

Abstract
Recently, a novel group of [NiFe]-hydrogenases has been defined that appear to have a great impact in the global hydrogen cycle. This so-called group 5 [NiFe]-hydrogenase is widespread in soil-living actinobacteria and can oxidize molecular hydrogen at atmospheric levels, which suggests a high affinity of the enzyme toward H2. Here, we provide a biochemical characterization of a group 5 hydrogenase from the betaproteobacterium Ralstonia eutropha H16. The hydrogenase was designated an actinobacterial hydrogenase (AH) and is catalytically active, as shown by the in vivo H2 uptake and by activity staining in native gels. However, the enzyme does not sustain autotrophic growth on H2. The AH was purified to homogeneity by affinity chromatography and consists of two subunits with molecular masses of 65 and 37 kDa. Among the electron acceptors tested, nitroblue tetrazolium chloride was reduced by the AH at highest rates. At 30°C and pH 8, the specific activity of the enzyme was 0.3 μmol of H2 per min and mg of protein. However, an unexpectedly high Michaelis constant (Km) for H2 of 3.6 ± 0.5 μM was determined, which is in contrast to the previously proposed low Km of group 5 hydrogenases and makes atmospheric H2 uptake by R. eutropha most unlikely. Amperometric activity measurements revealed that the AH maintains full H2 oxidation activity even at atmospheric oxygen concentrations, showing that the enzyme is insensitive toward O2.
INTRODUCTION
The global hydrogen cycle is based on numerous natural as well as man-made sources and sinks of molecular hydrogen (H2), which lead to an average tropospheric H2 concentration of 0.5 ppmv (1, 2). Soil has been identified as the greatest H2 sink, which removes approximately 50 Tg of H2 per year from the atmosphere. This represents ca. 80% of the annual total tropospheric H2 consumption (1, 3). However, until recently little was known about the nature and mechanism of the H2-scavenging capacity of soils. In 2008, spore-forming soil bacteria belonging to the genus Streptomyces were found to be capable of hydrogen uptake at very low H2 concentrations (4), and it was suggested that these ubiquitous bacterial species are responsible for the soil-based hydrogen sink (5).
The bacterial H2 metabolism is governed by metalloenzymes designated as hydrogenase. Hydrogenases catalyze the reversible cleavage of dihydrogen into two protons and two electrons. They either support the oxidation of H2 as a valuable energy source or function as an electron valve during fermentative growth leading to the evolution of H2 (6, 7). In the majority of cases, H2 uptake is catalyzed by the large class of [NiFe]-hydrogenases. According to their physiological role, subunit composition and cellular localization, [NiFe]-hydrogenases are further subdivided into four groups. Group 1 includes periplasmic and membrane-bound uptake hydrogenases. Group 2 comprises cyanobacterial uptake and cytoplasmic H2-sensing hydrogenases. Group 3 contains complex cytoplasmic enzymes, which interact reversibly with pyridine nucleotides or flavin derivatives, whereas group 4 consists of membrane-associated, H2-evolving multisubunit hydrogenases (6).
Interestingly, genome analysis of Streptomyces species that are capable of high-affinity H2 uptake revealed the presence of genes encoding a putative [NiFe]-hydrogenase, which is clearly distinct from members of groups 1 to 4 (8). Homologues of these genes have been found mainly in the genomes of members of the phylum Actinobacteria, but also in some Proteobacteria, Chloroflexi, and Acidobacteria. Due to particular structural features and the anticipated high affinity toward hydrogen, this group of [NiFe]-hydrogenases was classified as the novel group 5 hydrogenases (8).
Group 5 hydrogenases display the basic structural features of [NiFe]-hydrogenase: an active site-containing large subunit and a small subunit mediating the transfer of electrons from the active site to the protein surface and finally to an external electron acceptor of an as-yet-unknown nature. In almost all bacterial species encoding group 5 hydrogenases, the genes for the small and large subunits are clustered with a set of hydrogenase maturation genes (hypABCDEF and hupD), and three to four open reading frames coding for conserved proteins of unknown function (Fig. 1) (8). The maturation proteins HypABCDEF are mandatory for all [NiFe]-hydrogenases and are responsible for assembly and insertion of the [NiFe] active site into the large subunit (9, 10). The hupD gene product resembles the hydrogenase-specific endopeptidases, which remove the C-terminal extension of the large subunit precursor after the [NiFe] cofactor has been inserted (9, 11). In contrast to group 1 hydrogenases, a twin-arginine translocation signal peptide is missing in group 5 hydrogenases pointing to a cytoplasmic location of the enzyme (12).
Fig 1.

(A) Gene cluster encoding the actinobacterial hydrogenase (AH) of R. eutropha, which represents a typical group 5 hydrogenase. The cluster consists of genes encoding the two hydrogenase subunits, four proteins of unknown function, an endoprotease required for proteolytic maturation of the large hydrogenase subunit and a complete set of Hyp proteins responsible for assembly of the [NiFe] cofactor. (B) Schematic representation of a group 5 [NiFe]-hydrogenase. The [NiFe] center is embedded in the large subunit and three [FeS] clusters within the small subunit, which constitute an intramolecular electron transport pathway to an as-yet-unknown electron acceptor, which further transfers the electrons into the respiratory chain.
Until now, little is known about the fundamental biochemical properties of group 5 hydrogenases. Studies with crude extracts from aerobic sporulating Streptomyces strain PCB7 mycelia revealed similar hydrogen uptake rates under aerobic and anaerobic conditions (5). In Mycobacterium smegmatis, an upregulation of group 5 hydrogenase genes was observed under carbon-limited growth and under hypoxia. A corresponding M. smegmatis mutant, carrying a hydrogenase gene deletion, exhibited significantly slower growth rates in continuous culture and reached lower optical densities in batch cultures compared to the wild-type strain (13). However, thus far, no group 5 hydrogenase has been purified and characterized biochemically.
The genome sequence of the well-studied Knallgas bacterium Ralstonia eutropha H16 revealed the presence of a gene cluster with striking similarity to that of group 5 hydrogenases (8, 14, 15). R. eutropha H16 harbors three [NiFe]-hydrogenases involved in energy conversion and hydrogen sensing under aerobic conditions. These have been purified and characterized in great detail (10, 16, 17, 18). Here, we present the first purification and biochemical characterization of a group 5 hydrogenase, performed on the actinobacterial hydrogenase (AH) of Ralstonia eutropha.
MATERIALS AND METHODS
Strains and plasmids.
Bacterial strains and plasmids used in the present study are listed in Table 1. Primer sequences are listed in Table S1 in the supplemental material. Escherichia coli JM109 (19) was used as a host in standard cloning procedures and E. coli S17-1 (20) served as a donor in conjugative transfers. Strains carrying the letters HF are derivatives of the wild-type strain Ralstonia eutropha H16.
Table 1.
Bacterial strains and plasmids used in this study
| Strain or plasmid | Relevant characteristics and/or genotypea | Source or reference |
|---|---|---|
| Strains | ||
| R. eutropha | ||
| HF371 (RH+, AH+) | hoxGΔ hoxHΔ; MBH− SH− RH+ AH+ | 51 |
| HF500 (AH+) | hoxGΔ hoxHΔ hoxCΔ; MBH− SH− RH− AH+ | 26 |
| HF579 | hypA1B1F1C1D1E1XΔ hypA2B2F2Δ hoxGΔ hoxHΔ; MBH− SH− RH− AH+ | 52 |
| HF864 (AH++) | hoxGΔ hoxHΔ hoxCΔ, PMBH-hofK; MBH− SH− RH− AH++ | This study |
| HF866 (AH−) | hoxGΔ hoxHΔ hoxCΔ hofGΔ; MBH− SH− RH− AH− | This study |
| HF868 (AH−, RH+) | hoxGΔ hoxHΔ hofGΔ; MBH− SH− AH− RH+ | This study |
| HF901 (AHStrep) | hoxGΔ hoxHΔ hoxCΔ, PMBH-StrephofK; MBH− SH− RH− AH++ | This study |
| E. coli | ||
| JM109 | F′ traD36 lacIq Δ(lacZ)M15 proA+B+/e14− (McrA−) Δ(lac-proAB) thi gyrA96 (Nalr) endA1 hsdR17(rK− mK+) relA1 supE44 recA1 | 19 |
| S17-1 | Tra+ recA pro thi hsdR chr::RP4-2 | 20 |
| Plasmids | ||
| pET24a | Kmr PT7 expression vector | Novagen |
| pLO1 | Kmr sacB, RP4 oriT, ColE1 ori | 21 |
| pCH1676 | PMBH with flanking sequences of 0.8 kb upstream of the AH operon and N-terminal part of hofK in pET24a | This study |
| pGE814 | 3.0-kb EcoRV-SmaI fragment from pCH1676 in pLO1 | This study |
| pGE815 | 3.3-kb ′hofK-hofG-PHG066 fragment in pLO1 | This study |
| pGE816 | Religated 9.5-kb fragment from SexAI-digested pGE815 | This study |
| pGE817 | 2.2-kb ′hofK-Strep-hofG′ fragment in pLO1 | This study |
Kmr, kanamycin resistance.
