

Abstract
Lactobacillus plantarum is an attractive candidate for bioprocessing of lignocellulosic biomass due to its high metabolic variability, including its ability to ferment both pentoses and hexoses, as well as its high acid tolerance, a quality often utilized in industrial processes. This bacterium grows naturally on biomass; however, it lacks the inherent ability to deconstruct lignocellulosic substrates. As a first step toward engineering lignocellulose-converting lactobacilli, we have introduced genes coding for a GH6 cellulase and a GH11 xylanase from a highly active cellulolytic bacterium into L. plantarum. For this purpose, we employed the recently developed pSIP vectors for efficient secretion of heterologous proteins. Both enzymes were secreted by L. plantarum at levels estimated at 0.33 nM and 3.3 nM, for the cellulase and xylanase, respectively, in culture at an optical density at 600 nm (OD600) of 1. Transformed cells demonstrated the ability to degrade individually either cellulose or xylan and wheat straw. When mixed together to form a two-strain cell-based consortium secreting both cellulase and xylanase, they exhibited synergistic activity in the overall release of soluble sugar from wheat straw. This result paves the way toward metabolic harnessing of L. plantarum for novel biorefining applications, such as production of ethanol and polylactic acid directly from plant biomass.
INTRODUCTION
Plant cell wall fibers are composed of polymeric components, such as cellulose, lignin, pectins, and hemicelluloses, that collectively represent the most abundant renewable organic polymers on Earth (1). Despite its recalcitrant nature, the polysaccharides of the plant cell wall provide an exceptional source of carbon and energy, and a multitude of different microorganisms have evolved enzyme systems (notably glycoside hydrolases) which are capable of degrading plant cell wall polysaccharides. Exploiting these enzymes in a biotechnological process, e.g., via metabolic engineering, holds great environmental and applicative potential.
One attractive candidate for metabolic engineering toward plant mass bioprocessing is Lactobacillus plantarum, which is a common lactic acid bacterium with homolactic fermentation on hexose sugars and heterolactic (lactic and acetic acid) fermentation on pentoses. L. plantarum is used in a variety of industrial and agricultural applications and prospers in environments containing decomposed lignocellulosic plant biomass (2). In agriculture, the acidifying properties of these organisms are employed for conservation of plant biomass for use in animal feed (3). The ability to produce lactic acid in large amounts could also be used for the production of bio-based plastics (polylactic acid) from plant biomass. Interestingly, Lactobacillus species are predominant in contaminated ethanol fermentations (4, 5), and L. plantarum shows high ethanol tolerance (6), rendering it as a possible candidate for the production of biofuel by introduction of ethanol-producing enzymes into its genetic repertoire (7). In contrast to the commonly used ethanol-producing yeast Saccharomyces cerevisiae, L. plantarum is able to metabolize pentose sugars derived from lignocellulosic biomass (8–11). The production of acid and the bacterium's acid tolerance reduces the risk of contamination by other bacteria and fungi and may enable degradation of substrates directly after acid pretreatments that are commonly used for lignin deconstruction in plant biomass. L. plantarum contains 55 genes encoding 18 glycoside hydrolase families, but none are strict cellulases or xylanases (12). Consequently, the bacterium lacks the inherent ability to degrade cellulose and hemicelluloses. Therefore, we have studied the possibility to introduce secreted lignocellulolytic enzymes into this bacterium.
Around 1990, a few groups reported the expression of cellulases from Gram-positive bacteria in L. plantarum, using original (i.e., heterologous) promoters and secretion signals (12–14). Recent developments in the molecular biology of L. plantarum include novel protein expression systems (13–18) and the availability of its full genome sequence (9). Intracellular expression with the pSIP system (13) has recently been used for the expression of a recombinant Pyrococcus furiosus cellulase in both L. plantarum and L. casei strains (19). In an attempt to select potential homologous signal peptides for Sec-dependent secretion, Mathiesen et al. carried out a functional analysis of 76 of the 93 signal peptides from L. plantarum WCFS1, resulting in the construction of several pSIP derivatives that yielded efficient secretion of reporter enzymes at high levels (15). These pSIP derivatives have a modular nature, allowing for easy exchange of the reporter gene with a gene coding for a protein of interest (20). Here, we have used two of the selected signal peptides, originating from L. plantarum WCFS1 proteins pLp_2145s and pLp_3050s and designated herein as leader peptides 1 and 2, respectively (Lp1 and Lp2), for expression of potent lignocellulolytic enzymes. The enzymes expressed and secreted were an endoglucanase, Cel6A, and an endoxylanase, Xyn11A, both from the well-characterized cellulolytic bacterium Thermobifida fusca. The enzymatic activity of the individually secreted enzymes on cellulose and xylan was assessed. In addition, a cell consortium for synergistic interaction of the cellulase and the xylanase was established for optimal degradation of wheat straw.
MATERIALS AND METHODS
Cloning.
Wild-type enzymes Cel6A and Xyn11A were cloned from Thermobifida fusca genomic DNA as described previously (21, 22). The enzyme constructs in pET28a were designed to contain a His tag for subsequent purification.
For expression and secretion in L. plantarum, the glycoside hydrolases were cloned in the modular secretion plasmids pLp_2145sAmy and pLp_3050sAmy (15) by replacing the amylase gene in these plasmids by an appropriately amplified gene fragment, using either SalI or XhoI (SalI is compatible with XhoI) and HindIII restriction sites. For this purpose, the Cel6A-encoding gene was amplified using the forward primer 5′-TCTTctcgagATGGCATCCCCCAGACCTCT-3′ and reverse primer 5′-AATaagcttTCAGCTGGCGGCGCAGGTAAG-3′ (XhoI and HindIII sites are in lowercase letters). The Xyn11A-encoding gene amplified was cloned using 5′-TCTTgtcgacATGGCCGTGACCTCCAACGAG-3′ and 5′-AATaagcttCTAGTTGGCGCTGCAGGACA-3′ primers (SalI and HindIII sites are in lowercase letters). The pLp_2145s constructs are referred to as Lp1, whereas the pLP_3050s-containing constructs are referred to as Lp2.
pLp_2145sAmy and pLp_3050sAmy are part of the pSIP400 series (13). As a control, the two enzymes were also cloned into pSIP407 (referred to as No-Lp), which contains the same replicon and promoter as Lp1 and Lp2 but lacks a leader peptide (13). To make these constructs, the pepN gene present in pSIP407 was replaced by an NcoI-XbaI fragment containing the cel6A gene or a BspHI-XbaI fragment containing the xyn11A gene, which leads to the gene being translationally fused to the promoter (BspHI is compatible with NcoI). For this purpose, the Cel6A-encoding gene was amplified using the forward primer 5′-ATATATccatggATGGCATCCCCCAGACCTCTTCGC-3′ and reverse primer 5′-ATATATtctagaTCACTCCAGGCTGGCGGCGCAGG-3′ (NcoI and XbaI sites are in lowercase letters). The Xyn11A-encoding gene amplified was cloned using 5′-TCAGTCtcatgaATGGCCGTGACCTCCAACGAGACCGG-3′ and 5′-AGCGTAtctagaCTAGTTGGCGCTGCAGGACACC-3′ primers (BspHI and XbaI sites are in lowercase letters).
