

Abstract
The ability of Saccharomyces cerevisiae to efficiently produce high levels of ethanol through glycolysis has been the focus of much scientific and industrial activity. Despite the accumulated knowledge regarding glycolysis, the modification of flux through this pathway to modify ethanol yields has proved difficult. Here, we report on the systematic screening of 66 strains with deletion mutations of genes encoding enzymes involved in central carbohydrate metabolism for altered ethanol yields. Five of these strains showing the most prominent changes in carbon flux were selected for further investigation. The genes were representative of trehalose biosynthesis (TPS1, encoding trehalose-6-phosphate synthase), central glycolysis (TDH3, encoding glyceraldehyde-3-phosphate dehydrogenase), the oxidative pentose phosphate pathway (ZWF1, encoding glucose-6-phosphate dehydrogenase), and the tricarboxylic acid (TCA) cycle (ACO1 and ACO2, encoding aconitase isoforms 1 and 2). Two strains exhibited lower ethanol yields than the wild type (tps1Δ and tdh3Δ), while the remaining three showed higher ethanol yields. To validate these findings in an industrial yeast strain, the TPS1 gene was selected as a good candidate for genetic modification to alter flux to ethanol during alcoholic fermentation in wine. Using low-strength promoters active at different stages of fermentation, the expression of the TPS1 gene was slightly upregulated, resulting in a decrease in ethanol production and an increase in trehalose biosynthesis during fermentation. Thus, the mutant screening approach was successful in terms of identifying target genes for genetic modification in commercial yeast strains with the aim of producing lower-ethanol wines.
INTRODUCTION
Reducing the ethanol yields produced by yeast during the fermentation process has become an important biotechnological focus in the past decade, driven by consumer and industry demand for lower-alcohol wines (1, 2, 3). This demand stems from health issues linked to excessive alcohol consumption, as well as concerns related to wine quality, as high alcohol levels have a masking effect on the flavor and aroma bouquet of wine (4). Other practical problems arise as restrictions are placed on the ethanol content in wines in certain countries and additional taxes are levied according to ethanol concentration (2, 5).
The existing procedures for the removal or reduction of ethanol postfermentation, including spinning cone columns and reverse osmosis, are costly and have a negative impact on wine quality. An alternative solution is the development of yeast strains that produce lower levels of ethanol during fermentation. Several such studies have been attempted in the past, with measured success. Most of these studies have employed metabolic engineering strategies, and Varela et al. (3) have recently evaluated and compared several of these strategies.
Several applications have focused on increasing glycolytic flux to glycerol as opposed to ethanol (6). The target genes include GPD1 and -2 (7, 8, 9), the ADH gene family (10, 11), and the PDC gene family (12). However, fermentations conducted with these genetically modified yeast strains are sometimes sluggish and are typically characterized by the formation of unwanted by-products, mostly due to the disruption of redox balance imposed by the genetic modifications.
All approaches have been hampered by our limited knowledge of the global and specific regulation of fermentative metabolism in yeast. The available data are derived from investigations of selected gene expression (13), global gene expression (14, 15, 16), proteomic responses to wine fermentation (17), and metabolite profiling (18), as well as the integration of transcript and proteome data (19, 20) and transcriptome and aroma metabolite data sets (21, 22, 23). Very little information pertaining to the link between genetic regulation and metabolite yields, particularly the flux toward ethanol during fermentation, can be derived from these limited data sets. The most insightful gene expression studies in line with this area of interest, all conducted under simulated wine fermentation conditions, have shown that the bulk of glycolytic genes are slowly downregulated as fermentation progresses, with only a few exceptions where isoforms of the same protein are differentially expressed (15, 16). A comparison of gene expression levels of glycolytic genes at the same stage in fermentation showed large variations between genes in this pathway, confirming the complex regulation governing this central metabolic pathway (15). As would be expected under glucose-repressed fermentative conditions, the bulk of tricarboxylic acid (TCA) genes appear to be expressed at low levels during fermentation. Further investigation into metabolic fluxes under simulated wine fermentation conditions (24) drew attention to discrepancies between these fluxes and the corresponding gene expression patterns (15).
In our study, we opted for a novel, untargeted approach to screen for genes which may be candidates for genetic modification in the quest for low-ethanol fermentations. In this endeavor, we made use of the EUROSCARF deletion library, allowing us to select strains with a single deletion in genes involved in the various categories of central carbon metabolism. The 66 selected strains were used to ferment synthetic wine must, and the must was analyzed for key metabolites during and at the end of fermentation. For the purpose of this study, central carbohydrate metabolism was defined as the pathways of glycolysis, ethanol production, TCA cycle, oxidative pentose phosphate pathway (OPPP), trehalose and glycogen metabolism, glycerol synthesis and anapleurotic reactions of glycolysis, and the TCA cycle. By using this unbiased initial screen of the 66 deletion mutants, we were able to identify several candidate genes associated with the predetermined parameters of altered ethanol yield. Of these, 5 genes of interest were selected for validation and further in-depth analysis. Based on the results, one gene (TPS1) was selected for proof-of-concept validation in an industrial yeast strain. A novel strategy for modifying TPS1 expression levels in a growth phase-specific manner using promoters of medium strength was implemented. The impact of these constructs was assessed in an industrial wine yeast strain, VIN13. The data show that this strategy of moderately increasing TPS gene expression at selected stages during fermentation avoided the complications and unrelated phenotypic effects that are caused by the metabolic burden of multicopy plasmids and highly expressed transgenes. Most importantly, the modified strains also showed higher trehalose accumulation and significantly lower ethanol yields than the corresponding control strains. The strategy therefore adds a useful additional target in achieving lower ethanol yields in wine fermentations. The strategy can be expanded to include additional targets identified in the first- and second-round screens.
MATERIALS AND METHODS
Selection of deletion strains.
Saccharomyces cerevisiae strains from the EUROSCARF library were selected from the haploid BY4742 (MATa his3Δ1 leu2Δ0 lys2Δ0 ura3Δ0) background (Table 1). Homozygous deletion strains for essential genes were selected from the diploid BY4743 (MATa/MATa his3Δ1/his3Δ1 leu2Δ0/leu2Δ0 met15Δ0/MET15 LYS2/lys2Δ0 ura3Δ0/ura3Δ0) background. For the industrial yeast, the commercial strain VIN13 (Anchor yeast) was used.
Table 1.