The strain R. eutropha HF500 (AH+) carries the AH genes under the native promoter in a background free of all of the other hydrogenases. In R. eutropha HF864 (AH++), the native promoter of the AH was exchanged with the strong promoter from the membrane-bound hydrogenase (MBH) of R. eutropha (PMBH). A 0.8-kb fragment upstream from the AH operon (primers 3 and 4), a 0.8-kb fragment carrying the N-terminal part of hofK (primers 5 and 6) and a 0.3-kb fragment carrying the PMBH (primers 1 and 2) were amplified by PCR with the R. eutropha megaplasmid pHG1 as a template. The fragments were sequentially inserted into the pET24a vector resulting in plasmid pCH1676. A 3.0-kb EcoRV/SmaI fragment was cut from pCH1676 and subsequently ligated into the PmeI-linearized suicide vector pLO1, resulting in plasmid pGE814. The desired modification was inserted into R. eutropha HF500 via conjugative transfer with this vector.
A strain lacking the large subunits of all hydrogenases was constructed from R. eutropha HF500 (AH+) by isogenic deletion of a 1.1-kb fragment from the AH large subunit hofG. For this purpose, a 3.3-kb fragment carrying hofG and several hundred base pairs upstream and downstream of the gene was amplified from pHG1 using the primers 8 and 9. The fragment was digested with XbaI and ligated into XbaI-cut vector pLO1. From the resulting plasmid pGE815, a 1,131-bp SexAI fragment was excised, and the remaining fragment was religated, yielding plasmid pGE816. The hofG deletion allele was inserted into the strains R. eutropha HF500 (AH+) and HF371 (RH+ AH+) by double homologous recombination as described previously (21). This procedure yielded the strains R. eutropha HF866 (AH−) and HF868 (AH− RH+).
For facile purification of the AH, a Strep-Tag II-encoding sequence was fused to the 3′ end of hofK and inserted into the strain R. eutropha HF864 (AH++), resulting in strain HF901 (AHStrep). Therefore, two 1.1-kb fragments carrying the 3′ part of hofK (primers 12 and 13) and the 5′ region of hofG, respectively (primers 14 and 15), were amplified by PCR with pHG1 as a template. The first fragment was digested with NdeI and MluI and inserted into pLO1. The second fragment was inserted into the resulting plasmid by using the restriction sites XbaI and MluI, yielding pGE817. The resulting hofK allele was inserted into R. eutropha HF864 by double homologous recombination.
Media and growth conditions.
E. coli strains were grown in Luria-Bertani (LB) medium; solid media contained 1.5% (wt/vol) agar. Kanamycin was used at a concentration of 30 μg per ml of culture. Precultures of R. eutropha strains were grown in a mineral salts medium (21) containing 0.4% fructose (FN medium) for 48 h at 30°C and 120 rpm. Main cultures were grown in FGN medium (0.2% fructose and 0.2% glycerol) in baffled Erlenmeyer flasks (filled to 20% with medium) at 30°C and 120 rpm until an optical density at 436 nm (OD436) of approximately 11 was reached (approximately 48 h). For AH purification, cultures were grown in FGNmod medium (0.05% fructose and 0.4% glycerol [22]). An amount of 4 liters of culture medium was deposited in baffled 5-liter Erlenmeyer flasks, which were then shaken at 120 rpm at 30°C for approximately 7 days. Cells were harvested by centrifugation (6,800 × g, 4°C, 12 min). The resulting cell pellet was frozen in liquid N2 and stored at −80°C.
Purification.
Cell pellets were resuspended in 2.5 ml of resuspension buffer (50 mM potassium phosphate [pH 7.2]) per 1 g of cells (wet weight), containing protease inhibitor mixture (Complete EDTA-Free; Roche) and DNase I. The resuspended cells were disrupted in a French pressure cell (Constant Cell Disruption Systems). Cell debris and membranes were sedimented by ultracentrifugation (100,000 × g, 4°C, 45 min), yielding soluble protein extracts in the supernatant.
For fast purification of the AHStrep protein, with three remaining impurities, the soluble extract was loaded on a Strep-Tactin Superflow column (IBA, Göttingen, Germany). Then, 2 ml of bed volume was used for 25 ml of soluble extract. The column was washed with 10 bed volumes of washing buffer (50 mM potassium phosphate [pH 7.2], 150 mM NaCl); the AHStrep protein was eluted with six bed volumes of elution buffer (washing buffer with 2.5 mM desthiobiotin). The eluate was concentrated by centrifugation (3,300 × g, 4°C, 20 min) using a centrifugal filter device (Amicon Ultra Ultracel 30K; Millipore). The resulting concentrate was frozen in liquid nitrogen and stored at −80°C. Protein concentrations were determined with the BCA method, using bovine serum albumin as a standard (23). The samples were checked for purity by sodium dodecyl sulfate (SDS)-polyacrylamide gels stained with Coomassie brilliant blue G-250.
To obtain the AHStrep protein in high purity, the soluble extract was adjusted to pH 7.2 with diluted KOH and loaded on a Strep-Tactin Superflow column (IBA), using 1 ml of bed volume for 25 ml of soluble extract. The column was washed with 10 bed volumes of washing buffer II (50 mM potassium phosphate [pH 6.7], 150 mM NaCl), the washing fraction was adjusted to pH 7.2 and loaded on a second Strep-Tactin Superflow column with the same bed volume as the first column. After a washing step with 10 bed volumes washing buffer (50 mM potassium phosphate [pH 7.2], 150 mM NaCl), the pure AHStrep protein was obtained by elution with six bed volumes of elution buffer (washing buffer with 2.5 mM desthiobiotin) and treated as described above.
Reverse transcription-quantitative PCR (RT-qPCR).
For quantification of AH transcripts, cells of R. eutropha strains were grown in FGN medium to an OD436 of 8. Total RNA isolation and reverse transcription to cDNA was done as described previously (24). Quantitative PCR was performed on the obtained cDNA using SYBR green fluorescent dye and a 7500 Fast PCR Cycler (Applied Biosystems). The PCR was done in a total volume of 5 μl containing 5 ng of cDNA, 1× SYBR green reaction mix and 500 nM concentrations of each primer. The temperature profile was composed of 2 min at 50°C and 20 s at 95°C as initial steps, followed by 40 cycles of 3 s at 95°C and 30 s at 60°C. The primer pairs 391/392 and 105/106 were used for hofK and hoxK cDNA amplification, respectively (see Table S1 in the supplemental material). The amplified fragments had a length of 150 to 200 bp. Control runs with serially diluted cDNAs revealed PCR amplification efficiencies of 100% ± 10% for both primer pairs. Dissociation curves confirmed the uniformity of the PCR products. The gyrB gene, which is constitutively expressed in R. eutropha, was used as control for the relative quantification of the gene expression. The data were calculated from two biological replicates each with three technical replicates.
Size determination.
