct evolutionary properties of solo and clustered miRNAs
In our final analyses, we segregated canonical miRNAs according to whether they were “solo” or present in genomic clusters. Seventy-four of 201 canonical miRNAs within the D. melanogaster genome are clustered, with 12 of 74 melanogaster-subgroup miRNAs residing in clusters (Fig. 4C; Supplemental Table S10). In some clusters composed exclusively of well-conserved miRNAs, we observed individual members with atypical evolutionary dynamics. For example, dme-mir-309 in the mir-309 → 6-3 cluster (Fig. 4D) is an unusual well-conserved miRNA that contains sequence divergence on both miRNA and star arms. By comparison, few other conserved Drosophilid miRNAs exhibit divergence in their mature miRNA sequence, and nearly half lack divergence on either miRNA or star arms (Fig. 4A; Okamura et al. 2008). In total, 18 clustered miRNAs, but only eight canonical solo miRNAs, exhibit dual-arm divergence (Fig. 4A).
Other miRNA clusters contain a mix of “old” and “new” miRNAs. For example, the mir-959 → 964 cluster includes both pan-Drosophilid miRNAs and miRNAs specific to the Sophophoran group (Fig. 4F). Similarly, dme-mir-999 and dme-mir-4969 are a two-member cluster with a pan-Drosophilid and melanogaster-subgroup-specific miRNA (Supplemental Table S10). We also identified clusters consisting exclusively of melanogaster-subgroup miRNAs, suggesting that these miRNAs may have coevolved as de novo polycistronic transcripts. Notably, the mir-992/991/2498 cluster contains three melanogaster-subgroup-specific miRNAs that each exhibit divergent mature and star arms (Fig. 4E).
Apart from miRNA emergence within clusters, we observed miRNAs with peculiar species- or clade-specific functional ortholog death events. Of the 18 miRNAs with such unusual death events, 11 were located in genomic clusters (Supplemental Tables S6, S10). These also spanned varied evolutionary age depths. For example, the melanogaster-subgroup clustered miRNAs mir-303 and mir-982 lost D. erecta orthologs, despite confident Drosophila yakuba orthologs, and within the mir-959 → 964 cluster, mir-960 lost its Drosophila virilis ortholog, and mir-963 lost its obscura subgroup orthologs (Fig. 4F). Taken together, these observations suggest that miRNA genomic clusters may be unusually fertile grounds for both the gain and loss of miRNAs.
Comparison of the patterns of sequence conservation between solo and clustered canonical miRNAs revealed striking differences (see Materials and Methods) (Fig. 5). In particular, clustered miRNAs exhibited greater variance in phyloP scores across all pre-miRNA partitions, and this was true when comparing cohorts of deeply conserved canonical miRNAs as well as when examining melanogaster-subgroup-specific or Sophophoran-group-specific miRNAs (Fig. 5C). In fact, the clustered canonical miRNAs proved to be disproportionately responsible for the variability that we observed earlier among our aggregated analysis of canonical miRNAs (Fig. 3B,C). Moreover, the unusually fast evolution of mature-unpaired seed sites seen in the aggregate analysis of melanogaster-subgroup-specific canonical miRNAs was again primarily driven by clustered loci (Fig. 3C).
Overall, these analyses highlight the distinct evolutionary dynamics of miRNA clusters and genomically solo miRNAs, even when performing strict “apples-to-apples” comparisons of miRNA ages. Among deeply conserved miRNAs, we see that clustered loci harbor a disproportionately large group of miRNAs that exhibit sequence divergence on miRNA and star arms. Moreover, we observe frequent cases of newly emerging miRNAs in clusters, which are anchored by a more deeply conserved miRNA locus. Finally, we observe that among melanogaster-subgroup-specific miRNAs, members of genomic clusters diverge much more rapidly than solo loci and can exhibit divergence across the seed regions of both duplex arms. In particular, this final trend is inconsistent with purifying selection for miRNA function as we understand it, namely, to constrain for seed-matched targets. Instead, it may potentially signify adaptive evolution of these clusters of newly emerging miRNAs.
