

Abstract
Variovorax sp. strain WDL1, which mineralizes the phenylurea herbicide linuron, expresses a novel linuron-hydrolyzing enzyme, HylA, that converts linuron to 3,4-dichloroaniline (DCA). The enzyme is distinct from the linuron hydrolase LibA enzyme recently identified in other linuron-mineralizing Variovorax strains and from phenylurea-hydrolyzing enzymes (PuhA, PuhB) found in Gram-positive bacteria. The dimeric enzyme belongs to a separate family of hydrolases and differs in Km, temperature optimum, and phenylurea herbicide substrate range. Within the metal-dependent amidohydrolase superfamily, HylA and PuhA/PuhB belong to two distinct protein families, while LibA is a member of the unrelated amidase signature family. The hylA gene was identified in a draft genome sequence of strain WDL1. The involvement of hylA in linuron degradation by strain WDL1 is inferred from its absence in spontaneous WDL1 mutants defective in linuron hydrolysis and its presence in linuron-degrading Variovorax strains that lack libA. In strain WDL1, the hylA gene is combined with catabolic gene modules encoding the downstream pathways for DCA degradation, which are very similar to those present in Variovorax sp. SRS16, which contains libA. Our results show that the expansion of a DCA catabolic pathway toward linuron degradation in Variovorax can involve different but isofunctional linuron hydrolysis genes encoding proteins that belong to evolutionary unrelated hydrolase families. This may be explained by divergent evolution and the independent acquisition of the corresponding genetic modules.
INTRODUCTION
Linuron [3-(3,4-dichlorophenyl)-1-methoxy-1-methyl urea] is a phenylurea herbicide widely used in agriculture to control germinating and newly emerging grasses and broad-leafed weeds. Biodegradation contributes largely to the dissipation of linuron in the environment. Several single bacterial strains (1, 2) and consortia (3, 4) that degrade (3, 5) or even mineralize and use linuron as the sole source of carbon, nitrogen, and energy have been reported (1–4). Bacterial degradation of linuron is initiated by amide hydrolysis of linuron to 3,4-dichloroaniline (DCA) and N,O-dimethylhydroxylamine (N,O-DMHA). In the case of linuron mineralization, DCA is further converted to water and carbon dioxide (Fig. 1). Bacteria belonging to the genus Variovorax appear to play a crucial role in linuron biodegradation. In linuron-degrading consortia, they are almost always responsible for at least the initial hydrolysis step in linuron degradation, and most linuron-mineralizing single-strain isolates are of the genus Variovorax (1, 2). The genetic basis of linuron degradation in the linuron-mineralizing Variovorax sp. strain SRS16 was recently elucidated (6) and involves three major catabolic gene modules. In strain SRS16, conversion of linuron to DCA is catalyzed by the hydrolase LibA, encoded by the libA gene. Further mineralization of DCA involves a multicomponent dioxygenase complex encoded by dcaQTA1A2BR, which degrades DCA to a chlorocatechol intermediate. The latter is further degraded by a modified ortho-cleavage pathway encoded by ccdRCFDE. Apparently, the acquisition of the ability to mineralize linuron by strain SRS16 involved patchwork assembly of these three catabolic gene modules. A survey of the occurrence of libA in other linuron-degrading Variovorax strains revealed that some strains do not carry a libA homologue, suggesting that alternative enzymes for the initial linuron hydrolysis step exist in Variovorax (6). However, until now, it is not known if, or to what extent, various linuron hydrolases in Variovorax are evolutionarily related. To determine this relationship is important in order to understand the evolutionary adaptation toward linuron degradation in the genus Variovorax and the ecology of linuron-degrading Variovorax in linuron-treated ecosystems.
Fig 1.
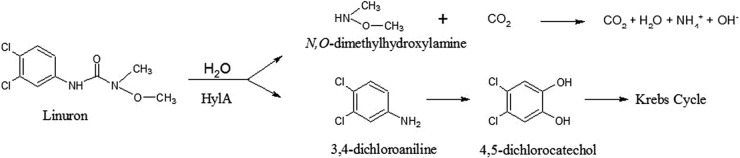
Proposed catabolic pathway of linuron degradation in Variovorax sp. WDL1. The catabolic step attributed to HylA is indicated.