Deletion mutations of strains from the EUROSCARF collection used to screen for lower ethanol yield under simulated wine fermentation conditions
| Category, protein | Standard name of gene | Systematic name of gene | EUROSCARF straina |
|---|---|---|---|
| Glycolysis | |||
| Hexokinase I | HXK1 | YFR053c | Y15867 |
| Hexokinase II | HXK2 | YGL253w | Y14620 |
| Phosphofructokinase I | PFK1 | YGR240c | Y15893 |
| Phosphofructokinase II | PFK2 | YMR205c | Y10791 |
| Glyceraldehyde-3-phosphate dehydrogenase I | TDH1 | YJL052w | Y11371 |
| Glyceraldehyde-3-phosphate dehydrogenase II | TDH2 | YJR009c | Y16806 |
| Glyceraldehyde-3-phosphate dehydrogenase III | TDH3 | YGR192c | Y14822 |
| Pyruvate kinase II | PYK2 | YOR347c | Y11644 |
| Anapleurotic reactions | |||
| Pyruvate decarboxylase I | PDC1 | YLR044c | Y12655 |
| Pyruvate decarboxylase V | PDC5 | YLR134w | Y14091 |
| Pyruvate decarboxylase VI | PDC6 | YGR087c | Y14717 |
| Pyruvate carboxylase 1 | PYC1 | YGL062w | Y14429 |
| Pyruvate carboxylase 2 | PYC2 | YBR218c | Y13358 |
| Phosphoenolpyruvate carboxykinase | PCK1 | YKR097w | Y16013 |
| Fermentation | |||
| Alcohol dehydrogenase I | ADH1 | YOL086c | Y16236 |
| Alcohol dehydrogenase II | ADH2 | YMR303c | Y10891 |
| Alcohol dehydrogenase III | ADH3 | YMR083w | Y16217 |
| Alcohol dehydrogenase IV | ADH4 | YGL256w | Y14623 |
| Alcohol dehydrogenase V | ADH5 | YBR145w | Y13284 |
| Aldehyde dehydrogenase VI (major cytoplasmic) | ALD6 | YPL061w | Y12767 |
| Aldehyde dehydrogenase IV (major mitochondrial) | ALD4/ALD3 | YMR169c | Y10752 |
| Aldehyde dehydrogenase V (minor mitochondrial) | ALD5/ALD2/ALD3 | YMR170c | Y10753 |
| Aldehyde dehydrogenase VII | ALD7 | YOR374w | Y11671 |
| Trehalose metabolism | |||
| Trehalose-6-phosphate synthase in TPS complex | TPS1 | YBR126c | Y13265 |
| Trehalose-6-phosphate phosphatase in TPS complex | TPS2 | YDR074w | Y16692 |
| Trehalose synthase long chain in TPS complex | TSL1 | YML100w | Y16498 |
| TPS regulatory unit | TPS3 | YMR261c | Y10847 |
| Neutral trehalase | NTH1 | YDR001c | Y13941 |
| Acid trehalase | ATH1 | YPR026w | Y17145 |
| Glycogen metabolism | |||
| Glycogen synthase initiator | GLG2 | YJL137c | Y17003 |
| Glycogen synthase 1 | GSY1 | YFR015c | Y15694 |
| Glycogen synthase 2 | GSY2 | YLR258w | Y15167 |
| Glycogen branching enzyme | GLC3 | YEL011w | Y16388 |
| Sporulation-specific glycoamylase | SGA1 | YIL099w | Y12258 |
| Glycogen phosphorylase 1 | GPH1 | YPR160w | Y15575 |
| Glycogen debranching enzyme 1 | GDB1 | YPR184w | Y15599 |
| Hexose phosphate and OPPP | |||
| Phosphoglucomutase 1 | PGM1 | YKL127w | Y14977 |
| Phosphoglucomutase 2 | PGM2 | YMR105c | Y16545 |
| Homologous to UDP-glucose pyrophosphorylase | YHL012W | Y10951 | |
| Glucose-6-phosphate dehydrogenase | ZWF1 | YNL241c | Y11971 |
| Transketolase | TKL1 | YPR074c | Y15493 |
| Glycerol metabolism | |||
| Glycerol kinase 1 | GUT1 | YHL032c | Y10931 |
| Glycerol kinase 2 | GUT2 | YIL155c | Y12314 |
| Glycerol-3-phosphate dehydrogenase 1 | GPD1 | YDL022w | Y13718 |
| Glycerol-3-phosphate dehydrogenase 2 | GPD2 | YOL059w | Y11751 |
| TCA metabolism | |||
| Citrate synthase 1 | CIT1 | YNR001c | Y15376 |
| Citrate synthase 2 | CIT2 | YCR005c | Y13485 |
| Citrate synthase 3 | CIT3 | YPR001w | Y12828 |
| Aconitase 1 | ACO1 | YLR304c | Y15212 |
| Aconitase 2 | ACO2 | YJL200C | Y17022 |
| Isocitrate dehydrogenase NAD+ subunit 1 | IDH1 | YNL037c | Y15362 |
| Isocitrate dehydrogenase NAD+ subunit 2 | IDH2 | YOR136w | Y12392 |
| α-Ketoglutarate dehydrogenase (KGDH) complex component | KGD1 | YIL125w | Y12284 |
| Dihydrolipoyl transsuccinylase α-KGDH complex component | KGD2 | YDR148c | Y13506 |
| α-Subunit of succinyl-coenzyme A (CoA) ligase | LSC1 | YOR142w | Y12398 |
| β-Subunit of succinyl-CoA ligase | LSC2 | YGR244c | Y15897 |
| Diploids (essential genes) | |||
| Glucose-6-phosphate isomerase | PGI1 | YBR196c | Y23336 |
| Aldolase | FBA1 | YKL060c | Y24909 |
| Triosephosphate isomerase | TPI1 | YDR050c | Y23986 |
| 3-Phosphoglycerate kinase | PGK1 | YCR012w | Y23492 |
| Phosphoglycerate mutase | GPM1 | YKL152c | Y25002 |
| Enolase I | ENO1 | YGR254w | Y27286 |
| Pyruvate kinase I | PYK1/CDC19 | YAL038w | Y20368 |
| Pyruvate decarboxylase | PDC2 | YDR081c | Y24016 |
| Transcription factor | TOA2 | YKL058w | Y24907 |
| UDP-glucose pyrophosphorylase | UGP1 | YKL035w | Y24884 |
Strains in all but the last category are all mutants in the haploid BY4742 background. Strains in the last category are mutants in the diploid BY4743 strain.
Deletion strain verification.
All strains from the EUROSCARF library were confirmed either by PCR with gene-specific primers and size graduation on an agarose gel or by sequencing the unique UPTAGs and DOWNTAGs coded in each deletion strain as detailed by the Saccharomyces Genome Deletion Project.
Construction of deletion and overexpression laboratory strains.