DISCUSSION
In this study, we sought to extract new features of miRNA evolution from the recently expanded catalog of D. melanogaster miRNA annotations. In previous studies, all Drosophilid miRNAs were grouped together for evolutionary analysis (Drosophila 12 Genomes Consortium et al. 2007; Ruby et al. 2007b; Stark et al. 2007a; Lu et al. 2008a,b; Nozawa et al. 2010; Price et al. 2011). Since the majority of Drosophila miRNAs annotated at the advent of the 12 Drosophila genome sequences (Drosophila 12 Genomes Consortium et al. 2007; Stark et al. 2007b) were deeply conserved, the features of miRNA evolution discerned in earlier studies were disproportionately representative of the well-conserved, canonical miRNA loci. In particular, the aggregate signatures were dominated by the robust signatures generated by the set of ancient miRNAs.
Critical to our approach was the careful curation of Drosophila miRNA orthologs (including culling of sequences aligned to Dmel miRNAs that did not exhibit hairpin potential), extensive fine-partitioning of pri-miRNA hairpins based on extensive small RNA data, and appropriate segregation of groups of miRNAs according to evolutionary depth, biogenesis strategy, and genomic locale. This allowed us to recover unsuspected features of miRNA evolution, even among the heavily studied, well-conserved Drosophilid miRNAs. In addition, we were able to discern characteristic properties of splicing-derived miRNAs and genomically clustered miRNAs.
The foundation of our work was a high-quality collection of Drosophila miRNA alignments. Essentially all previous analyses of Drosophila miRNA evolution used the global genome alignments available from the UCSC Genome Browser (http://genome.ucsc.edu). However, our exhaustive searches of genome sequences, shotgun trace data, available small RNA-seq data, and targeted PCR experiments allowed us to identify new bona fide orthologs and to correct discordant genomic bases across nearly 100 loci. These include a substantial number of loci that are completely missing or have the incorrect ortholog called in the global alignments, as well as many corrected miRNA bases that might otherwise be erroneously interpreted as divergence events or perhaps edited nucleotides. Our updated Drosophilid miRNA alignments substantially improve the accuracy of measuring many aspects of miRNA evolution, and we suggest that they supplant those downloadable from the UCSC Genome Browser for any future phylogenetic analyses of fly miRNAs.
Even though the evolutionary properties of pan-Drosophilid conserved miRNAs have been well studied (Drosophila 12 Genomes Consortium et al. 2007; Ruby et al. 2007b; Stark et al. 2007a; Nozawa et al. 2010), our analysis of a larger collection of such loci allowed us to update their characteristics. More significantly, our examination of melanogaster-subgroup miRNAs offered insight into the novel and shared evolutionary patterns of these recently evolved loci. Even though melanogaster-subgroup miRNAs are obviously under less constraint than pan-Drosophilid miRNAs, we still observed that they exhibit common patterns including higher sequence conservation within paired sites and greater constraint of lower- and upper-stem regions compared with the terminal loop. As well, normalized free-energy values and CCSRs of “young” and “old” miRNAs are similar. These features are collectively distinct from a substantial collection of well-conserved Drosophilid hairpins that lack evidence as small RNA-generating loci from ultra-deep-sequence analysis (Berezikov et al. 2011).
These findings affirm that not all well-conserved hairpins are miRNAs and reciprocally that miRNAs do not have to be well conserved to have evidence for regulatory potential. The generally very modest expression levels of newly evolved miRNAs have raised questions as to whether they can even impart sufficient regulation to be subject to selection. Indeed, it has been mostly an open question whether recently emerged miRNAs are incorporated into regulatory networks, since alignments of 3′ UTRs do not yield sufficient signal to evaluate the preferential conservation of seed matches to recently evolved miRNAs. Nevertheless, we observe that the scores of melanogaster-subgroup-specific miRNAs, which emerged within only the past ∼8 million yr, collectively exhibit evidence for evolutionary constraint as functional miRNA loci.
