

Abstract
Cheese fermentations involve the growth of complex microbial consortia, which often originate in the processing environment and drive the development of regional product qualities. However, the microbial milieus of cheesemaking facilities are largely unexplored and the true nature of the fermentation-facility relationship remains nebulous. Thus, a high-throughput sequencing approach was employed to investigate the microbial ecosystems of two artisanal cheesemaking plants, with the goal of elucidating how the processing environment influences microbial community assemblages. Results demonstrate that fermentation-associated microbes dominated most surfaces, primarily Debaryomyces and Lactococcus, indicating that establishment of these organisms on processing surfaces may play an important role in microbial transfer, beneficially directing the course of sequential fermentations. Environmental organisms detected in processing environments dominated the surface microbiota of washed-rind cheeses maturing in both facilities, demonstrating the importance of the processing environment for populating cheese microbial communities, even in inoculated cheeses. Spatial diversification within both facilities reflects the functional adaptations of microbial communities inhabiting different surfaces and the existence of facility-specific “house” microbiota, which may play a role in shaping site-specific product characteristics.
INTRODUCTION
For millennia, food fermentations have coevolved with their microbial agents, giving rise to products that were simultaneously safer and more stable, nutritious, delicious, and valuable than their raw ingredients. Microbial activities are inherent to all food fermentations, introduced by direct inoculation, raw materials (e.g., raw milk), and the processing environment, shaping final product qualities in addition to causing spoilage (1). Thus, food fermentations have always been a custodial process, intentionally or unintentionally managing microbial communities in the fermentation and the environment through direct and indirect interventions, including environmental conditioning (temperature, humidity, light), moisture control (brining and pressing of cheese), and cleaning procedures. However, this has historically been an “artisanal” process, developing management practices through empirical trials, and these traditional methods emerged long before knowledge of the microbial denizens of these fermentations.
During cheese production, the product encounters many equipment surfaces on its journey from milk to curd to cheese, all acting as potential vectors for microbes. Additionally, the product undergoes several changes in physiochemical composition, promoting the growth of a succession of microorganisms (2), and by extension a changing susceptibility to colonization by environmental microbes. Hence, the processing environment may serve as an important reservoir for bidirectional microbial transfer between fermentations, and microbial surveillance of this environment is critical to understanding the complete microbial ecosystem of cheese production. In modern cheese production facilities, biofilms of psychrotrophic bacteria (3, 4) and nonstarter lactic acid bacteria (5–8) can form on equipment surfaces, acting as a source of contamination in successive batches of cheese. Wooden processing surfaces, including aging boards (9, 10) and milk vats (11–13), are also rich sources of microbes that are important for cheese acidification and ripening. In traditional cheesemaking facilities, adventitious microbes inhabiting such equipment surfaces can represent a “house” microbiota important for the development of specific cheese characteristics (14). However, all previous studies of cheese processing environments focused on selected, isolated equipment surfaces and all employed culture-based approaches (3–9, 13, 14) or low-throughput molecular tools (10–12), which possess limitations for the study of food fermentations (1, 15), including throughput and sensitivity. More comprehensive facility monitoring is necessary to better establish the relationship between processing environment microbiota and cheese fermentations.
The advent of high-throughput sequencing (HTS) technology has revolutionized the study of microbial ecosystems, including food fermentations (1). HTS enables comprehensive microbial surveys with detection sensitivities and throughputs orders of magnitude greater than earlier molecular techniques through massively parallel sequencing of short amplicons of universally conserved DNA fragments, typically the 16S rRNA gene in bacteria and the internal transcribed spacer (ITS) domain in fungi (1). Comparisons to other methods and disadvantages of HTS have been reviewed elsewhere (1, 15, 16). The advantages of HTS tools are apparent in uncovering previously undetectable diversity in cheese (17–22), wine (23), beer (24), and other fermented foods (1). HTS also facilitates the investigation of microbial communities in indoor environments at a massive scale, including wineries (25), hospitals (26–28), office spaces (29), public restrooms (30), and domestic kitchens (31). Thus, in addition to advancing our knowledge of food fermentations, these tools hold great promise for describing the microbial diversity of food processing facilities, where microbes play critical roles in influencing both the chemosensory qualities of food products and their healthfulness for human consumption.
To explore the microbial ecosystems of cheese processing environments, an HTS approach was used to monitor the bacterial and fungal communities of surfaces and equipment across two different cheese production plants. Results illustrate that spatial diversification within each cheese plant reflects the functional adaptations of microbial communities inhabiting different surfaces and the existence of facility-specific “house” microbiota. Cheese-associated microbiota, especially Debaryomyces and Lactococcus, dominated most surfaces, indicating that establishment of these organisms on processing surfaces may play an important role in populating cheese microbial communities, beneficially directing the course of sequential fermentation.
MATERIALS AND METHODS
Facility description.
Samples were collected from two artisan cheesemaking facilities located in the United States on a single sampling day (14 November 2012). The two facilities produce similar ranges of products, consisting of fresh, bloomy-rind, and washed-rind (smear-ripened) cheeses, but each facility produces a distinct product line. Both facilities pasteurize their milk immediately prior to cheesemaking. Facility A is 6 years old and inoculates strains of Lactococcus lactis, Lactobacillus casei, Lactobacillus paracasei, Lactobacillus delbrueckii, Lactobacillus helveticus, Lactobacillus rhamnosus, Leuconostoc mesenteroides, and Streptococcus thermophilus for curd acidification and Staphylococcus xylosus, Debaryomyces hansenii, Penicillium camemberti, and Geotrichum candidum for surface ripening in different product lines. All cultures are added directly to cheese milk before coagulation, with the exception of surface-ripening cultures for washed-rind cheeses, which are applied directly to the cheeses in a wash solution (see cheese sample description). Facility B is 16 years old and inoculates only the lactic acid starters Lactococcus lactis, Lactobacillus casei, Lactobacillus paracasei, and Leuconostoc mesenteroides for curd acidification in different products. The only surface-ripening cultures ever inoculated in this facility are P. camemberti and G. candidum in washed-rind and bloomy-rind cheeses. All cultures are added directly to the cheese milk before coagulation. The floor plans of the facilities and a key to all of the surfaces sampled are presented in Fig. 1.
Fig 1.

Floor plan key to equipment surfaces in both cheesemaking facilities.
Cheese samples.
