

Abstract
Ceratocystis platani is the causal agent of canker stain of plane trees, a lethal disease able to kill mature trees in one or two successive growing seasons. The pathogen is a quarantine organism and has a negative impact on anthropogenic and natural populations of plane trees. Contaminated sawdust produced during pruning and sanitation fellings can contribute to disease spread. The goal of this study was to design a rapid, real-time quantitative PCR assay to detect a C. platani airborne inoculum. Airborne inoculum traps (AITs) were placed in an urban setting in the city of Florence, Italy, where the disease was present. Primers and TaqMan minor groove binder (MGB) probes were designed to target cerato-platanin (CP) and internal transcribed spacer 2 (ITS2) genes. The detection limits of the assay were 0.05 pg/μl and 2 fg/μl of fungal DNA for CP and ITS, respectively. Pathogen detection directly from AITs demonstrated specificity and high sensitivity for C. platani, detecting DNA concentrations as low as 1.2 × 10−2 to 1.4 × 10−2 pg/μl, corresponding to ∼10 conidia per ml. Airborne inoculum traps were able to detect the C. platani inoculum within 200 m of the closest symptomatic infected plane tree. The combination of airborne trapping and real-time quantitative PCR assay provides a rapid and sensitive method for the specific detection of a C. platani inoculum. This technique may be used to identify the period of highest risk of pathogen spread in a site, thus helping disease management.
INTRODUCTION
Over the last 20 years, biosecurity protocols for plant protection have been developed in order to prevent the diffusion of invasive plant pathogens and to assist in their eradication (1). Most invasive alien pests and pathogens that spread into a new environment are introduced by the commercial trade in plants (2, 3, 4, 5). Control of these pathogens is extremely difficult when an airborne dispersal phase is present, enabling disease to spread on a wider scale (6).
At a local scale, including in urban areas, the ability to detect an airborne inoculum is crucial for understanding disease spread and managing injurious plant pathogens, such as Ceratocystis platani (J. M. Walter) Engelbr. & T. C. Harr. (=Ceratocystis fimbriata Ellis & Halsted f. sp. platani Walter), the causal agent of canker stain of plane trees. This fungus causes a lethal disease on Platanus × acerifolia (Aiton) Willd (London plane), Platanus orientalis L. (Oriental plane), and to a lesser extent on Platanus occidentalis L. (American sycamore) in urban plantations, plantations for timber and fiber production, and natural forests, both in North America and in Europe (7, 8, 9, 10).
In North America, C. platani caused significant losses in London plane trees in urban areas during the 1930s (7) and in P. occidentalis plantations in the 1960s (8) and 1990s (9).
C. platani was introduced into Europe from the southeastern United States, probably on wood associated with military supplies during World War II (10). The first confirmation of the disease was in Tuscany, Italy, in 1972 (11), where the pathogen had already destroyed the monumental avenue connecting the Reggia di Caserta with Naples (12). In Europe, the pathogen is now present in Armenia, France, Switzerland (13), and Greece (14), where it is causing widespread serious losses in natural P. orientalis populations. Because of its heavy impact on plane trees, C. platani is considered a quarantine organism by the European and Mediterranean Plant Protection Organization (EPPO) (15).
The fungus is a wound parasite that colonizes the xylem tissues, killing the tree within a few years of infection (16). C. platani is naturally transmitted via root anastomosis, infected water, and, as suggested by recent research results, through an association with ambrosia beetles (14, 17). Nevertheless, the most important means of dispersal, especially in urban areas, are contaminated sawdust and equipment used for sanitation fellings (7). In this context, monitoring of the inoculum can lead to a better understanding of the dynamics of airborne spread in the environment.
In recent years, several methods have been developed to trap the airborne inoculum of invasive forest pathogens from environmental samples. Woody discs were used to catch the conidia of Heterobasidion irregulare in pine plantations (18, 19), while paper filters were used to trap the inoculum of Fusarium circinatum in sites infected with pine pitch canker (20). The use of reliable trapping methods, combined with sensitive molecular approaches, such as real-time quantitative PCR (qPCR) assays, allows for rapid and specific detection of fungal pathogens from these samples (21). The advent of this molecular technique has enabled faster and more sensitive diagnostic tools for the identification and quantification of disease-causing agents. The accuracy and reliability of qPCR may also enable the detection of latent fungal infections before symptoms occur (22, 23) and the detection of fungal pathogens that are difficult to culture (21).
The aim of the work reported here was to develop an accurate and reproducible method to detect C. platani in airborne environmental samples using a qPCR assay. This molecular approach was then used to study the small-scale epidemiology of this pathogen and to assess the spreading of the inoculum during sanitation cuttings.
MATERIALS AND METHODS
Fungal isolates, DNA extraction, and phylogenetic analysis.
Strains of Ceratocystis platani used in this study (Table 1) were obtained from the Central Bureau Voor Schimmelcaltures (CBS, The Netherlands) and from the Istituto per la Protezione delle Piante-Consiglio Nazionale delle Ricerche (IPP-CNR, Italy) collections. Fungal isolates were grown on 300PT cellophane discs (Celsa, Varese, Italy) on potato dextrose agar (PDA; Difco Laboratories, Detroit, MI) in 90-mm-diameter petri dishes and incubated in the dark at 20°C. After 7 days, the mycelium was scraped from the surface of the cellophane and stored in 1.5-ml microcentrifuge tubes (Sarstedt, Verona, Italy) at −20°C. DNA extraction was performed from mycelium using the E.Z.N.A. plant DNA minikit (Omega Bio-tek, Norcross, GA), following the manufacturer's instructions. The concentration of extracted DNA was measured using a Nanodrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, DE). To elucidate the phylogenetic relationships between C. platani and the other Ceratocystis species used (Table 1) and to test the specificity of the qPCR assay, sequence alignment of the internal transcribed spacer (ITS), β-tubulin, and cerato-platanin (CP) genes, was conducted. Cerato-platanin is a small hydrophobic protein from C. platani (24). The ITS2 region was amplified with the general primers ITS1 and ITS4 according to White et al. (25), while the β-tubulin gene was amplified using primers Bt2a and Bt2b, as described by Glass and Donaldson (26). Cerato-platanin was amplified with primer set CeraF (5′-ATGAAGTTCTCTATCCTACCC-3′) and CeraR (5′-AGGAGCTTCCGGAGAAGTCAC-3′) (this study) with the following cycling conditions: initial denaturation of 95°C for 2 min, followed by 40 cycles of 94°C for 30 s, 55°C for 45 s, and 72°C for 90 s, with a final elongation step at 72°C for 9 min. PCR products were purified and sequenced directly using a BigDye197 terminator cycle sequencing ready reaction kit (Applied Biosystems, Foster City, CA) and analyzed with an ABI-3130xl 48-capillary DNA sequencer (Applied Biosystems). Maximum likelihood (ML) phylogenetic trees were produced for ITS and for concatenated β-tubulin and CP sequences using the program RAxML (27) with the GTR model (28, 29). Bootstrap values were estimated from 1,000 replicates. Computations were performed at the Vital-IT Center (http://www.vital-it.ch) for high-performance computing of the Swiss Institute of Bioinformatics (SIB). Petrellia setifera (AF043596) from Witthuhn et al. (30) was used as an outgroup for the ITS tree, and Ceratocystis albifundus (EU244986) from Heath et al. (31) was used as the outgroup for the concatenated tree.
Table 1.
