

Abstract
Sulfate-reducing bacteria (SRB) participate in microbially induced corrosion (MIC) of equipment and H2S-driven reservoir souring in oil field sites. Successful management of industrial processes requires methods that allow robust monitoring of microbial communities. This study investigated the applicability of denaturing high-performance liquid chromatography (DHPLC) targeting the dissimilatory sulfite reductase ß-subunit (dsrB) gene for monitoring SRB communities in oil field samples from the North Sea, the United States, and Brazil. Fifteen of the 28 screened samples gave a positive result in real-time PCR assays, containing 9 × 101 to 6 × 105 dsrB gene copies ml−1. DHPLC and denaturing gradient gel electrophoresis (DGGE) community profiles of the PCR-positive samples shared an overall similarity; both methods revealed the same samples to have the lowest and highest diversity. The SRB communities were diverse, and different dsrB compositions were detected at different geographical locations. The identified dsrB gene sequences belonged to several phylogenetic groups, such as Desulfovibrio, Desulfococcus, Desulfomicrobium, Desulfobulbus, Desulfotignum, Desulfonatronovibrio, and Desulfonauticus. DHPLC showed an advantage over DGGE in that the community profiles were very reproducible from run to run, and the resolved gene fragments could be collected using an automated fraction collector and sequenced without a further purification step. DGGE, on the other hand, included casting of gradient gels, and several rounds of rerunning, excising, and reamplification of bands were needed for successful sequencing. In summary, DHPLC proved to be a suitable tool for routine monitoring of the diversity of SRB communities in oil field samples.
INTRODUCTION
Uncontrolled growth of microbes in oil field production systems may have severe negative impacts on productivity of the systems. Microbial activity may lead to increased frequency of equipment failure because of corrosion, elevated hydrogen sulfide (H2S) concentrations, reservoir souring, formation of metal sulfide scales, filter plugging, loss of injectivity, and inefficient heat exchange (1).
Many of the microbes are indigenous to the oil-bearing deep subsurface environments, but oil production operations also introduce microbes to the system (2, 3). One of the most common and problematic groups of bacteria present in oil and gas field systems is sulfate-reducing bacteria (SRB) (4, 5). The activity of SRB is one of the most significant sources of H2S in reservoir souring. The most frequently isolated mesophilic SRB from oil production waters belong to the genus Desulfovibrio, but members of the genera Desulfacinum, Desulfobacter, Desulfobacterium, Desulfomicrobium, and Desulfotomaculum have also been isolated (3). Thermophilic groups frequently detected from oil production waters include the bacterial genera Thermodesulfobacterium and Thermodesulforhabdus and the archaeal genus Archaeoglobus (6).
In order to effectively manage microbe-induced reservoir fouling, methods that allow robust monitoring of detrimental microbial communities are required. Currently, SRB in the oil field industry are mainly detected and quantified by using the culture-dependent most probable number (MPN) method which requires 28 days to obtain results (7). Furthermore, only a limited number of bacteria can be assessed by a culture-dependent approach (8). Direct culture-independent detection of SRB from community DNA using denaturing gradient gel electrophoresis (DGGE) or terminal restriction length polymorphism (TRFLP) analysis can significantly speed up microbial community profiling and process monitoring and simultaneously provide information about spatial and temporal changes in microbial community composition (9–12). However, these methods are labor-intensive, and gel-based analysis allows only a limited number of samples to be analyzed during the same assay under consistent conditions. As a result, new methods that allow automated and high-throughput analysis of process samples would be advantageous.
Denaturing high-performance liquid chromatography (DHPLC) is a method that has traditionally been used for DNA mutational analysis, especially for detection of single nucleotide polymorphisms, in medical applications. In recent years, the technique has been successfully applied to the study of bacterial diversity in various ecosystems, such as seawater (13), the gut (14), dairy products (15), soil, fermenter sludge and compost (16), sediments (17), and human infections (18–20), and to monitor fungal communities present in air (21), wood decay (22), cheese (23), and milk (24). Similar to DGGE, DHPLC can theoretically resolve DNA fragments with sequence and/or size differences. However, DHPLC is based on separation of partially heat-denatured DNA fragments by ion pair reverse-phase liquid chromatography instead of a denaturing chemical gradient. The DNA fragments are eluted from the column by an increasing gradient of acetonitrile, are detected by UV or fluorescence detector, and can be collected using an automated fraction collector for sequence analysis.
The aim of this study was to characterize SRB communities from oil field samples and to develop a DHPLC method for profiling SRB communities from oil field reservoirs.
MATERIALS AND METHODS
Strains and environmental samples.
Bacterial and archaeal strains or their DNA were obtained from the VTT Culture Collection (culturecollection.vtt.fi) or from the German Resource Centre for Biological Material (www.dsmz.de) (Table 1). In addition to these strains, Desulfomicrobium macestii DSM 4194 (VTT E-001444) was used in the DGGE ladder and quantitative PCR (qPCR) standards. The strains were chosen to represent microbial diversity detected at oil field sites or in marine environments. The strains were grown essentially as recommended by the culture collections. Injection water and samples of produced water (water recovered with the oil) (n = 28) were obtained from different oil field sites in the North Sea, the United States, and Brazil (Table 2). The water samples were filtered onto Sterivex-GP 0.22-μm-pore-size filter units (Millipore, Billerica, MA, USA) and shipped frozen to the laboratory.
Table 1.