Perhaps the most intriguing conclusions of this study regard the distinct evolutionary properties of miRNAs according to biogenesis class and genomic location. We confirmed and extended our previous proposition that splicing-derived miRNAs evolve more quickly than do canonical miRNAs (Berezikov et al. 2010), even though overall they exhibit similar overall evolutionary pressures along the hairpin structure. Therefore, this class of noncanonical miRNA substrate generally behaves like canonical miRNA substrates, but is subject to reduced purifying selection. It is plausible that their requirement for only a single RNase III cleavage event may render them both easier to emerge than canonical miRNAs, but as they are no more likely to obtain beneficial regulatory targets that outweigh their potentially detrimental, spurious targets, they may also be under pressure to disappear just as quickly.
Even more striking were the properties of genomically clustered canonical miRNAs. We observed many cases of miRNAs emerging in the vicinity of more established miRNAs. It is tempting to speculate that miRNA emergence is favorable in the vicinity of an existing miRNA hairpin, perhaps because of the local concentration of nuclear miRNA processing factors that have been proposed to operate cotranscriptionally and near chromatin (Kim and Kim 2007; Morlando et al. 2008). Moreover, clustered canonical miRNAs exhibited far greater variance of conservation scores than solo canonical miRNAs or mirtrons, and this property was true when comparing deeply conserved miRNA or recently emerged loci. In addition, the majority of miRNAs bearing diverged nucleotides on both mature and star arms were clustered, a pattern that is sufficiently rare in general as to indicate that miRNAs in genomic clusters are far less evolutionarily constrained than expected from solo miRNA loci. Among well-conserved, clustered miRNAs, such mutations still generally avoid seed regions. However, particularly unexpected was the recognition that several clusters of miRNAs contain loci with seed-disrupting mutations on both miRNA and star strands, even though their overall hairpin qualities were maintained. Such a pattern is not compatible with current notions of how miRNAs evolve and may suggest a class of rapidly evolving miRNAs that is subject to positive selection (Lu et al. 2008a). The potential regulatory effects of such rapidly evolving miRNAs should be a compelling topic of future study.
MATERIALS AND METHODS
Data files
We obtained the MULTIZ multiple sequence alignment files for 12 Drosophilid species from the UCSC Genome Browser (http://genome.ucsc.edu/) (Blanchette et al. 2004; Drosophila 12 Genomes Consortium et al. 2007). D. melanogaster miRNA annotations were from miRBase Release 19 (Kozomara and Griffiths-Jones 2011). We also analyzed a cohort of non-miRNA conserved hairpins, annotated in previous studies (Ruby et al. 2007b; Stark et al. 2007a; Lu et al. 2008b) but later shown either to lack small RNA evidence or read patterns indicative of RNase III cleavage in ultra-deep data (Berezikov et al. 2010, 2011). Annotations of D. melanogaster protein-coding genes, transfer RNAs (tRNAs), and small nuclear RNAs (snRNAs) were obtained from FlyBase (http://flybase.org, dmel rev 5.36) (Marygold et al. 2013). Genome assembly trace data for the 12 Drosophila species were downloaded from NCBI (ftp://ftp.ncbi.nih.gov/pub/TraceDB/), and small RNA data for Drosophila simulans (GSM343915) and Drosophila pseudoobscura (GSM343916, GSM444067) (Berezikov et al. 2010) were obtained from the Gene Expression Omnibus (http://www.ncbi.nlm.nih.gov/geo/).
Alignment correction
Starting from the global MULTIZ alignments (Drosophila 12 Genomes Consortium et al. 2007), we replaced low-confidence orthologs with higher-confidence ones and updated individual genomic bases with alternative bases supported by shotgun trace reads and small RNA data from D. simulans and D. pseudoobscura. To facilitate correct ortholog identification, we searched 11 genome assemblies for miRNA orthologs using LASTZ, a recent replacement for BLASTZ (Harris 2007) (http://www.bx.psu.edu/miller_lab). Since synteny can improve the identification of confidence of predicted orthologs, our query sequences included the D. melanogaster pre-miRNA sequence and 1 kb of flanking genomic sequence on each side. The same parameter settings were used as in the MULTIZ alignment (H = 2000, Y = 3400, L = 4000, K = 2200, Q = HoxD55.q). All predicted orthologs were ranked by their percent identity to the pre-miRNA query sequence after normalizing for percent coverage of the query sequence covered in the alignment. Higher-ranked orthologs than the MULTIZ-reported ortholog were substituted within the alignment. If no ortholog could confidently be identified, we searched the shotgun trace reads for orthologous sequences not incorporated into the assembly, in a similar fashion. Finally, we attempted to experimentally clone and sequence several loci whose orthologs were not recovered in either data search (see below).