Fungal isolates used in this study
| Fungal species | Strain identification no. | Host | Origin | Sourcea |
|---|---|---|---|---|
| Ceratocystis | ||||
| C. cacaofunesta | CBS15362 | Theobroma cacao | Ecuador | CBS |
| C. fimbriata | CBS115166 | Ficus carica | Brazil | CBS |
| C. fimbriata | CBS14653 | Coffea arabica | Suriname | CBS |
| C. fimbriata | CBS115167 | Syngonium | Florida | CBS |
| C. fimbriata | CBS115173 | Gmelina arborea | Brazil | CBS |
| C. fimbriata | CBS118126 | Syzygium aromaticum | Sulawesi | CBS |
| C. fimbriata | CBS114723 | Ipomoea batatas | NC | CBS |
| C. fimbriata | CBS115175 | Mangifera indica | Brazil | CBS |
| C. fimbriata | CBS115174 | Eucalyptus | Brazil | CBS |
| C. fimbriata | CBS115171 | Colocasia esculenta | Brazil | CBS |
| C. fimbriata | CBS74040 | Crotolaria juncea | Brazil | CBS |
| C. fimbriata | CBS114717 | Prunus dulcis | CA | CBS |
| C. populicola | CBS115161 | Populus tremuloides | CBS | |
| C. platani | CBS115162 | Platanus occidentalis | NC | CBS |
| C. platani | CBS117355 | Platanus sp. | France | CBS |
| C. variospora | CBS114714 | Quercus robur | Rhodes | CBS |
| C. platani | Cp1 | Platanus sp. | Greece | NAGREF |
| C. platani | CF0 | P. × acerifolia | Italy | IPP-CNR |
| C. platani | Cp3 | P. × acerifolia | Italy | IPP-CNR |
| C. platani | Cp5 | P. × acerifolia | Italy | IPP-CNR |
| C. platani | Cp6 | P. × acerifolia | Italy | IPP-CNR |
| C. platani | Cp8 | P. × acerifolia | Italy | IPP-CNR |
| C. platani | Cp9 | P. × acerifolia | Italy | IPP-CNR |
| Other species | ||||
| Alternaria alternata | 1259 | Spain | UPV | |
| A. alternata | A1 | F. ornus | Italy | IPP-CNR |
| Cladosporium sp. | C4/2 | F. ornus | Italy | IPP-CNR |
| Epicoccum niger | C8/3 | F. ornus | Italy | IPP-CNR |
| E. niger | C4/3 | F. ornus | Italy | IPP-CNR |
| Microsphaera platani | M1 | P. × acerifolia | Italy | IPP-CNR |
| M. platani | M2 | P. × acerifolia | Italy | IPP-CNR |
| Sarchodontia pachyodon | Sp9 | Quercus ilex | Italy | IPP-CNR |
| S. pachyodon | Sp5 | P. × acerifolia | Italy | IPP-CNR |
| S. pachyodon | Sp3 | Quercus ilex | Italy | IPP-CNR |
| S. pachyodon | Sp7 | P. × acerifolia | Italy | IPP-CNR |
CBS, Centraalbureau voor Schimmelcultures, Utrecht, The Netherlands; IPP-CNR, Istituto per la Protezione delle Piante, Consiglio Nazionale delel Ricerche, Florence, Italy; NAGREF, National Agricultural Research Foundation, Institute of Mediterranean Forest Ecosystems, Thermi, Thessaloniki, Greece; UPV, Universitat Politècnica de València, Instituto Agroforestal Mediterráneo, Valencia, Spain.
TaqMan MGB probes and primer design.
In order to obtain markers specific for C. platani, two sets of primers and TaqMan minor groove binding (MGB) probes were designed to amplify the CP single-copy and the ITS multiple-copy genes. Primers and probes were designed using Primer Express Software 3.0 (Applied Biosystems) on the basis of consensus sequences of ITS2 (EU426554) and CP (EF017218) derived from the NCBI database (www.ncbi.nlm.nih.gov). TaqMan MGB probes were labeled with 6-carboxyfluorescein (FAM) at the 5′ end and a nonfluorescent quencher (NFQ) with minor groove binder (MGB) ligands as the quencher at the 3′ end. The specificity of newly designed primers and probes was further tested with BLAST (32) using published sequences in GenBank. The specificity of the primers and probes was also tested by qPCR on DNA from axenic cultures of (i) Ceratocystis species strictly related to C. platani, (ii) common airborne fungi, and (iii) fungal species inhabiting plane trees (Table 1).
qPCR assay conditions.
A 25-μl PCR mixture containing 12.5 μl TaqMan Universal master mix (Applied Biosystems) was performed on an ABI Prism 7300 sequence detector (Applied Biosystems). The primer and probe final concentrations used were as follows: 300 nM forward primer (Eurofins MWG Operon, Ebersberg, Germany), 300 nM reverse primer (Eurofins MWG Operon), and 200 nM TaqMan MGB probe (Applied Biosystems) designed for amplification of ITS and CP genes. For each tube, 5 μl sample DNA was used.
All DNA samples were assayed in MicroAmp optical 96-well plates (Applied Biosystems), closed with MicroAmp optical caps, using the ABI Prism 7300 sequence detector (Applied Biosystems). Each DNA sample was assayed in three replicates. Four wells, each containing 5 μl sterile water, were used as the no-template control (NTC). The PCR protocol was 50°C (2 min), 95°C (10 min), 50 cycles of 95°C (30 s), and 60°C (1 min).
Results were analyzed using an SDS 1.9 sequence detection system (Applied Biosystems) after manual adjustment of the baseline and fluorescence threshold. Measurements of C. platani DNA in unknown samples were made by interpolation from a standard curve generated with a C. platani DNA standard, which was amplified in the same PCR run. The standard curve was generated from 5-fold serial dilutions (ranging from 100 ng/tube to 0.25 pg/tube) of a known concentration of C. platani DNA (strain Cp5) and analyzed in triplicate in seven independent assays. The amount of C. platani DNA was expressed as pg/μl.
AITs.
The effectiveness of the airborne inoculum trap (AIT) system was evaluated by inoculation of Whatman filter papers with a conidial suspension of C. platani, followed by molecular detection. AITs were prepared using 90-mm-diameter petri dishes (Sarstedt, Verona, Italy) containing a Whatman no. 1 filter paper (Sigma-Aldrich, Milan, Italy), moistened with 4× TE buffer (40 mM Tris HCl, 4 mM EDTA [pH 8.0]) (20). Each petri dish was attached to the base of a small aluminum tray (500 ml) using double-sided adhesive tape. The tray was covered with a plastic net (6- by 6-mm mesh). A conidial suspension (1 ml in sterile water) was obtained from C. platani isolate CF0 (Table 1), following incubation on 1.5% PDA for 10 days. The number of conidia was determined in a Burker chamber, and five 1:10 serial dilutions ranging from 10 to 105 conidia/ml were prepared. For each conidial dilution, a pellet was obtained after centrifugation at 16,100 × g for 5 min and used for DNA extraction. Two Whatman no. 1 filter papers were inoculated with each of the conidial dilutions. Filters were then washed in a 50-ml centrifuge tube (Sarstedt) containing 20 ml TE buffer (4×) at 65°C. Each tube was centrifuged at 289 × g for 90 min to precipitate the pellet. One milliliter of the suspension containing the pellet and TE buffer was then transferred to a 2-ml microcentrifuge tube (Sarstedt), and following centrifugation at 16,100 × g for 5 min, the pellet was used for DNA extraction using the E.Z.N.A. plant DNA kit, as previously described. Both the DNA from the conidial suspension and the inoculated filter papers were tested by qPCR, as described above.