Bacterial and archaeal strains used for method optimization and as reference strains
| Phylogenetic cluster and species | Strain | Isolation source (type and location) | GenBank accession no. | GC content (%) | Fragment length (bp) | DHPLC migration (%) | DGGE migration (%) |
|---|---|---|---|---|---|---|---|
| Deltaproteobacteria/Desulfobacterales/Desulfobacteraceae | |||||||
| Desulfobacter hydrogenophilus | DSM 3380 | Marine mud, Italy | NAa | NA | NA | 30 | 10 |
| Desulfatiferula olefinivorans | DSM 18843/VTT E-103143 | Oil-polluted sediment, France | DQ826725 | 61 | 381 | 61 | 72 |
| Desulfobacter curvatus | DSM 3379T/VTT E-001657T | Marine mud, Italy | AF418199 | 52 | 350 | NA | 57 |
| Desulfobacter vibrioformis | DSM 8776/VTT E-103147 | Water-oil from oil platform, Norway | AJ250472 | 52 | 381 | 36 | 28 |
| Desulfosarcina variabilis | DSM 2060T/VTT E-001656T | Marine black mud, France | AF191907 | 58 | 378 | 58 | 57 |
| Deltaproteobacteria/Desulfovibrionales/Desulfovibrionaceae | |||||||
| Desulfatibacillum alkenivorans | DSM 16219 | Oil-polluted sediment, France | AY504426 | 59 | 377 | 54 | 57 |
| Desulfovibrio vulgaris | DSM 644T/VTT E-001447T | Soil, UK | U16723 | 62 | 378 | NA | 57 |
| Desulfovibrio alaskensis | DSM 16109/VTT E-103145 | Gravel material from soured oil reservoir, USA | CP000112 | 61 | 353 | NA | 54 |
| Desulfovibrio desulfuricans | DSM 17464/VTT E-103144 | Oil well corrosion site, USA | AJ249777 | 61 | 378 | 57 | 54 |
| Deltaproteobacteria/Desulfovibrionales/Desulfohalobiaceae | |||||||
| Desulfonauticus autotrophicus | DSM 4206 | Oil production water, Germany | NA | 45 | 314 | 22 | 9 |
| Deltaproteobacteria/Desulfovibrionales/Desulfomicrobiaceae | |||||||
| Desulfomicrobium apsheronum | DSM 5918/VTT E-103146 | Water, oil-bearing deposits, Russia | AF418188 | 59 | 393 | 58 | 59 |
| Clostridia/Clostridiales/Peptococcaceae | |||||||
| Desulfotomaculum geothermicum | DSM 3669/VTT E-061476T | Anoxic geothermal ground water, France | AF273029 | 57b | NA | 52, 53c | 44 |
| Archaea/Euryarchaeota/Archaeoglobi/Archaeoglobales/Archaeoglobaceae | |||||||
| Archaeoglobus fulgidus | DSM 4304 | Submarine hot spring, Italy | NC_000917 | 51 | 360 | 31 | 39 |
NA, not available.
The GC content is an estimate as the GenBank sequence entry does not cover the complete amplicon used in this study.
The two values represent the two peaks in the chromatogram.
Table 2.
Description of oilfield samples and SRB quantification results from qPCR of dsrB gene fragment
| Sample location and code | Sample type and source | Sampling date (mo/yr) | Filtered vol (ml) | No. of dsrB gene copies/mla | Sample selected for identification |
|---|---|---|---|---|---|
| North Sea | |||||
| MOB1 | Injection water, well 1 | 01/2010 | 100 | 5 × 105 | Yes |
| MOB2 | Injection water, well 2 | 02/2010 | 100 | 4 × 104 | Yes |
| MOB3 | Injection water, well 2 | 04/2010 | 100 | 7 × 104 | |
| MOB4 | Injection water, well 3 | 05/2010 | 100 | ND | |
| MOB5 | Injection water, well 4 | 05/2010 | 100 | ND | |
| MOB6-1 | Produced water, well 5 | 01/2010 | 40 | ND | |
| MOB6-2 | Produced water, well 6 | 01/2010 | 40 | ND | |
| MOB6-3 | Produced water, well 7 | 01/2010 | 40 | ND | |
| MOB6-4 | Produced water, well 8 | 01/2010 | 40 | ND | |
| MOB6-5 | Produced water, well 9 | 01/2010 | 40 | 5 × 103 | Yes |
| MOB6-6 | Produced water, well 10 | 01/2010 | 40 | ND | |
| MOB6-7 | Produced water, well 11 | 01/2010 | 40 | ND | |
| MOB6-8 | Produced water, well 12 | 01/2010 | 40 | ND | |
| MOB7 | Injection water, well 13 | 11/2010 | 100 | 5 × 102 | |
| MOB8 | Produced water, well 13 | 12/2010 | 50 | 3 × 103 | |
| MOB9 | Produced water, well 13 | 12/2010 | 50 | 9 × 102 | Yes |
| MOB10 | Produced water, well 13 | 12/2010 | 50 | 1 × 103 | |
| Brazil | |||||
| MOB11 | Produced water, well 14 | 03/2011 | 1000 | 6 × 103 | Yes |
| MOB12 | Produced water, well 15 | 03/2011 | 1000 | 1 × 104 | Yes |
| USA | |||||
| MOB13A | Produced water, well 16 | 07/2010 | 20 | 2 × 104 | Yes |
| MOB13B | Produced water, well 16 | 07/2010 | 20 | 6 × 105 | |
| MOB14 | Produced water, well 17 | 11/2010 | 150 | 4 × 102 | Yes |
| MOB15 | Produced water, well 18 | 03/2011 | 100 | ND | |
| MOB16 | Produced water, well 19 | 03/2011 | 100 | ND | |
| MOB20 | Produced water, well 19 | 04/2011 | 20 | ND | |
| MOB17 | Produced water, well 20 | 03/2011 | 500 | 9 × 101* | Yes |
| MOB19 | Produced water, well 20 | 04/2011 | 500 | ND | |
| MOB18 | Produced water, well 21 | 04/2011 | 500 | 8 × 102 | Yes |
ND, not detected; *, extrapolated.
DNA isolation.
For the DNA analysis, Sterivex filter units containing the oil field samples were aseptically broken with a hammer, and the filters were cut into pieces (approximately 2.5 cm2) with a sterile scalpel and placed with sterile forceps into the lysing tube of a DNA extraction kit. Total DNA from pure cultures and environmental samples was isolated with a FastDNA Spin kit for soil (MP Biomedicals, Santa Ana, CA, USA) according to the manufacturer's instructions, except that the cells were lysed for 2 min in a FastPrep-24 instrument (MP Biomedicals). For Desulfobacter hydrogenophilus (DSM 3380), Desulfotomaculum geothermicum (DSM 3669), and Archaeoglobus fulgidus (DSM 4304), a PowerSoil DNA extraction kit (MoBio Laboratories Inc., Carlsbad, CA, USA) was used according to the manufacturer's instructions, with the exception that the cells were lysed by bead beating with a Ribolyser (Hybaid) device for 30 s at 6 m s−1.
Real-time quantitative PCR.
Quantification of the dsrB copy number in the extracted DNA was performed using a LightCycler 480 instrument (Roche Diagnostics, Basel, Switzerland) and SYBR green-based detection for double-stranded DNA. An approximately 350-bp fragment of the dsrB gene was amplified with primers dsr4R (5′-GTGTAGCAGTTACCGCA-3′) and DSRp2060F (5′-CAACATCGTYCAYACCCAGGG-3′) (11, 25). The amplification was done in a 20-μl reaction volume containing LightCycler 480 SYBR green I master mix (Roche Diagnostics), 0.5 μM each primer, and 2 μl of sample DNA. The amplification reaction program consisted of initial denaturation at 95°C for 5 min and 35 cycles with 10 s at 95°C (denaturation), 20 s at 57°C (annealing), and 20 s at 72°C (elongation). At the end of the run a melting curve analysis was performed from 65 to 97°C.