To identify base-call errors in the genome assemblies, we aligned long shotgun trace reads and small RNA reads to their respective genome sequences using BWA (Li and Durbin 2009) with default parameters. First, we flagged positions at which the nucleotide in the genome assembly was supported by less than half of the mapped reads, requiring a minimum coverage of three trace reads or, when available, 10 small RNA reads. The flagged bases were then visually inspected and further classified as genomic errors or ambiguous. Genomic errors were identified only when an alternative base from the aligned reads was concordant with the aligned bases of at least one of the two most closely related species in the phylogeny (Supplemental Table S1 for concordance scenarios). These putative genomic errors were corrected within the miRNA alignments. In contrast, positions were labeled as ambiguous when there were either multiple alternative bases, suggesting a tri-allelic state, or when an alternative base showed discordance with the aligned bases of related species. Ambiguous bases were not altered in the alignments. In general, we were cautious about modifying the alignments and replaced bases in only two of the six scenarios we considered (Supplemental Table S1).
We performed PCR experiments that identified four miRNA loci that were missing both from their cognate assembled genome and from the corresponding genomic trace reads (Supplemental Fig. S1; Supplemental Table S8). We also analyzed five miRNA loci bearing ambiguous bases within the miRNA/star duplex, that we could not confidently assign based on the available trace data (Supplemental Fig. S1; Supplemental Table S8). We obtained the isogenic Drosophila species used for genome sequencing from the Drosophila Species Stock Center (https://stockcenter.ucsd.edu). We extracted genomic DNA by homogenizing flies in 0.1 M Tris (pH 7.4), 0.1 M EDTA, and 1% SDS, heating the lysate for 20 min at 70°C, then adding potassium acetate to a final concentration of 0.4 M followed by incubation for 20 min on ice. DNA was precipitated by adding isopropanol to 60% of the total volume and centrifugation at maximum speed for 30 min at 4°C, followed by washing with 70% EtOH for 10 min at 4°C. Pellets were resuspended in 50 µL of water, and ∼50 ng of genomic DNA was used for PCR using the primers listed in Supplemental Table S2. In most cases, the amplified fragments were sequenced directly, generating consensus sequences through automatic base calls by a 3730XL/3730 sequencer. For Drosophila mojavensis mir-263b, due to the proximity of the miRNA hairpin to the end of the fragment, we cloned PCR products into pGEM-T Easy (Promega) and sequenced three clones to generate a consensus sequence.
Secondary structure prediction and partitions
miRNA secondary structures were predicted using the RNAfold and RNAsubopt programs within the Vienna RNA software package (v2.0.6) (Lorenz et al. 2011). RNAfold predicts a single, minimal free-energy structure, whereas RNAsubopt reports a collection of suboptimal structures. We ranked all predictions by thermodynamic free energy and examined up to six structures with the lowest free energy (see HTML documents).
We partitioned miRNA secondary structures into distinct regions defined by miRNA biogenesis and target regulation. The hairpin partitions studied by the Drosophila 12 Genomes Consortium et al. (2007) included the (1) terminal loop, (2) mature unpaired sites, (3) unpaired sites within the mature-complementary region, (4) mature/complementary paired sites, and (5) paired and (6) unpaired sites outside the mature:complementary double-stranded duplex region (Supplemental Fig. S2, diagram inset). We refined and expanded these partitions as follows: First, we added the pri-miRNA lower stem, believed to comprise ∼10 bp adjacent to the pre-miRNA hairpin (Han et al. 2006). We included 15 nt flanking Drosha crop sites to be inclusive of the conserved nucleotides, and this region contains a majority of paired residues, in contrast to distal nucleotides (Supplemental Fig. S3). The “lower stem” partition was excluded from mirtrons, which are not processed by Drosha. Next, we replaced the miRNA “complementary” regions used in partitions 4, 5, and 6 with the dominant expressed star sequences based on deep sequencing (Berezikov et al. 2011). Finally, we subpartitioned mature miRNA and star sequences into seed, mid, and distal regions, to study the functional constraint introduced by Drosha and Dicer processing, and miRNA–mRNA pairing.