In addition to facility surface swabs, samples were collected from the surfaces of washed-rind cheeses produced in each facility on a single sampling day (18 March 2013). Cheese A (from facility A) is produced by inoculation of Lactobacillus delbrueckii, Streptococcus thermophilus, Lactobacillus rhamnosus, Lactobacillus helveticus, and Lactobacillus paracasei directly into the cheese milk prior to coagulation. The formed cheeses are washed twice weekly with brine containing Kluyveromyces marxianus, Debaryomyces hansenii, and Staphylococcus xylosus and aged 75 to 90 days. Cheese B (from facility B) is produced by inoculation of Lactococcus lactis, Leuconostoc mesenteroides, P. camemberti, and G. candidum directly into the cheese milk prior to coagulation. The formed cheeses are washed with water and salt only (no inocula) three or four times during aging. Four maturing cheeses from each facility, representing two separate production batches in each facility, were sampled. These batches were all newly produced and immature at the time when facility swabs were collected, so the surface microbiota detected on these cheeses represents growth following exposure to the aging-room environments. The surface layer samples of each cheese were removed and preserved by swabbing as described below.
Sample collection and DNA extraction.
Surfaces were sampled with sterile cotton-tipped swabs (Puritan Medical, Guilford, ME). Swabs were moistened with sterile phosphate-buffered saline and streaked across a 4-in.2 (or equivalent) area of the target surface in two perpendicular series of firm, overlapping S strokes with rotation of the swab to ensure full contact of all parts of the swab tip and the surface. Swab tips were snapped off into sterile 1.5-ml polyethylene tubes against the inner edge of the tube without manual contact. Samples were placed on ice and frozen immediately in a −20°C freezer for storage. The cotton tip of each swab was aseptically removed from the shaft and placed directly into the 96-well lysis plate provided in the ZR-96 Fecal DNA extraction kit (Zymo Research, Irvine, CA). DNA was extracted by using the standard protocol for the ZR-96 kit, with bead beating with a Genogrinder high-throughput tissue homogenizer (SPEX SamplePrep, Metuchen, NJ) and stored at −20°C until further processing. A complete list of each sample collected is presented in Tables S1 and S2 in the supplemental material as mapping files formatted for sequencing analysis directly in QIIME (32).
Sequencing library construction.
Amplification and sequencing were performed as described previously for bacterial (23) and fungal communities (33). Briefly, the V4 domain of bacterial 16S rRNA genes was amplified with primers F515 (5′-NNNNNNNNGTGTGCCAGCMGCCGCGGTAA-3′) and R806 (5′-GGACTACHVGGGTWTCTAAT-3′) (34), with the forward primer modified to contain a unique 8 nt barcode (italicized poly-N section of primer above) and a 2-nucleotide (nt) linker sequence (bold, underlined) at the 5′ terminus. All of the F515 primer barcodes used are presented in Table S1 in the supplemental material. PCR mixtures contained 5 to 100 ng DNA template, 1× GoTaq Green Master Mix (Promega), 1 mM MgCl2, and 2 pmol of each primer. The reaction conditions consisted of an initial 94°C for 3 min, followed by 35 cycles of 94°C for 45 s, 50°C for 60 s, and 72°C for 90 s and a final extension of 72°C for 10 min. Fungal ITS 1 loci were amplified with primers BITS (5′-NNNNNNNNCTACCTGCGGARGGATCA-3′) and B58S3 (5′-GAGATCCRTTGYTRAAAGTT-3′) (33), with a unique 8-nt barcode and linker sequence incorporated in each forward primer. All of the BITS primer barcodes used are presented in Table S2 in the supplemental material. PCR mixtures contained 5 to 100 ng DNA template, 1× GoTaq Green Master Mix (Promega, Madison, WI), 1 mM MgCl2, and 2 pmol of each primer. The reaction conditions consisted of an initial 95°C for 2 min, followed by 40 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 60 s and a final extension of 72°C for 5 min. Amplicons were combined into two separate pooled samples (keeping bacterial and fungal amplicons separate) at roughly equal amplification intensity ratios, purified with the QIAquick spin kit (Qiagen), and submitted to the UC Davis Genome Center DNA Technologies Core for Illumina paired-end library preparation, cluster generation, and 250-bp paired-end sequencing on an Illumina MiSeq instrument in two separate runs. Cheese surface swab samples received identical treatment, but fungal and bacterial amplicons from these samples were sequenced in two additional, separate Illumina MiSeq runs. The bacterial 16S rRNA sequencing runs yielded 2,399,507 reads (post quality filtering, described below), with a mean read length of 240.0 (±17.3) nt. The fungal ITS sequencing runs yielded 2,828,508 reads (post quality filtering), with a mean read length of 232.3 (±35.9) nt. Per-sample sequence coverage is depicted via rarefaction curves in Fig S1 in the supplemental material.
Data analysis.
Raw fastq files were demultiplexed, quality filtered, and analyzed with QIIME 1.6.0 (32). The 250-bp reads were truncated at any site of more than three sequential bases receiving a Phred quality score of <Q20 and any read containing ambiguous base calls or barcode/primer errors were discarded, as were reads with <75% (of the total read length) consecutive high-quality base calls (35). For ITS sequences, primer sequences were trimmed from the ends of each sequence and operational taxonomic units (OTUs) were clustered de novo with the QIIME implementation of UCLUST (36), with a threshold of 97% pairwise identity. Bacterial 16S rRNA gene sequences were clustered with the QIIME subsampled reference OTU-picking pipeline with UCLUST-reference 36 against the Greengenes 16S rRNA database (February 2011 release) (37), clustered at 97% pairwise identity. OTUs were classified taxonomically with a QIIME-based wrapper of BLAST (38) against the Greengenes 16S rRNA gene database (for 16S rRNA gene sequences) or the UNITE (39, 40) database (for ITS sequences) modified as described previously (33). Any OTU comprising less than 0.0001% of the total sequences in each run were removed prior to further analysis, calibrating against defined mock communities included in both sequencing runs (35). Bacterial 16S rRNA gene sequences were aligned with PyNAST (41) against a reference alignment of the Greengenes core set (37). From this alignment, chimeric sequences were identified and removed with ChimeraSlayer (42) and a phylogenic tree was generated from the filtered alignment with FastTree (43). Sequences failing alignment or identified as chimeric were removed prior to downstream analysis.
Jackknifed beta diversity estimates (between-sample diversity comparisons) were calculated within QIIME with unweighted and weighted UniFrac (44) distance between samples for bacterial 16S rRNA gene reads (evenly sampled at 400 reads per sample) and Canberra distance for fungal ITS reads (evenly sampled at 400 reads per sample), subsampled 10 times without replacement. From these estimates, principal coordinates were computed to compress dimensionality into two-dimensional principal-coordinate analysis (PCoA) plots. In order to determine whether sample classifications (sample time, equipment type, location) contained differences in OTU diversity, permutational MANOVA (45) with 999 permutations was used to test the null hypothesis that sample groups were not statistically significant based on evenly sampled UniFrac and Canberra distance matrices, with the QIIME-wrapped R module Adonis. For all classifications rejecting this null hypothesis, one-way analysis of variance (ANOVA) was used to determine which taxa differed significantly (with Bonferroni error correction) between sample groups.
Environmental surveillance heat maps were generated on the basis of taxonomic abundance tables generated in QIIME and visualized with SitePainter 1.1 (46).
Quantitative PCR.