Five genes were selected for further investigation and knocked out in Saccharomyces cerevisiae BY4742. Deletion cassettes, including the KANMX4 cassette, were amplified from the corresponding EUROSCARF strain using the following gene-specific primers (5′-to-3′ direction): TPS1_f, GTCCAAGCACGTCAGCGCTG, and TPS1_r, GTCGCTGTTCACACCGCAT; TDH3_f, GAGAACAGGGGCACAAACAGG, and TDH3_r, GCGTTCCTATCGGTACAGCC; ZWF1_f, GATGCATACTCCGGCGGTCTT, and ZWF1_r, GGCATCTTCCCCCCACCAA; ACO1_f, AGAGCCGCAAAAGGGAGGTC, and ACO1_r, GACGTTCGGCTGGAGAAGTC; and ACO2_f, CATACAGCTCTCACATCGTAG, and ACO2_r, CAACTACGGCTTAACTCAAGG.
PCR products were integrated into the BY4742 background strain using a standard lithium acetate transformation protocol. Transformation mixes were plated onto agar plates containing yeast extract-peptone-dextrose (YPD) plus the aminoglycoside antibiotic G418 (300 μg · ml−1). Putative colonies were restreaked onto fresh selection plates before strain verification. Verification was done by genomic DNA (gDNA) PCR using a KANMX4-specific primer (ACAGTCTTGACGTGCGCAG) and the following gene-specific primers binding downstream from the integration site: TPS_i, GTCAGGGGTGATAGCCAT; TDH3_i, GGAGCCCGCTTTTTAAGCT; ZWF1_i, GTAGCGCTACTGGAAGCA; ACO1_i, GGTGCCTGATTCTCGATTGTG; and ACO2_i, CACGCTCTTGAGTCATCGC.
Overexpression of the five genes of interest was achieved by cloning into the multicopy 2μ episomal vector pPVD1. All genes were under the regulation of the PGK1 promoter. Transformations were carried out using a standard lithium acetate protocol with selection on minimal medium lacking histidine, including 2% (wt/vol) glucose as the carbon source.
Construction of TPS overexpression plasmids and transformation of industrial yeast.
A centromeric plasmid was constructed by inserting the centromere and ARS sequence from the Ycplac22 plasmid into the pTEF/Zeo expression vector (Invitrogen). Next, the two promoter regions (upstream from the DUT1 and GIP2 genes) and the gene of interest (TPS1) were cloned from the genomic DNA of the industrial S. cerevisiae strain VIN13 and inserted into the centromeric expression vector. The trehalose-6-phosphate synthase (TPS1) coding sequence and terminator regions were amplified from VIN13 genomic DNA. Construct integrity was confirmed by PCR and sequencing. The primers used to amplify target sequences were as follows: TPS1f, GATCCAGCTGATGACTACGGATAACGCTAAGG; TPS1r, GATCGGCGCCTAACAGCGCTACAGACAGGC; DUT1f, GATCGCATGCACTATGTACATACACACGCACC; DUT1r, GATCCAGCTGTTGGTTATTTTTTGGCTCGCTGTA; GIP2f, GATCGCATGCGCTGTCTAGAATGCATTTTTCCA; and GIP2r, GATCCAGCTGTGTTGCGTTGATGAAATCCTAA.
The final constructs were transformed into the VIN13 yeast strain by electroporation (25, 26). Positive transformants were selected by plating on selective medium containing 1 g · liter−1 zeocin. To verify transformants, plasmid isolation from yeast was performed using the Zymoprep yeast plasmid miniprep II kit (Inqaba Biotech, Johannesburg, South Africa). The plasmids isolated from the various transformed strains were used in a back transformation in Escherichia coli strain DH5α and plated on zeocin selective medium. Restriction digests of isolated plasmids were also performed to confirm the identities of constructs isolated from the yeast.
Four strains were generated in this part of the study. Two were control strains transformed with the plasmids containing only the DUT1 or GIP2 promoter regions (named DUT-control and GIP-control), and two were TPS1 overexpression strains transformed with plasmids containing the promoter regions as well as the TPS1 gene (named DUT-TPS and GIP-TPS).
Fermentation conditions.
All strains were maintained on YPD plates, and overnight cultures grown in YPD broth. For the initial screen, small-scale fermentations were conducted in 100-ml tubular bottles sealed with a rubber bung and an S-bend airlock. These fermentations were conducted in 80 ml synthetic wine must MS300 (27) with 100, 200, or 250 g · liter−1 initial hexoses as indicated in the text. Glucose and fructose were added in equimolar quantities. All fermentations were inoculated to an initial cell density of an optical density at 600 nm (OD600) of 0.1. All fermentations were conducted with at least three independent replicates. The progression of fermentations was monitored by weight loss (indicative of CO2 generation) and allowed to run to completion at room temperature.
Fermentations conducted with control and transformed industrial yeast strains were carried out in 250-ml Erlenmeyer flasks containing 200 ml MS300 (27) and sealed with rubber bungs and S-bend airlocks. Glucose and fructose were added in equal amounts (125 g · liter−1). Preinoculated cultures were grown overnight at 30°C in YPD broth containing 1 g · liter−1 zeocin and used to inoculate fermentations to a cell density (OD600) of 0.1. All fermentations were carried out in triplicate. Zeocin was not included in the must under fermentation to avoid metabolic impacts related to stress imposed by high antibiotic concentrations. Fermentations were monitored by weight loss, and cell proliferation was determined by OD600 readings (Shimadzu UV-1601PC UV-visible scanning spectrophotometer; Shimadzu, Japan).
Real-time PCR.
To verify gene expression in the transformed and control strains, RNA isolations were performed on samples taken at time points (corresponding to days) T2, T5, T11, and T18 to cover the range of different growth phases of the yeast during fermentation. RNA was extracted using the hot phenol extraction protocol (28). RNA was quantified using the Nanodrop ND-1000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, USA), and integrity assessed on denaturing and nondenaturing gels. The ImProm-II reverse transcription system was used to synthesize cDNA using a random primer set.
Primers were designed using Primer Express software, version 3 (Applied Biosystems), and a KAPA SYBR fast quantitative PCR (qPCR) kit was used to perform quantitative real-time (qRT)-PCR analysis. Spectral data were captured with the Applied Biosystems 7500 cycler (Life Technologies, Foster City, CA, USA). Data analyses were conducted using Signal Detection Software (SDS), version 1.3.1 (Applied Biosystems), to determine the corresponding cycle threshold (CT) values and PCR efficiencies for the samples analyzed (29). The primer sequences used for qRT-PCR were as follows: TPS1f, TTGCACGCCATGGAAGTG; TPS1r, AACAACCTTGCCCCTCCATT; ACTf, GCCGAAAGAATGCAAAAGGA; and ACTr, TCTGGAGGAGCAATGATCTTGAC.
Metabolite analysis.