The molecular mass of native AH complexes was determined by gel permeation chromatography. A total of 500 μg of purified protein was separated on a gel permeation column (Superdex 200 HR10/30, approximately 20 ml column volume) with 50 mM potassium phosphate (pH 7.0)–150 mM NaCl as the eluent. The absorption was monitored at 280 nm (protein absorption) and 405 nm ([FeS] cluster absorption). The molecular masses of the obtained maxima were determined by comparison with a standard curve of the proteins thyroglobulin (669 kDa), apoferritin (443 kDa), β-amylase (200 kDa), and bovine serum albumin (66 kDa). The protein composition of the absorption maxima was analyzed by SDS-PAGE.
Metal determination.
The content of the metals Fe, Ni, Cu, and Zn in the purified protein samples was analyzed by inductively coupled plasma optical emission spectroscopy (ICP-OES), as previously described (25).
Activity assays.
H2 uptake of living cells was measured from the headspace over a cell culture. For this, cells were grown heterotrophically in FGN medium for 48 h to an OD436 of ∼10. Volumes equivalent to 1,000 OD units were adjusted to a total volume of 115 ml and transferred in gas-tight bottles with a headspace of 1.03l air or N2 (headspace to liquid culture, 9:1). Depending on the experiment, 800 ppm of H2 (∼32 μM), 0.3% H2 (∼120 μM), or 2% H2 (∼800 μM) were added to the headspace; the bottles were incubated at 30°C and 160 rpm. The amount of H2 in the headspace was determined by thermal conductivity in a gas chromatograph (GC-2014; Shimadzu) over a period of several hours. The peak areas obtained upon injection of various volumes of H2 were used for calibration. To circumvent inaccuracies from the injection process, the peak areas for H2 were normalized to a constant peak area of N2. The detection limit was approximately 12 ppm of H2 (0.5 μM).
H2 uptake activities were also shown by chromogenic detection in native polyacrylamide gels. Soluble extracts or purified protein samples were resolved in 4 to 15% native gradient PAGE gels. The gels were then transferred to a sealed glass bottle containing H2-saturated 50 mM potassium phosphate buffer (pH 7.0). The headspace was filled with 100% H2 (26). Hydrogenase activity was visualized through addition of 0.15 mM nitroblue tetrazolium chloride (NBT) and subsequent incubation of the gel for approximately 3 h at 37°C.
A spectrophotometric assay was used to quantify H2-uptake activities of purified AH using NBT as artificial electron acceptor. The assay was performed in H2-saturated buffer with 75 μM NBT at 30°C. Reduction of NBT was monitored at 593 nm. One unit of enzyme activity is defined as the oxidation of 1 μmol of H2 or the reduction of 1 μmol of NBT, respectively, per min and per mg of enzyme. Most measurements were performed in 50 mM Tris-HCl (pH 8.0). For measurements at different pH values, the following buffers were used: 50 mM potassium phosphate for pH 5.5 to 8, 50 mM Tris-HCl for pH 8 to 10, 50 mM Capso [3-(cyclohexylamino)-2-hydroxy-1-propanesulfonic acid]-HCl for pH 9 to 12. For the determination of the acceptor specificity of the AH, the spectrophotometric assay was performed with different electron acceptors at specific wavelengths, as listed in Table 2.
Table 2.
Electron acceptors tested for the H2-oxidizing activity of AH
| Acceptor | Wavelength (nm) | Mean AH activity (U mg−1) ± SD |
|---|---|---|
| NAD+ | 365 | 0.00 |
| FMN | 450 | 0.07 ± 0.02 |
| FAD | 450 | 0.09 ± 0.03 |
| Duroquinone | 255 | 0.08 ± 0.01 |
| Ferricyanide | 405 | 0.12 ± 0.03 |
| Menadione | 360 | 0.13 ± 0.02 |
| Methylene blue | 570 | 0.17 ± 0.04 |
| Phenazine methosulfate | 388 | 0.18 ± 0.03 |
| Methyl viologen | 578 | 0.23 ± 0.03 |
| Nitroblue tetrazolium chloride | 593 | 0.28 ± 0.02 |
For the determination of oxygen tolerance, H2-uptake activities of the AH were quantified amperometrically with a Clark-type electrode (Oxygraph; Hansatech Instruments) adapted for H2 measurements (27). H2- and O2-saturated buffers (both 50 mM Tris-HCl [pH 8]) were mixed in ratios between 100:0% and 30:70% (O2 concentrations, 0 to 162 μM). A 75 μM concentration of NBT was applied as electron acceptor. All measurements were performed at 30°C.
RESULTS
The AH of R. eutropha is catalytically active.
The operon encoding the “actinobacterial hydrogenase” (AH) of R. eutropha H16 has been identified in the course of sequencing of the indigenous megaplasmid pHG1 (Fig. 1) (14). However, a mutant derivative lacking the membrane-bound hydrogenase (MBH), the soluble, NAD+-reducing hydrogenase (SH) and the H2-sensing regulatory hydrogenase (RH) was unable to grow autotrophically on H2 as the sole energy source indicating that the AH does not sustain lithoautotrophic growth. Soluble extract derived from these cells showed no H2 uptake activity with methylene blue as artificial electron acceptor (26). Moreover, proteome analyses of cell extracts derived from R. eutropha provided no evidence for the synthesis of AH-related gene products (24, 28). These observations suggested that the AH of R. eutropha is synthesized, if at all, only at very low levels.
To enhance expression of the AH gene cluster, the operon was placed under the control of the strong promoter of the MBH operon (PMBH). The native promoter was replaced by inserting the PMBH directly upstream of a plasmid-borne copy of the hofK gene, which encodes for the AH small subunit. A fragment containing PMBH-hofK was cut from this plasmid and inserted into the suicide vector pLO1. A derivative of R. eutropha (AH+), deleted for the large subunit genes of the three well-defined hydrogenases (MBH, SH, and RH), served as recipient for the resulting plasmid. The PMBH-hofK fragment was established on the megaplasmid pHG1 of R. eutropha AH+ by double homologous recombination yielding strain R. eutropha AH++. Furthermore, an isogenic deletion in the AH large subunit hofG was established in R. eutropha AH+ yielding strain R. eutropha AH−, which was used as negative control in the following experiments. The same deletion was also established in R. eutropha AH+ RH+.and the resulting strain was named R. eutropha AH− RH+.
The R. eutropha derivatives AH+, AH++, and AH− were cultivated under standard hydrogenase-derepressing conditions in fructose-glycerol minimal medium, which leads to high activity of the PMBH promoter (22, 29). Soluble extracts were prepared and separated on a native polyacrylamide gel. Subsequently, an in-gel hydrogenase activity staining was performed using NBT as artificial electron acceptor. As depicted in Fig. 2A, both the AH+ and the AH++ strains displayed signals that were not observed in the AH− strain, showing that the AH is functional in H2 oxidation. Furthermore, placing expression of the AH operon under the control of PMBH resulted in a considerable increase of activity compared to the AH+ strain. Nevertheless, the result clearly shows that the AH is not only active when synthesized under the control of the artificial PMBH promoter but also under its native promoter.
Fig 2.
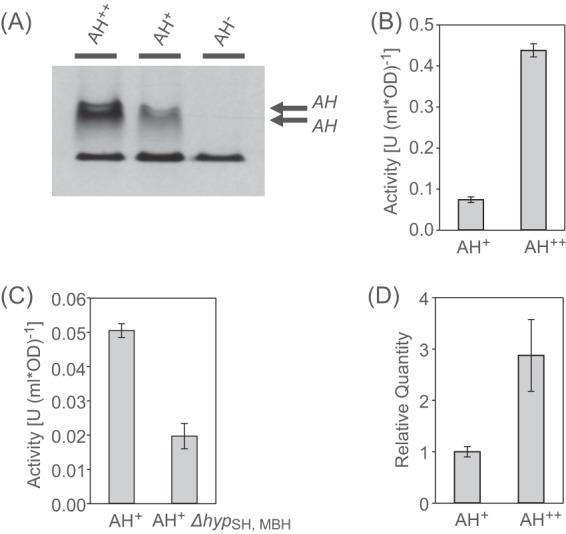
Activity of the R. eutropha AH. (A) Activities of the AH+ and AH++ strains, visualized by in-gel hydrogenase activity staining. Soluble cell extracts were separated electrophoretically in a native PAGE gel, which was subsequently incubated under H2. The reduction of NBT is documented by a dark blue color. The lower band in all lanes represents a H2-independent nonspecific reaction always present in R. eutropha soluble cell extracts. (B) In vivo H2 uptake activities of the AH+ and the AH++ strains. The hydrogen concentration in the headspace of different R. eutropha cultures was followed by gas chromatography, starting with 0.3% H2. (C) In vivo H2 uptake activities of the AH+ and the ΔhypSH, MBH cells. (D) Relative transcript quantities of the small subunit hofK in the AH+ and the AH++ cells as determined by RT-qPCR.