In order to quantify net microbial biomass on cheesemaking facility surfaces, quantitative PCR (qPCR) was used to enumerate total fungi and bacteria. qPCR was performed in 20-μl reaction mixtures containing 2 μl of DNA template, 5 pmol of each respective primer, and 10 μl of TaKaRa SYBR 2× Perfect Real Time Master Mix (TaKaRa Bio Inc.). For quantification of total fungi, the primers YEASTF (5′-GAGTCGAGTTGTTTGGGAATGC-3′) and YEASTR (5′-TCTCTTTCCAAAGTTCTTTTCATCTT-3′) were used (47). The reaction conditions involved an initial step of 95°C for 10 min, followed by 40 cycles of 15 s at 95°C, 1 min at 60°C, and 30 s at 72°C. For amplification of total bacteria, the primers Uni334F (5′-ACTCCTACGGGAGGCAGCAGT-3′) and Uni514R (5′-ATTACCGCGGCTGCTGGC-3′) (48) were used. The reaction conditions consisted of an initial hold at 95°C for 20 s, followed by 40 cycles of 4 s at 95°C and 25 s at 65.5°C. All reactions were performed in triplicate in optical-grade 96-well plates on an ABI Prism 7500 Fast Real-Time PCR System (Applied Biosystems). The instrument automatically calculated the cycle threshold (CT), efficiency (E), confidence intervals, and cell concentration (fungi) or 16S rRNA gene copy number (bacteria) by comparing sample CT values to a standard curve of serially diluted genomic DNA extracted from a known concentration of Saccharomyces cerevisiae or Escherichia coli cells. One-way ANOVA and protected least significant difference (PLSD) with Bonferroni correction were calculated with R software to test significant differences between individual sample classifications. qPCR microbial biomass heat maps were generated with SitePainter 1.1 (46).
Data availability.
Sequence data generated in this study are publicly available on the QIIME database (www.microbio.me/qiime/) as studies 1884 (bacterial 16S rRNA sequencing run data) and 1919 (fungal ITS sequencing run data).
RESULTS
Microbial landscape of artisan cheesemaking facilities.
To better elaborate the role that processing environments may play in directing the microbial communities of cheese fermentations, an HTS approach was used to explore the microbial ecosystems of two artisan cheesemaking plants (Fig. 1). Both facilities produce distinct product lines but produce the same styles of cheese, including fresh, bloomy-rind, and washed-rind cheeses. Facility B is older and does not employ any surface-ripening cultures in their washed-rind production process (though P. camemberti and G. candidum are inoculated into milk for washed-rind cheeses prior to coagulation). Facility A is newer and utilizes several bacterial and fungal cultures for surface ripening. In spite of this, both facilities displayed the involvement of similar genera of fungi (Fig. 2) and bacteria (Fig. 3) in distinct processing areas of each facility. Overall, both facilities were dominated by the yeast Debaryomyces (most likely D. hansenii because of its involvement in dairy fermentations and inoculation in facility A) and the fungus Penicillium commune (Fig. 2 and 4), whereas the bacterial communities were dominated by Lactococcus (Fig. 3 and 4). Preprocessing areas related to milk handling exhibited higher relative population densities of the yeast Lachancea thermotolerans (Fig. 5) and the filamentous fungi P. commune, Botrytis fuckeliana, and Aureobasidium pullulans in both facilities (Fig. 2 and 4). These sites also contained higher relative abundances of the bacterial taxa Comamonadaceae, Moraxellaceae, Aeromonas, Brevundimonas, and Limnohabitans than did downstream processing sites (Fig. 3, 4, and 6) but the lowest absolute quantities of bacteria and fungi of any surface type (Fig. 7). Curd-making and brining surfaces predominantly comprised Lactococcus, Lactobacillus, and Debaryomyces (Fig. 2 to 4 and 7). In facility A, bloomy-rind maturation room surfaces were populated by Lactococcus and Pseudoalteromonas, in addition to higher relative levels of Pseudomonas and Psychrobacter, whereas washed-rind maturation room surfaces in both facilities contained higher populations of Brevibacterium, Staphylococcus, Corynebacterium, Halomonas, and Halomonadaceae (Fig. 3 and 4). Debaryomyces dominated the fungal communities of all maturation surfaces in both facilities (Fig. 2, 4, and 5). In both facilities, the mean bacterial and fungal loads were significantly higher on brining and aging surfaces than on milk- and curd-handling surfaces (Fig. 2, 3, and 7). Tables S3 and S4 in the supplemental material contain comprehensive lists of the bacterial and fungal taxa detected in all of the samples.
Fig 2.
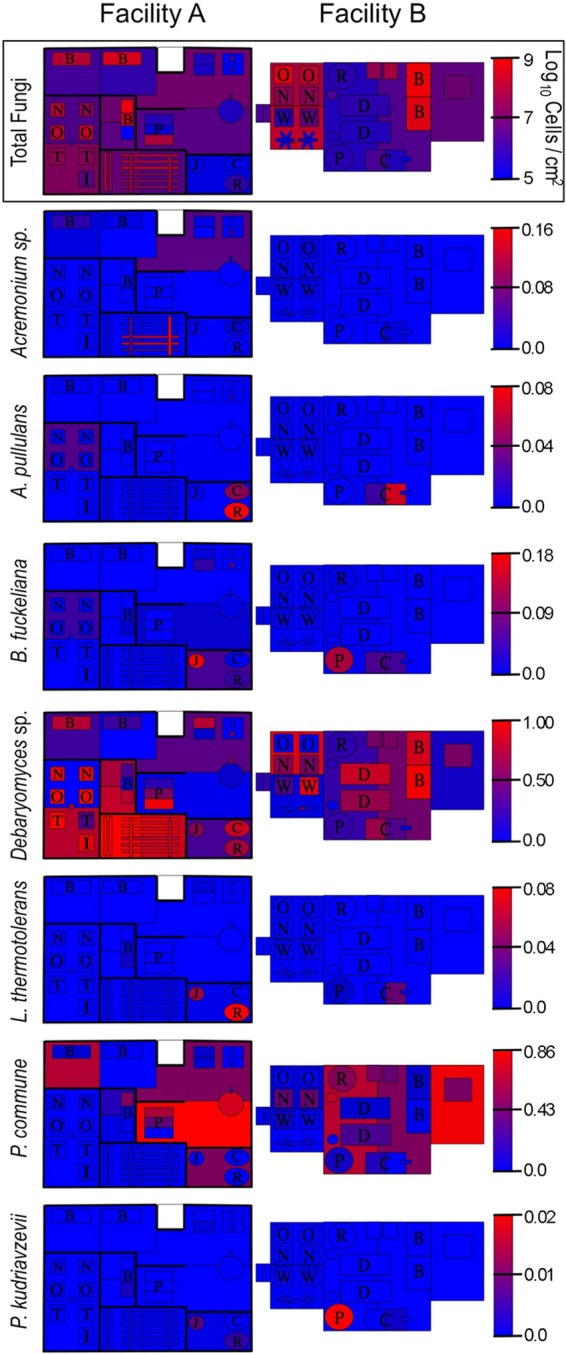
Fungal landscape of the processing environment of two artisan cheesemakers. Fungal HTS heat maps indicate relative genus abundances at each sample site. Gradient keys are in units of fractional species abundance. The qPCR heat map (outlined) indicates fungal biomass (as cells/cm2) across facility surfaces.