For preparation of standards for real-time PCR, the dsrB gene fragments from Desulfovibrio desulfuricans DSM 17464, Desulfobacter vibrioformis DSM 8776, and Desulfomicrobium macestii DSM 4194 were cloned into the pGEM-T vector (Promega, Madison, WI, USA) and transformed into Escherichia coli JM109 cells according to the manufacturer's instructions. The plasmids were purified with a QIAprep Spin Miniprep kit (Qiagen, Hilden, Germany), and the dsrB copy number of the plasmid preparations was calculated based on their DNA concentration, measured by a NanoDrop 2000 spectrophotometer (Thermo Scientific, Wilmington, DE, USA). In every run, a dilution series of a mixture of these circular plasmids was incorporated as a standard curve. DNAs from Pyrobaculum islandicum DSM 4184 and Allochromatium vinosum DSM 180 were included as negative controls in each run (11). Each standard and sample were run as at least three replicates.
PCR amplification of dsrB gene fragments for DHPLC and DGGE.
The same dsrB gene fragment as targeted in qPCR was amplified for community profiling by DHPLC and DGGE with primers dsr4R and DSRp2060F. In order to separate the dsrB gene fragments in DGGE, a 40-bp GC clamp was attached to the forward primer (5′-CGCCCGCCGCGCGCGGCGGGCGGGGCGGGGGCACGGGGGG-3′) (26). In the DHPLC analysis, gene fragments amplified both with and without the GC clamp were evaluated. The PCR amplification was performed in 50-μl reaction mixtures containing 1× DynaZyme II buffer (10 mM Tris-HCl, pH 8.8, 1.5 mM MgCl2, 50 mM KCl, and 1% Triton X-100), 0.2 mM each deoxynucleoside triphosphate, 50 pmol of each primer, 1 U of Dynazyme II DNA polymerase (Finnzymes, Espoo, Finland), and 2 μl of template DNA. The PCR was performed in a MasterCycler thermal cycler (Eppendorf, Germany). The PCR program consisted of a 5-min initial denaturation at 94°C followed by 35 cycles of 1 min at 94°C, 1 min at 55°C, and 1 min at 72°C, with a final elongation step at 72°C for 10 min. The amplification products were run in a 1.5% agarose gel and purified with a QIAquick PCR purification kit (Qiagen) according to the instructions of the manufacturer. The concentration of the purified PCR products was determined with a Nanodrop ND-1000 spectrophotometer (Thermo Scientific).
DHPLC.
The DHPLC analysis was performed with a WAVE 4500 microbial analysis system composed of a DNASep cartridge, high-precision Peltier oven, quaternary gradient solvent delivery system, WAVE autosampler model 7200, and UV/visible light (UV/VIS) and fluorescence detectors (Transgenomic, Inc., Omaha, NE, USA). DHPLC analysis was optimized with individual and mixed dsrB gene products from pure cultures (Table 1) adjusted to a final concentration of 10 ng μl−1 by dilution in PCR-grade water. The separation of gene fragments was optimized by varying the temperature (from 62°C to 70°C), acetonitrile gradient composition (from 46 to 70%), gradient rate (from 0.5% min−1 to 2% min−1), and flow rate (from 0.3 ml min−1 to 1.2 ml min−1). For pure cultures, a constant amount of 50 ng of each PCR amplification product was injected into the column, and for oil field samples 5 μl of undiluted amplification product was used. The amplification products were stained with WAVE Optimized HS staining solution during separation, and the elution of dsrB gene fragments was recorded with a fluorescence detector and visualized as chromatograms using Navigator software, version 3.0.0 (build 31) (Transgenomic, Inc.). All buffers and dyes were obtained from Transgenomic, Inc. Buffer A contained 0.1 M triethylammonium acetate (TEAA), and buffer B contained 0.1 M TEAA and 25% acetonitrile.
In the DHPLC analysis, the dsrB gene fragments were collected to chilled 96-well plates for sequencing by using an FCW 180 WAVE fragment collector (Transgenomic, Inc.) and the automated threshold function of the Navigator software (Transgenomic, Inc.). The unpurified fractions containing the collected peaks were then directly reamplified from the fractions with the corresponding primers under the same conditions as used in the original PCRs.
DGGE.
DGGE analysis was performed with a DCode universal mutation detection system (Bio-Rad Laboratories GmbH, Germany). The PCR-amplified dsrB gene fragments containing the GC clamp were separated in a DGGE gel (160 mm by 160 mm by 1 mm) containing 8% acrylamide and 35 to 70% denaturing gradient (100% denaturing gradient contains 7 M urea and 40% formamide) in 0.5× TAE buffer (20 mM Tris, 10 mM acetate, 0.5 mM EDTA, pH 8.0) at 85 V and 60°C for 16 h. The DGGE gels were stained with SYBR green II (New England BioLabs, Ipswich, MA, USA) and imaged with a GelDoc imager (Bio-Rad, Hercules, CA, USA) under UV light. A sequence ladder consisting of PCR products from selected reference strains was run in parallel with the samples. The SRB ladder was a mixture of PCR products from Desulfobacter hydrogenophilus DSM 3380, Desulfonauticus autotrophicus DSM 4206, Desulfobacter vibrioformis DSM 8776, Archaeoglobus fulgidus DSM 4304, Desulfotomaculum geothermicum DSM 3669, Desulfatiferula olefinivorans DSM 18843, Desulfovibrio desulfuricans DSM 17464, Desulfatibacillum alkenivorans DSM 16219, Desulfomicrobium macestii DSM 4194, and Desulfomicrobium apsheronum DSM 5918 (Table 1, Fig. 1).
Fig 1.

Separation of dsrB gene fragments PCR amplified from pure culture bacterial and archaeal cultures in a DHPLC chromatogram (a) and DGGE gel (b). DSM numbers are identified in Table 1.
The bands corresponding to different dsrB gene fragments were excised from the DGGE gel using a Pasteur pipette and kept in 20 μl of PCR-grade water overnight at +4°C. The obtained gene fragments were then reamplified for sequencing with the corresponding primers under the same conditions as used in the original PCR. This procedure was repeated until a single band was obtained in the DGGE gel. The reamplification products were subsequently purified with a QIAquick PCR purification kit (Qiagen).
Sequencing.
Sequencing of DHPLC fragments and DGGE bands was performed from both ends of the PCR amplicon with a BigDye Terminator, version 3.1, cycle sequencing kit (Applied Biosystems, California, USA) in an ABI Prism 310 genetic analyzer (Applied Biosystems);alternatively, samples were sent to Macrogen, Inc., for custom DNA sequencing using the EZ-purification service (Amsterdam, The Netherlands).
Community profiles and sequence analysis.