In order to assess whether the activity of the AH plays a physiological role, the H2 uptake activity of living cells from the R. eutropha strains AH+, AH++, AH−, and AH− RH+ was monitored by gas chromatography. Cells were grown under standard conditions to an optical density (OD) at 436 nm of about 10. A culture volume equivalent to 1,000 OD · ml was placed in a sealed flask and incubated under an atmosphere consisting of air supplemented with 0.3% (vol/vol) H2. H2 consumption was detected for both the AH+ and the AH++ strains, although, as expected, at different levels. The AH+ strain exhibited an activity of 0.074 ± 0.007 U (1,000 OD · ml)−1, whereas the AH++ strain showed an ∼6-fold higher activity of 0.438 ± 0.016 U (1,000 OD · ml)−1 (Fig. 2B and see Fig. S1 in the supplemental material). Both strains consumed H2 beyond the detection limit of the gas chromatograph (∼12 ppm). Interestingly, no H2 uptake was detected when cultures synthesizing active AH were incubated under hydrogen in the absence of oxygen (0.3% [vol/vol] H2 in N2). H2 consumption was restored, however, by injecting oxygen into the headspace of the culture vessels. (see Fig. S2 in the supplemental material). As expected, the AH− strain showed no H2 uptake activity under any conditions. Notably, strain R. eutropha AH−RH+, which contains only regulatory hydrogenase, did not show any H2 uptake activity (see Fig. S3 in the supplemental material). These observations indicate a physiological role of the AH that is distinct from the regulatory task of the RH. They suggest that the AH oxidizes hydrogen and transfers the resulting electrons via a native electron acceptor of unknown nature into the respiratory chain in which oxygen serves as an terminal electron acceptor.
Subsequently, the H2 consumption of the AH+ strain was compared to that of an R. eutropha derivative lacking all hyp genes located in the gene clusters of MBH, RH, and SH (ΔhypMBH,RH,SH). The ΔhypMBH,RH,SH strain displayed H2 consumption of ca. 40% of the AH+ level (Fig. 2C; see Fig. S4 in the supplemental material). This result supports the conclusion that the hyp genes of the AH gene cluster are coexpressed and instrumental in AH maturation.
To quantify the enhancement on AH gene expression mediated by the PMBH promoter, the transcript level of the AH small subunit gene was determined by quantitative reverse transcription-PCR (RT-qPCR). Transcripts of hofK were detected in both the AH+ and the AH++ strain. When under the control of the native promoter, the hofK transcript level amounted to ca. 5% of the transcript level of the MBH small subunit hoxK (see Table S2 in the supplemental material). The presence of PMBH resulted in an increase of the hofK transcript level by a factor of 3 (Fig. 2D).
Purification and structural features of the AH.
For a detailed characterization of the AH, the protein was purified by affinity chromatography. A Strep-tag II-encoding sequence was fused to the 3′ end of the AH small subunit hofK (designated hofKStrep) as described in Materials and Methods. The resulting hofK allele was transferred to the vector pLO1, which was used to introduce the hofKStrep copy into the megaplasmid pHG1 of R. eutropha by homologous recombination.
Strep-tagged AH (AHStrep) was purified from soluble extract by means of Strep-Tactin affinity chromatography. The resulting protein fraction was subjected to SDS-polyacrylamide electrophoresis. Five bands corresponding to molecular masses of approximately 130, 70, 65, 60, and 37 kDa were identified (Fig. 3). From the amino acid sequences of the small and large subunits, molecular masses of 37.8 kDa for HofK and 67.9 kDa for HofG were extrapolated (apo-proteins). The identity of the contaminants with molecular masses of 60 and 70 kDa has been described previously (30). These proteins bind to the Strep-Tactin matrix if the protein of interest is produced only at low concentration.
Fig 3.
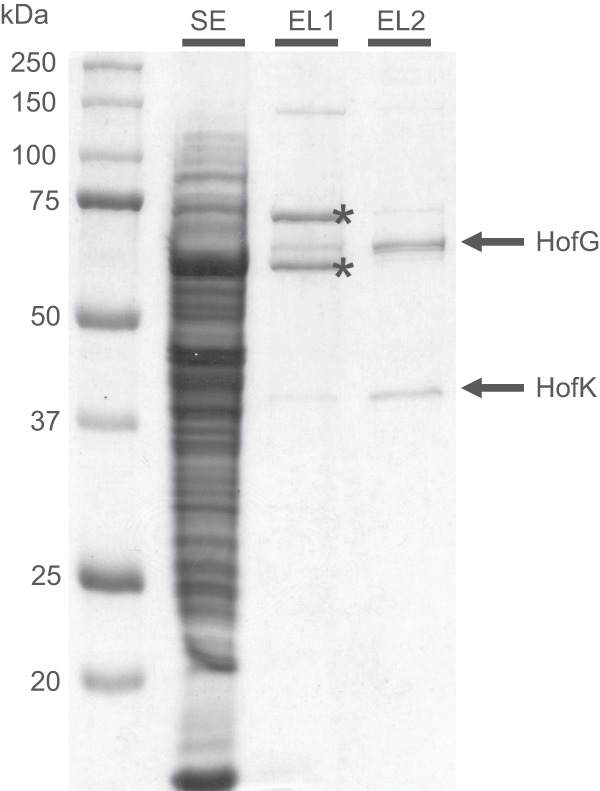
Purification of the AH by Strep-Tactin affinity chromatography. Soluble extract (lane SE, 25 μg of protein) was applied to a Strep-Tactin column and bound protein was eluted with a slightly acidic (pH 6.7) buffer (lane EL1, 2 μg of protein). The elution fraction was adjusted to pH 7.2 and again applied to a Strep-Tactin column. The rebound AH was then eluted with desthiobiotin-containing buffer (lane EL2, 2 μg of protein). The fractions were separated by SDS-PAGE and proteins were visualized by Coomassie blue staining. The identity of the major contaminant protein bands in lane EL1 (marked with asterisks) was described in reference 30.
Almost homogeneous heterodimeric AH was obtained by the following procedure. AHStrep-containing soluble extract was adjusted to a pH of 7.2 and applied to a Strep-Tactin column, which was subsequently washed with buffer at pH 7.2. Elution was conducted by washing the column with buffer that had been adjusted to pH 6.7. The pH of the resulting eluate was readjusted to pH 7.2 and subjected to “conventional” Strep-Tactin affinity chromatography. The analysis by SDS-PAGE is presented in Fig. 3.
The molecular mass and the oligomeric state of purified AH were determined by size-exclusion chromatography, revealing peaks with apparent molecular masses of 144 ± 7 kDa and 290 ± 44 kDa (Fig. 4). The latter peak occurred at variable intensities in the individual runs. Analysis of the peak constituents by SDS polyacrylamide gel revealed an almost identical protein pattern with major bands at 37 and 65 kDa in the elution fractions of both peaks (Fig. 4). This suggests the presence of monomeric and dimeric forms of the AH heterodimer. However, the molecular masses obtained from the size-exclusion chromatography are higher than the calculated molecular masses of 103 and 206 kDa corresponding to monomeric and dimeric AH, respectively.
Fig 4.