For generation of empty pLP_2145s and pLP_3050s, the Amy gene was excised using SalI and EcoRI restriction enzymes. The linearized plasmid was purified and blunted using the Quick blunting kit (NEB, Massachusetts). Blunt fragments were self-ligated to create the empty plasmids.
PCRs were performed using Phusion high-fidelity DNA polymerase F530-S (New England BioLabs, Inc.), and DNA samples were purified using a HiYield gel/PCR fragment extraction kit (Real Biotech Corporation [RBC], Taiwan). Restriction enzymes were purchased from New England BioLabs (Beverly, MA) and the T4 DNA ligase from Fermentas (Vilnius, Lithuania). L. plantarum plasmids were subcloned in Escherichia coli TG1 competent cells (Lucigen Corporation, Wisconsin). L. plantarum strain WCFS1 was transformed according to the protocol of Aukrust and Blom (23). Erythromycin concentrations used for positive clone selection and added in media were 10 μg/ml and 200 μg/ml for L. plantarum and E. coli, respectively.
Protein expression in E. coli and purification.
The plasmids pCel6A and pXyn11A were expressed in E. coli BL21(lDE3)/pLysS cells, and the His-tagged enzymes were purified on a nickel-nitrilotriacetic acid (Ni-NTA) column (Qiagen), as reported earlier (24). Purity of the recombinant proteins was tested by SDS-PAGE on 10% acrylamide gels, and fractions containing the pure recombinant protein were pooled and concentrated using Amicon centrifugal filters (Millipore, France). Protein concentrations were determined by measuring absorbance at 280 nm, using theoretical extinction coefficients calculated with the Protparam tool (http://www.expasy.org/tools/protparam.html). Proteins were stored in 50% (vol/vol) glycerol at −20°C.
Activity assay for the pure enzymes.
The activities of purified recombinant Cel6A and Xyn11A were tested in reaction mixtures containing 0.5 μM enzyme and 7.5 g/liter phosphoric acid-swollen cellulose (PASC; prepared as described by Lamed et al. [25]) or 2% oat spelt xylan (Sigma Chem. Co, St. Louis, MO) in 50 mM citrate buffer, pH 5 or 6. Samples were incubated for 30 min at 37 or 50°C, cooled to 0°C by being placed on ice, and then centrifuged for 5 min at 14,000 rpm at 4°C. The amount of soluble reducing sugars in the supernatants was determined by the dinitrosalicylic acid (DNS) method as described below.
Protein expression in L. plantarum.
Freshly inoculated cultures of L. plantarum WCFS1 harboring a pSIP-derived expression plasmid were grown at 37°C in MRS broth (BD Difco, Franklin Lakes, NJ) containing 10 μg/ml erythromycin. Gene expression was induced at an optical density at 600 nm (OD600) of 0.3 by adding the inducing peptide for sakacin P production (Caslo Laboratory, Denmark) (26) to a final concentration of 25 ng/ml, and cultures were then incubated for another 3 h at 37°C. For coculture experiments, strains producing either the cellulase or the xylanase were mixed at equal ODs or at various ratios and then grown and induced in the same manner.
Western blotting.
Proteins from the culture supernatants were separated on SDS-PAGE gels (10% acrylamide) and transferred to a nitrocellulose membrane using Mini Trans-Blot cells (Bio-Rad Laboratories Ltd., Israel). Nonspecific protein interactions were blocked by incubating the membrane for 1 h with 5% bovine serum albumin (BSA; prepared in Tris-buffered saline–Tween 20 [TBS-T]). The membrane was then rinsed twice (1 min) with TBS-T. Rabbit antibody against each enzyme (prepared by Sigma, Israel) was incubated with the appropriate membrane for 1 h in TBS-T, containing 1% BSA. The membrane was again rinsed twice (1 min) with TBS-T and then incubated for 1 h with secondary antibody, goat anti-rabbit antibody labeled with horseradish peroxidase (HRP), at a dilution of 1:10,000. The membrane was rinsed as described above and then rinsed twice (30 min) with TBS and 1% Triton X-100. Blots were developed by incubating the membrane for 1 min with equal amounts of solutions A and B (ECL, Ornat, Israel). Chemiluminescence was quantified using a luminescent image analyzer (ImageQuant LAS 4000 Mini; Danyel Biotech, Israel).
Dot blotting.
A volume of 50 ml of supernatant from culture at an OD600 of 1, expressing the Cel6A enzyme (Lp1, Lp2, or No-Lp), was concentrated 50 times using Amicon centrifugal filters (Millipore, France). For the Xyn11A enzyme, 1 ml of supernatant from each culture at an OD600 of 1 (Lp1, Lp2, or No-Lp) was dialyzed in TBS to remove MRS medium. Purified enzymes were blotted in concentrations ranging from 0.5 to 20 nM for the cellulase or 0.1 to 10 nM for the xylanase by applying 2 μl of an appropriate solution (in TBS) to a nitrocellulose membrane (Whatman). Concentrated and/or dialyzed culture supernatants were blotted by applying 2 μl of the supernatant to a nitrocellulose membrane. The above-described protocol for Western blotting was then followed.
Congo red assay.
The protocol of Anbar was followed with modifications (27). Oat spelt xylan (0.3%) was used instead of carboxymethyl cellulose (CMC) for xylanase activity detection. Transformed L. plantarum cells were spread onto MRS plates containing erythromycin (10 μg/ml) and incubated overnight at 37°C. The plates were overlaid with 20 ml soft agar containing 0.3% (wt/vol) CMC or oat spelt xylan (for cellulase or xylanase activity detection), 0.7% agar, and 200 μl of 0.1 μg/ml pSIP induction peptide in citrate buffer (25 mM, pH 5.0). The plates were incubated for 2 h at 37°C to induce enzyme expression and activity. The plates were then stained for 10 min with fresh Congo red solution (0.25%) and destained in 1 M NaCl. Formation of halos around the colonies indicated production of endoglucanase or endoxylanase activity.
Activity assay.
PASC degradation was assayed by mixing pure recombinant Cel6A varying from 0 to 100 nM (final concentration) or a volume of 30 μl of concentrated supernatants of the cultures (as described above) with 150 μl of 7.5 g/liter PASC in a final volume of 200 μl of 50 mM acetate buffer, pH 5.0. Samples were incubated at 37°C for 18 h, and the reactions were terminated by immersing the sample tubes in ice water. The samples were then centrifuged for 2 min at 14,000 rpm to remove the substrate.