Glucose, fructose, glycerol, and ethanol were monitored by high-performance liquid chromatography (HPLC). Components were separated on a Bio-Rad Aminex HPX-87H (300- by 7.8-mm) column at 55°C with 0.5 mM H2SO4 as the mobile phase at a flow rate of 0.5 ml · min−1. Data are represented as either concentration (g · liter−1) or yield (g · gsugar consumed−1 × 100). All data are reported as the means of at least three replicates ± standard deviations.
Trehalose extraction (30) and quantification were performed at selected time points during fermentation by harvesting 5 ml of fermentation culture. The cells were dried and weighed. For the extraction, 500 μl of 0.25 M Na2CO3 was added for every 25 mg of cells. The buffer/cell mixture was vortexed and incubated at 95°C for 20 min, followed by centrifugation to pellet cell resides. Supernatants were analyzed for trehalose using the Megazyme trehalose assay kit (Megazyme International, Ireland) according to the supplier's specifications.
RESULTS
Initial screen for altered ethanol yield.
Sixty-six single-deletion mutants (in the haploid background BY4742 with the exception of essential genes, in which case, the diploid BY4743 background deletion was used; all from EUROSCARF) were selected with the aim of identifying steps in central carbohydrate metabolism that could potentially cause changes in carbon flux and metabolite yields under simulated wine fermentation conditions (synthetic grape must containing 100 g · liter−1 glucose and 100 g · liter−1 fructose) (Table 1). Of particular interest were strains with dual abilities to consume all sugars and increase flux either toward or away from ethanol. Deletion mutants in the BY7472 genetic background showed huge variations in improved or inferior abilities to yield ethanol (Fig. 1) and utilize sugars (Fig. 2). The diploid strains (BY4743) performed well with regard to consuming glucose and fructose.
Fig 1.

Ethanol yield (gethanol · g−1sugar consumed × 100) of EUROSCARF deletion strains fermented in synthetic grape must (100 g · liter−1 glucose and 100 g · liter−1 fructose). (A to H) Results for indicated deletion mutant strains in the haploid BY4742 background. The carbon metabolism category is indicated for each panel. (I) Results for indicated deletion mutant strains in the diploid BY4743 background.
Fig 2.

Residual sugar (g · liter−1) in EUROSCARF deletion strains fermented in synthetic grape must (100 g · liter−1 glucose and 100 g · liter−1 fructose). (A to H) Results for indicated deletion mutant strains in the haploid BY4742 background. The carbon metabolism category is indicated for each panel. (I) Results for indicated deletion mutant strains in the diploid BY4743 background.
A number of knockouts led to higher ethanol yields. Of these, three genes showing the most extreme yield increases were selected for further analysis (zwf1Δ, aco1Δ, and aco2Δ). Only 7 gene knockouts led to fermentations with lower ethanol yields. Of these, the most interesting two (tdh3Δ and tps1Δ) were selected for preliminary follow-up. All five strains fermented more sugars than the wild type, with the tdh3Δ strain fermenting completely to dryness (Fig. 1 and 2). For these genes, knockouts were generated in the wild-type BY4742 background to confirm phenotypes. Fermentations with these strains were repeated, and the results shown in Fig. 1 and 2 were reproduced using the newly generated knockout strains (see Table S1 in the supplemental material).
Overexpression of selected genes of interest.
Overexpression of the five selected genes of interest (TDH3, ZWF1, ACO1, ACO2, and TPS1) was carried out in wild-type BY4742 and BY4742 Δtps1::KANMX4 genetic backgrounds. As the aco1, aco2, and zwf1 deletions led to increased ethanol yields, it was necessary to investigate whether their overexpression could present the opposite phenotype of decreased ethanol yields. For this reason, these genes were overexpressed in the wild-type BY4742 genetic background, as well as in the tps1 and thd2 deletion mutants (which showed the lowest ethanol yields). It was hypothesized that the combined effect of the overexpression and deletion mutations, both of which potentially have a reductive effect on ethanol yields, could reduce the ethanol yield even further. The overexpression of the TPS1 and THD3 genes in their respective null backgrounds served as a control. Only TDH3 and TPS1 were overexpressed in the BY4742 Δtdh3::KANMX4 strain. Genes were overexpressed under the control of the PGK1 promoter in a multicopy 2μ plasmid (pPVD1). This system was chosen to represent the extreme opposite of a complete deletion of the gene. Fermentations were performed with an initial sugar concentration of 100 g · liter−1 and allowed to run to completion before analysis of the final parameters (Table 2). All data were calculated relative to the results for the empty-plasmid control.
Table 2.
Final fermentation characteristics of overexpressing strains
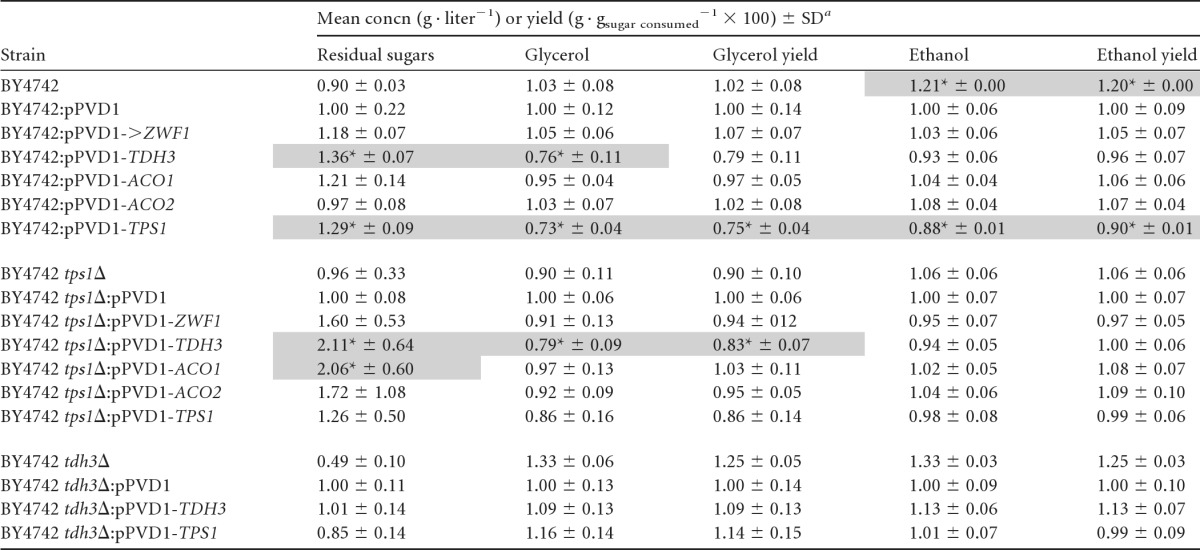
Fermentations were started at 100 g · liter−1 sugar in a synthetic grape must. All results were calculated relative to the results for the empty plasmid control (pPVD1). Values were normalized to those of the empty-plasmid control. *, value is significantly different from the value for the empty plasmid control.