Fig 3.

Bacterial landscape of the processing environment of two artisan cheesemakers. Bacterial heat maps indicate relative genus abundances at each sample site. Gradient keys are in units of fractional species abundance. The qPCR heat map (outlined) indicates bacterial biomass (as 16S rRNA copy number/cm2) across facility surfaces.
Fig 4.

Processing area type modulates bacterial and fungal community structure. Mean relative abundance of bacteria (A) and fungal species (B) across surfaces classified by equipment type. Jackknifed weighted UniFrac distance PCoA of bacterial communities (C) and jackknifed Canberra distance PCoA of fungal communities (D) on surfaces classified by equipment type. P values indicate Bonferroni-corrected P values for permutational MANOVA significance between surface types. Milk, milk-handling surfaces; Curd, curd processing; Mixed, mixed-use facility (facility B); Brine, brining surfaces; Bloomy, bloomy-rind aging space; Wash, washed-rind aging space; Pack, packaging area surfaces (Fig. 1).
Fig 5.
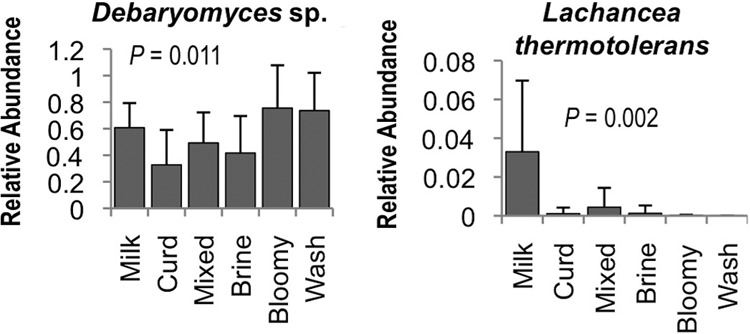
Processing area type modulates yeast species relative abundance. Bonferroni-corrected one-way ANOVA P values are shown. Milk, milk-handling surfaces; Curd, curd processing; Mixed, mixed-use facility (facility B); Brine, brining surfaces; Bloomy, bloomy-rind aging space; Wash, washed-rind aging space (Fig. 1). Error bars represent standard deviations.
Fig 6.
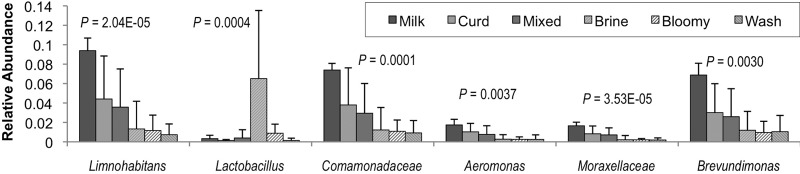
Processing area type modulates bacterial relative abundance. Bonferroni-corrected one-way ANOVA P values are shown. Milk, milk-handling surfaces; Curd, curd processing; Mixed, mixed-use facility (facility B); Brine, brining surfaces; Bloomy, bloomy-rind aging space; Wash, washed-rind aging space (Fig. 1). Error bars represent standard deviations.
Fig 7.
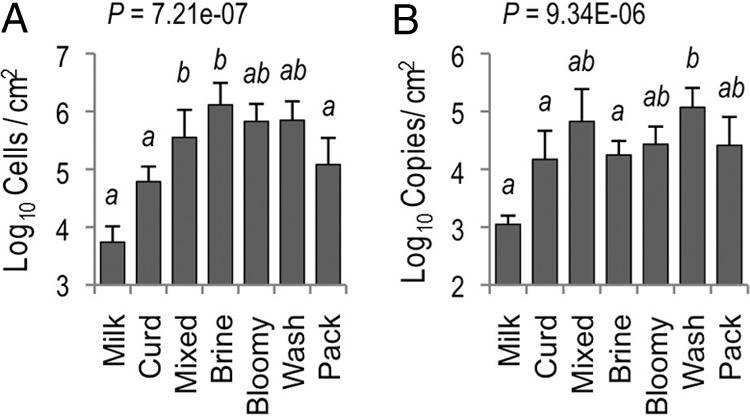
Microbial biomass of processing surfaces increases during cheese fermentation stage. Average fungal (A) and bacterial (B) biomass across surface types in both cheesemaking facilities, determined by qPCR. One-way ANOVA P values and Bonferroni-corrected PLSD are shown. Means with the same letter are not significantly different. Brine, brining surfaces; Bloomy, bloomy-rind aging space; Wash, washed-rind aging space (Fig. 1). Error bars represent standard deviations.
In addition to facility surface swabs, samples from washed-rind cheese surfaces at both facilities were collected and analyzed to elucidate the link between indigenous, environmental microbiota and the microbiota actually present on washed-rind cheese surfaces during maturation. The cheeses from each facility exhibited markedly different bacterial communities, primarily composed of noninoculated taxa (see Table S3 in the supplemental material), closely reflecting the surface microbiota detected in the aging environments where they were matured (Fig. 8). Cheese A was dominated by Brevibacterium, Psychrobacter, Brachybacterium, Corynebacterium, and Staphylococcus (the only genus among these that represents an inoculated culture in this facility). Cheese B was dominated by Pseudoalteromonas, with minor populations of Psychrobacter, Vibrio, and other members of the family Vibrionaceae, none of which are inoculated in this facility. Surface fungal communities on both cheeses were predominantly composed of Debaryomyces with secondary populations of P. commune (see Table S4). All of these cheese surface microbiota were also detected in the aging-room environments.
Fig 8.
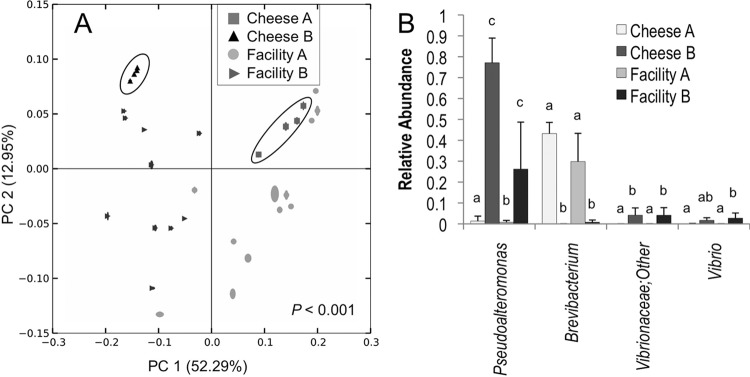
Facility-specific bacterial communities emerge in aging environments. Jackknifed weighted UniFrac distance PCoA (A) of bacterial communities on cheese surfaces and aging-room surfaces in facility A and facility B indicates significant separation between facilities. Cheese surface samples are circled to highlight clustering in a facility-dependent fashion. Bonferroni-corrected permutational MANOVA P values are shown. (B) Bacterial taxa exhibiting significant differences in relative abundance between aging-room surfaces in facility A and facility B and washed-rind cheese surfaces in facilities A and B. Error bars represent standard deviations. Groups with different lowercase letters are significantly different, based on Bonferroni-corrected least significant difference multiple-comparisons tests (α = 0.05).