For comparisons of the similarity of community profiles generated by DHPLC and DGGE, the chromatogram and gel data were transferred into BioNumerics, version 5.10, software (Applied Maths, Sint-Martens-Latem, Belgium). Pearson's curve-based correlations were calculated, and clustering was done with the unweighted pair group method with arithmetic mean (UPGMA). The Shannon-Wiener diversity index was calculated to compare DHPLC and DGGE profiles (27), and they were compared by one-way analysis of variance (ANOVA) using SPSS, version 19 (IBM, USA). The dsrB gene sequences were checked, assembled, and manually edited using the Geneious Pro software package (Biomatters, Inc., Auckland, New Zealand) or Kodon, version 3.61 (Applied Maths). The edited dsrB sequences (138 to 357 bp) were aligned to reference sequences (357 bp) using the MUSCLE (multiple sequence comparison by log expectation) alignment feature in Geneious Pro. The alignment was edited manually, and a maximum-likelihood phylogenetic tree was calculated on the nucleic acid sequence alignment using PhyLM (28) and a Jukes-Cantor substitution model (29). Bootstrap support for the nodes was calculated with 1,000 random repeats.
Nucleotide sequence accession numbers.
The dsrB gene fragment nucleotide sequences of >200 bp were deposited in GenBank under the accession numbers KF269027 to KF269070.
RESULTS
SRB detection with quantitative real-time PCR.
The SRB were detected with qPCR targeting the dsrB gene from 15 of the 28 water samples obtained from oil fields in the North Sea, the United States, and Brazil (Table 2). The qPCR assay was linear in the range of 2 × 102 to 2 × 107 dsrB copies per PCR. The dsrB gene copy numbers in the samples ranged from 102 to 105 ml−1. Based on the qPCR results, 15 positive samples were selected for community analysis.
Optimization of the DHPLC protocol.
Optimal conditions for the analysis of PCR-amplified dsrB gene fragments by DHPLC were determined with PCR amplicons from 12 sulfate-reducing bacterial strains and one archaeal strain (Table 1). Varying oven temperature, buffer flow rate, and strength and duration of the acetonitrile gradient showed that the best separation of dsrB gene fragments could be achieved by using an oven temperature of 65°C, a 54 to 70% acetonitrile gradient that changed 1.5% min−1, and a 0.6-ml min−1 flow rate. The best peak separation was obtained with PCR amplicons containing the GC-rich clamp, while very poor separation was observed with gene fragments amplified without the GC clamp (data not shown).
Under the final optimized conditions, 10 strains out of the 13 tested yielded one or two major peaks in the chromatogram (Fig. 1). The first dsrB gene fragments to elute from the column 3 to 6 min after injection belonged to Desulfobacter and Desulfonauticus species as well as to Archaeoglobus fulgidus. These species had relatively low GC contents (45 to 52%) (Table 1). The gene fragments derived from Desulfotomaculum, Desulfomicrobium, Desulfovibrio, Desulfatibacillum, Desulfatiferula, and Desulfosarcina strains eluted 8 to 10 min after injection and were characterized by higher (57 to 61%) GC contents (Table 1). A significant (P ≤ 0.01) positive correlation was observed between percent GC and retention time (r = 0.97). In general, the strains were distinguishable from each other based on the retention time of their corresponding dsrB amplicons. However, some strains had overlapping retention times. These included Archaeoglobus fulgidus DSM 4304 and Desulfobacter hydrogenophilus DSM 3380, Desulfotomaculum geothermicum DSM 3669 and Desulfatibacillum alkenivorans DSM 16219, and Desulfosarcina variabilis DSM 2060 and Desulfomicrobium apsheronum DSM 5918. The results were consistent across different runs, independent of whether the PCR-amplified gene fragments were injected to the column separately or as part of a mixture (data not shown). Three strains, Desulfobacter curvatus DSM 3379, Desulfovibrio alaskensis DSM 16109, and Desulfovibrio vulgaris DSM 644, could not be successfully separated by DHPLC. They produced smeared peaks in the chromatograms independent of different running parameters.
The separation of dsrB gene fragments in DHPLC was consistent with DGGE analysis (Fig. 1). The dsrB gene fragments showing low retention times on DHPLC were the ones to denature first in the DGGE analysis, whereas the ones possessing higher GC contents and retention times also migrated further in the denaturing gradient of the DGGE. However, among the dsrB gene fragments having lower GC contents, there were some discrepancies between the two methods in the order in which the dsrB fragments were denatured/eluted. For instance, the dsrB gene fragment PCR amplified from Archaeoglobus fulgidus DSM 4304 and Desulfobacter hydrogenophilus DSM 3380 eluted simultaneously in the DHPLC analysis but migrated far apart in the denaturing gel. In DGGE, strains Desulfatibacillum alkenivorans DSM 16219, Desulfobacter curvatus DSM 3379, Desulfosarcina variabilis DSM 2060, Desulfovibrio vulgaris DSM 644, and Desulfomicrobium macestii DSM 4194 could not be separated from each other, whereas Desulfatibacillum alkenivorans DSM 16219 and Desulfosarcina variabilis DSM 2060 were clearly distinguishable by DHPLC. Thus, the overall resolution was similar with both community profiling methods.
Community profiles of oil field samples.
As shown in Fig. 2, DHPLC analysis of the dsrB gene profiles of the oil field samples was reproducible between different runs. Even though peak intensities varied across different runs, this had no effect on peak retention times. DHPLC and DGGE community profiles shared an overall similarity; samples with the lowest and highest diversity were detected by both methods (Fig. 3). The mean Shannon-Wiener diversity indexes were 1.17 (standard deviation [SD], 0.60) and 1.27 (SD, 0.36) for DHPLC and DGGE, respectively, and did not differ statistically significantly (P ≤ 0.1) from each other. The clustering of SRB community profiles determined with DHPLC and DGGE shared an overall similarity with some exceptions (Fig. 4). The grouping followed the geographical location: samples MOB7 to MOB10 collected from the same North Sea site grouped together with both methods, as well as MOB2 and MOB3 from another North Sea site and MOB13A and -B from the same produced water in the United States.
Fig 2.
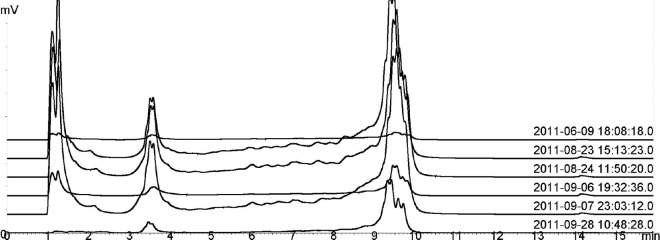
Replicate measurements of sample MOB3 in DHPLC. Samples are identified by date and time on the right side of the graph.