This study reports on the identification of the enzyme and gene involved in linuron hydrolysis in the linuron-mineralizing strain Variovorax sp. WDL1, a Variovorax strain that carries no libA homologue. The enzyme responsible for linuron hydrolysis in strain WDL1 was purified and characterized, and the distribution of the corresponding gene in other linuron-degrading strains was analyzed.
MATERIALS AND METHODS
Bacterial strains, cultivation conditions, and chemicals.
Variovorax sp. strains WDL-1 (4), PBS-H4 (3), SRS16 (2), PBL-H6 (3), and PBL-E5 (3) and Hydrogenophaga sp. strain PBL-H3 (3) were routinely cultivated on R2A medium (solid agar or broth) supplemented with 20 mg liter−1 of linuron at 25°C. Mutants of strain WDL1 lacking the capacity for linuron hydrolysis arose spontaneously when strain WDL1 was plated onto R2A medium without linuron. Degradation of linuron was assessed in MMO broth supplemented with 160 μM linuron. R2A and MMO media were prepared as described previously (3, 4). Linuron [3-(3,4-dichlorophenyl)-1-methoxy-1-methyl urea] (99.5%), diuron [3-(3,4-dichlorophenyl)-1,1-dimethyl urea] (99.5%), isoproturon [3-(4-isopropylphenyl)-1,1-dimethyl urea] (99%), metobromuron [3-(4-bromophenyl)-1-methoxy-1-methyl urea] (99.9%), monolinuron [3-(4-chlorophenyl)-1-methoxy-1-methyl urea] (99%), and 3,4-dichloroaniline (DCA) (98%) were purchased from Sigma-Aldrich, Belgium.
HPLC analysis.
Reverse-phase high-pressure liquid chromatography (HPLC) was used to detect and quantify phenylurea herbicides and their metabolites in the supernatants of bacterial cultures. HPLC analyses were performed at 21°C with a LaChrom (Merck Hitachi) HPLC system equipped with a reversed-phase C18 Alltima column (100 mm by 4.6 mm by 3 μm) using CH3CN-H2O (65:35, vol/vol) as the mobile phase at a flow rate of 0.8 ml min−1. The injection volume was 20 μl (for concentrations of >100 μM) or 100 μl (for concentrations of <100 μM). UV absorption at a wavelength of 210 nm was used to detect the compounds that were identified and quantified based on retention times and peak areas derived from corresponding standard solutions of known concentrations.
Purification of the linuron hydrolase.
Strain WDL1 was cultured in five replicates in 1 liter of R2A supplemented with 20 mg liter−1 of linuron for 2 days under agitation in the dark at 25°C until an optical density at 600 nm (OD600) of 4.5 was reached. After centrifugation of the cultures (15 min, 3,400 × g), the pellets were resuspended in the same volume of MMO containing linuron (40 mg liter−1). Every 90 min, samples were taken for HPLC-based analysis of the residual linuron concentration. After 3 h, when about 65% of the linuron was degraded, the cultures were centrifuged (3,400 × g, 15 min, 4°C) and the pellets washed with phosphate-buffered saline (150 mM NaCl, 7 mM K2HPO4, 2.35 mM KH2PO4). The pellets were suspended in 10 ml of morpholinepropanesulfonic acid (MOPS) buffer (25 mM MOPS, 1 mM dithiothreitol, 5% glycerol [pH 7.6]) and stored at −80°C. The concentrated cell suspension was defrosted, and the cells were lysed in a French press (Thermo). The crude cell extract was centrifuged (8,000 × g, 30 min, 4°C) and concentrated by ultrafiltration on a Sartorius Stedim Vivaspin 6 column (molecular mass cutoff, 3 kDa; 4,000 × g, 4°C). The pH of the concentrated protein extract was adjusted to 7.6 by addition of Tris-HCl (20 mM, pH 7.6), and the solution was incubated with 1 μl of recombinant Benzonase nuclease (25 KU; Sigma) for 45 min at 37°C. The obtained protein extract was then concentrated as described above and loaded on an anion-exchange column (AIEX Q Sepharose HP), equilibrated with Tris-HCl buffer (20 mM, pH 7.6). The proteins were eluted over a linear gradient from 0 to 30% NaCl in an elution volume of 200 ml with a flow rate of 1 ml min−1. Active fractions were pooled, concentrated by ultrafiltration as described above, and dissolved in a 1 M (NH4)2SO4 solution in Tris-HCl (50 mM, pH 7) before being subjected to hydrophobic interaction chromatography (Phenyl Sepharose