Beta-diversity (between-sample community comparison) distance estimates are important ecological metrics for comparison of the similarity of different samples on the basis of species diversity. Abundance-weighted UniFrac PCoA of bacterial communities revealed that samples cluster by processing operation type in both facilities (Fig. 4C), indicating that phylogenically similar bacterial communities inhabit surfaces encountering the same production media (e.g., milk, curd, brine, cheese). Canberra distance PCoA of fungal communities revealed the same clustering pattern, albeit with looser clusters and lower explained variance (because of the high abundance of Debaryomyces on most surfaces), indicating that the fungal communities of these surfaces are also shaped by processing stage (Fig. 4D). Thus, milk-handling and curd-production surfaces cluster together away from brining and maturation surfaces. The mixed-use production area surfaces in facility B bridge the gap between these upstream- and downstream-processing surface clusters, as milk handling, curd production, and brining all occur in the same room. Interestingly, these clusters formed in a facility-independent fashion (i.e., each cluster category includes samples from each facility), revealing that the process type shapes surface microbiota across different cheese plants and independently of inoculation. However, weighted UniFrac PCoA of aging-room bacterial communities revealed facility-specific clusters, indicating the presence of phylogenically distinct subpopulations creating unique communities in each habitat (Fig. 8A). Further analysis revealed that Pseuodoalteromonas, Vibrio, and other members of the family Vibrionaceae were significantly more abundant in facility B and cheese B than in facility A and cheese A, whereas Brevibacterium was significantly more abundant in facility A and cheese A, underlining the existence of facility-specific microcosms (Fig. 8B). The Vibrio and Vibrionaceae sequences were most closely related to Vibrio casei, as determined by a manual NCBI BLAST search (data not shown). V. casei is a recently described species originally isolated from French washed-rind cheeses (49).
DISCUSSION
Microbial diversification of functional habitats.
Within each cheesemaking facility, similar communities of microbes occupied the same surface types, reflecting the selection for distinct communities on the basis of the production stage. In general, milk-handling surfaces were populated by Gram-negative Proteobacteria and filamentous fungi, which declined immediately following curd production, giving way to Gram-positive Lactobacillales and Actinomycetales. This trend continued postbrining, and the Gram-negative bacteria detected on maturation surfaces were primarily halotolerant Gammaproteobacteria (Pseudoalteromonas, Halomonas, Vibrio casei). Fungal communities were dominated by Debaryomyces sp. and P. commune throughout both facilities, with other minor fungal populations. The two key stages at which the largest shifts were observed were curd production and maturation, highlighting that the most obvious selective forces in these environments are milk acidification during curd production and salt content postbrining. Using a culture-based approach, Mounier et al. (14) detected a similar “house” microbiota in a washed-rind cheesemaking plant, dominated by Debaryomyces hansenii, Corynebacterium spp., and Staphylococcus saprophyticus, suggesting that this same functional community may generalize across facilities producing similar styles of cheese. Several of the halotolerant Gammaproteobacteria detected on aging surfaces and cheeses in this study have been isolated previously from European washed-rind cheeses, including Pseudoalteromonas (50), Halomonas (50–53), and Vibrio (49, 52), so likewise appear to be part of the style-specific microbiome enriched by washed-rind processing techniques and likely play a role in the maturation phenomena of these cheeses. The coryneform Actinobacteria detected on aging-room and cheese surfaces, particularly in facility A, have likewise been isolated from washed-rind cheeses previously, including Brevibacterium (50–52, 54), Corynebacterium (50–52, 54), and Brachybacterium (50, 54, 55).
Interestingly, the same dominant taxa were detected on each surface type in each facility independently of inoculation. In both facilities, where Lactococcus is inoculated, this appears to be evidence that inocula become established in the environment, facilitating transfer between fermentations, though whether these taxa comprise the same inoculated strains was not confirmed. However, uninoculated microbial communities populated downstream processing surfaces in both facilities, suggesting that the processing environment forms distinct functional niches, selecting for the species that best perform in that environment regardless of inoculation. These data corroborate the prior evidence of other authors (52, 56), who found that the majority of the bacteria and yeasts isolated from ripening washed-rind cheese surfaces were strains different from those inoculated into the milk, suggesting that facility-resident strains were overwhelmingly dominant and that commercial strains did not establish themselves in the processing environment in this system. Such establishment by indigenous microbiota may help direct the sensory characteristics of cheeses produced at that site and suggests that in some cases inoculation may have only a limited influence on driving cheese microbiota under a given set of conditions. Strain level dynamics were not tested in the present study, but these data suggest that at the genus and species levels these processing environments are dominated by resident microbiota, and many of the primary inocula could not be detected on equipment surfaces, including Leuconostoc, P. camemberti, and Geotrichum candidum. Thus, even in these relatively new facilities (e.g., compared to farmhouse cheesemakers in Europe), house microbial communities become established, most likely shaped by the production practices employed, providing continuous transfer to successive batches of cheese. These communities appear to be established on these surfaces in spite of frequent cleaning and sanitation, and cleaning practices are likely essential for maintaining this low-diversity environment, preventing the colonization of other species that are potentially detrimental in this environment. However, P. commune was detected in these production facilities and since it has an ITS1 domain highly similar to that of P. camemberti, this taxonomic assignment may represent a reference sequence misannotation, incorrect BLAST classification, or possibly even a misidentification of the inoculum.
Emergence of house microbiota.