Fig 3.


Community profiling of oil field samples. (a) Samples MOB1 to MOB11; (b) samples MOB12 to MOB18. DHPLC chromatograms are shown on the left, and DGGE gel lanes are on the right. Fragments collected from DHPLC or cut from DGGE gels and sequenced are marked with letters and numbers. ∗, sequencing was not successful; not included in the phylogenetic tree.
Fig 4.
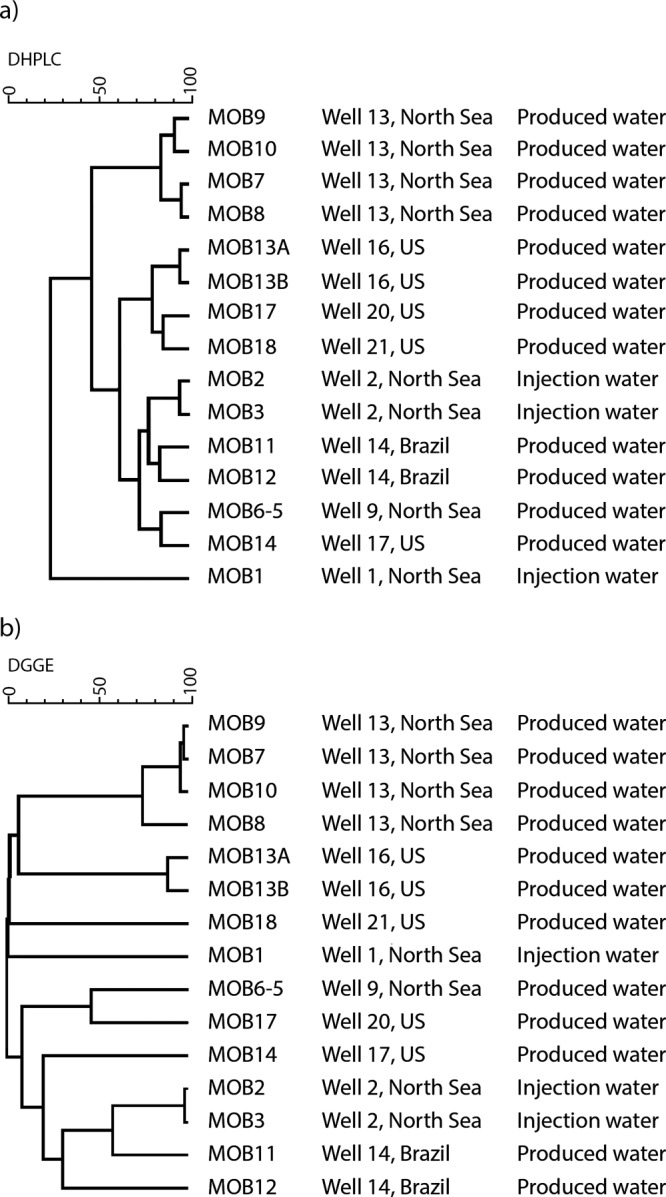
Cluster analysis of DHPLC (a) and DGGE (b) profiles generated from dsrB PCR-positive oil field samples. Pearson's correlation and UPGMA were used to construct the dendrograms.
One sample from each of these sample sets having similar profiles was selected for further analysis; i.e., altogether 10 samples were selected for identification of DHPLC fractions and DGGE bands by sequencing (Table 2). Sequencing of a total of 37 DHPLC fragments was attempted, and 19 of them were successfully sequenced without purification steps (51%) (Fig. 3). From DGGE gels 54 bands were excised, out of which 27 (50%) produced readable sequences after several reamplification and purification rounds.
Based on the phylogenetic analysis, the SRB found from the oil field water samples fell within several taxa (Fig. 5). The dsrB fragments amplified from North Sea water samples and detected by both DHPLC and DGGE were most closely related to Archaeoglobus (MOB1), Desulfovibrio longus (MOB2), and Desulfotomaculum acetoxidans (MOB2). In North Sea produced-water samples the fragments from DGGE and DHPLC were most similar to Desulfovibrio aespoeensis (MOB6-5) and Archaeoglobus fulgidus (MOB9). In addition, three other fragments closest to Desulfotignum phosphitoxidans, Desulfovibrio aespoeensis, and Syntrophobacteraceae were identified with DGGE. In the produced water from Brazil (MOB11) no fragments from DGGE could be identified, and all four fragments from DHPLC fell close to Desulfovibrio longus in the phylogenetic analysis. In the other sample from Brazil (MOB12), the four fragments identified with DHPLC and two identified by DGGE fell within the same cluster close to Desulfococcus multivorans. The DGGE analysis of the MOB12 sample showed three additional fragments belonging to Desulfomicrobium sp., Desulfovibrio aespoeensis, and Desulfovibrio desulfuricans. Produced-water sample MOB13 from the United States had the highest SRB diversity. Both methods identified dsrB fragments close to Desulfovibrio sp. In addition, a DHPLC fragment close to Desulfococcus multivorans, as well DGGE fragments close to Desulfobulbus rhabdoformis, Desulfotignum phosphitoxidans, and Desulforhopalus singaporensis, was identified. In another produced-water sample from the United States (MOB14), a fragment close to Desulfovibrio aespoeensis was detected by both methods. DGGE also showed additional fragments close to Desulfobacter curvatus and Desulfovibrio zosterae. The fragments identified from the well water samples MOB17 and MOB18 detected by both DHPLC and DGGE all fell within the same cluster close to Desulfonatronovibrio hydrogenovorans.
Fig 5.