The xylanase assay mixture consisted of 100 μl buffer (50 mM citrate buffer, pH 6.0) with purified Xyn11A enzyme (0 to 5 nM) or a volume of 30 μl of dialyzed supernatants of the cultures in 50 mM of the same buffer. The reaction was commenced by adding 100 μl of 2% oat spelt xylan and continued for 2 h at 37°C. The reaction was stopped by transferring the tubes to an ice-water bath followed by centrifugation for 2 min at 14,000 rpm.
Wheat straw (0.2 to 0.8 mm) provided by Valagro (Poitiers, France) was washed as described previously (28, 29). The material was then subjected to sodium hypochlorite (12%) pretreatment at room temperature for 1 h (30). The degradation assay was conducted in 200 μl of 50 mM acetate-citrate buffer, pH 5 to 6.0, containing 3.5 g/liter of pretreated wheat straw and 30 μl of concentrated or dialyzed culture supernatants. Supernatants from cocultures (50 ml at an OD600 of 1) of strains secreting the Cel6A and Xyn11A enzymes were concentrated 50 times using Amicon centrifugal filters (Millipore, France). Reaction mixtures were incubated for 24 h at 37°C.
All assays were performed in triplicate. Enzymatic activity was determined quantitatively by measuring the soluble reducing sugars released from the polysaccharide substrates by the DNS method (31, 32). DNS solution (150 μl) was added to 100 μl of sample, and after the reaction mixture was boiled for 10 min, absorbance at 540 nm was measured. Sugar concentrations were determined using a glucose standard curve.
Evaluation of synergism.
For determination of theoretical enzymatic activity in cocultures (additive effect), enzymatic activities were calculated from two different assays. In each assay, a coculture of one of the enzyme-secreting strains together with the respective empty plasmid-bearing control strain was grown (and induced as described above), and its supernatant was analyzed for enzymatic activity. The theoretical additive activity was calculated by computing the sum of activities for each of the individually measured enzymes. For example, for the 1:500 ratio, one volume of the Cel6A-secreting strain (Lp1) and 500 volumes of the empty pLp_3050s plasmid-bearing strain (as a replacement for the Xyn11A-secreting strain [Lp2]) were cocultured. In parallel, one volume of the empty pLp_2145s plasmid-bearing strain (as a replacement for the Cel6A-secreting strain [Lp1]) and 500 volumes of the Xyn11A-secreting strain (Lp2) were cocultured. The enzymatic activities on wheat straw substrate of 30 μl of concentrated supernatants (as described above for the coculture experiments) from each of the cocultures were determined individually, added together, and defined as the theoretical additive effect. These values were then compared with those of the corresponding combined cocultures of the cellulase- and xylanase-secreting strains.
Plasmid extraction.
Cocultures of cellulase- and xylanase-secreting strains were grown as described above. At an OD600 of 1, cells were pelleted from 5 ml of culture by centrifugation at 5,000 × g for 10 min at 4°C and resuspended in 200 μl of PD1 buffer from a high-speed plasmid minikit (Geneaid, New Taipei City, Taiwan). Lysozyme was added to the suspensions to a final concentration of 3 mg/ml. Suspensions were incubated at 37°C for 15 min and then subjected to five freeze-thaw cycles as follows: the samples were submerged in liquid nitrogen for 3 min, transferred to a 70°C water bath for an additional 3 min, and then mixed gently but thoroughly. Following this step, the protocol was carried out according to the manufacturer's instructions.
Real-time PCR.
Quantitative real-time PCR analysis was performed to verify the ratios between the cellulase- and xylanase-secreting strains in the bacterial consortium. A specific fragment of each plasmid (140 and 124 bp for pLP_2145s and pLP_3050s, respectively) was amplified using the forward primer 5′-ATTTAGCTGGCTGGCGTAAAGTATG-3′ for both plasmids and the reverse primers 5′-TCATTTCAGGATTGATCATTGTTGC-3′ for pLP_2145s (Lp1) and 5′-GACGACCCCGAAGACACAACTAG-3′ for pLP_3050s (Lp2). Individual standard curves suitable for the quantification of each plasmid were generated by amplifying serial 10-fold dilutions of quantified gel-extracted PCR products obtained by the amplification of each fragment. The standard curves were obtained using four dilution points and were calculated using the Rotorgene 6000 series software (Qiagen, Hilden, Germany). Subsequent quantifications were calculated with the same program using the standard curves generated. As a positive control, one purified product with known concentration that was used for the standard curve was added to each quantification reaction. This also served to assess the reproducibility of the reactions and to fit the results to the standard curve. Two negative controls were performed; the first contained the purified product of one of the plasmids and the primers of the other. This was done in order to eliminate the possibility of primer cross-reactivity. The second control did not contain any DNA template. All obtained standard curves met the required standards of efficiency (R2 [coefficient of correlation of the standard curve] > 0.99, 90% < E [efficiency of the PCR] < 115%). The number of copies of each plasmid in the cultures was assessed, and the ratio between the plasmids was determined. Real-time PCR was performed in a 10-μl reaction mixture containing 5 μl Absolute blue SYBR green master mix (Thermo Scientific, Massachusetts), 0.5 μl of each primer (10 μM working concentration), 2 μl nuclease-free water, and 2 μl of 10 ng/μl DNA template. Amplification involved one hold cycle at 95°C for 15 min for initial denaturation and activation of the hot-start polymerase system and then 30 cycles at 95°C for 10 s, followed by annealing for 20 s at 53.3°C and extension at 72°C for 20 s. To determine the specificity of amplification, a melting curve of PCR products was monitored by slow heating, with fluorescence collection at 1°C increments, from 45 to 99°C.
RESULTS
Choice of lignocellulolytic enzymes.
The selected enzymes for L. plantarum transformation originate from the very well-characterized cellulolytic bacterium Thermobifida fusca. This bacterium produces a set of only six cellulases and four xylanases. In previous studies, we have successfully employed eight of these enzymes in the construction of designer cellulosomes (21, 24, 30, 33–35), indicating their suitability for engineering and heterologous expression. These moderately thermophilic enzymes are known to have a broad temperature activity and pH activity (36), which might be compatible with the conditions expected during Lactobacillus fermentation.