In addition to the core, style-associated microbiota apparently enriched by processing methods in both cheesemaking facilities, several bacterial taxa distinguished the aging-room surfaces and washed-rind cheese communities. In facility A, cheese and aging-room surfaces displayed similar bacterial community assemblages, dominated by Brevibacterium, Staphylococcus, Psychrobacter, and Corynebacterium. In facility B, Pseuodoalteromonas, Vibrio, and Vibrionaceae were similarly abundant on aging-room and cheese surfaces, illustrating a close relationship between environmental and cheese surface microbiota. Vibrio casei, the closest match for the Vibrionaceae sequences, was originally isolated from a French washed-rind cheese surface (49), and its appearance here suggests a wider association with cheeses of this type. These and other halotolerant Gammaproteobacteria (especially Pseudoalteromonas) dominated the washed-rind maturation room surfaces in facility B in lieu of Brevibacterium and Staphylococcus, both of which dominated these surfaces and cheese surfaces in facility A. Thus, while a common process-driven microbiome is shared between the maturation rooms, a location-specific house microbiota also appears to be established, differentiating these two facilities and the cheeses made there. In facility A, this is possibly partially driven by inoculation (species of Staphylococcus being inoculated onto cheeses in this plant), but in facility B, this is entirely due to indigenous taxa establishing themselves on these surfaces. This study did not explicitly explore whether these house microbiota populating aging-room environments are actually responsible for sensory transformations in these cheeses, but these taxa dominate the surfaces of the washed-rind cheeses maturing here, most likely impacting chemosensory properties and contributing to the site-specific characteristics of these cheeses.
Historically, all cheeses were produced—and very many continue to be produced—without direct inoculation of defined starter cultures, and the microbial agents involved in these fermentations were unquestionably enriched from environmental sources. Together with the influence of regional practices, ingredients, and technological innovations, the distinct microbial consortia cultivated in geographically disparate processing environments would have given rise to the vast diversity of cheese varieties appreciated today. Subtle variations in these style-driven communities then could develop site-specific “house” microbiomes, possibly explaining the added diversity of cheese characteristics expressed within a given style of cheese and the unique idiosyncrasies observed in artisanal cheeses from different production facilities. House microbiota have been implicated in the production of many traditional cheeses (10, 57–63), and regional, site-specific microbial patterns have been linked to the chemosensory properties of water buffalo mozzarella (64) and Pecorino Crotonese (65), suggesting that house microbiota may partially drive the sensory characteristics of cheeses from different facilities. The establishment of such a consortium appears to happen fairly quickly, as the older cheesemaking plant analyzed in this study (facility B) is only 16 years old. Even in facilities incorporating defined commercial inocula (such as the facilities in this study), the production environment remains a pertinent source of microbes throughout the course of fermentation, most likely subtly shaping product qualities. Given the importance of processing environment microbiota in conducting aspects of cheese fermentations, facility ecosystem surveillance may become a new approach for the study of fermentation microbiota in cheese and other food fermentation systems. Following this model, fermentations and their surrounding environments would be analyzed together with the goal of describing microbial transactions within the fermentation ecosystem. In addition, high-density facility monitoring may become a valuable tool in food processing facilities, where routine microbial surveillance can monitor populations at critical control points for improved process control and sanitation management.
Supplementary Material
ACKNOWLEDGMENTS
We thank Mariya Ryazantseva and Chad Masarweh for technical support with DNA extractions.
N.A.B. was supported by the 2012-2013 Dannon Probiotics Fellow Program (The Dannon Company, Inc.), the Brian Williams Scholarship Fund (American Society of Brewing Chemists Foundation), a 2012-2013 American Wine Society Educational Foundation scholarship, and a Wine Spectator scholarship during the completion of this work. D.A.M. acknowledges the support of the Shields Endowed Chair in Dairy Food Science.
Footnotes
Published ahead of print 21 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.00934-13.
REFERENCES
- 1.Bokulich NA, Mills DA. 2012. Next-generation approaches to the microbial ecology of food fermentations. BMB Rep. 45:377–389 [DOI] [PubMed] [Google Scholar]
- 2.Irlinger F, Mounier J. 2009. Microbial interactions in cheese: implications for cheese quality and safety. Curr. Opin. Biotechnol. 20:142–148 [DOI] [PubMed] [Google Scholar]
- 3.Suárez B, Ferreiros CM, Criado MT. 1992. Adherence of psychrotrophic bacteria to dairy equipment surfaces. J. Dairy Res. 59:381–888 [DOI] [PubMed] [Google Scholar]
- 4.Lewis SJ, Gilmour A. 1987. Microflora associated with the internal surfaces of rubber and stainless steel milk transfer pipeline. J. Appl. Bacteriol. 62:327–333 [DOI] [PubMed] [Google Scholar]
- 5.Somers EB, Johnson ME, Wong ACL. 2001. Biofilm formation and contamination of cheese by nonstarter lactic acid bacteria in the dairy environment. J. Dairy Sci. 84:1926–1936 [DOI] [PubMed] [Google Scholar]
- 6.Broadbent JR, Houck K, Johnson ME, Oberg CJ. 2003. Influence of adjunct use and cheese microenvironment on nonstarter bacteria in reduced-fat cheddar-type cheese. J. Dairy Sci. 86:2773–2782 [DOI] [PubMed] [Google Scholar]
- 7.Agarwal S, Sharma K, Swanson BG, Yuksel GU, Clark S. 2006. Nonstarter lactic acid bacteria biofilms and calcium lactate crystals in cheddar cheese. J. Dairy Sci. 89:1452–1466 [DOI] [PubMed] [Google Scholar]
- 8.Steele J, Budinich MF, Cai H, Curtis SC, Broadbent J. 2006. Diversity and metabolic activity of Lactobacillus casei in ripening cheddar cheese. Aust. J. Dairy Technol. 61:53–60 [Google Scholar]
- 9.Mariani C, Briandet R, Chamba JF, Notz E, Carnet-Pantiez A, Eyoug RN, Oulahal N. 2007. Biofilm ecology of wooden shelves used in ripening the French raw milk smear cheese Reblochon de Savoie. J. Dairy Sci. 90:1653–1661 [DOI] [PubMed] [Google Scholar]
- 10.Feligini M, Panelli S, Buffoni JN, Bonacina C, Andrighetto C, Lombardi A. 2012. Identification of microbiota present on the surface of Taleggio cheese using PCR-DGGE and RAPD-PCR. J. Food Sci. 77:M609–M615 [DOI] [PubMed] [Google Scholar]