It was evident that the plasmid itself had an effect on fermentation characteristics, as seen by the difference between the results for the background strain and the empty-plasmid control. For this reason, comparisons were made between overexpressing strains and the empty-plasmid control as an indication of the effect of the overexpressed gene on metabolism. The two most-significant phenotypes from the overexpression set were those of the TDH3 and TPS1 mutants. Both strains demonstrated a reduced fermentation capacity (see Fig. S1 in the supplemental material) as defined by lower rates of CO2 production. The BY4742:pPVD1-TPS1 strain was the only one to render a significantly lower ethanol yield (Table 2). All overexpression strains in the wild-type and tps1Δ background, with the exception of BY4742:pPVD1-ACO2, had higher residual sugar contents at the end of fermentation. None of the overexpression strains in the tdh3Δ background showed significant changes in carbohydrate levels at the completion of fermentation.
Overexpression of the TPS1 gene in commercial yeast.
Based on the results of the mutant screen in the haploid laboratory yeast, several genes would be potential targets for future modification in wine yeast. TPS1 was selected for overexpression in an industrial yeast genetic background (strain VIN13) as proof of concept to extend the results from experiments conducted with the laboratory strains to diploid wine yeast strains used in industry. For this purpose, two different low-strength promoters were selected to drive the expression of the transgene. The promoters (DUT1 and GIP1) drive gene expression during early exponential growth of the yeast and early stationary phase, respectively. The relative levels of gene expression were determined at four time points during fermentation: T2, T5, T11, and T14 (Fig. 3). The first time point, T2, is representative of the exponential growth phase. A significant (>2-fold) overexpression of the TPS1 gene under the control of the DUT1 promoter was observed during the exponential growth phase compared to its expression in the wild type (VIN13) and the DUT control (plasmid containing promoter only). The second time point, T5, is representative of early stationary phase, and a significant overexpression of the TPS1 gene under the control of the GIP2 promoter is observed. As the fermentations were conducted in the absence of selection pressure (i.e., without zeocin), the plasmid retention was assayed at the end of fermentation and found to be upward of 95%.
Fig 3.

Relative gene expression levels at different stages of wine fermentation. Values are the average of three biological repeats ± standard deviation. *, P < 0.05 (95% confidence).
Primary fermentation kinetics of TPS1-overexpressing VIN13 strains.
Samples of the fermentation must were taken at various time points during fermentation to determine the fermentation kinetics of transformed strains. The glucose consumption curves for all transformed and control strains appeared similar, while the transformed strains displayed slightly lower rates of fructose utilization (see Table S2 in the supplemental material). All TPS1-overexpressing strains, as well as the plasmid-only transformed strains, showed significant increases in glycerol levels (Fig. 4). The DUT-TPS and GIP-TPS test strains showed significantly reduced ethanol levels, as well as reduced ethanol yields, at the end of fermentation (Fig. 4). While the ethanol yields were close to or slightly over the theoretical maximum ethanol yield of 0.5 for some data points, the trends were consistent between independent biological repeats within experiments and between sequential repeats of entire fermentation sets.
Fig 4.

Ethanol, acetic acid, and glycerol concentrations at the end of fermentation (T18). Sugar utilization and ethanol yield as determined at the end of fermentation are also depicted. Values are the average of three biological repeats ± standard deviation. The Student t test was used to establish significant differences between fermentations conducted with the TPS1-overexpressing strains and their respective empty plasmid controls. *, P < 0.05 (95% confidence).
Trehalose production by TPS1-overexpressing strains.
The trehalose levels showed typical trends throughout fermentation, increasing as fermentation progressed through to the stationary growth phase of the yeast and subsequently declining toward the end of fermentation. Sampling for trehalose was also carried out at time points T2, T5, T11, and T18. At T2, the DUT-TPS strain showed significantly higher levels of trehalose production than all other strains and the wild-type control (Fig. 5). This increase in trehalose in the DUT-TPS strain persisted throughout fermentation, though the difference was less obvious and not statistically significant for the T18 time point. The trehalose levels in the GIP-TPS strain increased beyond the wild-type levels by T5 (Fig. 5), aligning with the increase in TPS gene expression observed for this time point (Fig. 3). This increase appeared to be maintained throughout fermentation, as the increases in trehalose levels for this strain (relative to the levels in the wild-type and empty plasmid controls) were statistically significant at all subsequent sampling points.
Fig 5.
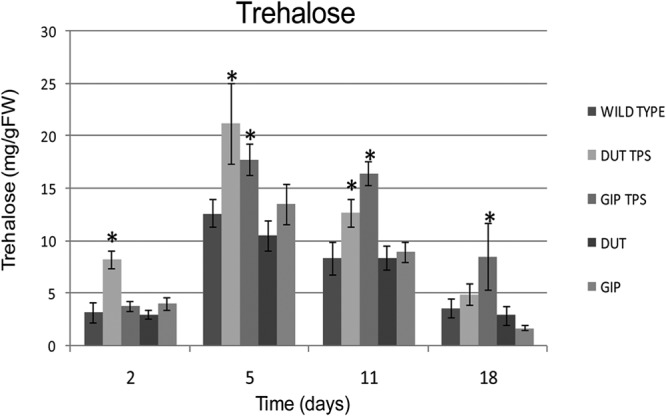
Trehalose levels (normalized relative to cell biomass) at different stages of wine fermentation. Values, calculated as milligrams of trehalose per gram of fresh weight, are the average of three biological repeats ± standard deviation. *, P < 0.05 (95% confidence).
DISCUSSION
Screening for reduced ethanol yield.
Identification of key steps in the pathways of central carbohydrate metabolism contributing to ethanol yield could be achieved by various approaches. As a first attempt, one could choose genes whose transcripts or enzymes have been highlighted by one of the -omics approaches mentioned in the introduction. However, these are not without their challenges, particularly considering that comparing data sets from studies of different yeast strains may result in incorrect selections. Second, in this in silico age, it is appealing to pursue a modeling and prediction technique. A kinetic model of yeast glycolysis has been successfully constructed and applied to aerobic, glucose-limited cultures (31). Although this model is of great use, its experimental growth conditions are far removed from those in wine fermentations. Multiple parameters would need to be determined experimentally and substituted into the model toward the purpose of identifying key steps in the regulation of flux toward ethanol in wine fermentations. Therefore, the current study followed a different strategy: the availability of the EUROSCARF deletion library has allowed for a significant amount of research to be carried out in the global yeast community. Strains with gene deletions in central carbohydrate metabolism (Table 1) were systematically selected with a view to identifying key steps regulating flux toward ethanol under simulated wine fermentation conditions. Of particular interest were strains that fermented more sugars than the wild type and had altered ethanol yields.