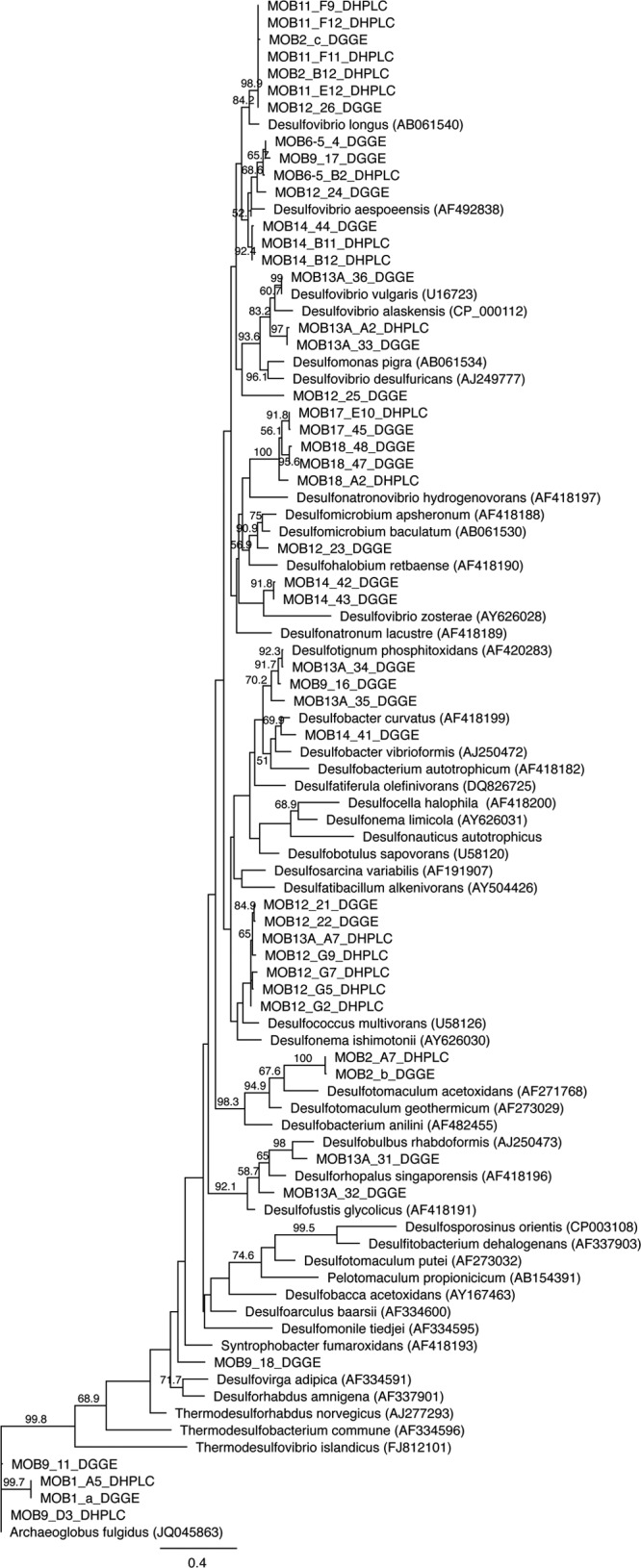
A maximum-likelihood phylogenetic tree of the dsrB sequences obtained by DHPLC and DGGE. Bootstrap probabilities (>50%) are indicated at branch nodes. Accession numbers are given in parentheses. Sequenced fragments are identified in Fig. 3.
DISCUSSION
Uncontrolled microbial growth can have detrimental effects on production efficiency in oil production systems. SRB are one of the most common and problematic group of bacteria found in oil field systems (4, 5). These organisms are notoriously difficult to cultivate. MPN-based methods routinely applied in the oil and gas industry mainly reveal the easily cultivable species. Culture-independent methods for detecting and monitoring the presence and diversity of detrimental microbes are needed for enhanced process control, as well as for increasing knowledge about SRB ecology in oil fields. In this study, we evaluated the applicability of DHPLC to monitoring SRB in injection and produced waters from oil production sites in comparison with DGGE. To our knowledge, this is the first study in which DHPLC has been applied to SRB community profiling. The SRB were first detected by quantitative real-time PCR (qPCR). The analysis was based on the dissimilatory sulfite reductase (dsr) gene that encodes the enzyme catalyzing the conversion of sulfite to sulfide during sulfate reduction. Because this gene is required by all sulfate reducers, it has frequently been used as a functional marker both in qPCR and in community profiling by DGGE (9, 11, 30). Twenty-eight water samples were obtained from oil fields from very distinct geographic locations, 15 of which contained SRB based on qPCR quantification of dsrB gene fragments. These samples were analyzed by DHPLC and DGGE targeting the same functional gene (Table 2).
The qPCR assay targeting the dsrB fragment was shown to be applicable for the detection and quantification of SRB in environmental samples. The linear range of the assay, 2 × 102 to 2 × 107 dsrB copies per PCR, was well in the range found in other studies in which dsrB gene fragments were quantified (9, 30, 31). When qPCR is used, it should be borne in mind that the result is influenced by copy number, which may vary among species (Ribosomal RNA Operon Copy Number Database [http://rrndb.mmg.msu.edu/]). Bacteria exhibit great variation in 16S rRNA gene copy numbers, but the variation in the dsrAB copy number seems to be more restricted, which may make it a better candidate for quantitative applications. Notably Desulfobulbus rhabdoformis, Desulfovibrio vulgaris, Desulfitobacterium hafniense, and Archaeoglobus fulgidus have only a single copy of the dsr gene although more than one copy of dsr has been detected in some Desulfovibrio species (32).
DHPLC and DGGE showed similar powers of discrimination with pure cultures of SRB when the dsrB gene fragment was targeted. The addition of the GC clamp was needed for discriminating genetic differences, as found also by Barlaan et al. (13). Elution of low-GC-content (45 to 52%) dsrB gene fragments before gene fragments with high GC contents (57 to 61%) in DHPLC showed that the separation was dependent on GC content, as in DGGE. A general agreement between the GC content and the behavior of gene fragments in DHPLC has also been reported in previous studies (13, 16, 33). Three SRB, however, produced irresolvable smears in DHPLC which could not be explained by the GC contents of the amplicons (52 to 62%). Troedsson et al. (33) reported that the elution behavior of DNA fragments in DHPLC is correlated with their DNA helicity at the assay temperature.
DHPLC and DGGE gave, in general, similar results for SRB diversity in the oil field samples (Fig. 3 and 4). Similar DHPLC and DGGE profiles have also been obtained for intestinal bacterial communities by Goldenberg et al. (14) and for natural whey cultures by Ercolini et al. (15). In comparison to DGGE, DHPLC provided benefits in sample throughput, reproducibility, robustness, and flexibility. Analyzing a single sample by DHPLC took 16 min, making possible analysis of 90 samples in 24 h, whereas analysis by DGGE required 1.5 working days with a maximum capacity for analyzing 48 samples. The automated fraction collection in DHPLC successfully identified eluted gene fragments and allowed them to be collected for sequencing without a further purification step. In DGGE the preparation of gels is labor-intensive, and three to four rounds of excising the bands from the gels and reamplifying them were necessary in order to obtain readable sequences from them. The success rates of DNA sequencing were similar in DHPLC and DGGE, i.e., approximately 50%.
Our results showed that DHPLC is suited for studying the diversity of a specific group of organisms by targeting functional group-specific genes. In most bacterial DHPLC studies, this technique has been used for 16S rRNA gene analysis (13–15). For SRBs, no 16S rRNA gene-based primer is available to detect all known SRB species. Wagner et al. (25) amplified the 1.9-kb dsrAB gene fragment with DSR1F and DSR4 primers, but later a shorter dsrB sequence was shown to be adequate for distinguishing between different species of SRB (11, 30). It is obvious that some dsrB fragments, despite their sequence divergence, may comigrate in both DHPLC and DGGE. Therefore, the DHPLC or DGGE profiles do not necessarily entirely reflect the true diversity in the field.