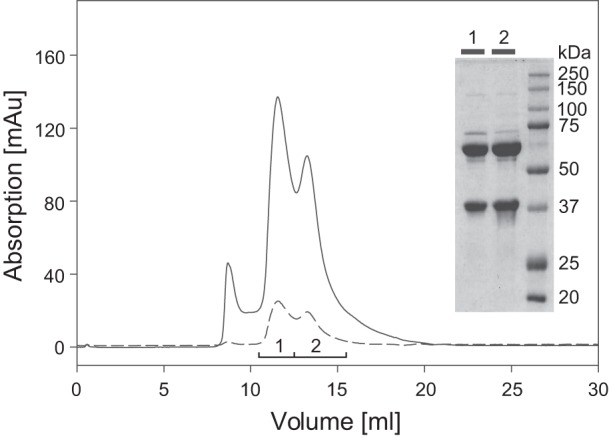
Size-exclusion chromatography of purified AH protein. AH protein purified by Strep-Tactin chromatography was applied to a Superdex 200 HR10/30 column and eluted with 50 mM KPO4 (pH 7.0)–150 mM NaCl,. Three protein peaks (A280, solid line) were observed; two of them contained FeS clusters (A405, dashed line). The two AH subunits were present in both peak fractions as revealed by SDS-PAGE analysis (inset).
The AH enzyme contains 1.2 ± 0.4 nickel atoms and 9.0 ± 4.0 iron atoms per AH molecule as determined by ICP-OES. Copper and zinc were not detected in significant amounts. This analysis confirms that the AH is a member of the widespread [NiFe]-hydrogenases.
Catalytic activity of purified AH.
The H2 uptake capacity of the purified AH was measured in a spectrophotometric assay using various artificial and natural electron acceptors. H2 uptake activity was observed with a number of compounds (Table 2). However, the highest H2 oxidation activity was measured with NBT. Therefore, NBT was used as electron acceptor for the subsequent determination of the kinetic parameters of the AH. The extinction coefficients of NBT were determined at 593 nm for various pH values (see Table S3 in the supplemental material). An extinction coefficient of 17.7 mM−1 cm−1 (pH 8) served as the basis for determination of the Km value for NBT (KmNBT). With H2 as reductant, the AH revealed a KmNBT of 7.8 ± 0.9 μM (pH 8) (see Fig. S5 in the supplemental material). An initial concentration of 75 μM NBT was used throughout the following measurements. NBT was nonspecifically reduced by the soluble extract (see also Fig. 2A) even in the absence of H2. Therefore, quantitative AH activity measurements were only possible with purified protein. The H2 oxidation activity of the AH with NBT as the sole electron acceptor was 0.28 ± 0.02 U per mg of protein at pH 8 and 30°C. Assuming a heterodimeric composition and a calculated molecular mass of 103.2 kDa (mature protein), the activity corresponds to a turnover rate of 0.48 s−1.
In order to determine the pH optimum of the purified AH, H2-dependent NBT reduction activities were determined within a pH range from 5.5 to 12 (Fig. 5). The AH activity remained stable between pH 7 and 9. At pH values below 7, the activity started to decrease, while it increased steadily at pH values above 9. At pH 11 the AH showed a 2-fold higher activity than at pH 9. At pH values higher than 11, the AH protein started to degrade. Since the pH optimum of pH 11 is definitely out of the physiological range and unlikely reflects the interaction of the AH with its native electron acceptor, all further experiments were done at pH 8.
Fig 5.
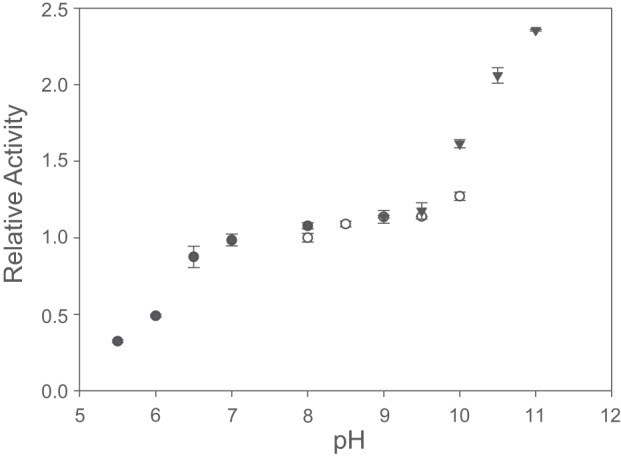
pH dependence of the H2 oxidation activity of the AH. The H2-dependent reduction of NBT was measured at pH values between 5.5 and 11. The following buffers were used: 50 mM KPO4 (●) 50 mM Tris-HCl (○), and 50 mM Capso [3-(cyclohexylamino)-2-hydroxy-1-propanesulfonic acid, ▼]. All activities were normalized to Tris-HCl buffer (pH 8). The data points represent the mean of three independent measurements. The respective standard deviations are indicated as error bars.
Temperature dependence was an additional parameter tested for the AH (Fig. 6). At temperatures below 30°C, the AH displayed a very low activity. However, the activity rose exponentially within a temperature range between 30 and 80°C. At 80°C the AH activity was ∼20-fold higher than at 30°C. Due to technical limitations, measurements above 80°C were not possible. Remarkably, even at 80°C, the AH activity remained relatively stable during the measurement period of about 3 min.
Fig 6.
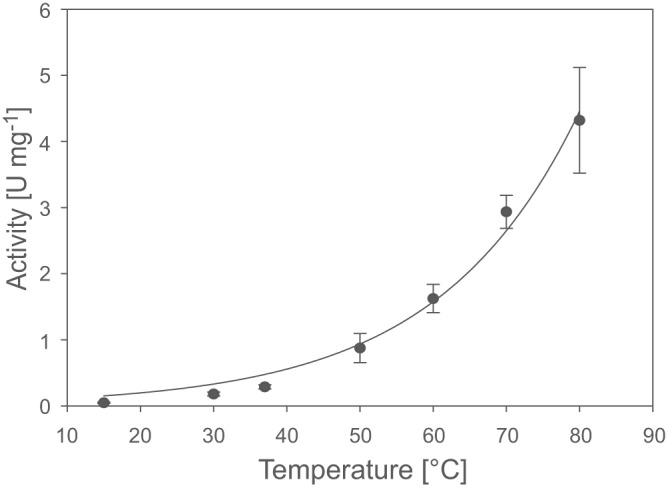
Temperature dependence of the H2-oxidizing activity of the AH. The H2-dependent reduction of NBT was measured in the temperature range of 15 to 80°C. The data points represent the mean of three independent measurements. The respective standard deviations are indicated as error bars.
In order to determine the half-life of the AH at different temperatures, the enzyme was incubated for various time periods at the desired temperature, and the remaining activity was measured subsequently at 30°C. At 30°C the AH was stable for several hours. At 80 and 60°C the AH showed a half-life of 3.5 and 80 min, respectively.
Substrate affinity and oxygen tolerance of the AH.
As a basic kinetic parameter, we determined the Km for H2 (KmH2) of the AH, which was 3.6 ± 0.5 μM at 30°C (see Fig. S6 and S7 in the supplemental material). R. eutropha has been isolated from soil with an average temperature presumably far below 30°C (31). Therefore, we determined the KmH2 also at 15°C. In case of the R. eutropha MBH, a significant decrease in the KmH2 was observed upon shifting the temperature from 30 to 15°C (32). However, at 15°C the AH displayed a KmH2 of 4.9 ± 1.2 μM, which was even slightly higher than that determined at 30°C.
All R. eutropha hydrogenases that have been characterized thus far are catalytically active in the presence of O2 (10, 29). To test whether the AH exhibits the same feature, O2 tolerance of the AH was determined amperometrically with a modified Clark-electrode (27). The AH-mediated H2 consumption in the presence of NBT was followed in buffer containing variable H2-O2 mixtures (Fig. 7). Even in the presence of 162 μM O2 in the reaction mixture (70% O2), the AH activity showed no decrease in activity compared to anoxic conditions. This result indicates that the enzyme is insensitive toward O2.
Fig 7.

H2-oxidizing activity of the AH at different oxygen concentrations. Activities were measured amperometrically at 30°C in a modified Clark electrode with NBT as electron acceptor. For each O2 concentration H2- and O2-saturated buffers were mixed at different ratios. The data points represent the mean of at least three independent measurements. The respective standard deviations are indicated as error bars.