Under these conditions of high initial sugar concentration, the wild-type haploid BY4742 laboratory strain was unable to ferment to dryness (Fig. 2). These poorer fermentation characteristics of BY4742 provided an appropriate background for screening selected single-deletion mutants for improved fermentation characteristics and performance.
The results from small-scale fermentations of all 66 strains showed their differing abilities to yield ethanol (Fig. 1) and ferment hexoses (Fig. 2). Of the haploid strains, there were 5 with significantly lower and 25 with significantly higher ethanol yields. Forty-three fermented significantly more sugar and 5 significantly less sugar than BY4742. The deletion strain yielding the lowest ethanol was the adh1Δ knockout. However, this strain could not complete fermentation and was stuck at a residual sugar level of 124.6 ± 2.8 g · liter−1 residual sugars. This is easily explained by the facts that ADH1 is the major cytosolic alcohol dehydrogenase responsible for the production of ethanol during fermentation (11) and that, when it is deleted, the strain grows slowly under anaerobic growth on glucose. Knockout strains with mutations of TPS1 and TDH3 had the next-lowest ethanol yields after the adh1Δ knockout (Fig. 1). The combined phenotype of lower ethanol yield and improved ability to utilize sugars made these strains attractive for further study. The glycerol levels also differed between the wild-type and knockout strains. The tps1Δ strain produced nearly double the amount of glycerol as BY4742 at the end of fermentation (see Table S1 in the supplemental material). This property could also be an advantage in the wine-making context, as increased glycerol levels could contribute to an overall improvement in sensory quality (32).
Apart from the strains yielding lower ethanol, those yielding higher ethanol were also of interest. It was tentatively hypothesized that overexpressing these genes could reverse the ethanol yield phenotype. Strains selected toward this purpose were representative of the oxidative pentose phosphate pathway (ZWF1, encoding glucose-6-phosphate dehydrogenase) and the TCA cycle (ACO1, encoding aconitase 1, and its putative isozyme, ACO2). Interestingly, both ZWF1 and ACO1 had been identified in transcript expression studies (15, 17) as being upregulated under simulated wine fermentation conditions. The phenotypes were confirmed in independent knockout experiments (see Table S1 in the supplemental material), which was particularly pertinent for the tps1Δ deletion as this phenotype reportedly varies in different genetic backgrounds (33).
Overexpression of five genes of interest.
The five genes selected from the initial screen (ZWF1, TDH3, ACO1, ACO2, and TPS1) were overexpressed in BY4742 under the control of the PGK1 promoter in a multicopy plasmid (pPVD1). The empty-plasmid control showed altered fermentation characteristics compared to those of the wild type. This may be a result of the metabolic load of such a multicopy expression system, and in consequence, all comparisons will be discussed relative to the results for this control. None of the overexpression strains presented an opposite ethanol yield phenotype in comparison to the corresponding deletion phenotype (Table 2). In fact, the overexpression strains for ZWF1, ACO1, and ACO2 showed no significant changes in any measured fermentation characteristic in the wild-type BY4742 background. This could be attributed to strict posttranscriptional and posttranslational regulation of these metabolic steps. However, both the TPS1 and TDH3 overexpression strains fermented at reduced rates and exhibited higher residual sugar levels than the control (Table 2). This was in contrast to their deletion counterparts' improved ability to utilize sugars.
Consequences of altering TDH3 expression.
Tdh3p catalyzes the reduction of glyceraldehyde-3-phosphate and is the first reductive step in glycolysis, using NAD+ as the reducing equivalent. It is one of three isozymes and accounts for between 50 and 60% of glyceraldehyde-3-phosphate dehydrogenase activity (34). It is conceivable that Vmax was increased due to the overexpression of TDH3 perturbing the redox balance. The glycerol levels in the TDH3 overexpression strain were lower than in the control (perhaps as a result of substrate competition at the triose-phosphate level), and the ethanol levels were unchanged (Table 2), indicating that these two pathways were not employed to restore the NAD+/NADH ratio. This potential redox imbalance may explain the higher residual sugars at the end of fermentation, caused by a slowdown in glycolytic flux. These results highlight the fine control of glycolytic flux by the expression and regulation of the pathway's enzymes in their endogenous state and that perturbations to any one step may affect other steps in an unpredictable manner.
Altered TPS1 expression causes modifications in flux and carbohydrate distribution.
The overexpression of TPS1 yielded a more-predictable fermentation phenotype. Trehalose-phosphate-synthase 1 (TPS1) catalyzes the first step in trehalose biosynthesis, converting UDP-glucose and glucose-6-phosphate to trehalose-6-phosphate (35). Trehalose-6-phosphate is responsible for the inhibition of hexokinase-mediated phosphorylation of glucose at the entry point of glycolysis. It has been proposed that this regulation is crucial in the control of flux through glycolysis (34). The reduced fermentative capacity of the TPS1 overexpression strain would suggest that glycolytic flux has been partially inhibited. Although we did not measure trehalose-6-phosphate levels, we hypothesize that they were increased, causing inhibition of Hxkp2 (the major isoform of hexokinase) and, thus, an attenuation in the amount of glucose entering glycolysis. The effect was not purely in reducing overall flux through glycolysis but also in the distribution of carbohydrates. This is evident by reductions in the amounts of both absolute and yielded ethanol and glycerol, both metabolites representing the endpoint of a pathway under fermentative conditions remote from the initial step of hexose assimilation (Table 2).
Regulated TPS1 overexpression in a commercial yeast.
The part of the study discussed here focused on the slight overexpression of the TPS1 gene under the control of a low-strength promoter (of the DUT1 gene) that has been linked to gene expression during the exponential growth phase of yeast, as well as the promoter of the GIP2 gene (linked to gene expression during the stationary growth phase of yeast). The aim was to slightly increase TPS1 gene expression and enzyme activity without burdening the yeast cell and challenging the redox balance. The expression of TPS1 under the control of the DUT1 promoter was up to 2-fold higher than in the wild-type strain (VIN13) during early exponential growth, whereas the expression of TPS1 under the control of the GIP2 promoter was up to 60% higher than in the wild type during early stationary phase (Fig. 3). It appears that the expression of TPS1 both under the control of the DUT1 promoter during the early exponential growth phase and of the GIP2 promoter in early stationary phase has a significant metabolic impact in terms of decreasing total ethanol and ethanol yield (Fig. 4). Expression under the control of the GIP2 promoter seems to reduce the impact on fermentation capacity and the slower sugar utilization to a lesser extent than DUT1 promoter-driven expression. This might suggest that the expression of the TPS1 gene during early stationary phase (GIP2 promoter induced) is more effective and causes less stress to the cell than expression during early exponential growth.