The SRB communities in the oil field samples were diverse, and the sequences identified belonged to several dsrB gene clusters (Fig. 5). Desulfovibrio-related sequences were the most common and were found from 7 of the 10 identified samples. Sequences close to Desulfococcus, Desulfomicrobium, Desulfobulbus, Desulfotignum, Desulfonatronovibrio, and Desulfonauticus were also detected. A wide range of Desulfovibrionaceae, Desulfobacteraceae, and Desulfotomaculum-related sequences have previously been found from oil field samples (34–38). Archaeoglobus fulgidus-like sequences were found from samples MOB1 and MOB9, which were both from the North Sea. Archaeoglobus sp. has previously been selectively enriched and immunomagnetically captured from three different platforms in the North Sea by Beeder et al. (6). Sample MOB7 was injection seawater, and samples MOB8 to MOB10 were produced waters from the same site, obtained 1 month after injection. The similarity of SRB profiles of these samples shows that the SRB injected into the well come back up, indicating that a continuous flow of SRB in the injection water to the reservoir may increase the risk of microbiological H2S production, resulting in reservoir souring.
Care must be taken, however, in the interpretation of which species are indigenous in the oil field sites and which are introduced during reservoir development or sampling procedures. The aim of this study was not a systematic screening of oil field bacterial communities, for which the current sample set is not suitable. The sample set in this study included only water samples with planktonic bacteria even though the majority of bacteria in nature are attached to surfaces and form biofilms. For better understanding of the microbiology of oil reservoirs, improved sampling procedures of both planktonic and biofilm SRB communities would be needed (2, 39).
In this study, the DHPLC method was optimized and successfully applied for the profiling of SRB communities in oil field samples. Amplified dsrB fragments could be separated and collected by DHPLC. The results were consistent with DGGE analysis, which showed the applicability of the technique for studying the diversity of SRB based on dsrB gene sequence divergence. The advantage of DHPLC was that it provided a reproducible and automated method of analysis with a high sample throughput capability and flexibility, which are important for routine process monitoring in the oil sector. It is anticipated that the application described here also has broader applicability in the environmental diversity analysis of SRB.
ACKNOWLEDGMENTS
We thank Merja Salmijärvi and Tarja Nordenstedt for skillful technical assistance. We are grateful to Kemira Oyj for providing the oil field samples.
The research was funded by Kemira Oyj, VTT Technical Research Centre of Finland, the Finnish Funding Agency for Technology and Innovation, and the Academy of Finland. The support of these organizations is gratefully acknowledged.
Footnotes
Published ahead of print 21 June 2013
REFERENCES
- 1.Sanders P, Sturman P. 2005. Biofouling in the oil industry, p 171–198 Ollivier B, Magot M. (ed), Petroleum microbiology. ASM Press, Washington, DC [Google Scholar]
- 2.Dahle H, Garshol F, Madsen M, Birkeland N-K. 2008. Microbial community structure analysis of produced water from a high-temperature North Sea oil-field. Antonie Van Leeuwenhoek 93:37–49 [DOI] [PubMed] [Google Scholar]
- 3.Magot M, Ollivier B, Patel BKC. 2000. Microbiology of petroleum reservoirs. Antonie Van Leeuwenhoek 77:103–116 [DOI] [PubMed] [Google Scholar]
- 4.Bass C, Lappin-Scott H. 1997. The bad guys and the good guys in petroleum microbiology. Oilfield Rev. 9:17–25 [Google Scholar]
- 5.Birkeland N-K. 2005. Sulfate-reducing bacteria and Archaea, p 35–54 In Ollivier B, Magot M. (ed), Petroleum microbiology. ASM Press, Washington, DC [Google Scholar]
- 6.Beeder J, Nilsen RK, Rosnes JT, Torsvik T, Lien T. 1994. Archaeoglobus fulgidus isolated from hot North Sea oil field waters. Appl. Environ. Microbiol. 60:1227–1231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.NACE International 2004. Field monitoring of bacterial growth in oil and gas systems. Standard TM0194-2004 NACE International, Houston, TX [Google Scholar]
- 8.Amann RI, Ludwig W, Schleifer K-H. 1995. Phylogenetic identification and in situ detection of individual microbial cells without cultivation. Microbiol. Rev. 59:143–169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Foti M, Sorokin DY, Lomans B, Mussman M, Zacharaova EE, Pimenov NV, Kuenen JG, Muyzer G. 2007. Diversity, activity, and abundance of sulfate-reducing bacteria in saline and hypersaline soda lakes. Appl. Environ. Microbiol. 73:2093–2100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Fourcans A, Ranchou-Peyruse A, Caumette P, Duran R. 2008. Molecular analysis of the spatio-temporal distribution of sulfate-reducing bacteria (SRB) in Camargue (France) hypersaline microbial mat. Microb. Ecol. 56:90–100 [DOI] [PubMed] [Google Scholar]
- 11.Geets J, Borremans B, Diels L, Springael D, Vangronsveld J, van der Lelie D, Vanbroekhoven K. 2006. DsrB gene-based DGGE for community and diversity surveys of sulfate-reducing bacteria. J. Microbiol. Methods 66:194–205 [DOI] [PubMed] [Google Scholar]
- 12.Pérez-Jiménez JR, Kerkhof LJ. 2005. Phylogeography of sulfate-reducing bacteria among disturbed sediments, disclosed by analysis of the dissimilatory sulfite reductase genes (dsrAB). Appl. Environ. Microbiol. 71:1004–1011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barlaan EA, Sugimori M, Furukawa S, Takeuchi K. 2005. Profiling and monitoring microbial populations by denaturing high-performance liquid chromatography. J. Microbiol. Methods 61:399–412 [DOI] [PubMed] [Google Scholar]
- 14.Goldenberg O, Herrmann S, Marjoram G, Noyer-Weidner M, Hong G, Bereswill S, Gobel UB. 2007. Molecular monitoring of the intestinal flora by denaturing high performance liquid chromatography. J. Microbiol. Methods 68:94–105 [DOI] [PubMed] [Google Scholar]