DISCUSSION
The AH from Ralstonia eutropha represents the first group 5 [NiFe]-hydrogenase accessible to purification and detailed biochemical characterization. The presence of a gene cluster potentially coding for a fourth [NiFe]-hydrogenase in the genome of R. eutropha was uncovered 10 years ago (14), (15), leaving questions concerning its expression and possible physiological function to be solved.
As also observed in previous studies (26), our data confirm that the AH does not sustain lithoautotrophic growth of R. eutropha when the remaining hydrogenases are missing. The AH genes in R. eutropha are only weakly expressed under their native promoter. According to the RT-PCR analysis, the genes of the energy-conserving membrane-bound hydrogenase (MBH) of R. eutropha are expressed to a 20-fold-higher level than the AH encoding genes. Moreover, the specific activity of purified AH is two orders of magnitude lower than the activities determined for the MBH and the soluble, NAD+-reducing hydrogenase (SH) (22, 33). A summary of the kinetic parameters of the four hydrogenases in R. eutropha is presented in Table S4 in the supplemental material.
The low activity of the AH is reminiscent to that of the regulatory hydrogenase of R. eutropha. The RH acts as an H2 sensor in conjunction with a cytoplasmic histidine protein kinase and a response regulator. The interaction between RH and its cognate histidine protein kinase is mediated by a specific peptide at the C terminus of the small subunit (34). This peptide is absent in the AH. Furthermore, the H2 uptake experiments conducted with living cells in the present study revealed that H2 was consumed only by cells containing functional AH, but not by cells synthesizing solely the RH. Moreover, AH-driven H2 consumption was exclusively observed in the presence of O2. These results suggest that the AH is no H2 sensor, but an energy-conserving enzyme, which is connected in some way to the respiratory chain. This is consistent with previous assumptions made for Streptomyces strain PCB7, for which H2 uptake was only observed in sporulating aerial mycelia (4, 5). The presence of group 5 hydrogenases in spores is in accordance with comparatively high temperature tolerance of the AH. In conclusion, group 5 hydrogenases might play a role in maintaining basic metabolic functions under challenging conditions such as nutrient deprivation. However, in case of R. eutropha, the extremely low catalytic activity of the AH is not sufficient to support lithoautotrophic growth.
In search of the natural electron acceptor of the AH, a number of compounds have been investigated, including NAD+, NADP+, FMN, and FAD, spinach ferredoxin, cytochrome c (bovine heart and horse), cytochrome c6 from Synechocystis sp. strain PCC6803, and quinone derivatives, including ubiquinone Q0, duroquinone, and menadione. However, reasonable H2 oxidation activities were not detected with any of these natural acceptors. Therefore, also a number of artificial electron acceptors were tested. Highest H2 oxidation activity was obtained with NBT, which only recently has been described as electron acceptor for Hyd-1 from Escherichia coli (35). NBT serves also as acceptor for several other oxidoreductases (36–39) and has a midpoint potential of E0 = +50 mV (40). Except for methyl viologen, the midpoint potentials of the best electron acceptors for the AH lie in the range between 0 mV and +100 mV. Despite having a very negative redox potential of −446 mV, methyl viologen obviously can serve as electron acceptor for a number of hydrogenases (41, 42). It might extract the electrons at a low-potential site close to the catalytic center, e.g., the proximal iron-sulfur cluster. Nevertheless, the nature of the physiological electron acceptor of the AH remains elusive. Possible candidates are dedicated cytochromes that have not been identified yet. The genome of R. eutropha has coding capacity for several cytochromes (14, 15). However, electron transfer might be mediated by one of the four unknown conserved proteins encoded in the AH operon as suggested previously (8). The product of gene PHG067 of R. eutropha represents a candidate since it contains motifs for coordination of one or two iron-sulfur clusters (Fig. 1). Another gene presumably encoding an iron-sulfur cluster-containing protein is present in most group 5 hydrogenase operons. However, the product of the corresponding R. eutropha gene PHG066 does not contain the motifs necessary for iron-sulfur cluster coordination. In order to test the function of these genes, hydrogen uptake by living cells should be measured for appropriate knockout strains of R. eutropha.
The amino acid sequence deduced from the genome analysis suggests that the heterodimeric AH protein harbors a [NiFe] center in the large subunit and three [FeS] clusters in the small subunit (see Fig. S8 in the supplemental material). Three cysteines and one histidine are predicted to coordinate a [4Fe-4S] cluster in the distal position relative to the active site. In most [NiFe]-hydrogenases, the medial cluster in the iron-sulfur cluster relay is represented by a [3Fe-4S] center. The amino acid sequence of the AH suggests rather a [4Fe-4S] cluster species coordinated by four cysteines. The predicted coordination sphere of the cluster proximal to the active site encompasses three cysteines and one aspartate. Such a configuration might also coordinate a [4Fe-4S] cluster, as observed for a ferredoxin from Pyrococcus furiosus (43) and the transcriptional regulator Fnr of Bacillus subtilis (44). Metal determination revealed 1.2 ± 0.4 nickel atoms and 9.0 ± 4.0 iron atoms per AH heterodimer, which supports the presence of at least two [4Fe-4S] clusters.
The operons of almost all group 5 [NiFe]-hydrogenases contain six genes involved in maturation of the nickel-iron catalytic center (8). In R. eutropha, these are designated hypA3, hypB3, hypC2, hypD2, hypE2, and hypF3, since a copy of all of these hyp genes is present in the MBH operon and additional copies of the hypA, hypB, and hypF genes are constituents of the SH operon of R. eutropha (14). Former studies revealed that the products from hypABF genes of the SH and MBH operons can mutually substitute each other (45), (46). However, mutants lacking both copies of these genes still exhibited trace activities of the SH (46). This suggests that the HypABF copies encoded in the AH operon are partially functional in SH maturation. Whether the active site of the AH can be fully assembled by the Hyp proteins encoded in the SH and MBH operons is still unknown. However, a mutant carrying deletions in the MBH- and SH-related hyp genes shows comparable AH activities as the AH+ strain, in which all hyp genes of R. eutropha are functional. This shows that the hyp genes of the AH operon are sufficient for AH maturation.
Group 5 [NiFe]-hydrogenases have been shown to be responsible for H2 uptake in soils (5). It is thus necessary for these enzymes to consume hydrogen at concentrations of 1 to 10 nM, as typically found in soils (47). Cell cultures of several Streptomyces species containing group 5 hydrogenases show H2 uptake activities with Km values in the nanomolar range and thus are potentially able to use hydrogen at ambient concentration in the air (48). However, up to now this high affinity for H2 has not been shown for a hydrogenase purified from these species. Sharing similar habitats and thus being faced with comparable environmental conditions, the possibility of high-affinity hydrogen uptake would be advantageous also for Ralstonia eutropha. However, the AH from R. eutropha, which was purified to homogeneity, does not display a Km in the nanomolar range as determined spectrophotometrically in a solution assay using the artificial electron acceptor NBT. Thus, uptake of atmospheric hydrogen by the AH is unlikely. Notably, this conclusion is in full agreement with the previous finding that R. eutropha cells are not capable of high-affinity hydrogen uptake (49). In an earlier study, a lateral gene transfer of the group 5 hydrogenase operon into R. eutropha has been suggested (14). Our results allow the interpretation that the AH might not have accomplished full functionality in its new host. However, a high affinity toward hydrogen seems not to be obligatory for hydrogenases of the group 5 type.