For initial studies, we focused on the T. fusca endoglucanase Cel6A, which is highly induced by cellobiose (37), and endoxylanase Xyn11A, which is the most abundant xylanase produced during growth on xylan (38). In addition to their catalytic modules, Cel6A has a C-terminal family 2 carbohydrate binding module (CBM) which binds selectively to cellulose, and Xyn11A contains a C-terminal family 2 CBM that binds both cellulose and xylan. The molecular masses of the enzymes are 46,980 Da and 33,168 Da for Cel6A and Xyn11A, respectively. The selection of Cel6A and Xyn11A was also based on their simple modular architecture and their considerable residual activity under acidic conditions (activity at pH 5.0 is >90% of that at pH 6) and at 37°C (∼40% and ∼70% of the activity at 50°C for Cel6A and Xyn11A, respectively) (Fig. 1), consistent with normal growth of L. plantarum.
Fig 1.
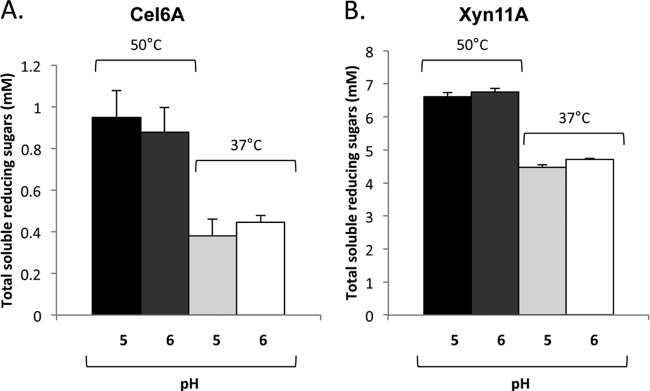
Comparative enzymatic activity of purified recombinant Cel6A (A) and Xyn11A (B) enzymes on PASC or xylan, at 37°C or 50°C and at pH 5 or 6. Enzymatic activity is defined as mM total reducing sugars following a 30-min reaction period. Each reaction was performed in triplicate, and standard deviations are indicated.
Enzyme secretion by L. plantarum.
The presence of secreted enzymes in the culture medium was observed by Western blotting using specific antibodies against each enzyme (Fig. 2). The enzymes were visible in the extracellular fraction of the strains carrying the Lp1 and Lp2 secretion plasmids, and the observed bands corresponded well to their theoretical masses. Degradation products, i.e., smaller bands, were also observed. No extracellular enzymes were detected in the supernatants of strains carrying the expression plasmid lacking the secretion peptide (Fig. 2, lane 3).
Fig 2.
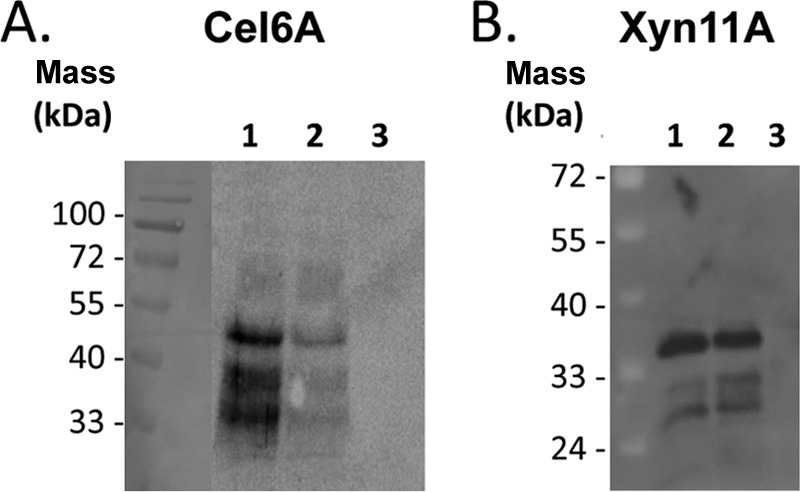
Western blot analysis of culture supernatants from transformed lactobacilli. (A) Lanes 1 to 3, Cel6A expressed with the Lp1, Lp2, and No-Lp plasmids, respectively; (B) lanes 1 to 3, Xyn11A expressed with the Lp1, Lp2, and No-Lp plasmids, respectively. The calculated masses of secreted Cel6A and Xyn11A are 46.9 kDa and 33.2 kDa, respectively. The lane of the prestained molecular mass markers in panel A was manually inserted as a reference onto the chemiluminescent image of the blot.
Extracellular cellulase and xylanase activities in transformed colonies were detected by the Congo red method (data not shown) and by activity assays of culture supernatants (Fig. 3; see below). Control cultures with intracellular expression of the respective enzymes did not exhibit any activity using the Congo red assay, and their supernatants did not show hydrolytic activity on xylan or PASC.
Fig 3.

Quantification of the secreted enzymes. (A) Dot blot analysis of increasing concentrations of purified Cel6A in nM and 2 μl of concentrated culture supernatant fluids. The graph shows the mean intensity of each spot for the calibration curve in black, and the white circles represent the intensity of the spot for Cel6A cultures (No-Lp, Lp1, and Lp2). (B) Dot blot analysis of increasing concentrations of purified Xyn11A in nM and 2 μl of dialyzed culture supernatant fluids. The irrelevant spots between the samples of Lp2 and No-Lp were cropped in the panel. (C) Enzymatic activity on PASC. Reactions were conducted with increasing concentrations of purified Cel6A and with 30 μl of concentrated culture supernatant fluid. Enzymatic activity is defined as mM soluble reducing sugars released following an 18-h reaction period. Each reaction was performed in triplicate, and standard deviations are indicated. (D) Enzymatic activity on xylan. Reactions were conducted with increasing concentrations of purified Xyn11A and with 30 μl dialyzed culture supernatant fluids. Enzymatic activity is defined as mM soluble reducing sugars following a 2-h reaction period. Each reaction was performed in triplicate, and standard deviations for xylan hydrolysis are indicated.
The concentrations of the secreted enzymes in the different cultures were calculated by comparing the extracellular fraction to serial dilutions of purified enzymes, either by dot blot analysis or by measuring reducing sugar formation on PASC or xylan substrates. The cellulase concentrations at an OD600 of 1 were estimated at 0.33 nM and 0.27 nM for the Lp1 and Lp2 secretion plasmids, respectively. For xylanase, these values were estimated at 2.7 nM and 3.3 nM, respectively (Fig. 3C and D). The concentrations, calculated either by the dot blot quantification or enzymatic activity method, were similar for both enzymes, suggesting that the major portion of the secreted enzymes is functional and that the expression and secretion processes do not substantially affect their activity. The culture supernatants retained full cellulase/xylanase activity after storage for several days at 4°C without added protease inhibitors.
The fact that culture supernatants from strains with intracellular expression did not exhibit enzymatic activity (Fig. 3C and D) indicates that the detected activities for the Lp1 and Lp2 constructs reflect properly secreted enzymes and do not originate from cell lysis.
Wheat straw degradation.