C. platani environmental survey.
The environmental survey was conducted in October 2011 on street plane trees in Florence, Italy (43°47′59″N, 11°13′94″E; 51 m above sea level), to assess the presence of the C. platani inoculum under urban conditions using AITs. The presence of the pathogen was determined in two sampling areas: (i) an infection area with Platanus × acerifolia showing symptoms of canker stain and (ii) the surrounding area, with healthy plane trees. Sampling was conducted in areas at least 50 m apart from each other. In the infection area, a sanitation cutting had been planned to remove infected plane trees. The presence of C. platani in the infection area on symptomatic planes was confirmed by isolations onto PDA (33). Briefly, isolations were carried out from samples collected from the boundary between necrotic and healthy tissues. Small fragments were collected from the margin of the necrotic tissue and placed in 90-mm-diameter petri dishes on 1.5% PDA and incubated at 20°C for 10 days. In addition, DNA was extracted from both necrotic and healthy tissues and analyzed by qPCR, as described above. In total, 25 AITs were prepared: 11 were placed at the infected site, and the remaining 14 were placed in the surrounding area. Each AIT was placed 2 m above the soil level at a 10-m distance from each other, on each streetlight. In the surrounding area, traps were placed 7 days before the sanitation fellings and wetted daily with 5 ml of 4× TE buffer (pH 8.0) on to the filter. To avoid contamination of the traps by C. platani before felling began, traps were placed in the infection site in the early morning and removed on the same day, once the felling operation concluded. AITs were transported to the laboratory, DNA was extracted from the filters as described above, and qPCR was carried out to determine the presence of C. platani.
Pathogenicity tests.
To assess the minimal amount of C. platani able to cause damage on plane trees, inoculation tests were carried out on 18 5-year-old potted Platanus × acerifolia ramets obtained by rooted cuttings of an individual plane tree. (Ramets are vegetatively reproduced copies of a plant; each ramet has the same genotype as the original parent tree.) Inoculation was performed in May 2012 by wounding 40 cm above the soil (stem diameter, 1 cm), with a single wound per plant, using a knife blade carrying a drop of a 0.2 ml of five 1:10 serial dilutions, previously used for AIT inoculations (conidial concentrations ranging from 10 to 105 conidia/ml). Three ramets for each conidial concentration were inoculated, and three wounded noninoculated ramets were used as control plants. Ramets were wrapped in Parafilm at the inoculation sites to reduce contamination and prevent excessive drying. After 4 months, C. platani was detected by the presence of specific symptoms and mycelium reisolation from inoculated plane tissue on PDA (33).
Data analysis.
PCR efficiency was calculated from the slope of the standard curve: efficiency = 10(−1/slope) − 1 (34). Reproducibility of the qPCR assay was assessed by computing the coefficient of variation (%CV) among the mean values in seven independent assays. The threshold value was 0.15, and the baseline emission was calculated from cycles 3 to 15. The accuracy of the AITs in intercepting the aerial inoculum of C. platani at various conidial concentrations was tested by quantifying the ITS region by qPCR, with the DNA extracted from a large series of dilutions of fungal conidia, obtained either from inoculated Whatman filter papers or directly from distilled water. A log-log exponential function on the form y = a + bex was fitted to the data to describe the relationship between conidial dilution (x) and quantity of fungal DNA (y) in both sample types, and the parameter estimates were compared. Model comparison involved testing the hypothesis that both data sets (extraction from conidia in water or from conidia on Whatman filter papers) shared the same curve. Parameter estimates obtained by separately applying the model to two data sets were tested (F test) against estimates obtained by global fitting of the same model to both data sets. Factorial analysis of variance (ANOVA) (substrate type × conidial concentration interaction) was applied to further test for differences between the two techniques. The logit model (generalized linear model [GLZ]) was applied to test the diffusion of the inoculum (both its presence and quantity) as a function of the distance from the closest symptomatic tree and of the number of symptomatic trees in the surroundings of the traps. The relationships between presence or absence or quantity of the inoculum in AITs and distance from symptomatic trees was described by fitting an inverse exponential model shown by the equation (y = a + bex) to these data. GLZ analyses, nonlinear estimation (least-square curve-fitting procedure, Levenberg-Marquardt algorithm), and ANOVA were performed in Statistica 6.0 (35).
Nucleotide sequence accession numbers.
Newly determined sequences (for ITS1-5.8S-ITS2, CP, and β-tubulin DNA sequences) have been deposited in GenBank under accession numbers KC493159 to KC493171, KF302676 to KF302687, and KF302688 to KF302703 (for more details, see Table S1 in the supplemental material).
RESULTS
Phylogenetic analysis of Ceratocystis isolates.
Phylogenetic topology, reconstructed with ITS sequences and from concatenated β-tubulin and CP sequences, showed that C. platani occurred in the main cluster of C. fimbriata isolates (see Fig. S2 and S3 in the supplemental material). Classification of Ceratocystis cacaofunesta in the same cluster confirmed a close phylogenetic relationship between these strains. In contrast, C. variospora and C. populicola differentiated into a separate clade (see Fig. S2 and S3).
TaqMan MGB probes and primer specificity.
For each target gene (ITS and CP) the positions of the upstream primer, reverse primer, and the internal probe are shown in Fig. 1. The total lengths of the amplicons were 55 bp and 59 bp for the ITS and CP target genes, respectively. Characteristics of the primers and probe are given in Table 2. The nucleotide-nucleotide BLAST search showed complete homology (100%) between the CP amplicon sequence designed for both C. platani target genes (CP and ITS). Only C. platani isolates were positively amplified by qPCR on fungal DNA, using both primer-probe combinations designed for multicopy (ITS) and single-copy (CP) genes. DNAs from isolates of other Ceratocystis species and fungi in other genera were not amplified (Table 3).
Fig 1.
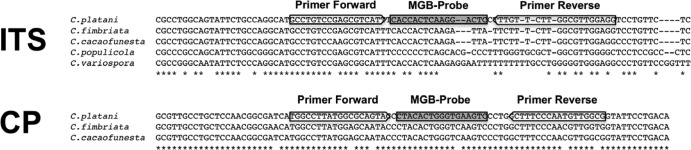
Alignment of two primers and the TaqMan MGB probe sequences against the internal transcribed spacer (ITS) and cerato-platanin (CP) gene for which the assay was designed. Primer sequences are indicated by the arrow boxes, and the probe sequences are shown in the shaded boxes. The Ceratocystis platani reference sequences were EU426554 for ITS and EF017218 for CP. The sequences of C. cacaofunesta (CBS15362), C. fimbriata (114723), C. populicola (115161), and C. variospora (114719) were obtained in this study.
Table 2.
Primers and probes designed for Ceratocystis platani by using Primer Express software 3.0
| Primer/TaqMan MGB probe | Target gene | Sequence (5′→3′) | Positionsa | Amplicon size (bp) |
|---|---|---|---|---|
| CP-F | CP | TGGCCTTATGGCGCAGTAC | 192–210 | 59 |
| CP-R | CGCCAACATTGGGAAAGC | 250–233 | ||
| CP-Pr | FAM-CTACACTGGGTGAAGTC-MGB | 212–228 | ||
| CpITS-F | ITS | GCCTGTCCGAGCGTCATT | 299–316 | 55 |
| CpITS-R | CCTCCAACGCCAAGAACAAA | 354–335 | ||
| CpITS-Pr | FAM-CACCACTCAAGGACTC-MGB | 318–333 |
Table 3.