In its natural environment, R. eutropha has to cope with variable O2 concentrations. To allow the lithoautotrophic growth mode even in the presence of oxygen, tolerance toward oxygen is a prerequisite for the hydrogenases. The mechanism(s) for oxygen-tolerance of a number of [NiFe]-hydrogenases has been subject of many investigations in the past years. For example, the MBH from R. eutropha, a paradigm of O2-tolerant hydrogenases, sustains H2 conversion in the presence of O2 (10, 29). This characteristic is attributed to a novel [4Fe-3S] cluster in the small subunit at the proximal position to the active site (22, 50). However, the activity of the MBH decreases with increasing O2 concentrations. The behavior of the AH toward oxygen, however, is different, since no decrease of activity was observed even in the presence high oxygen concentrations. In contrast to O2-tolerant hydrogenases of the MBH-type, the AH and generally all group 5 hydrogenases do not contain additional cysteines that could be involved in the coordination of a [4Fe-3S] cluster. Instead, only three cysteines appear to coordinate the proximal cluster in the AH. Furthermore, an aspartate, which replaces a fourth cysteine in group 5 hydrogenases is probably in binding distance, suggesting the formation of a nonstandard [FeS] cluster. Whether this property contributes to the insensitivity of the AH toward oxygen remains to be elucidated.
Supplementary Material
ACKNOWLEDGMENTS
We gratefully thank Alexander Schwarze for valuable advice in designing the expression constructs, Anne Pohlmann for the introduction into quantitative RT-PCR techniques and assistance in data evaluation, Silke Leimkühler (University of Potsdam) for ICP-OES measurements, and Angelika Strack for skillful technical assistance.
This study was supported by the German Federal Ministry of Education and Research (BMBF project H2-Designcells), a Cluster of Excellence “Unifying Concepts in Catalysis” grant (to O.L.), and the Fonds der Chemischen Industrie (to B.F.).
Footnotes
Published ahead of print 21 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01576-13.
REFERENCES
- 1.Constant P, Poissant L, Villemur R. 2009. Tropospheric H2 budget and the response of its soil uptake under the changing environment. Sci. Total Environ. 407:1809–1823 [DOI] [PubMed] [Google Scholar]
- 2.Thauer RK. 2011. Hydrogenases and the global H2 cycle. Eur. J. Inorg. Chem. 2011:919–921 [Google Scholar]
- 3.Guo RB, Conrad R. 2008. Extraction and characterization of soil hydrogenases oxidizing atmospheric hydrogen. Soil Biol. Biochem. 40:1149–1154 [Google Scholar]
- 4.Constant P, Poissant L, Villemur R. 2008. Isolation of Streptomyces sp. PCB7, the first microorganism demonstrating high-affinity uptake of tropospheric H2. ISME J. 2:1066–1076 [DOI] [PubMed] [Google Scholar]
- 5.Constant P, Chowdhury SP, Hesse L, Conrad R. 2011. Co-localization of atmospheric H2 oxidation activity and high affinity H2-oxidizing bacteria in non-axenic soil and sterile soil amended with Streptomyces sp. PCB7. Soil Biol. Biochem. 43:1888–1893 [Google Scholar]
- 6.Vignais PM, Billoud B. 2007. Occurrence, classification, and biological function of hydrogenases: an overview. Chem. Rev. 107:4206–4272 [DOI] [PubMed] [Google Scholar]
- 7.Schwartz E, Fritsch J, Friedrich B. 2013. H2-metabolizing prokaryotes, p 119–199 In Rosenberg E, DeLong E, Lory S, Stackebrandt E, Thompson F. (ed), The prokaryotes: prokaryotic physiology and biochemistry, 4th ed Springer, Berlin, Germany [Google Scholar]
- 8.Constant P, Chowdhury SP, Hesse L, Pratscher J, Conrad R. 2011. Genome data mining and soil survey for the novel group 5 [NiFe]-hydrogenase to explore the diversity and ecological importance of presumptive high-affinity H2-oxidizing bacteria. Appl. Environ. Microbiol. 77:6027–6035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Böck A, King PW, Blokesch M, Posewitz MC. 2006. Maturation of hydrogenases. Adv. Microb. Physiol. 51:1–71 [DOI] [PubMed] [Google Scholar]
- 10.Lenz O, Ludwig M, Schubert T, Bürstel I, Ganskow S, Goris T, Schwarze A, Friedrich B. 2010. H2 conversion in the presence of O2 as performed by the membrane-bound [NiFe]-hydrogenase of Ralstonia eutropha. Chem. Phys. Chem. 11:1107–1119 [DOI] [PubMed] [Google Scholar]
- 11.Blokesch M, Paschos A, Theodoratou E, Bauer E, Hube M, Huth S, Böck A. 2002. Metal insertion into [NiFe]-hydrogenases. Biochem. Soc. Trans. 30:674–680 [DOI] [PubMed] [Google Scholar]
- 12.Palmer T, Berks BC. 2012. The twin-arginine translocation (Tat) protein export pathway. Nat. Rev. Microbiol. 10:483–496 [DOI] [PubMed] [Google Scholar]
- 13.Berney M, Cook GM. 2010. Unique flexibility in energy metabolism allows mycobacteria to combat starvation and hypoxia. PLoS One 5:e8614. 10.1371/journal.pone.0008614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwartz E, Henne A, Cramm R, Eitinger T, Friedrich B, Gottschalk G. 2003. Complete nucleotide sequence of pHG1: a Ralstonia eutropha H16 megaplasmid encoding key enzymes of H2-based lithoautotrophy and anaerobiosis. J. Mol. Biol. 332:369–383 [DOI] [PubMed] [Google Scholar]
- 15.Pohlmann A, Fricke WF, Reinecke F, Kusian B, Liesegang H, Cramm R, Eitinger T, Ewering C, Potter M, Schwartz E, Strittmatter A, Voss I, Gottschalk G, Steinbüchel A, Friedrich B, Bowien B. 2006. Genome sequence of the bioplastic-producing “Knallgas” bacterium Ralstonia eutropha H16. Nat. Biotechnol. 24:1257–1262 [DOI] [PubMed] [Google Scholar]
- 16.Lenz O, Bernhard M, Buhrke T, Schwartz E, Friedrich B. 2002. The hydrogen-sensing apparatus in Ralstonia eutropha. J. Mol. Microbiol. Biotechnol. 4:255–262 [PubMed] [Google Scholar]
- 17.Burgdorf T, Löscher S, Liebisch P, Van der Linden E, Galander M, Lendzian F, Meyer-Klaucke W, Albracht SPJ, Friedrich B, Dau H, Haumann M. 2005. Structural and oxidation-state changes at its nonstandard [NiFe] site during activation of the NAD-reducing hydrogenase from Ralstonia eutropha detected by X-ray absorption, EPR, and FTIR spectroscopy. J. Am. Chem. Soc. 127:576–592 [DOI] [PubMed] [Google Scholar]
- 18.Fritsch J, Lenz O, Friedrich B. 2013. Structure, function and biosynthesis of O2-tolerant hydrogenases. Nat. Rev. Microbiol. 11:106–114 [DOI] [PubMed] [Google Scholar]
- 19.Yanisch-Perron C, Vieira J, Messing J. 1985. Improved M13 phage cloning vectors and host strains: nucleotide sequences of the M13mp18 and pUC19 vectors. Gene 33:103–119 [DOI] [PubMed] [Google Scholar]
- 20.Simon R, Priefer U, Pühler A. 1983. A broad host range mobilization system for in vivo genetic engineering: transposon mutagenesis in Gram-negative bacteria. Biotechnology 1:784–791 [Google Scholar]
- 21.Lenz O, Schwartz E, Dernedde J, Eitinger M, Friedrich B. 1994. The Alcaligenes eutrophus H16 hoxX gene participates in hydrogenase regulation. J. Bacteriol. 176:4385–4393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Goris T, Wait AF, Saggu M, Fritsch J, Heidary N, Stein M, Zebger I, Lendzian F, Armstrong FA, Friedrich B, Lenz O. 2011. A unique iron-sulfur cluster is crucial for oxygen tolerance of a [NiFe]-hydrogenase. Nat. Chem. Biol. 7:310–318 [DOI] [PubMed] [Google Scholar]