Prior to enzymatic degradation, wheat straw was subjected to chemical pretreatment with sodium hypochlorite that served to reduce the lignin content while preserving the cellulose/hemicellulose fractions in order to promote enzymatic degradation. The chemical composition of the pretreated wheat straw was 63% cellulose, 31% hemicellulose, and 3% lignin (30). Both the secreted cellulase and the secreted xylanase exhibited enzymatic activity on the pretreated wheat straw (Fig. 4).
Fig 4.
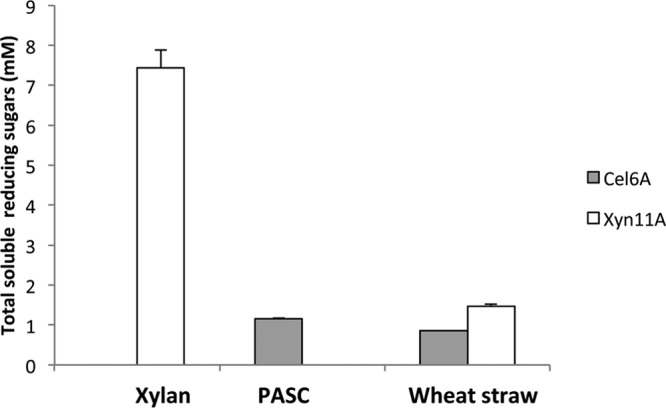
Activity of secreted enzymes on various substrates. Comparative enzymatic activity of supernatants derived from cultures producing either the cellulase (gray bars) or the xylanase (white bars). The substrates, PASC, xylan, or pretreated wheat straw, were incubated with 30 μl of supernatant fluids (concentrated to approximately 16.5 nM enzyme). The enzymatic activity of Cel6A is represented by gray bars and Xyn11A by white bars. Enzymatic activity is defined as mM soluble reducing sugars following a 2-h reaction period for xylan, an 18-h incubation for PASC, or a 24-h incubation for wheat straw at pH 5 and 37°C. Each reaction was performed in triplicate, and standard deviations are indicated.
Supernatants of cocultures of a Cel6A-secreting strain (Lp1) and an Xyn11A-secreting strain (Lp2) exhibited activity when incubated on wheat straw (Fig. 5). Synergistic activities (>1) were observed for ratios of 1:50 and greater, in favor of bacteria secreting either enzyme. The highest overall activities and the largest synergistic effect were observed in reactions with a strong dominance of the Xyn11A-secreting strain and yielded up to 27.6% of available sugars (Fig. 5), suggesting that xylan degradation by Xyn11A is a faster process than cellulose degradation by Cel6A. This observation further suggests that xylan degradation is more beneficial for cellulose accessibility than cellulose degradation is for xylan accessibility. Real-time (RT)-PCR of the different plasmids at the end of the growth period revealed that the ratios of the bacterial strains remained similar to the inoculation ratios (Fig. 6), thus indicating that expression and secretion of the two proteins did not have a differential effect on the growth rates of the bacteria.
Fig 5.
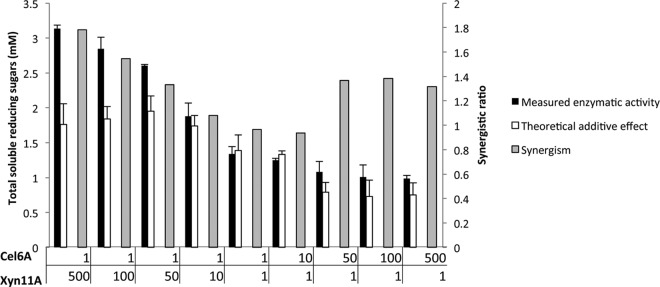
Enzymatic activity in supernatants of cocultures producing the cellulase and the xylanase. The substrate was pretreated wheat straw, and the measured activities are compared with the corresponding theoretical additive effect. Cells were inoculated using various ratios (Cel6A/Xyn11A): 1:500, 1:100, 1:50, 1:10, 1:1, 10:1, 50:1, 100:1, and 500:1 (where the 10:1 cell ratio corresponds to an approximate 1:1 molar ratio of the secreted enzymes, since cellulase production is approximately 10-fold lower; see the text and Fig. 3). The concentration of pretreated wheat straw (dry matter) in the reactions was 3.5 g/liter. Assuming that all detected reducing ends belong to dimers, the highest detected product concentration (1:500 ratio) represents 27.6% polysaccharide conversion. Enzymatic activity is defined as mM soluble reducing sugars following a 24-h reaction period at 37°C and pH 5. Each reaction was performed in triplicate, and standard deviations are indicated. The theoretical additive effect is defined as the sum of the activities of the individual Cel6A- and Xyn11A-secreting cultures (see Materials and Methods for a detailed explanation), and synergism was calculated as the ratio between the measured activity and the theoretical activity, assuming additivity.
Fig 6.
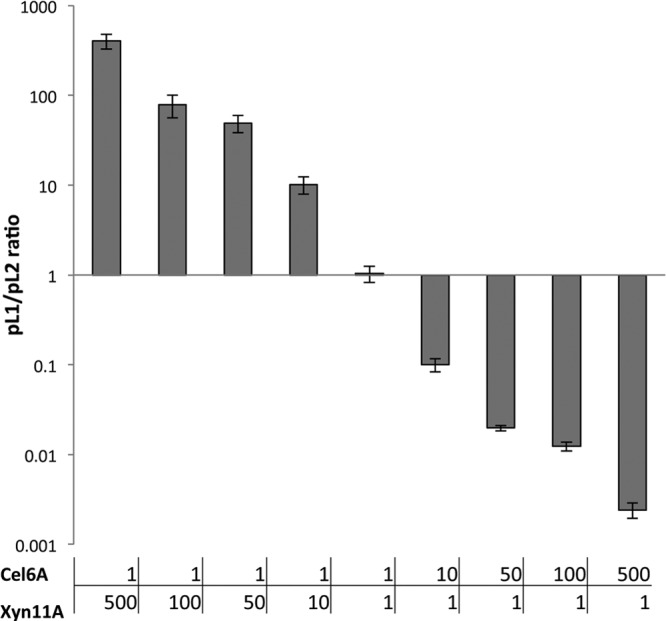
Ratios between Lp1-Cel6A and Lp2-Xyn11A in cocultures after the growth period, as determined by RT-PCR. Cultures were inoculated using various ratios (Cel6A/Xyn11A): 1:500, 1:100, 1:50, 1:10, 1:1, 10:1, 50:1, 100:1, and 500:1. Total copy numbers of each plasmid were determined for each coculture, and ratios were calculated.