Real-time PCR assays for the detection of Ceratocystis platani from mycelium, AITs, and Platanus tissue by using ITS and CP target genes
| Sample type (total no.)a | Result for MGB probe for: |
|||
|---|---|---|---|---|
| ITS geneb |
CP genec |
|||
| No. (%) of samples positive | CT ± SD | No. (%) of samples positive | CT ± SD | |
| Mycelium | ||||
| Ceratocystis platani (9) | 9 (100) | 15.75 ± 0.75 | 9 (100) | 14.25 ± 0.46 |
| C. cacaofunesta (1), C. fimbriata (11), C. populicola (1), C. variospora (1), Alternaria alternata (2), Cladosporium sp. (1), Epicoccum niger (2), Microsphaera platani (2), Sarchodontia pachyodon (4) | 0 (0) | >40 | 0 (0) | >40 |
| AITs | ||||
| Infected area (10) | 9 (90) | 34.89 ± 3.01 | 3 (30) | 37.82 ± 1.30 |
| Surrounding area (14) | 4 (28.5) | 36.74 ± 1.37 | 0 (0) | >40 |
| Plane tissue | ||||
| Infected (10) | 10 (100) | 26.45 ± 0.93 | 10 (100) | 26.59 ± 0.80 |
| Healthy (10) | 0 (0) | >40 | 0 (0) | >40 |
Each sample was processed in triplicate.
Real-time PCR assay based on MGB probe and primers designed on the ITS region.
Real-time PCR assay based on MGB probe and primers designed on the CP region.
Reproducibility and sensitivity of the qPCR assay.
Quality of the standard curves was evaluated by comparison of the cycle thresholds (CT) for each point of the curve, since each value should hypothetically be the same in replicate samples run under the same conditions. Differences were observed between the standard curves constructed with the CP and ITS genes (Table 4, Fig. 2). The reproducibility of the standard curves, however, was high as the variation coefficient for each point was below 5% for both C. platani target genes (Table 4). For each target gene, measurements of linearity such as slope, y intercept, and correlation coefficient (r2) did not vary significantly among the seven assays (%CV < 2%). Comparison between the two CP and ITS primer-probe combinations showed differences in sensitivity: CT values of molecular markers designed for the CP gene were higher than those obtained for the ITS genes (Fig. 2). The difference (ΔCT) between these two target genes was 6.22 ± 0.2 (mean ± standard deviation [SD]). The detection limits of the qPCR assay were 0.05 pg/μl and 2 fg/μl DNA for the CP and ITS primer-probe combinations, respectively. Due to the higher sensitivity, the ITS primer-probe combination sets were used subsequently to detect and quantify C. platani from environmental AIT samples.
Table 4.
Reproducibility of ITS and CP standard curves
| Parameter | Standard curve result fora: |
|||
|---|---|---|---|---|
| ITS |
CP |
|||
| Serial dilution (pg/tube) (n = 24) | CT ± SD | %CV | CT ± SD | %CV |
| 2 × 104 | 14.22 ± 0.05 | 0.39 | 18.12 ± 0.38 | 2.12 |
| 4 × 103 | 15.29 ± 0.13 | 0.85 | 20.16 ± 0.67 | 3.33 |
| 800 | 17.09 ± 0.07 | 0.39 | 22.72 ± 0.71 | 2.85 |
| 160 | 19.45 ± 0.19 | 0.96 | 25.02 ± 0.71 | 2.85 |
| 32 | 21.87 ± 0.25 | 1.15 | 27.31 ± 0.80 | 2.94 |
| 6.4 | 24.60 ± 0.23 | 0.95 | 29.70 ± 0.89 | 3.00 |
| 1.28 | 26.87 ± 0.28 | 1.05 | 32.25 ± 0.96 | 2.98 |
| 0.25 | 29.50 ± 0.56 | 1.90 | 35.05 ± 1.08 | 3.09 |
| Other parameters (n = 7) | Mean ± SD | %CV | Mean ± SD | %CV |
| Slope | 3.22 ± 0.01 | 0.39 | 3.30 ± 0.03 | 0.94 |
| y-intercept | 27.01 ± 0.06 | 0.21 | 32.23 ± 0.62 | 1.91 |
| Value of fit (r2) | 0.987 ± 0.005 | 0.55 | 0.993 ± 0.01 | 0.59 |
| Efficiency | 1.03 ± 0.01 | 0.55 | 1.02 ± 0.00 | 0.30 |
Standard curves for eight 1:5 serial dilutions were constructed with Cp5 reference DNA. The DNA control was diluted in water to cover a linear range of five orders of magnitude (from 2 × 104 to 0.25 pg/tube per sample). Each curve was run in triplicate in seven independent assays. Reproducibility was assessed by computing the coefficient of variation (%CV) among the mean values in seven independent assays. The efficiency of the standard curves was calculated based on the slope from seven independent experiments.
Fig 2.

Amplification plots of Ceratocystis platani DNA showing comparison between two different TaqMan MGB probes. The fluorescence (normalized reporter signal, Rn) of the TaqMan MGB probes for the CP target gene (broken line) appears later than the fluorescence of the probe designed for the ITS region (continuous lines). For each target gene, seven different 1:5 serial dilutions (ranged from 100 ng to 0.25 pg/tube) of C. platani strain Cp5 DNA were carried out. The inset shows the standard curves for the absolute quantification of C. platani using TaqMan MGB probes designed based on (i) the CP gene (broken line) and (ii) the ITS region (continuous line). Standard curves were generated by plotting threshold cycle (CT) numbers versus the logarithmic genomic DNA concentration of each dilution series.
Detection of C. platani in environmental samples.
C. platani isolates (Cp3, Cp5, Cp6, Cp8, and Cp9) (Table 1) obtained from symptomatic plane trees were identified on the basis of morphological characteristics and by using qPCR tested on DNA extracted from mycelium.
qPCR was also used to identify and quantify C. platani in DNA extracted from symptomatic plane tissues (Table 3). The quantity of C. platani DNA detected in these samples using both primer and probes (sets for CP and ITS) ranged between 2.0 × 103 and 5.0 × 103 pg DNA per μg of total DNA extracted. C. platani DNA was absent in asymptomatic plane tissues, used as negative controls (Table 3). Selected amplification plots of PCRs obtained from the samples tested are shown in Fig. 3.
Fig 3.

Ceratocystis platani detection by TaqMan MGB probe on ITS2. Selection of amplification plots from different DNA samples: (i) C. platani mycelium and infected plane tissues (continuous lines), (ii) airborne inoculum traps (AITs) from sampling area (broken lines with dots), and (iii) C. platani conidial suspension (broken lines with long segments). The fluorescent signal (normalized reporter signal, Rn) was absent from the no-template control (NTC), from other Ceratocystis species and other fungi, and from healthy plane tissue (horizontal dotted line).
Using the ITS primer-probe combination, the amount of C. platani DNA present in conidial suspensions was between 0.5 × 10−2 and 1.8 × 102 pg/μl total DNA (∼10 to 105conidia/ml, respectively). Detection of the pathogen from AITs differed according to the molecular marker used. When the ITS primer-probe combination was used, all the 11 AITs from the infection site included C. platani, whereas 4 out of 14 were positive in the surrounding area. The quantity of fungal DNA on these AITs was between 1.2 × 10−2 and 5.3 pg/μl. In contrast, the CP primer-probe combination gave positive results for only 3 out of 11 AITs from the infected site, while traps located from the surrounding area were all negative. The quantity of C. platani DNA ranged between 4.5 × 10−2 and 1.1 pg/μl.