- 15.Ercolini D, Frisso G, Mauriello G, Salvatore F, Coppola S. 2008. Microbial diversity in natural whey cultures used for the production of Caciocavallo Silano PDO cheese. Int. J. Food Microbiol. 124:164–170 [DOI] [PubMed] [Google Scholar]
- 16.Wagner AO, Malin C, Illmer P. 2009. Application of denaturing high-performance liquid chromatography in microbial ecology: fermentor sludge, compost, and soil community profiling. Appl. Environ. Microbiol. 75:956–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kjellerup BV, Sun X, Ghosh U, May HD, Sowers KR. 2008. Site-specific microbial communities in three PCB-impacted sediments are associated with different in situ dechlorinating activities. Environ. Microbiol. 10:1296–1309 [DOI] [PubMed] [Google Scholar]
- 18.Domann E, Hong G, Imirzalioglu C, Turschner S, Kühle J, Watzel C, Hain T, Hossain T, Chakraborty T. 2003. Culture-independent identification of pathogenic bacteria and polymicrobial infections in the genitourinary tracts of renal transplant recipients. J. Clin. Microbiol. 41:5500–5510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Imirzalioglu C, Hain T, Chakraborty T, Domann E. 2008. Hidden pathogens uncovered: metagenomic analysis of urinary tract infections. Andrologia 40:66–71 [DOI] [PubMed] [Google Scholar]
- 20.Jacinto RC, Gomes BP, Desai M, Rajenfram D, Shah HN. 2007. Bacterial examination of endodontic infections by clonal analysis in concert with denaturing high-performance liquid chromatography. Oral Microbiol. Immunol. 22:403–410 [DOI] [PubMed] [Google Scholar]
- 21.Nieguitsila A, Goldenberg O, Deville M, Arne P, Benoit-Valiergue H, Chermette R, Latouche-Cottenot S, Pissard S, Guillot J. 2010. Molecular monitoring of fungal communities in air samples by denaturing high-performance liquid chromatography (D-HPLC). J. Appl. Microbiol. 109:910–917 [DOI] [PubMed] [Google Scholar]
- 22.Maurice S, Le Floch G, Bras-Quéré M, Barbier G. 2011. Improved molecular methods to characterise Serpula lacrymans and other Basidiomycetes involved in wood decay. J. Microbiol. Methods 84:208–215 [DOI] [PubMed] [Google Scholar]
- 23.Mounier J, Le Blay G, Vasseur V, Le Floch G, Jany JL, Barbier G. 2010. Application of denaturing high-performance liquid chromatography (DHPLC) for yeasts identification in red smear cheese surfaces. Lett. Appl. Microbiol. 51:18–23 [DOI] [PubMed] [Google Scholar]
- 24.Delavenne E, Mounier J, Asmani K, Jany J-L, Barbier G, Le Blay G. 2011. Fungal diversity in cow, goat and ewe milk. Int. J. Food Microbiol. 151:247–251 [DOI] [PubMed] [Google Scholar]
- 25.Wagner M, Roger AJ, Flax JL, Brusseau GA, Stahl DA. 1998. Phylogeny of dissimilatory sulfite reductases supports an early origin of sulfate respiration. J. Bacteriol. 180:2975–2982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Muyzer G, De Waal EC, Uitterlinden AG. 1993. Profiling of complex microbial populations by denaturing gradient gel electrophoresis analysis of polymerase chain reaction-amplified genes encoding for 16S rRNA. Appl. Environ. Microbiol. 59:695–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Boon N, De Windt W, Verstraete W, Top EM. 2002. Evaluation of nested PCR-DGGE (denaturing gradient gel electrophoresis) with group-specific 16S rRNA primers for the analysis of bacterial communities from different wastewater treatment plants. FEMS Microbiol. Ecol. 39:101–112 [DOI] [PubMed] [Google Scholar]
- 28.Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52:696–704 [DOI] [PubMed] [Google Scholar]
- 29.Jukes TH, Cantor CR. 1969. Evolution of protein molecules, p 21–123 In Munro HN. (ed), Mammalian protein metabolism. Academic Press, New York, NY [Google Scholar]
- 30.Agrawal A, Lal B. 2009. Rapid detection and quantification of bisulfite reductase genes in oil field samples using real-time PCR. FEMS Microbiol. Ecol. 69:301–312 [DOI] [PubMed] [Google Scholar]
- 31.Ben-Dov E, Brenner A, Kushmaro A. 2007. Quantification of sulfate-reducing bacteria in industrial wastewater, by real-time polymerase chain reaction (PCR) using dsrA and apsA genes. Microb. Ecol. 54:439–451 [DOI] [PubMed] [Google Scholar]
- 32.Kondo R, Nedwell DB, Purdy KJ, de Queiroz Silva S. 2004. Detection and enumeration of sulphate-reducing bacteria in estuarine sediments by competitive PCR. Geomicrobiol. J. 21:145–157 [Google Scholar]
- 33.Troedsson C, Lee RF, Stokes V, Walters TL, Simonelli P, Frischer ME. 2008. Development of a denaturing high-performance liquid chromatography method for detection of protest parasites of metazoans. Appl. Environ. Microbiol. 74:4336–4345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leu J-Y, McGovern-Traa CP, Porter AJR, Harris WJ, Hamilton WA. 1998. Identification and phylogenetic analysis of thermophilic sulfate-reducing bacteria in oil field samples by 16S rDNA gene cloning and sequencing. Anaerobe 4:165–174 [DOI] [PubMed] [Google Scholar]
- 35.Magot M, Caumette P, Desperrier JM, Matheron R, Dauga C, Grimont F, Carreau L. 1992. Desulfovibrio longus sp. nov., a sulphate-reducing bacterium isolated from an oil-producing well. Int. J. Syst. Bacteriol. 42:398–403 [DOI] [PubMed] [Google Scholar]
- 36.Ommedal H, Torsvik T. 2007. Desulfotignum toluenicum sp. nov., a novel toluene-degrading, sulphate-reducing bacterium isolated from an oil-reservoir model column. Int. J. Syst. Evol. Microbiol. 57:2865–2869 [DOI] [PubMed] [Google Scholar]
- 37.Tardy-Jacquenod C, Magot M, Patel BKC, Matheron R, Caumette P. 1998. Desulfotomaculum halophilum sp. nov., a halophilic sulfate-reducing bacterium isolated from oil production facilities. Int. J. Syst. Bacteriol. 48:333–338 [DOI] [PubMed] [Google Scholar]
- 38.Voordouw G, Armstrong SM, Reimer MF, Fouts B, Telang AJ, Shen Y, Gevertz D. 1996. Characterization of 16S rRNA genes from oil field microbial communities indicates the presence of a variety of sulfate-reducing, fermentative, and sulfide-oxidizing bacteria. Appl. Environ. Microbiol. 62:1623–1629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Magot M. 2005. Indigenous microbial communities in oil fields, p 21–33 In Ollivier B, Magot M. (ed), Petroleum microbiology. ASM Press, Washington, DC [Google Scholar]