DISCUSSION
In this article, we report the successful individual production and secretion of a cellulase and a xylanase by using the biotechnologically interesting bacterium Lactobacillus plantarum. Despite the use of identical cloning strategies, the enzymes were produced at different levels, which is not uncommon, even when expressing similar genes (39). An optimized cell consortium comprising two of the resulting strains was established using the efficiency of wheat straw degradation as the output parameter. These results provide a proof of principle for the engineering of lactobacilli for advanced biomass conversions. The T. fusca enzymes exhibit temperature optima ranging from 50 to 60°C (22, 40) but were nevertheless selected due to their considerable residual activities at 37°C and pH 5 (Fig. 1), i.e., conditions that are common in L. plantarum cultures (41). Nevertheless, at a more advanced stage of L. plantarum development as a possible cellular vehicle for consolidated bioprocessing, it could be advisable to focus on highly active enzymes from mesophilic cellulolytic bacteria (such as Ruminococcus flavefaciens, Clostridium cellulolyticum, or Fibrobacter succinogenes).
As a first step toward more complex biotransformations, we studied cocultures of recombinant bacteria secreting the two enzymes. This rather simple approach was possible because the expression of the heterologous enzymes did not affect the bacterial growth, meaning that strain ratios remained rather stable during the growth period. Clearly, in terms of genetic engineering, more advanced, alternative strategies can be envisaged. One future prospect would be to integrate each gene directly into the genome of L. plantarum. Additionally, it may be appropriate to construct a synthetic operon comprising two or more enzymes. The gene regulation system and the inducible promoters used in this study naturally drive expression of a large array of proteins (e.g., see reference 42), and it should thus be possible to use this system to drive expression of a carefully designed enzyme array.
An advantage of using cocultures is that a cell consortium can easily be optimized by adjusting the ratio of each cell type during inoculation. In a recent publication, a mixture of S. cerevisiae cells with an optimized endoglucanase/exoglucanase/β-glucosidase ratio produced 1.3-fold more ethanol than cells composed of an equal amount of each cell type, suggesting the usefulness of a consortium of bacteria for lignocellulose bioprocessing (43). Such an approach can also be used to balance production levels, which may differ, as observed for Cel6A and Xyn11A in the present study.
The transformed L. plantarum cells were able to degrade either xylan or cellulose and wheat straw. Interestingly, coculturing revealed clear synergistic effects, with the synergy factor reaching 1.8 for combinations with a large excess of the xylanase. These results suggest that the action of the xylanase in deconstructing the substrate renders the cellulose accessible to the cellulase, as described in previous studies (44–46).
Several studies on other bacteria illustrate that L. plantarum producing these lignocellulolytic enzymes could have attractive applications. For example, integration of a cellulase from Bacillus sp. ATCC 21833 into the genome of L. plantarum led to increased efficiency in alfalfa silage fermentation (47). A similar result was reported for L. lactis strains transformed with a Neocallimastix sp. cellulase (48). The expression of genes coding for fibrolytic enzymes in lactobacilli is also of interest for the development of intestinal probiotic strains (49–51). Recently, coexpression of a β-glucanase and a xylanase in Lactobacillus reuteri has been reported (51), and the transformed strain exhibited enzymatic activity on soluble β-glucan and xylan.
Providing L. plantarum cells with highly secreted lignocellulolytic enzymes is a step toward metabolically engineered bacteria that may be used for the production of industrial products, such as polylactic acid or ethanol directly from plant biomass. The concept of engineering L. plantarum to produce ethanol from plant biomass is very tempting, as this bacterium possesses high tolerance to ethanol (up to 13% [vol/vol]) under conditions of low pH (in the range of 3.2 to 4) (6). These traits, along with the ability to utilize hexose and pentose sugars, may render this bacterium a competitive alternative to other types of microbial systems (e.g., Clostridium thermocellum, Saccharomyces cerevisiae, or Escherichia coli) engineered for this purpose (52–54).
The development of a novel bioprocessing system in L. plantarum for converting biomass to biofuels could thus be of major importance to the field of green energy, which will have a tremendous impact on global economic and environmental concerns.
ACKNOWLEDGMENTS
We appreciate the technical assistance of Nili Caspi and thank David B. Wilson and Maxim Kostylev of Cornell University for providing Thermobifida fusca genomic DNA.
This research was supported by a grant number 3620320151 from the Israel Ministry of Science (IMOS). Additional support was obtained by a grant (no. 24/11) issued to R.L. by The Sidney E. Frank Foundation through the Israel Science Foundation (ISF), by the Research Fund of Israel Dairy Board grant no. 362-0348-12362-0316-11, and by a grant (no. 966/09) to E.A.B., also from the ISF. This research was also supported by the establishment of an Israeli Center of Research Excellence (I-CORE Center grant no. 152/11, to E.A.B.) managed by the Israel Science Foundation, by the United States-Israel Binational Science Foundation (BSF), Jerusalem, Israel, by the Weizmann Institute of Science Alternative Energy Research Initiative (AERI), and the Helmsley Foundation. E.A.B. is the incumbent of The Maynard I. and Elaine Wishner Chair of Bio-Organic Chemistry.