- 11.Lortal S, Di Blasi A, Madec MN, Pediliggieri C, Tuminello L, Tanguy G, Fauquant J, Lecuona Y, Campo P, Carpino S, Licitra G. 2009. Tina wooden vat biofilm: a safe and highly efficient lactic acid bacteria delivering system in PDO Ragusano cheese making. Int. J. Food Microbiol. 132:1–8 [DOI] [PubMed] [Google Scholar]
- 12.Licitra G, Ogier JC, Parayre S, Pediliggieri C, Carnemolla TM, Falentin H, Madec MN, Carpino S, Lortal S. 2007. Variability of bacterial biofilms of the “tina” wood vats used in the Ragusano cheese-making process. Appl. Environ. Microbiol. 73:6980–6987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Didienne R, Defargues C, Callon C, Meylheuc T, Hulin S, Montel MC. 2012. Characteristics of microbial biofilm on wooden vats (‘gerles') in PDO Salers cheese. Int. J. Food Microbiol. 156:91–101 [DOI] [PubMed] [Google Scholar]
- 14.Mounier J, Goerges S, Gelsomino R, Vancanneyt M, Vandemeulebroecke K, Hoste B, Brennan NM, Scherer S, Swings J, Fitzgerald GF, Cogan TM. 2006. Sources of the adventitious microflora of a smear-ripened cheese. J. Appl. Microbiol. 101:668–681 [DOI] [PubMed] [Google Scholar]
- 15.Bokulich NA, Bamforth CW, Mills DA. 2012. A review of molecular methods for microbial community profiling of beer and wine. J. Am. Soc. Brew. Chem. 70:150–162 [Google Scholar]
- 16.Ercolini D. 2013. High-throughput sequencing and metagenomics: steps ahead in the culture-independent analysis of food microbial ecology. Appl. Environ. Microbiol. 79:3148–3155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Masoud W, Takamiya M, Vogensen FK, Lillevang S, Abu Al-Soud W, Sorensen SJ, Jakobsen M. 2011. Characterization of bacterial populations in Danish raw milk cheeses made with different starter cultures by denaturating gradient gel electrophoresis and pyrosequencing. Int. Dairy J. 21:142–148 [Google Scholar]
- 18.Lusk TS, Ottesen AR, White JR, Allard MW, Brown EQ, Kase JA. 2012. Characterization of microflora in Latin-style cheeses by next-generation sequencing technology. BMC Microbiol. 12:254. 10.1186/1471-2180-12-254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quigley L, O'Sullivan O, Beresford TP, Ross RP, Fitzgerald GF, Cotter PD. 2012. High-throughput sequencing for detection of subpopulations of bacteria not previously associated with artisanal cheeses. Appl. Environ. Microbiol. 78:5717–5723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Masoud W, Vogensen FK, Lillevang S, Abu Al-Soud W, Sørensen SJ, Jakobsen M. 2012. The fate of indigenous microbiota, starter cultures, Escherichia coli, Listeria innocua and Staphylococcus aureus in Danish raw milk and cheeses determined by pyrosequencing and quantitative real time (qRT)-PCR. Int. J. Food Microbiol. 153:192–202 [DOI] [PubMed] [Google Scholar]
- 21.Alegría A, Szczesny P, Mayo B, Bardowski J, Kowalczyk M. 2012. Biodiversity in Oscypek, a traditional Polish cheese, determined by culture-dependent and -independent approaches. Appl. Environ. Microbiol. 78:1890–1898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ercolini D, De Filippis F, La Storia A, Iacono M. 2012. “Remake” by high-throughput sequencing of the microbiota involved in the production of water buffalo mozzarella cheese. Appl. Environ. Microbiol. 78:8142–8145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bokulich NA, Joseph CML, Allen GR, Benson A, Mills DA. 2012. Next-generation sequencing reveals significant bacterial diversity of botrytized wine. PLoS One 7:e36357. 10.1371/journal.pone.0036357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bokulich NA, Bamforth CW, Mills DA. 2012. Brewhouse-resident microbiota are responsible for multi-stage fermentation of American coolship ale. PLoS One 7:e35507. 10.1371/journal.pone.0035507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bokulich NA, Ohta M, Richardson P, Mills DA. 2013. Monitoring seasonal changes in winery-resident microbiota. PLoS One 8:e66437. 10.1371/journal.pone.0066437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hewitt KM, Mannino FL, Gonzalez A, Chase JH, Caporaso JG, Knight R, Kelley ST. 2013. Bacterial diversity in two neonatal intensive care units (NICUs). PLoS One 8:e54703. 10.1371/journal.pone.0054703.t001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kembel SW, Jones E, Kline J, Northcutt D, Stenson J, Womack AM, Bohannan BJ, Brown GZ, Green JL. 2012. Architectural design influences the diversity and structure of the built environment microbiome. ISME J. 6:1469–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bokulich NA, Mills DA, Underwood M. 5 January 2013, posting date. Surface microbes in the neonatal intensive care unit: changes with routine cleaning and over time. J. Clin. Microbiol. [Epub ahead of print.] 10.1128/JCM.00898-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hewitt KM, Gerba CP, Maxwell SL, Kelley ST. 2012. Office space bacterial abundance and diversity in three metropolitan areas. PLoS One 7:e37849. 10.1371/journal.pone.0037849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Flores GE, Bates ST, Knights D, Lauber CL, Stombaugh J, Knight R, Fierer N. 2011. Microbial biogeography of public restroom surfaces. PLoS One 6:e28132. 10.1371/journal.pone.0028132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flores GE, Bates ST, Caporaso JG, Lauber CL, Leff JW, Knight R, Fierer N. 2013. Diversity, distribution and sources of bacteria in residential kitchens. Environ. Microbiol. 15:588–596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caporaso JG, Kuczynski J, Stombaugh J, Bittinger K, Bushman FD, Costello EK, Fierer N, Gonzalez Pena A, Goodrich JK, Gordon JI, Huttley GA, Kelley ST, Knights D, Koenig JE, Ley RE, Lozupone CA, McDonald D, Muegge BD, Pirrung M, Reeder J, Sevinsky JR, Turnbaugh PJ, Walters WA, Widmann J, Yatsunenko T, Zaneveld J, Knight R. 2010. Qiime allows analysis of high-throughput community sequence data. Nat. Methods 7:335–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bokulich NA, Mills DA. 2013. Improved selection of internal transcribed spacer-specific primers enables quantitative, ultra-high-throughput profiling of fungal communities. Appl. Environ. Microbiol. 79:2519–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Lozupone CA, Turnbaugh PJ, Fierer N, Knight R. 2011. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. U. S. A. 108:4516–4522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bokulich NA, Subramanian S, Faith JJ, Gevers D, Gordon JI, Knight R, Mills DA, Caporaso JG. 2013. Quality-filtering vastly improves diversity estimates from Illumina amplicon sequencing. Nat. Methods 10:57–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Edgar RC. 2010. Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26:2460–2461 [DOI] [PubMed] [Google Scholar]