Assignment of miRNA presence–depth
To group D. melanogaster miRNAs by age, we identified all confident orthologs across the Drosophilid phylogeny, based on the presence of aligned sequences with the capacity to adopt a pri-miRNA-like hairpin (Supplemental Table S9; Supplementary HTML documents). miRNAs were classified as melanogaster-subgroup miRNAs (which contain at least one confident hairpin ortholog among the five melanogaster-subgroup species), Sophophoran miRNAs (which contain at least one confident ortholog within a Sophophoran species), and pan-Drosophilid miRNAs (which contain confident hairpin orthologs within at least one species of the Drosophila group) (Fig. 2; Supplemental Table S9).
We took especial care to include only likely miRNA-generating orthologs in our studies. Consequently, we excluded orthologs whose secondary structure predictions were non-hairpins, since their sequences likely do not report on the evolution of miRNA-encoding potential. In 18 cases, we could confidently infer that an ancestral miRNA ortholog was lost in a derived species (Fig. 2; Supplemental Table S6). The majority of these death events were consistent across adjacent miRNAs in clusters, were lineage-specific, or occurred within confident regions of the genome assembly bearing no gaps. Because the set of Sophophoran-specific miRNAs and mirtrons was limited (10 total), we excluded them from the main analyses, but we do present a separate analysis of these loci (Supplemental Fig. S9).
Throughout our analysis, several additional miRNAs were also excluded (Supplemental Table S11). We removed dme-mir-280, dme-mir-287, dme-mir-288, and dme-mir-289, which are well conserved but lack experimental support for miRNA-generating potential from more than 1 billion small RNA reads (Berezikov et al. 2011). Additionally, eight CDS miRNAs (Berezikov et al. 2011) were removed because their evolutionary signatures are likely to be strongly influenced by protein-coding constraints.
Conservation estimation
Conservation scores were computed using our confident miRNA alignments described above. Each collection of orthologous miRNAs was aligned with FSA (Fast Statistical Aligner) (Bradley et al. 2009). Conservation scores were computed using the phyloP function within the RPHAST software package (Pollard et al. 2010; Hubisz et al. 2011). We used the “CONACC” method, which measures both conservation (reduced substitution rate) and acceleration (elevated substitution rate) relative to a model of neutral evolution. The neutral phylogenetic tree was inferred from fourfold degenerate sites using the genome-wide MULTIZ alignment and D. melanogaster gene annotations. Since the phyloP conservation scores depend on the number of sequences in the alignment, the scores were compared only for miRNAs with similar species presence and alignment depth (presence–depth). PhyloP reports a separate conservation score per nucleotide site; therefore, for each miRNA hairpin partition and presence–depth group, we aggregated the scores for all sites and all miRNAs and report the mean and one standard error. This approach allowed us to compare conservation scores across partitions of the same presence–depth and to compare conservation profile patterns across presence–depth groups.
We further grouped miRNAs by additional criteria, such as solo and clustered canonical miRNAs and mirtrons, and compared conservation profiles. In comparisons across miRNA classes, we removed miRNAs and mirtrons without duplex substitutions in order to visualize conservation variability with greater resolution. Because the “lower-stem” regions of mirtrons correspond to flanking exons, which do not appear to impact mirtron biogenesis substantially (Okamura et al. 2007; Chung et al. 2011), we removed these from conservation estimates.
Structure-evolution analysis
In addition to miRNA primary sequence evolution, we also investigated properties of structure evolution across miRNAs of several presence–depths and other structured RNA classes. We achieved this by inspecting conservation differences between paired and unpaired sites within each stem-region partition, computing normalized free-energy values, and measuring consistent-to-compensatory substitution ratios (CCSRs) along the branches of the phylogeny. We defined consistent events as individual substitutions that maintained base-pairing at a given paired position of the hairpin structure, such as an A →U substitution in a G-A →G-U paired-dinucleotide scenario. We defined compensatory events as pairs of substitutions that together maintained base-pairing, as in a A-G →G-U scenario. Paired and unpaired residues and free energy measures were directly obtained from RNAfold and RNAsubopt. Because the minimum free energy depends strongly on sequence length, we normalized free-energy scores by dividing them by sequence length to enable comparisons.