Footnotes
Published ahead of print 28 June 2013
REFERENCES
- 1.Bayer EA, Lamed R, White BA, Flint HJ. 2008. From cellulosomes to cellulosomics. Chem. Rec. 8:364–377 [DOI] [PubMed] [Google Scholar]
- 2.Teusink B, Wiersma A, Molenaar D, Francke C, de Vos WM, Siezen RJ, Smid EJ. 2006. Analysis of growth of Lactobacillus plantarum WCFS1 on a complex medium using a genome-scale metabolic model. J. Biol. Chem. 281:40041–40048 [DOI] [PubMed] [Google Scholar]
- 3.Klocke M, Mundt K, Idler C, McEniry J, O'Kiely P, Barth S. 2006. Monitoring Lactobacillus plantarum in grass silages with the aid of 16S rDNA-based quantitative real-time PCR assays. Syst. Appl. Microbiol. 29:49–58 [DOI] [PubMed] [Google Scholar]
- 4.Roach DR, Khatibi PA, Bischoff KM, Hughes SR, Donovan DM. 2013. Bacteriophage-encoded lytic enzymes control growth of contaminating Lactobacillus found in fuel ethanol fermentations. Biotechnol. Biofuels 6:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Limayem A, Hanning IB, Muthaiyan A, Illeghems K, Kim JW, Crandall PG, O'Bryan CA, Ricke SC. 2011. Alternative antimicrobial compounds to control potential Lactobacillus contamination in bioethanol fermentations. J. Environ. Sci. Health B 46:709–714 [DOI] [PubMed] [Google Scholar]
- 6.Alegria EG, Lopez I, Ruiz JI, Saenz J, Fernandez E, Zarazaga M, Dizy M, Torres C, Ruiz-Larrea F. 2004. High tolerance of wild Lactobacillus plantarum and Oenococcus oeni strains to lyophilisation and stress environmental conditions of acid pH and ethanol. FEMS Microbiol. Lett. 230:53–61 [DOI] [PubMed] [Google Scholar]
- 7.Nichols NN, Dien BS, Bothast RJ. 2003. Engineering lactic acid bacteria with pyruvate decarboxylase and alcohol dehydrogenase genes for ethanol production from Zymomonas mobilis. J. Ind. Microbiol. Biotechnol. 30:315–321 [DOI] [PubMed] [Google Scholar]
- 8.Domagk GF, Horecker BL. 1958. Pentose fermentation by Lactobacillus plantarum. V. Fermentation of 2-deoxy-d-ribose. J. Biol. Chem. 233:283–286 [PubMed] [Google Scholar]
- 9.Kleerebezem M, Boekhorst J, van Kranenburg R, Molenaar D, Kuipers OP, Leer R, Tarchini R, Peters SA, Sandbrink HM, Fiers MW, Stiekema W, Lankhorst RM, Bron PA, Hoffer SM, Groot MN, Kerkhoven R, de Vries M, Ursing B, de Vos WM, Siezen RJ. 2003. Complete genome sequence of Lactobacillus plantarum WCFS1. Proc. Natl. Acad. Sci. U. S. A. 100:1990–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ganzle MG, Vermeulen N, Vogel RF. 2007. Carbohydrate, peptide and lipid metabolism of lactic acid bacteria in sourdough. Food Microbiol. 24:128–138 [DOI] [PubMed] [Google Scholar]
- 11.Okano K, Yoshida S, Yamada R, Tanaka T, Ogino C, Fukuda H, Kondo A. 2009. Improved production of homo-d-lactic acid via xylose fermentation by introduction of xylose assimilation genes and redirection of the phosphoketolase pathway to the pentose phosphate pathway in l-lactate dehydrogenase gene-deficient Lactobacillus plantarum. Appl. Environ. Microbiol. 75:7858–7861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, Henrissat B. 2009. The carbohydrate-active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37:D233–D238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sorvig E, Mathiesen G, Naterstad K, Eijsink VGH, Axelsson L. 2005. High-level, inducible gene expression in Lactobacillus sakei and Lactobacillus plantarum using versatile expression vectors. Microbiology 151:2439–2449 [DOI] [PubMed] [Google Scholar]
- 14.Sorvig E, Gronqvist S, Naterstad K, Mathiesen G, Eijsink VGH, Axelsson L. 2003. Construction of vectors for inducible gene expression in Lactobacillus sakei and L. plantarum. FEMS Microbiol. Lett. 229:119–126 [DOI] [PubMed] [Google Scholar]
- 15.Mathiesen G, Sveen A, Brurberg MB, Fredriksen L, Axelsson L, Eijsink VGH. 2009. Genome-wide analysis of signal peptide functionality in Lactobacillus plantarum WCFS1. BMC Genomics 10:425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mierau I, Kleerebezem M. 2005. 10 years of the nisin-controlled gene expression system (NICE) in Lactococcus lactis. Appl. Microbiol. Biotechnol. 68:705–717 [DOI] [PubMed] [Google Scholar]
- 17.Kleerebezem M, Beerthuyzen MM, Vaughan EE, de Vos WM, Kuipers OP. 1997. Controlled gene expression systems for lactic acid bacteria: transferable nisin-inducible expression cassettes for Lactococcus, Leuconostoc, and Lactobacillus spp. Appl. Environ. Microbiol. 63:4581–4584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pavan S, Hols P, Delcour J, Geoffroy MC, Grangette C, Kleerebezem M, Mercenier A. 2000. Adaptation of the nisin-controlled expression system in Lactobacillus plantarum: a tool to study in vivo biological effects. Appl. Environ. Microbiol. 66:4427–4432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bohmer N, Lutz-Wahl S, Fischer L. 2012. Recombinant production of hyperthermostable CelB from Pyrococcus furiosus in Lactobacillus sp. Appl. Microbiol. Biotechnol. 96:903–912 [DOI] [PubMed] [Google Scholar]
- 20.Mathiesen G, Sveen A, Piard JC, Axelsson L, Eijsink VGH. 2008. Heterologous protein secretion by Lactobacillus plantarum using homologous signal peptides. J. Appl. Microbiol. 105:215–226 [DOI] [PubMed] [Google Scholar]
- 21.Moraïs S, Barak Y, Caspi J, Hadar Y, Lamed R, Shoham Y, Wilson DB, Bayer EA. 2010. Contribution of a xylan-binding module to the degradation of a complex cellulosic substrate by designer cellulosomes. Appl. Environ. Microbiol. 76:3787–3796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ghangas GS, Wilson DB. 1988. Cloning of the Thermomonospora fusca endoglucanase E2 gene in Streptomyces lividans: affinity purification and functional domains of the cloned gene product. Appl. Environ. Microbiol. 54:2521–2526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aukrust T, Blom H. 1992. Transformation of Lactobacillus strains used in meat and vegetable fermentations. Food Res. Int. 25:253–261 [Google Scholar]
- 24.Caspi J, Irwin D, Lamed R, Shoham Y, Fierobe H-P, Wilson DB, Bayer EA. 2006. Thermobifida fusca family-6 cellulases as potential designer cellulosome components. Biocatal. Biotransform. 24:3–12 [Google Scholar]
- 25.Lamed R, Kenig R, Setter E, Bayer EA. 1985. Major characteristics of the cellulolytic system of Clostridium thermocellum coincide with those of the purified cellulosome. Enzyme Microb. Technol. 7:37–41 [Google Scholar]
- 26.Eijsink VGH, Brurberg MB, Middelhoven PH, Nes IF. 1996. Induction of bacteriocin production in Lactobacillus sakei by a secreted peptide. J. Bacteriol. 178:2232–2237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anbar M, Lamed R, Bayer EA. 2010. Thermostability enhancement of Clostridium thermocellum cellulosomal endoglucanase Cel8A by a single glycine substitution. ChemCatChem 2:997–1003 [Google Scholar]