HP) [gradient from 0.7 M to 0 M (NH4)2SO4; flow rate of 1 ml min−1]. Finally, active fractions were subjected to gel permeation chromatography (column HiLoad Superdex 200 pg 16/60; flow rate of 1 ml min−1) using an elution buffer consisting of 50 mM NaCl in Tris-HCl 20 mM (pH 7.2). Protein purity was assessed by means of 10% SDS-PAGE and Lumitein Protein gel staining (Biotium). To assess the linuron amidohydrolase activity of the different fractions at each purification step, first the production of DCA from linuron was measured using a colorimetric assay consisting of a diazotization-coupling reaction that detects DCA and measurement of absorption at 500 nm according to the method of Pease (7). Then, the activity was checked and quantified by monitoring the degradation of linuron and production of DCA by HPLC as described above. For both the colorimetric and HPLC-based assays, 3 μl (about 180 ng) of purified enzyme solution in MOPS buffer was supplemented with 140 μl of linuron solution (50 mg liter−1 [i.e., 200 μM]) and incubated at room temperature for 60 min. In the case of HPLC-based measurement of linuron and DCA, 50 μl of 4 M HCl was added to stop the reaction after 1 h of incubation.
Characterization of the linuron hydrolase.
The oligomeric state of the enzyme was estimated by comparing the gel permeation chromatography elution time of active fractions with the elution times of the following proteins (size standards in parentheses): aprotinin (6.5 kDa), RNase A (13.7 kDa), carbonic anhydrase (29 kDa), conalbumin (75 kDa), and aldolase (158 kDa). Protein concentrations were estimated using a spectrophotometer (Nanodrop ND-1000). Tryptic peptides generated from the purified protein, excised from polyacrylamide gels, were sequenced using liquid chromatography-electrospray ionization–tandem mass spectrometry (LC-ESI-MS/MS), as previously described (8). N-terminal amino acid sequencing was performed as described previously (9). The kinetic parameters Km, Vmax, and kcat of the purified enzyme for linuron were calculated by determining the hydrolytic activity in the presence of a range of linuron concentrations (80, 60, 45, 20, 10, and 5 μM). The amount of linuron hydrolyzed was determined after 20 min of incubation at 22°C. For each concentration, the linuron degradation rate (V) was calculated as moles of linuron degraded per time and volume unit (mol linuron min−1 ml−1). These values were used to determine the kinetic parameters Km and Vmax of the purified enzyme for linuron based on the Michaelis-Menten kinetics. Vmax was normalized for the enzyme concentration (mol linuron min−1 mol of HylA−1), resulting in the turnover number, kcat. The activity of the linuron hydrolase at different temperatures (10, 15, 20, 25, 30, 35, 40, 45, and 60°C) was analyzed after 1 h of incubation using a linuron concentration of 50 mg liter−1 (200 μM). The substrate specificity of the purified linuron hydrolase was determined at 35°C by assessing the degradation of linuron, diuron, isoproturon, monolinuron, and metobromuron (100 μM each) after 1 h and 24 h of incubation. All above-mentioned tests were performed in a reaction volume consisting of 350 μl MOPS buffer and 350 μl of the herbicide of interest diluted in sterile water and using HPLC to determine residual phenylurea herbicide concentrations. All tests were performed in triplicate.
Sequence analyses.
ORF finder (NCBI) and GeneMark (10) were used to identify open reading frames (ORFs) in contigs of the draft genome sequence of Variovorax sp. WDL1 (P. Albers and D. Springael, unpublished results). TBlastN search was applied to identify the gene coding for the HylA-derived peptide sequences in the draft WDL1 genome sequence. BLASTP was used to identify proteins with significant sequence similarity to the deduced gene product (11). The presence of a signal peptide was predicted with SignalP 4.0 (12). Phylogenetic analysis of HylA and related protein sequences was performed with Geneious Pro (version 5.6.3) (13) using PHYML (JTT matrix) (14).
DNA extraction.