With the exception of the highest concentration of C. platani conidia, the quantity of DNA obtained did not vary between extraction substrates, i.e., conidial suspensions in water or suspension applied to filter papers (ANOVA, significant substrate × concentration interaction, P < 0.001; honestly significant difference [HSD] post hoc test, P > 0.1). At 105 conidia per ml, a significantly greater quantity of DNA was obtained from the conidia suspension in water (HSD post hoc test, P < 0.001) than from the AITs. The relationship between conidial dilution (x) and the quantity of C. platani DNA detected by qPCR using the ITS primer-probe combination (y) produced an exponential curve (as shown in the equation y = a + bex). Curves obtained by fitting the model to the quantities of DNA obtained from inoculated filter papers (y = −3.97 + 0.078ex) and from conidial suspensions in water (y = −4.22 + 0.093ex) were similar (Fig. 4). Estimates for both a and b in the equation y = a + bex had overlapping confidence intervals (Fig. 4, insert) and did not differ significantly between the two curves (a F value = 0.41; P = 0.532; b F value = 4.21, P = 0.063).
Fig 4.
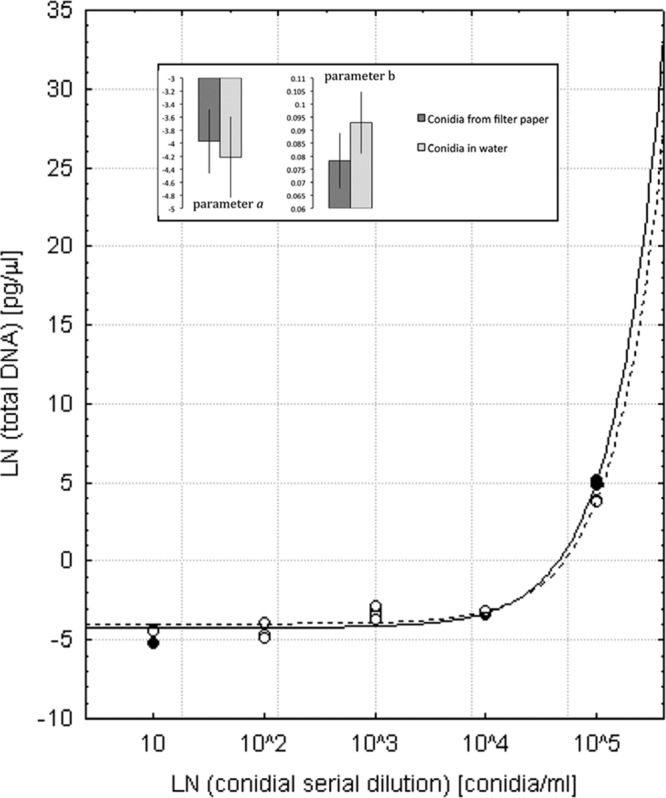
Detection of Ceratocystis platani in Whatman no. 1 filter papers artificially inoculated with conidial suspensions. The quantity of C. platani DNA detected by real-time PCR for the ITS region was related to the conidial dilution. A nonlinear regression (y = a + bex) was fitted to the natural logarithm (ln)-transformed data, where x = ln(conidial dilution) and y = ln(total DNA) extracted from conidia. The results obtained from the conidial suspensions applied to the Whatman filter papers (open symbols and broken line; y = −3.97 + 0.078ex) or from conidial suspensions in distilled water (closed symbols and continuous line; y = −4.22 + 0.093ex) were compared. Insets report parameter value estimates (a and b) ± 95% confidence interval.
C. platani was detected in AITs within 200 m of the closest symptomatic tree. Compared to negative AITs, positive AITs were surrounded by a higher number of symptomatic trees in a 100-m radius and were significantly closer to infected trees. The frequency of positive AITs (FS) increased significantly with higher numbers of symptomatic trees (NST) located within a 100-m radius (Fig. 5A), and decreased with increasing distance (D) from the closest symptomatic tree (Fig. 5B). Logit regression showed that both NST [χ2(1) = 8.26, P = 0.004] and D [χ2(1) = 10.33, P = 0.001] had a significant effect on the detection of the fungus on AITs. The dependence of successful detection on D was described by an inverse exponential function in the form FS = a + ebD (Fig. 5C) (a = 1.002, b = 0.008, P ≤ 0.05; explained variance = 81.6%). A similar curve efficiently described the reduction of the quantity of the DNA (QDNA) of C. platani retrieved from AITs when the distance between the trap and the closest symptomatic tree increased (Fig. 5D) (QDNA = a + ebD; a = 4.612, b = 0.016, P ≤ 0.05; explained variance = 95.3%).
Fig 5.

Airborne dispersal of a Ceratocystis platani inoculum during sanitation felling. Shown is variation in (A) the number of symptomatic trees within a 100-m distance and (B) the distance from the closest symptomatic tree in negative or positive airborne inoculum traps (AIT) for detection of C. platani. The horizontal line in each box is the mean value; the box represents the standard error, and the whisker indicates the standard deviation. Open circles and asterisks indicate outliers and extremes, respectively. (C and D) Decrease in the frequency of pathogen interception on AITs (successful detection) (C) and decrease in C. platani DNA quantities (D) as inverse exponential functions of the distance to the closest symptomatic tree.
Pathogenicity tests.
After 4 months, all ramets inoculated with a conidial concentration ranging from 102 to 105 conidia/ml were dead. These plants showed typical symptoms of blue stain canker consisting of stem cankers around the infection site and wood discolorations. Isolation on PDA medium showed the presence of fungal mycelium that was identified as C. platani. No symptoms were observed, and no successful pathogen isolation was possible from control plants and from plants inoculated with 10 conidia/ml.
DISCUSSION
The work reported here demonstrated the use of a sensitive and reliable method to detect and quantify a C. platani airborne inoculum involved in pathogen dispersal. The method combines a simple and cost-effective trapping technique with a molecular approach based on a qPCR assay.
Application of the qPCR technique combined with effective inoculum trapping methods made it possible to identify and quantify the pathogen from the air. The extraction of DNA directly from filters removes the need for laborious microscopy and culturing and enables direct detection of target microorganisms. The integration of airborne sampling and molecular diagnostic methods has provided sensitive, specific, and quantitative data for several pathogens (21). The use of qPCR for molecular diagnostics is attractive because of the high sensitivity and throughput capability (36, 37). Moreover, this approach allows the detection and quantitation of very small quantities of fungal DNA and is hence a powerful tool for early surveillance and detection of fungal pathogens in healthy plant tissue, before symptom expression in the host (22, 23).
Most reports on detection of airborne microorganisms by qPCR come from clinical microbiology (21). However, qPCR has been also used to quantify the airborne inoculum of fungal pathogens, such as Sclerotinia sclerotiorum, Leptosphaeria spp., Puccinia striiformis, and Botrytis squamosa (38, 39, 40, 41), which cause serious disease in arable crops, and also for the forest tree pathogen Fusarium circinatum (20, 42).
Many fungal diseases are initiated by airborne inoculum that lands on susceptible hosts under favorable environmental conditions. Ceratocystis platani poses a significant threat, especially in urban areas since it can infect healthy plane trees when, during sanitation pruning and fellings, the inoculum can be carried by wind to fresh wounds that are occasionally present on surrounding trees (16). For this reason, among others, the detection of the C. platani inoculum is important to improve the understanding of the mechanisms and dynamics of pathogen dispersal.