- 28.Fierobe H-P, Mingardon F, Mechaly A, Belaich A, Rincon MT, Lamed R, Tardif C, Belaich J-P, Bayer EA. 2005. Action of designer cellulosomes on homogeneous versus complex substrates: controlled incorporation of three distinct enzymes into a defined tri-functional scaffoldin. J. Biol. Chem. 280:16325–16334 [DOI] [PubMed] [Google Scholar]
- 29.Tabka MG, Herpoël-Gimbert I, Monod F, Asther M, Sigoillot JC. 2006. Enzymatic saccharification of wheat straw for bioethanol production by a combined cellulase xylanase and feruloyl esterase treatment. Enzyme Microb. Technol. 39:897–902 [Google Scholar]
- 30.Morais S, Morag E, Barak Y, Goldman D, Hadar Y, Lamed R, Shoham Y, Wilson DB, Bayer EA. 2012. Deconstruction of lignocellulose into soluble sugars by native and designer cellulosomes. mBio 3(6):e00508-12. 10.1128/mBio.00508-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Miller GL. 1959. Use of dinitrosalicylic acid reagent for determination of reducing sugar. Anal. Biochem. 31:426–428 [Google Scholar]
- 32.Ghose TK. 1987. Measurements of cellulase activity. Pure Appl. Chem. 59:257–268 [Google Scholar]
- 33.Moraïs S, Barak Y, Caspi J, Hadar Y, Lamed R, Shoham Y, Wilson DB, Bayer EA. 2010. Cellulase-xylanase synergy in designer cellulosomes for enhanced degradation of a complex cellulosic substrate. mBio 1(5):e00285-10. 10.1128/mBio.00285-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Caspi J, Barak Y, Haimovitz R, Irwin D, Lamed R, Wilson DB, Bayer EA. 2009. Effect of linker length and dockerin position on conversion of a Thermobifida fusca endoglucanase to the cellulosomal mode. Appl. Environ. Microbiol. 75:7335–7342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Caspi J, Irwin D, Lamed R, Fierobe H-P, Wilson DB, Bayer EA. 2008. Conversion of noncellulosomal Thermobifida fusca free exoglucanases into cellulosomal components: comparative impact on cellulose-degrading activity. J. Biotechnol. 135:351–357 [DOI] [PubMed] [Google Scholar]
- 36.Wilson DB. 2004. Studies of Thermobifida fusca plant cell wall degrading enzymes. Chem. Rec. 4:72–82 [DOI] [PubMed] [Google Scholar]
- 37.Chen S, Wilson DB. 2007. Proteomic and transcriptomic analysis of extracellular proteins and mRNA levels in Thermobifida fusca grown on cellobiose and glucose. J. Bacteriol. 189:6260–6265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim JH, Irwin D, Wilson DB. 2004. Purification and characterization of Thermobifida fusca xylanase 10B. Can. J. Microbiol. 50:835–843 [DOI] [PubMed] [Google Scholar]
- 39.Nguyen TT, Nguyen TH, Maischberger T, Schmelzer P, Mathiesen G, Eijsink VG, Haltrich D, Peterbauer CK. 2011. Quantitative transcript analysis of the inducible expression system pSIP: comparison of the overexpression of Lactobacillus spp. beta-galactosidases in Lactobacillus plantarum. Microb. Cell Fact. 10:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Irwin D, Jung ED, Wilson DB. 1994. Characterization and sequence of a Thermomonospora fusca xylanase. Appl. Environ. Microbiol. 60:763–770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McDonald LC, Fleming HP, Hassan HM. 1990. Acid tolerance of Leuconostoc mesenteroides and Lactobacillus plantarum. Appl. Environ. Microbiol. 56:2120–2124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mathiesen G, Huehne K, Kroeckel L, Axelsson L, Eijsink VGH. 2005. Characterization of a new bacteriocin operon in sakacin P-producing Lactobacillus sakei, showing strong translational coupling between the bacteriocin and immunity genes. Appl. Environ. Microbiol. 71:3565–3574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Baek SH, Kim S, Lee K, Lee JK, Hahn JS. 2012. Cellulosic ethanol production by combination of cellulase-displaying yeast cells. Enzyme Microb. Technol. 51:366–372 [DOI] [PubMed] [Google Scholar]
- 44.Hu J, Arantes V, Saddler JN. 2011. The enhancement of enzymatic hydrolysis of lignocellulosic substrates by the addition of accessory enzymes such as xylanase: is it an additive or synergistic effect? Biotechnol. Biofuels 4:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Qing Q, Wyman CE. 2011. Supplementation with xylanase and beta-xylosidase to reduce xylo-oligomer and xylan inhibition of enzymatic hydrolysis of cellulose and pretreated corn stover. Biotechnol. Biofuels 4:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang J, Tuomainen P, Siika-Aho M, Viikari L. 2011. Comparison of the synergistic action of two thermostable xylanases from GH families 10 and 11 with thermostable cellulases in lignocellulose hydrolysis. Bioresour. Technol. 102:9090–9095 [DOI] [PubMed] [Google Scholar]
- 47.Rossi F, Rudella A, Marzotto M, Dellaglio F. 2001. Vector-free cloning of a bacterial endo-1,4-beta-glucanase in Lactobacillus plantarum and its effect on the acidifying activity in silage: use of recombinant cellulolytic Lactobacillus plantarum as silage inoculant. Antonie Van Leeuwenhoek 80:139–147 [DOI] [PubMed] [Google Scholar]
- 48.Ozkose E, Akyol I, Kar B, Comlekcioglu U, Ekinci MS. 2009. Expression of fungal cellulase gene in Lactococcus lactis to construct novel recombinant silage inoculants. Folia Microbiol. 54:335–342 [DOI] [PubMed] [Google Scholar]
- 49.Cho JS, Choi YJ, Chung DK. 2000. Expression of Clostridium thermocellum endoglucanase gene in Lactobacillus gasseri and Lactobacillus johnsonii and characterization of the genetically modified probiotic lactobacilli. Curr. Microbiol. 40:257–263 [DOI] [PubMed] [Google Scholar]
- 50.Liu JR, Yu B, Liu FH, Cheng KJ, Zhao X. 2005. Expression of rumen microbial fibrolytic enzyme genes in probiotic Lactobacillus reuteri. Appl. Environ. Microbiol. 71:6769–6775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu JR, Yu B, Zhao X, Cheng KJ. 2007. Coexpression of rumen microbial beta-glucanase and xylanase genes in Lactobacillus reuteri. Appl. Microbiol. Biotechnol. 77:117–124 [DOI] [PubMed] [Google Scholar]
- 52.Wood BE, Beall DS, Ingram LO. 1997. Production of recombinant bacterial endoglucanase as a co-product with ethanol during fermentation using derivatives of Escherichia coli KO11. Biotechnol. Bioeng. 55:547–555 [DOI] [PubMed] [Google Scholar]
- 53.Balusu R, Paduru RM, Seenayya G, Reddy G. 2004. Production of ethanol from cellulosic biomass by Clostridium thermocellum SS19 in submerged fermentation: screening of nutrients using Plackett-Burman design. Appl. Biochem. Biotechnol. 117:133–141 [DOI] [PubMed] [Google Scholar]
- 54.Tsai SL, Oh J, Singh S, Chen R, Chen W. 2009. Functional assembly of minicellulosomes on the Saccharomyces cerevisiae cell surface for cellulose hydrolysis and ethanol production. Appl. Environ. Microbiol. 75:6087–6093 [DOI] [PMC free article] [PubMed] [Google Scholar]