- 37.McDonald D, Price MN, Goodrich J, Nawrocki EP, DeSantis TZ, Probst A, Andersen GL, Knight R, Hugenholtz P. 2012. An improved Greengenes taxonomy with explicit ranks for ecological and evolutionary analyses of bacteria and archaea. ISME J. 6:610–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 39.Kõljalg U, Larsson KH, Abarenkov K, Nilsson RH, Alexander IJ, Eberhardt U, Erland S, Hoiland K, Kjoller R, Larsson E, Pennanen T, Sen R, Taylor AF, Tedersoo L, Vralstad T, Ursing BM. 2005. UNITE: a database providing web-based methods for the molecular identification of ectomycorrhizal fungi. New Phytol. 166:1063–1068 [DOI] [PubMed] [Google Scholar]
- 40.Abarenkov K, Nilsson RH, Larsson K-H, Alexander IJ, Eberhardt U, Erland S, Høiland K, Kjøller R, Larsson E, Pennanen T, Sen R, Taylor AFS, Tedersoo L, Ursing BM, Vrålstad T, Liimatainen K, Peintner U, Kõljalg U. 2010. The UNITE database for molecular identification of fungi—recent updates and future perspectives. New Phytol. 186:281–285 [DOI] [PubMed] [Google Scholar]
- 41.Caporaso JG, Bittinger K, Bushman FD, DeSantis T, Andersen GL, Knight R. 2010. PyNAST: a flexible tool for aligning sequences to a template alignment. Bioinformatics 26:266–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, Ciulla DM, Tabbaa D, Highlander SK, Sodergren E, Methe B, DeSantis TZ, Petrosino JF, Knight R, Birren BW. 2011. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 21:494–504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. 10.1371/journal.pone.0009490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lozupone CA, Knight R. 2005. UniFrac: a new phylogenetic method for comparing microbial communities. Appl. Environ. Microbiol. 71:8228–8235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Anderson MJ. 2001. A new method for non-parametric multivariate analysis of variance. Austral Ecol. 26:32–46 [Google Scholar]
- 46.Gonzalez A, Stombaugh J, Lauber CL, Fierer N, Knight R. 2012. SitePainter: a tool for exploring biogeographical patterns. Bioinformatics 28:436–438 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hierro N, Esteve-Zarzoso B, Gonzalez A, Mas A, Guillamon JM. 2006. Real-time quantitative PCR (qPCR) and reverse transcription-qPCR for detection and enumeration of total yeasts in wine. Appl. Environ. Microbiol. 72:7148–7155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hartmann AL, Lough DM, Barupal DK, Fiehn O, Fishbein T, Zasloff M, Eisen JA. 2009. Human gut microbiome adopts an alternative state following small bowel transplantation. Proc. Natl. Acad. Sci. U. S. A. 106:17187–17192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bleicher A, Neuhaus K, Scherer S. 2010. Vibrio casei sp. nov., isolated from the surfaces of two French red smear soft cheeses. Int. J. Syst. Evol. Microbiol. 60:1745–1749 [DOI] [PubMed] [Google Scholar]
- 50.Ogier JC, Lafarge V, Girard V, Rault A, Maladen V, Gruss A, Leveau JY, Delacroix-Buchet A. 2004. Molecular fingerprinting of dairy microbial ecosystems by use of temporal temperature and denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 70:5628–5643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Maoz A, Mayr R, Scherer S. 2003. Temporal stability and biodiversity of two complex antilisterial cheese-ripening microbial consortia. Appl. Environ. Microbiol. 69:4012–4018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mounier J, Gelsomino R, Goerges S, Vancanneyt M, Vandemeulebroecke K, Hoste B, Scherer S, Swings J, Fitzgerald GF, Cogan TM. 2005. Surface microflora of four smear-ripened cheeses. Appl. Environ. Microbiol. 71:6489–6500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coton M, Delbes-Paus C, Irlinger F, Desmasures N, Le Fleche A, Stahl V, Montel MC, Coton E. 2012. Diversity and assessment of potential risk factors of Gram-negative isolates associated with French cheeses. Food Microbiol. 29:88–98 [DOI] [PubMed] [Google Scholar]
- 54.Gori K, Ryssel M, Arneborg N, Jespersen L. 2013. Isolation and identification of the microbiota of Danish farmhouse and industrially produced surface-ripened cheeses. Microb. Ecol. 65:602–615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schubert K, Ludwig W, Springer N, Kroppenstedt RM, Accolas JP, Fiedler F. 1996. Two coryneform bacteria isolated from the surface of French Gruyère and Beaufort cheeses are new species of the genus Brachybacterium: Brachybacterium alimentarium sp. nov. and Brachybacterium tyrofermentans sp. nov. Int. J. Syst. Bacteriol. 46:81–87 [DOI] [PubMed] [Google Scholar]
- 56.Goerges S, Mounier J, Rea MC, Gelsomino R, Heise V, Beduhn R, Cogan TM, Vancanneyt M, Scherer S. 2008. Commercial ripening starter microorganisms inoculated into cheese milk do not successfully establish themselves in the resident microbial ripening consortia of a South German red smear cheese. Appl. Environ. Microbiol. 74:2210–2217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dolci P, Barmaz A, Zenato S, Pramotton R, Alessandria V, Cocolin L, Rantsiou K, Ambrosoli R. 2009. Maturing dynamics of surface microflora in Fontina PDO cheese studied by culture-dependent and -independent methods. J. Appl. Microbiol. 106:278–287 [DOI] [PubMed] [Google Scholar]
- 58.Bonetta S, Carraro E, Rantsiou K, Cocolin L. 2008. Microbiological characterisation of Robiola di Roccaverano cheese using PCR-DGGE. Food Microbiol. 25:786–792 [DOI] [PubMed] [Google Scholar]
- 59.Poznanski E, Cavazza A, Cappa F, Cocconcelli PS. 2004. Indigenous raw milk microbiota influences the bacterial development in traditional cheese from an alpine natural park. Int. J. Food Microbiol. 92:141–151 [DOI] [PubMed] [Google Scholar]
- 60.El-Baradei G, Delacroix-Buchet A, Ogier JC. 2007. Biodiversity of bacterial ecosystems in traditional Egyptian Domiati cheese. Appl. Environ. Microbiol. 73:1248–1255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Randazzo CL, Vaughan EE, Caggia C. 2006. Artisanal and experimental Pecorino Siciliano cheese. Microbial dynamics during manufacture assessed by culturing and PCR-DGGE analyses. Int. J. Food Microbiol. 109:1–8 [DOI] [PubMed] [Google Scholar]
- 62.Van Hoorde K, Heyndrickx M, Vandamme P, Huys G. 2010. Influence of pasteurization, brining conditions and production environment on the microbiota of artisan Gouda-type cheeses. Food Microbiol. 27:425–433 [DOI] [PubMed] [Google Scholar]
- 63.Bonizzi I, Feligini M, Aleandri R, Enne G. 2007. Genetic traceability of the geographical origin of typical Italian water buffalo mozzarella cheese: a preliminary approach. J. Appl. Microbiol. 102:667–673 [DOI] [PubMed] [Google Scholar]
- 64.Mauriello G, Moio L, Genovese A, Ercolini D. 2003. Relationships between flavoring capabilities, bacterial composition, and geographical origin of natural whey cultures used for traditional water-buffalo mozzarella cheese manufacture. J. Dairy Sci. 86:486–497 [DOI] [PubMed] [Google Scholar]
- 65.Randazzo CL, Pitino I, Ribbera A, Caggia C. 2010. Pecorino Crotonese cheese: study of bacterial population and flavour compounds. Food Microbiol. 27:363–374 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequence data generated in this study are publicly available on the QIIME database (www.microbio.me/qiime/) as studies 1884 (bacterial 16S rRNA sequencing run data) and 1919 (fungal ITS sequencing run data).