In this study, both the CP and ITS primer-probe combination set could accurately detect C. platani from cultured isolates and showed no cross-reactivity with other phylogenetically related Ceratocystis species. The molecular markers developed here were specific and did not amplify DNA of common airborne fungi, such as Alternaria sp., Cladosporium sp., or Epicoccum (43), which are found in urban areas and of other species hosted by plane trees, such as Microsphaera platani (44) and Sarchodontia pachyodon (45). Several methods have been used to collect airborne spores, but the identification of different fungal species has usually relied on use of traditional methods, such as microscopy or culture on artificial media (21). These methods are time-consuming and require a high level of expertise to accurately identify the organisms, especially if different fungal species have similar spore morphologies. Furthermore, for C. platani the airborne inoculum is also represented by infected sawdust and woody debris that are spread in the environment during sanitation operations (7).
Detection systems available for qPCR can also be nonspecific where the intercalating dyes (e.g., SYBR green I dye) generate fluorescence bonding to PCR fragments (46). In this work, the qPCR assay was based on the TaqMan minor groove binder (MGB) probes that incorporate a 5′ reporter dye and a 3′ nonfluorescent quencher (NFQ). The NFQ offers the advantage of lower background signal, which results in better precision in quantitation, stabilizing the hybridization of the probe with single-stranded DNA targets and leading to improved specificity over conventional TaqMan probes (47).
Different results were observed, however, in terms of sensitivity when different marker genes designed based on CP or ITS regions were used. Although both primer-probe sets were specific to C. platani, differences in pathogen sensitivity were observed. Our results are in accordance with those reported for Aspergillus fumigatus, where an ∼5-fold difference in CT was found between the FKS1 gene, used as a single-copy control gene, against an 18S multicopy rRNA gene (48). Comparable differences (6-fold) were also found with F. circinatum in comparisons between the multicopy ribosomal IGS gene and the single-copy mating-type genes (20). These results highlight an advantage in using the ribosomal DNA (rDNA) multicopy gene, since amplification of the target gene can be 10 to 100 times more sensitive than that of single-copy genes (49, 50).
Although the single-copy CP gene represents a specific target for C. platani, the sensitivity of the primer-probe combination designed here was very low, and its use for detecting the fungus in environmental samples risks underestimating the quantity of airborne inoculum.
The differences we observed between single-copy and multiple-copy genes reflect differences in the detection limits of the qPCR assay that detected 0.05 pg and 2 fg C. platani DNA/μl for CP and ITS, respectively. These values are much lower than those for F. circinatum found by Schweigkofler et al. (20), who used a qPCR assay based on SYBR green chemistry. The reasons for these differences are unknown. While DNA extraction was carried out from Whatman filter papers, several other factors could explain differences in DNA quantitation—for instance, the DNA extraction methods but also the less sensitive qPCR chemistry (SYBR green versus TaqMan) (46).
Currently, the identification of C. platani is mainly based on visual inspections for symptoms followed by isolation in the laboratory on medium for confirmation (15). A trap technique was described previously for isolation of C. platani from soil and from infected wood (51). A serological assay has also been optimized to confirm the presence of the CP protein from C. platani ascospores and mycelium (52), but it never has been used for diagnostic purposes. A molecular approach based on qPCR to detect C. platani from artificially and naturally infected plane wood has been previously reported (53). However, the qPCR method reported in this study is more sensitive. The detection limit of the assay described here, 2 fg C. platani DNA/μl, was lower than that reported by Pilotti et al., 10 fg/μl (53).
The qPCR assay tested in the present article has been successfully used to detect C. platani directly from naturally infected plane tissues, making it a useful tool for the diagnosis of canker plane stain. However, differently from Pilotti et al. (53), the main goal of this study was to optimize a qPCR assay able to detect and quantify even small amounts of airborne C. platani DNA. For this reason, shorter probes were designed, such as the TaqMan MGB probe, providing increased sensitivity and specificity compared to a conventional TaqMan probe (47) and ensuring more effective fungal detection.
In this work, the method used to intercept the inoculum was similar to that described by Schweigkofler et al. (20). In this study, the lowest reliable detection limit for conidial suspensions was ∼10 conidia/ml (0.5 × 10−2 pg C. platani DNA/μl), while the highest tested amount was 105 conidia/ml (180 pg C. platani DNA/μl). Therefore, the minimum amount detected in this study was much lower than that reported by Schweigkofler et al. (20), who were able to detect a minimum of 102 conidia per 100 μl (i.e., 103 conidia per ml). Interception and quantitation of C. platani inoculum were also effective on AITs, where the minimal amount of fungal DNA was 1.2 × 10−2 to 1.4 × 10−2 pg/μl, corresponding to an amount of detected propagules close to 10 conidia/ml.
The pathogenicity tests carried out in this study showed that the minimal amount of fungal inoculum required to cause disease was 102 conidia/ml. Above these conidial concentrations, all inoculated plants showed symptoms of blue stain canker disease, resulting in death of the plant. These concentrations are even lower than those reported by Vigouroux (54), who showed that a minimum of 200 C. platani spores were required per wound for the development of consistent and reproducible symptoms. Moreover, these results showed that the qPCR technique presented here allows the detection of inoculum concentrations lower than those able to cause disease. Therefore, this technique can be used to detect latent infections by the pathogen in asymptomatic vegetative tissue or natural plantings, improving early detection of the disease. This feature is of primary importance in phytosanitary controls in plant trading, preventing or reducing the risk of spread of the disease.
Global trade in plants is the main cause of introduction of alien species, allowing long distance dispersal of pests and pathogens (1, 3, 5). However, human-mediated dispersal of pathogens also has an impact at the local scale, favoring the spread of pathogens in areas where they have not previously been detected. This process was the cause of spread of Fusarium oxysporum f. sp. canariensis that causes a lethal vascular disease of Canary Island date palms (Phoenix canariensis) (55). The fungus can spread indirectly during felling of infected plants, and in this case, contaminated sawdust was dispersed up to a distance of 100 ft (ca. 30 m) (56).
C. platani appears to be spread primarily by infected pruning tools and root anastomosis (16), although an airborne inoculum, including spores, sawdust, and woody debris, represents a serious risk for exposed fresh wounds of healthy plane trees. In our study, we found that using AITs enabled the detection of airborne inoculum of C. platani within 200 m of the closest symptomatic infected plane tree. In addition, qPCR was able to detect the presence of C. platani from AITs in the surrounding area with healthy plane trees, closest to the infected site.
For these reasons, the study of dispersal mechanisms for the airborne inoculum is important in developing greater understanding of the epidemiology of these pathogens. Moreover, the development of highly accurate and reliable molecular detection techniques could help in avoiding the invasion of uncontaminated areas, enhancing the activity of the National Plant Protection Organizations (NPPO). The use of these tools also proved to be useful in contrasting the dispersal across Europe of alien pathogens already present in restricted areas, as with C. platani. The life cycle and dissemination of C. platani are strictly related to human activities. For effective early surveillance of this pathogen and to prevent its diffusion into new environments, there is a real need for rapid, simple, and robust detection method. The use of airborne trapping methods combined with effective routine molecular detection tools can provide more accurate forecasts of the risk of pathogen spread and help the management of the disease.