There is also a slight, though not statistically significant increase in glycerol production observed for all transformed strains (Fig. 4). In S. cerevisiae, glycerol production plays an important role in stress tolerance and maintaining the intracellular redox balance (36, 37). No increase in acetic acid levels was observed for either of the test strains compared to the levels in their empty plasmid controls (Fig. 4). This is a positive outcome, suggesting that the slight overexpression of the TPS1 gene by these specific promoters can successfully shift carbon flux with minimal impact on redox balance and without any negative impact in terms of unwanted fermentation by-products.
The trehalose data (Fig. 5) align well with the patterns of TPS1 overexpression by the transformed strains (Fig. 3). The increases in trehalose levels in these strains show that carbon was likely redirected to this alternative carbon sink in the transformed strains, accounting for the decrease in ethanol yield (Fig. 4). Furthermore, the increased trehalose levels may have inhibited hexokinase activity, thus restricting associated flux through glycolysis. This would explain the reduced fermentation rate and slightly higher residual sugar levels in fermentations conducted with these strains.
In terms of carbon balance, the difference in ethanol levels is not accounted for entirely by the increase in trehalose. In the first place, it is unlikely that the particular discrete time points that we selected for sampling represent the points of maximum, or optimum, trehalose accumulation. Detailed flux balance analysis is also not feasible when extracellular metabolite measurements (ethanol) need to be compared with intracellular metabolite (trehalose) levels, particularly when the trehalose levels are in constant flux, in contrast to the ethanol levels, which represent a “pooled” end product. In the present study, we did not attempt to determine how much trehalose was reutilized, converted to glycogen and other sinks, or leaked into the medium toward the end of fermentation.
Conclusions.
This study has shown that selective screening of a deletion library for altered ethanol yields under simulated wine fermentation conditions is an ideal starting point for the identification and development of S. cerevisiae strains with altered flux in central carbohydrate metabolism. The results highlighted that key steps in multiple parts of this network of reactions are responsible for the regulation of flux and the final accumulated metabolite pools. Of the five genes selected for further investigation, alterations in TDH3 and TPS1 expression proved to be the most stable, reproducible, and insightful. TPS1 overexpression was extended to an industrial yeast genetic background, using graded expression in a growth stage-specific vector system. The results from fermentations conducted with these strains show successful (moderate and stage-specific) overexpression of the TPS1 gene leading to increased trehalose accumulation by these strains. The downstream effect of this modification was the desired metabolic target of reduced final ethanol and ethanol yield. This corroborates the findings of the deletion screen and provides the first example of a successful commercial wine yeast engineering strategy targeting trehalose as a means for lowering ethanol levels. The significant decreases in ethanol in these fermentations were not associated with any of the usual pitfalls related to redox disruption and stuck fermentations, confirming the suitability of the controlled promoter strategy for genetic engineering strategies in wine yeast. The current study was thus successful in terms of validating the screening method for the identification of genes that affect carbohydrate flux, the identification of deletion strains that alter final fermented ethanol yield, and the provision of a platform for application to industrial yeast tailored for lower-ethanol fermentations. The data also suggest that specifically targeting trehalose metabolism as part of a breeding and selection strategy is an option for generating non-genetically modified wine yeast strains with lower ethanol yields. The present study can be extended to include other potential targets identified in the initial mutant screens.
Supplementary Material
ACKNOWLEDGMENTS
Funding for this study was provided by WineTech, the National Research Foundation, Stellenbosch University, and the Claude Leon Foundation.
Footnotes
Published ahead of print 21 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00964-13.
REFERENCES
- 1.Hoffmann D. 1990. The European wine market up to the year 2000—trends and marketing efforts, p 138–153 Proceedings of the 9th International Oenological Symposium, Cascais, Portugal, 24 to 26 May 1990 [Google Scholar]
- 2.Pickering GJ, Heatherbell DA, Barnes MF. 1998. Optimising glucose conversion in the production of reduced alcohol wine using glucose oxidase. Food Res. Int. 31:685–692 [Google Scholar]
- 3.Varela C, Kutyna DR, Solomon MR, Black CA, Borneman A, Henschke PA, Pretorius IS, Chambers PJ. 2012. Evaluation of gene modification strategies for the development of low-alcohol-wine yeasts. Appl. Environ. Microbiol. 78:6068–6077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Guth H, Sies A. 2002. Flavour of wines: towards an understanding by reconstitution experiments and an analysis of ethanol's effect on odour activity of key compounds, p 128–139 In Blair RJ, Williams PJHø, j PB. (ed), Proceedings of the 11th Australian Wine Industry Technical Conference Australian Wine Industry Technical Conference, Inc., Adelaide, Australia [Google Scholar]
- 5.de Barros Lopes M, Eglinton J, Henschke P, Høj P, Pretorius I. 2003. The connection between yeast and alcohol: managing the double-edged sword of bottled sunshine. Aust. N. Z. Wine Ind. J. 18:17–22 [Google Scholar]
- 6.Cordier H, Mendes F, Vasconcelos I, Francois JM. 2007. A metabolic and genomic study of engineered Saccharomyces cerevisiae strains for high glycerol production. Metab. Eng. 9:364–378 [DOI] [PubMed] [Google Scholar]
- 7.Luyten K, Albertyn J, Skibbe W, Prior B, Ramos J, Thevelein J. 1995. Fps1, a yeast member of the MIP family of channel proteins, is a facilitator for glycerol uptake and efflux and is inactive under osmotic stress. EMBO J. 14:1360–1371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Barros Lopes M, Rehman A, Gockowiak H, Heinrich A, Langridge P, Henschke P. 2000. Fermentation properties of a wine yeast overexpressing the Saccharomyces cerevisiae glycerol 3-phosphate dehydrogenase gene (GPD2). Aust. J. Grape Wine Res. 6:208–215 [Google Scholar]