We wrote custom software for traversing the phylogeny in order to tally paired-to-paired substitutions for classifying consistent and complementary substitutions (see Supplemental Fig. S6 for illustration). For comparison to consistent substitutions, we counted each compensatory substitution as one event even though each resulted in two single-nucleotide substitutions. In this framework, we inferred ancestral sequences using the program prequel from the PHAST software package (Hubisz et al. 2011) conditioned on the neutral phylogenetic tree and a collection of extant miRNA sequences, and predicted optimal and suboptimal secondary structures for all sequences. Next, we traversed the species tree in level order, and at each edge, we structurally aligned all pairs of predicted parent and child structures using RNAforester (Hochsmann et al. 2003, 2004). Structures that maximized RNAforester’s reported score, which is based on the similarity of the structure shapes rather than primary sequences, were chosen. This method did not always choose minimum free-energy structures; however, it minimized the number of mismatches within the structure alignment and subsequent error in our analysis. To compare estimates across all structured RNA classes, only melanogaster-subgroup branches were examined across all classes.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article. Additionally, we provide online supplemental HTML documents available at http://compgen.bscb.cornell.edu/mirna/conservation/index.html detailing our miRNA alignment correction work and for illustrating reduced alignments of putative functional orthologs based on secondary-structure predictions.
ACKNOWLEDGMENTS
J.M. was supported in part by the Tri-Institutional Training Program in Computational Biology and Medicine (via NIH training grant 1T32GM083937). Work in E.C.L.’s group was supported by R01-GM083300, U01-HG004261, and RC2-HG005639. Work in A.S.’s group was supported by a David and Lucile Packard Fellowship for Science and Engineering and NIH grant R01-GM102192.
REFERENCES
- Altuvia Y, Landgraf P, Lithwick G, Elefant N, Pfeffer S, Aravin A, Brownstein MJ, Tuschl T, Margalit H 2005. Clustering and conservation patterns of human microRNAs. Nucleic Acids Res 33: 2697–2706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aravin A, Lagos-Quintana M, Yalcin A, Zavolan M, Marks D, Snyder B, Gaasterland T, Meyer J, Tuschl T 2003. The small RNA profile during Drosophila melanogaster development. Dev Cell 5: 337–350 [DOI] [PubMed] [Google Scholar]
- Auyeung VC, Ulitsky I, McGeary SE, Bartel DP 2013. Beyond secondary structure: Primary-sequence determinants license Pri-miRNA hairpins for processing. Cell 152: 844–858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Axtell MJ, Westholm JO, Lai EC 2011. Vive la différence: Biogenesis and evolution of microRNAs in plants and animals. Genome Biol 12: 221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batzoglou S, Jaffe DB, Stanley K, Butler J, Gnerre S, Mauceli E, Berger B, Mesirov JP, Lander ES 2002. ARACHNE: A whole-genome shotgun assembler. Genome Res 12: 177–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bentwich I, Avniel A, Karov Y, Aharonov R, Gilad S, Barad O, Barzilai A, Einat P, Einav U, Meiri E, et al. 2005. Identification of hundreds of conserved and nonconserved human microRNAs. Nat Genet 37: 766–770 [DOI] [PubMed] [Google Scholar]
- Berezikov E, Liu N, Flynt AS, Hodges E, Rooks M, Hannon GJ, Lai EC 2010. Evolutionary flux of canonical microRNAs and mirtrons in Drosophila. Nat Genet 42: 6–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berezikov E, Robine N, Samsonova A, Westholm JO, Naqvi A, Hung JH, Okamura K, Dai Q, Bortolamiol-Becet D, Martin R, et al. 2011. Deep annotation of Drosophila melanogaster microRNAs yields insights into their processing, modification, and emergence. Genome Res 21: 203–215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al. 2004. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res 14: 708–715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley RK, Roberts A, Smoot M, Juvekar S, Do J, Dewey C, Holmes I, Pachter L 2009. Fast statistical alignment. PLoS Comput Biol 5: e1000392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang HR, Schoenfeld LW, Ruby JG, Auyeung VC, Spies N, Baek D, Johnston WK, Russ C, Luo S, Babiarz JE, et al. 2010. Mammalian microRNAs: Experimental evaluation of novel and previously annotated genes. Genes Dev 24: 992–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung WJ, Agius P, Westholm JO, Chen M, Okamura K, Robine N, Leslie CS, Lai EC 2011. Computational and experimental identification of mirtrons in Drosophila melanogaster and Caenorhabditis elegans. Genome Res 21: 286–300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czech B, Hannon GJ 2010. Small RNA sorting: Matchmaking for Argonautes. Nat Rev Genet 12: 19–31 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drosophila 12 Genomes Consortium Clark AG, Eisen MB, Smith DR, Bergman CM, Oliver B, Markow TA, Kaufman TC, Kellis M, Gelbart W, et al. 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450: 203–218 [DOI] [PubMed] [Google Scholar]
- Flynt AS, Lai EC 2008. Biological principles of microRNA-mediated regulation: Shared themes amid diversity. Nat Rev Genet 9: 831–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynt AS, Chung WJ, Greimann JC, Lima CD, Lai EC 2010. microRNA biogenesis via splicing and exosome-mediated trimming in Drosophila. Mol Cell 38: 900–907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedlander MR, Mackowiak SD, Li N, Chen W, Rajewsky N 2012. miRDeep2 accurately identifies known and hundreds of novel microRNA genes in seven animal clades. Nucleic Acids Res 40: 37–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman RC, Farh KK, Burge CB, Bartel DP 2009. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19: 92–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Lee Y, Yeom KH, Nam JW, Heo I, Rhee JK, Sohn SY, Cho Y, Zhang BT, Kim VN 2006. Molecular basis for the recognition of primary microRNAs by the Drosha–DGCR8 complex. Cell 125: 887–901 [DOI] [PubMed] [Google Scholar]
- Harris RS 2007. Improved pairwise alignment of genomic DNA. Center for Comparative Genomics and Bioinformatics. Pennsylvania State University, University Park, PA [Google Scholar]
- Hochsmann M, Toller T, Giegerich R, Kurtz S 2003. Local similarity in RNA secondary structures. Proc IEEE Comput Soc Bioinform Conf 2: 159–168 [PubMed] [Google Scholar]
- Hochsmann M, Voss B, Giegerich R 2004. Pure multiple RNA secondary structure alignments: A progressive profile approach. IEEE/ACM Trans Comput Biol Bioinform 1: 53–62 [DOI] [PubMed] [Google Scholar]
- Hubisz MJ, Pollard KS, Siepel A 2011. PHAST and RPHAST: Phylogenetic analysis with space/time models. Brief Bioinform 12: 41–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim YK, Kim VN 2007. Processing of intronic microRNAs. EMBO J 26: 775–783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozomara A, Griffiths-Jones S 2011. miRBase: Integrating microRNA annotation and deep-sequencing data. Nucleic Acids Res 39: D152–D157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladewig E, Okamura K, Flynt AS, Westholm JO, Lai EC 2012. Discovery of hundreds of mirtrons in mouse and human small RNA data. Genome Res 22: 1634–1645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai EC, Tomancak P, Williams RW, Rubin GM 2003. Computational identification of Drosophila microRNA genes. Genome Biol 4: R42.41–R42.20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R 2009. Fast and accurate short read alignment with Burrows–Wheeler transform. Bioinformatics 25: 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim L, Lau N, Weinstein E, Abdelhakim A, Yekta S, Rhoades M, Burge C, Bartel D 2003a. The microRNAs of Caenorhabditis elegans. Genes Dev 17: 991–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim LP, Glasner ME, Yekta S, Burge CB, Bartel DP 2003b. Vertebrate microRNA genes. Science 299: 1540. [DOI] [PubMed] [Google Scholar]