Supplementary Material
ACKNOWLEDGMENTS
We are grateful to C. Comparini (University of Florence, Italy), A. Perez-Sierra (Universitat Politècnica de València, Spain), and N. Soulioti (NAGREF—Institute of Mediterranean Forest Ecosystems, Greece) for providing some isolates of Ceratocystis and Alternaria. We thank N. Casini (Comune di Firenze) for assistance.
This work was supported by the EU project ISEFOR (“Increasing Sustainability of European Forests: Modeling for Security against Invasive Pests and Pathogens under Climate Change”), funded by European Union Seventh Framework Programme FP7 2007-2013 KBBE 2009-3 under grant agreement 255268.
We thank the anonymous referees for detailed suggestions and constructive criticisms that greatly improved the manuscript.
Footnotes
Published ahead of print 28 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01484-13.
REFERENCES
- 1.Pyšek P, Richardson DM. 2010. Invasive species, environmental change and management, and health. Annu. Rev. Environ. Resour. 35:25–55 [Google Scholar]
- 2.Smith RM, Baker RHA, Malumphy CP, Hockland S, Hammon RP, Ostoja-Starzewski JC, Collins DW. 2007. Recent non-native invertebrate plant pest establishments in Great Britain: origins, pathways, and trends. Agric. For. Entomol. 9:307–326 [Google Scholar]
- 3.Santini A, Ghelardini L, De Pace C, Desprez-Loustau ML, Capretti P, Chandelier A, Cech T, Chira D, Diamandis S, Gaitniekis T, Hantula J, Holdenrieder O, Jankovsky L, Jung T, Jurc D, Kirisits T, Kunca A, Lygis V, Malecka M, Marcais B, Schmitz S, Schumacher J, Solheim H, Solla A, Szabò I, Tsopelas P, Vannini A, Vettraino AM, Webber J, Woodward S, Stenlid J. 2013. Biogeographical patterns and determinants of invasion by forest pathogens in Europe. New Phytol. 197:238–250 [DOI] [PubMed] [Google Scholar]
- 4.Kenis M, Rabitsch W, Auger-Rozenberg M-A, Roques A. 2007. How can alien species inventories and interception data help us prevent insect invasions? Bull. Entomol. Res. 97:489–502 [DOI] [PubMed] [Google Scholar]
- 5.Liebhold AM, Brockerhoff EG, Garrett LJ, Parke JL, Britton KO. 2012. Live plant imports: the major pathway for forest insect and pathogen invasions of the US. Front. Ecol. Environ. 10:135–143 [Google Scholar]
- 6.Brown JKM, Hovmøller MS. 2002. Aerial dispersal of pathogens on the global and continental scales and its impact on plant disease. Science 297:537–554 [DOI] [PubMed] [Google Scholar]
- 7.Walter JM. 1946. Canker stain of plane trees. USDA circular, no. 742. US Department of Agriculture, Washington, DC [Google Scholar]
- 8.Ross EW. 1971. Diplodia theobromae and Ceratocystis fimbriata f. platani found in silage sycamore plantings. Plant Dis. Rep. 55:741–743 [Google Scholar]
- 9.Britton OK, Leininger T, Chang CJ, Harrington TC. 1998. Association of Xylella fastidiosa, Ceratocystis fimbriata f. platani and Botryosphaeria rhodina with declining sycamore plantations in the southeastern USA, abstr 3.7.50. In Proceedings of the 7th International Congress of Plant Pathology (ICPP98). British Society for Plant Pathology, Edinburgh, United Kingdom [Google Scholar]
- 10.Engelbrecht CJB, Harrington TC, Steimel J, Capretti P. 2004. Genetic variation in eastern North American and putatively introduced populations of Ceratocystis fimbriata f. platani. Mol. Ecol. 13:2995–3005 [DOI] [PubMed] [Google Scholar]
- 11.Panconesi A. 1972. I nostri platani sono in pericolo. Inf. Fitopatol. 22:10–13 [Google Scholar]
- 12.Cristinzio M, Marziano F, Vernau R. 1973. La moria del platano in Campania. Riv. Patol. Veg. SIV 9:189–214 [Google Scholar]
- 13.EPPO Global Database 2013. Map for Ceratocystis platani. European and Mediterranean Plant Protection Organization, Paris, France: http://gd.eppo.int [Google Scholar]
- 14.Ocasio-Morales RG, Tsopelas P, Harrington TC. 2007. Origin of Ceratocystis platani on native Platanus orientalis in Greece and its impact on natural forests. Plant Dis. 91:901–904 [DOI] [PubMed] [Google Scholar]
- 15.OEPP/EPPO 1986. Data sheets on quarantine organisms no. 136, Ceratocystis fimbriata f.sp. platani. Bull. OEPP 16:21–24 [Google Scholar]
- 16.Panconesi A. 1999. Canker stain of plane trees: a serious danger to urban plantings. J. Plant Pathol. 81:3–15 [Google Scholar]
- 17.Vigouroux PA, Stojadinovic B. 1990. Possibilités d'infection du platane par Ceratocystis fimbriata f. platani après contamination de l'eau où se développent des racines blessées. Eur. J. For. Pathol. 20:118–121 [Google Scholar]
- 18.D'Amico L, Motta E, Annesi T, Scirè M, Luchi N, Hantula J, Korhonen K, Capretti P. 2007. The North American P group of Heterobasidion annosum s.l. is widely distributed in Pinus pinea forests of the western coast of central Italy. For. Pathol. 37:303–320 [Google Scholar]
- 19.Gonthier P, Nicolotti G, Linzer R, Guglielmo F, Garbelotto M. 2007. Invasion of European pine stands by a North American forest pathogen and its hybridization with a native interfertile taxon. Mol. Ecol. 16:1389–1400 [DOI] [PubMed] [Google Scholar]
- 20.Schweigkofler W, O'Donnell K, Garbelotto M. 2004. Detection and quantification of airborne conidia of Fusarium circinatum, the causal agent of pine pitch canker, from two California sites by using a real-time PCR approach combined with a simple spore trapping method. Appl. Environ. Microbiol. 76:3512–3520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.West JS, Atkins SD, Emberlin J, Fitt BD. 2008. PCR to predict risk of airborne disease. Trends Microbiol. 16:380–387 [DOI] [PubMed] [Google Scholar]
- 22.Luchi N, Capretti P, Vettraino AM, Vannini A, Pinzani P, Pazzagli M. 2006. Early detection of Biscogniauxia nummularia in symptomless European beech (Fagus sylvatica L.) by TaqMan real-time PCR. Lett. Appl. Microbiol. 43:33–38 [DOI] [PubMed] [Google Scholar]
- 23.Luchi N, Capretti P, Pinzani P, Orlando C, Pazzagli M. 2005. Real-time PCR detection of Biscogniauxia mediterranea in symptomless oak tissue. Lett. Appl. Microbiol. 41:61–68 [DOI] [PubMed] [Google Scholar]
- 24.Pazzagli L, Cappugi G, Manao G, Camici G, Santini A, Scala A. 1999. Purification, characterization and amino acid sequence of cerato-platanin, a new phytotoxic protein from Ceratocystis fimbriata f.sp. platani. J. Biol. Chem. 274:24959–24964 [DOI] [PubMed] [Google Scholar]
- 25.White TJ, Bruns T, Lee S, Taylor JW. 1990. Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics, p 315–322 In Innis MA, Gelfand DH, Sninsky JJ, White TJ. (ed), PCR protocols: a guide to methods and applications. Academic Press, Inc., New York, NY [Google Scholar]