Extraction of genomic DNA from bacterial cultures was performed with the DNeasy blood and tissue kit (Qiagen) using the protocol provided by the manufacturer but with an extra mechanical lysis step after enzymatic lysis. Mechanical lysis was performed by adding 0.3 g of glass beads to the sample, vortexing at full speed for 30 s, and heating the sample for 1 h at 60°C.
PCR analysis.
Primers for PCR detection of hylA were designed using Primer BLAST. BLAST and RDPII (http://rdp.cme.msu.edu/; July 2012) were used to confirm primer specificity. Primers were obtained from Thermo Fisher Scientific (Belgium). The presence of hylA in the different strains was examined by using two primer sets. The first primer set consisted of primers linhydrWDL1_S_Fw (5′-GACGCGACGTTCCGCCACT-3′) and linhydrWDL1_S_Rv (5′-AGGAAGCGCCTGCCGCATTA-3′), which amplify a genomic fragment of 1,919 bp that contains the complete hylA gene (1,755 bp). The second primer set consisted of primers linhydrWDL1_comp_Fw (5′-TCATTTCGCCGGCTCGCCAA-3′) and linhydrWDL1_comp_Rv (5′-ATGCCGATGCATAGGGCCATAT-3′), which amplify the hylA gene from the start to the stop codons. In both cases, PCRs were performed in a total reaction volume of 25 μl containing 200 μM each deoxyribonucleotide triphosphate (dNTP), 0.5 μM each primer, 1× PCR buffer containing 1.5 mM MgCl2, 0.625 U of Taq polymerase (Qiagen), and 1 μl of genomic DNA. PCRs were performed in an Eppendorf MasterCycler (Eppendorf, Germany) with the following reaction scheme: an initial denaturation at 95°C for 5 min, followed by 30 cycles of denaturation at 94°C for 1 min, primer annealing at 60°C for 1 min, and elongation at 72°C for 1 min, and a final extension step at 72°C for 10 min. Amplification products were analyzed by electrophoresis in a 1.5% agarose gel (wt/vol) in 1× Tris-borate EDTA (TBE) containing 0.02% ethidium bromide (90 V, 75 min).
Sequencing of PCR amplicons.
Amplicons were purified with the Qiaquick PCR purification kit (Qiagen) according to the manufacturer's instructions and sequenced using the BigDye Terminator cycle sequencing kit (Applied Biosystems) as described by the manufacturer. The sequencing reaction was performed on a thermocycler UNOII (Biometra, Germany). ClustalOmega (15) was used to align the obtained nucleotide sequences with the hylA sequence of Variovorax sp. WDL1.
Expression of hylA in Escherichia coli.
The amidohydrolase gene was amplified with primers linhydrWDL1_comp_Nostop_Fw (5′-TTTCGCCGGCTCGCCAA-3′) and linhydrWDL1_comp_Rv, and the amplification product was cloned into expression vector pEXP5-CT/TOPO following the manufacturer's recommendations (Invitrogen). A poly-His tag was added at the C-terminal end. Electroporation was used to transform E. coli BL21(DE3)(pLys) with the recombinant vector (pEXP5-CT_HylA). The transformed E. coli BL21(DE3)(pLys) was grown in 50 ml of LB with ampicillin (100 μg ml−1) at 37°C until an OD600 of 0.8 was reached. At this point, the cultures were split into two subcultures of 25 ml. In one subculture, 1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was added to induce expression of the amidohydrolase. During the first 5 h of incubation, each hour two 1-ml samples were taken from each subculture to determine residual linuron concentrations by HPLC and for SDS-PAGE analysis. After centrifugation (10,000 × g, 5 min), the bacterial pellet (1 ml) was dissolved in 80 μl of SDS-PAGE loading buffer and boiled at 100°C for 5 min. After centrifugation (10,000 × g, 5 min), 5 μl of the supernatant was used for SDS-PAGE on a 10% polyacrylamide gel. In parallel, the linuron degradation capacity was evaluated et each sampling time on 1 ml of both cultures by adding linuron at a final concentration of 10 mg liter−1 (40 μM) and incubation at 25°C. Linuron degradation was measured by HPLC after 1 h and after 24 h.
Nucleotide sequence accession numbers.
The nucleotide sequences of contig 1, contig 10, and contig 100 of the draft genome of WDL1 and the nucleotide sequence of the hylA gene of Variovorax sp. PBS-H4 recovered by PCR were deposited in GenBank under accession numbers KC146403, KC146405, KC146404, and KC146406, respectively.