- 9.Cambon B, Monteil V, Remize F, Camarasa C, Dequin S. 2006. Effects of GPD1 overexpression in Saccharomyces cerevisiae commercial wine yeast strains lacking ALD6 genes. Appl. Environ. Microbiol. 72:4688–4694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Drewke C, Thielen J, Ciriacy M. 1990. Ethanol formation in adh0 mutants reveals the existence of a novel acetaldehyde-reducing activity in Saccharomyces cerevisiae. J. Bacteriol. 172:3909–3917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leskovac V, Trivic S, Pericin D. 2002. The three zinc-containing alcohol dehydrogenases from baker's yeast, Saccharomyces cerevisiae. FEMS Yeast Res. 2:481–494 [DOI] [PubMed] [Google Scholar]
- 12.Nevoigt E, Stahl U. 1996. Reduced pyruvate decarboxylase and increased glycerol-3-phosphate dehydrogenase [NAD+] levels enhance glycerol production in Saccharomyces cerevisiae. Yeast 12:1331–1337 [DOI] [PubMed] [Google Scholar]
- 13.Riou C, Nicaud J-M, Barre P, Gaillardin C. 1997. Stationary-phase gene expression in Saccharomyces cerevisiae during wine fermentation. Yeast 13:903–915 [DOI] [PubMed] [Google Scholar]
- 14.Rossignol T, Dulua L, Julien A, Blondin B. 2003. Genome-wide monitoring of wine yeast gene expression during alcoholic fermentation. Yeast 20:1369–1385 [DOI] [PubMed] [Google Scholar]
- 15.Varela C, Cardenas J, Melo F, Agosin E. 2005. Quantitative analysis of wine yeast gene expression profiles under winemaking conditions. Yeast 22:369–383 [DOI] [PubMed] [Google Scholar]
- 16.Marks VD, Sui SJH, Erasmus D, Van der Merwe GK, Brumm J, Wasserman WW, Bryan J, van Vuuren HJJ. 2008. Dynamics of the yeast transcriptome during wine fermentation reveals a novel fermentation stress response. FEMS Yeast Res. 8:35–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trabalzini L, Paffetti A, Scaloni A, Talamo F, Ferro E, Coratza G, Bovalini L, Lusini P, Martelli P, Santucci A. 2003. Proteomic response to physiological fermentation stresses in a wild-type wine strain of Saccharomyces cerevisiae. Biochem. J. 370:35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Howell KS, Cozzolino D, Bartowsky EJ, Fleet GH, Henschke PA. 2006. Metabolic profiling as a tool for revealing Saccharomyces interactions during wine fermentation. FEMS Yeast Res. 6:91–101 [DOI] [PubMed] [Google Scholar]
- 19.Zuzuarregui A, Monteoliva L, Gil C, del Olmo M. 2006. Transcriptomic and proteomic approach for understanding the molecular basis of adaptation of Saccharomyces cerevisiae to wine fermentation. Appl. Environ. Microbiol. 72:836–847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rossouw D, van den Dool AH, Jacobson D, Bauer FF. 2010. Comparative transcriptomic and proteomic profiling of industrial wine yeast strains. Appl. Environ. Microbiol. 76:3911–3923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Beltran G, Novo M, Leberre V, Sokol S, Labourdette D, Guillamon JM, Mas A, Francois J, Rozes N. 2006. Integration of transcriptomic and metabolic analyses for understanding the global responses of low-temperature winemaking fermentations. FEMS Yeast Res. 6:1167–1183 [DOI] [PubMed] [Google Scholar]
- 22.Rossouw D, Naes T, Bauer FF. 2008. Linking gene regulation and the exo-metabolome: a comparative transcriptomics approach to identify genes that impact on the production of volatile aroma compounds in yeast. BMC Genomics 9:530–548. 10.1186/1471-2164-9-530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rossouw D, Olivares-Hernandes R, Nielsen J, Bauer FF. 2009. Comparative transcriptomic approach to investigate differences in wine yeast physiology and metabolism during fermentation. Appl. Environ. Microbiol. 75:6600–6612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Varela C, Pizarro F, Agosin E. 2004. Biomass content governs fermentation rate in nitrogen-deficient wine musts. Appl. Environ. Microbiol. 70:3392–3400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wenzel TJ, Migliazza A, Steensma HY, van den Berg JA. 1992. Efficient selection of phleomycin-resistant Saccharomyces cerevisiae transformants. Yeast 8:667–668 [DOI] [PubMed] [Google Scholar]
- 26.Lilly M, Bauer FF, Styger G, Lambrechts MG, Pretorius IS. 2006. The effect of increased branched-chain amino acid transaminase activity in yeast on the production of higher alcohols and on the flavour profiles of wine and distillates. FEMS Yeast Res. 6:726–743 [DOI] [PubMed] [Google Scholar]
- 27.Bely M, Sablayrolles JM, Barre P. 1990. Automatic detection of assimilable nitrogen deficiencies during alcoholic fermentation in enological conditions. J. Ferment. Bioeng. 70:246–252 [Google Scholar]
- 28.Schmitt ME, Brown TA, Trumpower BL. 1990. A rapid and simple method for preparation of RNA from Saccharomyces cerevisiae. Nucleic Acids Res. 18:3091–3092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ramakers C, Ruijter JM, Deprez RH, Moorman AF. 2003. Assumption-free analysis of quantitative real-time polymerase chain reaction (PCR) data. Neurosci. Lett. 339:62–66 [DOI] [PubMed] [Google Scholar]
- 30.Yoshikawa Y, Matsumoto K, Nagata K, Sato T. 1994. Extraction of trehalose from thermally-treated bakers' yeast. Biosci. Biotechnol. Biochem. 58:1226–1230 [Google Scholar]
- 31.Teusink B, Passarge J, Reijenga CA, Esgalhado E, van der Weijden CC, Schepper M, Walsh MC, Bakker BM, van Dam K, Westerhoff HV, Snoep JL. 2000. Can yeast glycolysis be understood in terms of in vitro kinetics of the constituent enzymes? Testing biochemistry. Eur. J. Biochem. 267:5313–5329 [DOI] [PubMed] [Google Scholar]
- 32.Noble AC, Bursick GF. 1984. The contribution of glycerol to perceived viscosity and sweetness in white wine. Am. J. Enol. Vitic. 35:110–112 [Google Scholar]
- 33.Xu Z, Tsurugi K. 2007. Destabilization of energy-metabolism oscillation in the absence of trehalose synthesis in the chemostat culture of yeast. Arch. Biochem. Biophys. 464:350–358 [DOI] [PubMed] [Google Scholar]
- 34.McAlister L, Holland MJ. 1985. Differential expression of the three yeast glyceraldehyde-3-phosphate dehydrogenase genes. J. Biol. Chem. 260:15019–15027 [PubMed] [Google Scholar]
- 35.Francois J, Parrou JL. 2001. Reserve carbohydrates metabolism in the yeast Saccharomyces cerevisiae. FEMS Microbiol. Rev. 25:125–145 [DOI] [PubMed] [Google Scholar]
- 36.Blomberg A, Adler L. 1992. Physiology of osmotolerance in fungi. Adv. Microb. Physiol. 33:145–212 [DOI] [PubMed] [Google Scholar]
- 37.Hohmann S. 2002. Osmotic stress signalling and osmoadaptation in yeasts. Microbiol. Mol. Biol. Rev. 66:300–372 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.