- 26.Glass NL, Donaldson GC. 1995. Development of primer sets designed for use with the PCR to amplify conserved genes from filamentous ascomycetes. Appl. Environ. Microbiol. 61:1323–1330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stamatakis A, Hoover P, Rougemont J. 2008. A rapid bootstrap algorithm for the RAxML Web servers. Syst. Biol. 57:758–771 [DOI] [PubMed] [Google Scholar]
- 28.Lanave C, Preparata G, Saccone C, Serio G. 1984. A new method for calculating evolutionary substitution rates. J. Mol. Evol. 20:86–93 [DOI] [PubMed] [Google Scholar]
- 29.Rodríguez F, Oliver JL, Marín A, Medina JR. 1990. The general stochastic model of nucleotide substitution. J. Theor. Biol. 142:485–501 [DOI] [PubMed] [Google Scholar]
- 30.Witthuhn RC, Wingfield BD, Wingfield MJ, Harrington TC. 1999. PCR-based identification and phylogeny of species of Ceratocystis sensu stricto. Mycol. Res. 103:743–749 [Google Scholar]
- 31.Heath RN, Wingfield MJ, Wingfield BD, Meke G, Mbaga A, Roux J. 2009. Ceratocystis species on Acacia mearnsii and Eucalyptus spp. in eastern and southern Africa including six new species. Fungal Divers. 34:41–67 [Google Scholar]
- 32.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 33.OEPP/EPPO 2013. Diagnostic protocols for regulated pests: Ceratocystis fimbriata f sp. platani (2003). Bull. OEPP/EPPO Bull. 33:245–247 [Google Scholar]
- 34.Bustin SA, Benes V, Garson JA, Hellemans J, Huggett J, Kubista M, Mueller R, Nolan T, Pfaffl MW, Shipley GL, Vandesompele J, Wittwer CT. 2009. The MIQE guidelines: minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 55:611–622 [DOI] [PubMed] [Google Scholar]
- 35.StatSoftInc 2001. STATISTICA (data analysis software systems), version 6 StatSoft, Inc, Tulsa, OK [Google Scholar]
- 36.Miller SA, Beed FD, Harmon CL. 2009. Plant disease diagnostic capabilities and networks. Annu. Rev. Phytopathol. 47:15–38 [DOI] [PubMed] [Google Scholar]
- 37.Valasek MA, Repa JJ. 2005. The power of real-time PCR. Adv. Physiol. Educ. 29:151–159 [DOI] [PubMed] [Google Scholar]
- 38.Rogers SL, Atkins SD, West JS. 2008. Detection and quantification of airborne inoculum of Sclerotinia sclerotiorum using quantitative PCR. Plant Pathol. 58:324–331 [Google Scholar]
- 39.Kaczmarek J, Jedryczka M, Cools HJ, Fitt BDL, Lucas JA, Latunde-Dada AO. 2012. Quantitative PCR analysis of abundance of airborne propagules of Leptosphaeria species in air samples from different regions of Poland. Aerobiologia 28:199–212 [Google Scholar]
- 40.Dedeurwaerder G, Duvivier M, Mvuyenkure SM, Renard ME, Van Hese V, Marchal G, Moreau JM, Legrève A. 2011. Spore traps network: a new tool for predicting epidemics of wheat yellow rust. Commun. Agric. Appl. Biol. Sci. 76:667–670 [PubMed] [Google Scholar]
- 41.Carisse O, Tremblay DM, Lévesque CA, Gindro K, Ward P, Houde A. 2009. Development of a TaqMan real-time PCR assay for quantification of airborne conidia of Botrytis squamosa and management of botrytis leaf blight of onion. Phytopathology 99:1273–1280 [DOI] [PubMed] [Google Scholar]
- 42.Garbelotto M, Smith T, Schweigkofler W. 2008. Variation in rates of spore deposition of Fusarium circinatum, the causal agent of pine pitch canker, over a 12-month-period at two locations in northern California. Phytopathology 98:137–143 [DOI] [PubMed] [Google Scholar]
- 43.Lee SH, Lee HJ, Kim SJ, Lee HM, Kang H, Kim YP. 2010. Identification of airborne bacterial and fungal community structures in an urban area by T-RFLP analysis and quantitative real-time PCR. Sci. Total Environ. 408:1349–1357 [DOI] [PubMed] [Google Scholar]
- 44.Anselmi N, Cardin L, Nicolotti G. 1994. Plane decline in European and Mediterranean countries: associated pests and their interactions. EPPO Bull. 24:159–171 [Google Scholar]
- 45.Luchi N, Pepori A, Capretti P, Santini A. 2011. Sarcodontia pachyodon: a canker and white-rot agent of plane-trees. J. Phytopathol. 159:117–119 [Google Scholar]
- 46.Orlando C, Pinzani P, Pazzagli M. 1998. Developments in quantitative PCR. Clin. Chem. Lab. Med. 36:255–269 [DOI] [PubMed] [Google Scholar]
- 47.Kutyavin IV, Afonina IA, Mills A, Gorn VV, Lukhtanov EA, Belousov ES, Singer MJ, Walburger DK, Lokhov SG, Gall AA, Dempcy R, Reed MW, Meyer RB, Hedgpeth J. 2000. 3′-minor groove binder-DNA probes increase sequence specificity at PCR extension temperatures. Nucleic Acids Res. 28:655–661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Herrera ML, Vallor AC, Gelfond JA, Patterson TF, Wickes BL. 2009. Strain-dependent variation in 18S ribosomal DNA copy numbers in Aspergillus fumigatus. J. Clin. Microbiol. 47:1325–1332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Maleszka R, Clark-Walker GD. 1990. Magnification of the rDNA cluster in Kluyveromyces lactis. Mol. Gen. Genet. 223:342–344 [DOI] [PubMed] [Google Scholar]
- 50.Maleszka R, Clark-Walker GD. 1993. Yeasts have a four-fold variation in ribosomal DNA copy number. Yeast 9:53–58 [DOI] [PubMed] [Google Scholar]
- 51.Grosclaude C, Olivier R, Pizzuto JC, Romiti C, Madec S. 1988. Detection of Ceratocystis fimbriata f. platani by trapping. Application to the study of the persistence of the parasite in infected wood. Eur. J. For. Pathol. 18:385–390 [Google Scholar]
- 52.Boddi S, Comparini C, Calamassi R, Pazzagli L, Cappugi G, Scala A. 2004. Cerato-platanin protein is located in the cell walls of ascospores, conidia and hyphae of Ceratocystis fimbriata f. sp. platani. FEMS Microbiol. Lett. 233:341–346 [DOI] [PubMed] [Google Scholar]
- 53.Pilotti M, Lumia V, Di Lernia G, Brunetti A. 2012. Development of real-time PCR for in wood-detection of Ceratocystis platani, the agent of canker stain of Platanus spp. Eur. J. Plant Pathol. 134:61–79 [Google Scholar]
- 54.Vigouroux A. 1992. Preliminary results for obtaining a plane tree resistant to canker stain and adapted to European conditions. Acta Hortic. 320:91–96 [Google Scholar]
- 55.Downer AJ, Uchida JY, Hodel DR, Elliott ML. 2009. Lethal palm diseases common in the United States. Horttechnology 19:710–716 [Google Scholar]
- 56.Hodel DR. 2009. Pest notes: palm diseases in the landscape. ANR publication no. 74148.1-6. University of California Statewide Integrated Pest Management Program, Division of Agriculture and National Resources, University of California, Davis, CA [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.