- 23.Smith PK, Krohn RI, Hermanson GT, Mallia AK, Gartner FH, Provenzano MD, Fujimoto EK, Goeke NM, Olson BJ, Klenk DC. 1985. Measurement of protein using bicinchoninic acid. Anal. Biochem. 150:76–85 [DOI] [PubMed] [Google Scholar]
- 24.Schwartz E, Voigt B, Zühlke D, Pohlmann A, Lenz O, Albrecht D, Schwarze A, Kohlmann Y, Krause C, Hecker M, Friedrich B. 2009. A proteomic view of the facultatively chemolithoautotrophic lifestyle of Ralstonia eutropha H16. Proteomics 9:5132–5142 [DOI] [PubMed] [Google Scholar]
- 25.Neumann M, Mittelstädt G, Seduk F, Iobbi-Nivol C, Leimkühler S. 2009. MocA is a specific cytidylyltransferase involved in molybdopterin cytosine dinucleotide biosynthesis in Escherichia coli. J. Biol. Chem. 284:21891–21898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kleihues L, Lenz O, Bernhard M, Buhrke T, Friedrich B. 2000. The H2 sensor of Ralstonia eutropha is a member of the subclass of regulatory [NiFe] hydrogenases. J. Bacteriol. 182:2716–2724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang R, Healey FP, Myers J. 1971. Amperometric measurement of hydrogen evolution in Chlamydomonas. Plant Physiol. 48:108–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kohlmann Y, Pohlmann A, Otto A, Becher D, Cramm R, Lütte S, Schwartz E, Hecker M, Friedrich B. 2011. Analyses of soluble and membrane proteomes of Ralstonia eutropha H16 reveal major changes in the protein complement in adaptation to lithoautotrophy. J. Proteome Res. 10:2767–2776 [DOI] [PubMed] [Google Scholar]
- 29.Ludwig M, Cracknell JA, Vincent KA, Armstrong FA, Lenz O. 2009. Oxygen-tolerant H2 oxidation by membrane-bound [NiFe] hydrogenases of Ralstonia species: coping with low level H2 in air. J. Biol. Chem. 284:465–477 [DOI] [PubMed] [Google Scholar]
- 30.Jones A, Lenz O, Strack A, Buhrke T, Friedrich B. 2004. [NiFe] hydrogenase active site biosynthesis: identification of Hyp protein complexes in Ralstonia eutropha. Biochemistry 43:13467–13477 [DOI] [PubMed] [Google Scholar]
- 31.Wilde E. 1962. Untersuchungen über Wachstum und Speicherstoffsynthese von Hydrogenomonas. Archiv. Mikrobiol. 43:109–137 [Google Scholar]
- 32.Cracknell JA, Wait AF, Lenz O, Friedrich B, Armstrong FA. 2009. A kinetic and thermodynamic understanding of O2 tolerance in [NiFe]-hydrogenases. Proc. Natl. Acad. Sci. U. S. A. 106:20681–20686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lauterbach L, Lenz O, Vincent KA. 2013. H2-driven cofactor regeneration using NAD(P)+-reducing hydrogenases. FEBS J. 280:3058–3068 [DOI] [PubMed] [Google Scholar]
- 34.Buhrke T, Lenz O, Porthun A, Friedrich B. 2004. The H2-sensing complex of Ralstonia eutropha: interaction between a regulatory [NiFe] hydrogenase and a histidine protein kinase. Mol. Microbiol. 51:1677–1689 [DOI] [PubMed] [Google Scholar]
- 35.Pinske C, Jaroschinsky M, Sargent F, Sawers G. 2012. Zymographic differentiation of [NiFe]-hydrogenases 1, 2, and 3 of Escherichia coli K-12. BMC Microbiol. 12:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liochev S, Hausladen A, Beyer W, Fridovich I. 1994. NADPH:ferredoxin oxidoreductase acts as a paraquat diaphorase and is a member of the soxRS regulon. Proc. Natl. Acad. Sci. U. S. A. 91:1328–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Brown DM, Upcroft JA, Upcroft P. 1996. A H2O-producing NADH oxidase from the protozoan parasite Giardia duodenalis. Eur. J. Biochem. 241:155–161 [DOI] [PubMed] [Google Scholar]
- 38.Schreiner ME, Eikmanns BJ. 2005. Pyruvate: quinone oxidoreductase from Corynebacterium glutamicum—purification and biochemical characterization. J. Bacteriol. 187:862–871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Niraula NP, Shrestha P, Oh TJ, Sohng JK. 2010. Identification and characterization of a NADH oxidoreductase involved in phenylacetic acid degradation pathway from Streptomyces peucetius. Microbiol. Res. 165:649–656 [DOI] [PubMed] [Google Scholar]
- 40.Michal G, Möllering H, Siedel J. 1983. Chemical design for indicator reactions in the visible range, p 197–232 In Bergmeyer H-U. (ed), Methods of enzymatic analysis, 3rd ed, vol 1 Verlag Chemie, Weinheim, Germany [Google Scholar]
- 41.Schneider K, Schlegel HG. 1976. Purification and properties of soluble hydrogenase from Alcaligenes eutrophus H 16. Biochim. Biophys. Acta 452:66–80 [DOI] [PubMed] [Google Scholar]
- 42.Hedderich R, Albracht SP, Linder D, Koch J, Thauer RK. 1992. Isolation and characterization of polyferredoxin from Methanobacterium thermoautotrophicum: the mvhB gene product of the methylviologen-reducing hydrogenase operon. FEBS Lett. 298:65–68 [DOI] [PubMed] [Google Scholar]
- 43.Calzolai L, Gorst CM, Zhao ZH, Teng Q, Adams MW, La Mar GN. 1995. 1H-NMR investigation of the electronic and molecular structure of the four-iron cluster ferredoxin from the hyperthermophile Pyrococcus furiosus: identification of Asp14 as a cluster ligand in each of the four redox states. Biochemistry 34:11373–11384 [DOI] [PubMed] [Google Scholar]
- 44.Gruner I, Frädrich C, Böttger LH, Trautwein AX, Jahn D, Härtig E. 2011. Aspartate 141 is the fourth ligand of the oxygen-sensing [4Fe-4S]2+ cluster of Bacillus subtilis transcriptional regulator Fnr. J. Biol. Chem. 286:2017–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dernedde J, Eitinger T, Patenge N, Friedrich B. 1996. hyp gene products in Alcaligenes eutrophus are part of a hydrogenase-maturation system. Eur. J. Biochem. 235:351–358 [DOI] [PubMed] [Google Scholar]
- 46.Wolf I, Buhrke T, Dernedde J, Pohlmann A, Friedrich B. 1998. Duplication of hyp genes involved in maturation of [NiFe] hydrogenases in Alcaligenes eutrophus H16. Arch. Microbiol. 170:451–459 [DOI] [PubMed] [Google Scholar]
- 47.Conrad R. 1996. Soil microorganisms as controllers of atmospheric trace gases (H2, CO, CH4, OCS, N2O, and NO). Microbiol. Rev. 60:609–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Constant P, Chowdhury SP, Pratscher J, Conrad R. 2010. Streptomycetes contributing to atmospheric molecular hydrogen soil uptake are widespread and encode a putative high-affinity [NiFe]-hydrogenase. Environ. Microbiol. 12:821–829 [DOI] [PubMed] [Google Scholar]
- 49.Conrad R, Aragno M, Seiler W. 1983. The inability of hydrogen bacteria to utilize atmospheric hydrogen is due to threshold and affinity for hydrogen. FEMS Microbiol. Lett. 18:207–210 [Google Scholar]
- 50.Fritsch J, Scheerer P, Frielingsdorf S, Kroschinsky S, Friedrich B, Lenz O, Spahn CMT. 2011. The crystal structure of an oxygen-tolerant hydrogenase uncovers a novel iron-sulphur centre. Nature 479:249–252 [DOI] [PubMed] [Google Scholar]
- 51.Massanz C, Fernandez VM, Friedrich B. 1997. C-terminal extension of the H2-activating subunit, HoxH, directs maturation of the NAD-reducing hydrogenase in Alcaligenes eutrophus. Eur. J. Biochem. 245:441–448 [DOI] [PubMed] [Google Scholar]
- 52.Buhrke T. 2002. Der H2-Sensor von Ralstonia eutropha: Struktur-Funktions-Beziehungen einer Neuartigen [NiFe]-Hydrogenase. Ph.D. thesis. Humboldt-Universität zu Berlin, Berlin, Germany [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.