- Lorenz R, Bernhart SH, Honer Zu Siederdissen C, Tafer H, Flamm C, Stadler PF, Hofacker IL 2011. ViennaRNA Package 2.0. Algorithms Mol Biol 6: 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Fu Y, Kumar S, Shen Y, Zeng K, Carthew R, Wu CI 2008a. Adaptive evolution of newly emerged micro-RNA genes in Drosophila. Mol Biol Evol 25: 929–938 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Shen Y, Wu Q, Kumar S, He B, Shi S, Carthew RW, Wang SM, Wu CI 2008b. The birth and death of microRNA genes in Drosophila. Nat Genet 40: 351–355 [DOI] [PubMed] [Google Scholar]
- Marygold SJ, Leyland PC, Seal RL, Goodman JL, Thurmond J, Strelets VB, Wilson RJ 2013. FlyBase: Improvements to the bibliography. Nucleic Acids Res 41: D751–D757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morlando M, Ballarino M, Gromak N, Pagano F, Bozzoni I, Proudfoot NJ 2008. Primary microRNA transcripts are processed co-transcriptionally. Nat Struct Mol Biol 15: 902–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobrega MA, Ovcharenko I, Afzal V, Rubin EM 2003. Scanning human gene deserts for long-range enhancers. Science 302: 413. [DOI] [PubMed] [Google Scholar]
- Nozawa M, Miura S, Nei M 2010. Origins and evolution of microRNA genes in Drosophila species. Genome Biol Evol 2: 180–189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Hagen JW, Duan H, Tyler DM, Lai EC 2007. The mirtron pathway generates microRNA-class regulatory RNAs in Drosophila. Cell 130: 89–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamura K, Phillips MD, Tyler DM, Duan H, Chou YT, Lai EC 2008. The regulatory activity of microRNA* species has substantial influence on microRNA and 3′ UTR evolution. Nat Struct Mol Biol 15: 354–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard KS, Hubisz MJ, Rosenbloom KR, Siepel A 2010. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 20: 110–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price N, Cartwright RA, Sabath N, Graur D, Azevedo RB 2011. Neutral evolution of robustness in Drosophila microRNA precursors. Mol Biol Evol 28: 2115–2123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby JG, Jan CH, Bartel DP 2007a. Intronic microRNA precursors that bypass Drosha processing. Nature 448: 83–86 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruby JG, Stark A, Johnston WK, Kellis M, Bartel DP, Lai EC 2007b. Evolution, biogenesis, expression, and target predictions of a substantially expanded set of Drosophila microRNAs. Genome Res 17: 1850–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siepel A, Diekhans M, Brejova B, Langton L, Stevens M, Comstock CL, Davis C, Ewing B, Oommen S, Lau C, et al. 2007. Targeted discovery of novel human exons by comparative genomics. Genome Res 17: 1763–1773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava A, Cai L, Mrazek J, Malmberg RL 2011. Mutational patterns in RNA secondary structure evolution examined in three RNA families. PLoS ONE 6: e20484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A, Kheradpour P, Parts L, Brennecke J, Hodges E, Hannon GJ, Kellis M 2007a. Systematic discovery and characterization of fly microRNAs using 12 Drosophila genomes. Genome Res 17: 1865–1879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stark A, Lin MF, Kheradpour P, Pedersen JS, Parts L, Carlson JW, Crosby MA, Rasmussen MD, Roy S, Deoras AN, et al. 2007b. Discovery of functional elements in 12 Drosophila genomes using evolutionary signatures. Nature 450: 219–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uva P, Da Sacco L, Del Corno M, Baldassarre A, Sestili P, Orsini M, Palma A, Gessani S, Masotti A 2013. Rat mir-155 generated from the lncRNA Bic is ‘hidden’ in the alternate genomic assembly and reveals the existence of novel mammalian miRNAs and clusters. RNA 19: 365–379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Lai EC 2011. Alternative miRNA biogenesis pathways and the interpretation of core miRNA pathway mutants. Mol Cell 43: 892–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JS, Phillips MD, Betel D, Mu P, Ventura A, Siepel AC, Chen KC, Lai EC 2011. Widespread regulatory activity of vertebrate microRNA* species. RNA 17: 312–326 [DOI] [PMC free article] [PubMed] [Google Scholar]