RESULTS
Purification and characterization of a linuron hydrolase in Variovorax sp. WDL1.
A protein with linuron hydrolase activity was purified from a crude protein extract of linuron-degrading WDL1 cells (see Fig. S1A in the supplemental material). Based on gel permeation chromatography, the size of the purified protein was estimated to be ∼118 kDa (data not shown). However, SDS-PAGE revealed a single protein of about ∼56 kDa (see Fig. S1A in the supplemental material). This suggests that the linuron amidohydrolase from Variovorax sp. strain WDL-1 is a homodimer in its active form. Tandem mass spectrometry analysis showed that the peptide amino acid sequences of the purified protein have homology to the sequences of amidohydrolase-like proteins that catalyze the hydrolysis of amide or ester functional groups at carbon and phosphorus centers. The rate of degradation of linuron and production of DCA by the purified enzyme was determined at different initial linuron concentrations to determine kinetic parameters. Stoichiometric production of DCA was observed until a concentration of linuron of 45 μM, but at higher concentrations less DCA was produced than expected (see Fig. S2 in the supplemental material), which may have been due to impurities in the enzyme solution. Km, Vmax, and kcat values were calculated based on the recorded linuron degradation rates (see Fig. S2 in the supplemental material). The novel amidase, designated HylA (hydrolase of linuron A), displayed a Km for linuron of 14.97 ± 0.81 μM, a Vmax of 2.66 10−4 ± 7.6 10−6 μmol min−1 ml−1, and a turnover number, kcat, of 9.00 104 ± 0.24 104 min−1. The temperature range for activity and the phenylurea herbicide substrate range of HylA were determined using a linuron concentration of 100 μM. As observed in the kinetic analysis, less DCA was produced than expected based on the linuron degradation extent. Maximum linuron hydrolase activity of HylA was around 35°C (see Fig. S3 in the supplemental material). The enzyme showed significant activity toward the N-methoxy-N-methyl phenylurea herbicides monolinuron and metobromuron, although at rates much lower than toward linuron (Fig. 2). On the other hand, HylA did not hydrolyze the N,N-dimethyl phenylurea herbicides diuron and isoproturon (Fig. 2), not even after prolonged incubation (for 24 h) (data not shown).
Fig 2.

Phenylurea herbicide substrate specificity of HylA. The purified enzyme was incubated with different phenylurea compounds (100 μM) at 35°C, and the residual herbicide concentration was quantified by HPLC after 1 h. Only for linuron was the simultaneous production of the hydrolysis product (DCA) monitored. HylA activity is calculated as the amount of pesticide hydrolyzed per unit of time (dark gray) or the amount of degradation product formed per unit of time (pale gray). All tests were realized in triplicate (n = 3). Data shown are average values. Standard deviations are indicated.
Identification of the hylA gene in Variovorax sp. WDL1.
The identified peptide sequences of the purified HylA enzyme were assigned to ORF 1.1 on contig 1 (7,530 bp) in the draft genome sequence of strain WDL1 (Fig. 3; see Table S1 in the supplemental material), covering all together about 75% of the deduced amino acid sequence. The hylA gene is absent in the genome of Variovorax paradoxus S110 (16) and V. paradoxus EPS (NC_014931). HylA shows homology with members of the YtcJ-like family (Pfam PF07969, amidohydrolase_3 family) that is part of the metal-dependent amidohydrolase superfamily (see Fig. S4 in the supplemental material). Although HylA and LibA both perform linuron hydrolysis in two closely related linuron-mineralizing Variovorax strains, HylA lacks amino acid homology with LibA (Pfam PF01425, amidase family). Furthermore, no significant amino acid sequence homology was found with other linuron-degrading metal-dependent phenylurea hydrolases (Pfam PF01979, amidohydrolase_1 family), i.e., the orthologues PuhA and PuhB from the actinomycetes Arthrobacter globiformis D47 (17) and Mycobacterium brisbanense JK1 (12), respectively. These enzymes are most similar to the molinate hydrolase MolA (∼50% amino acid sequence identity) mediating thiocarbamate herbicide degradation by the actinomycete Gulosibacter molinativorax ON4 (18). Until now, no enzymes closely related to HylA have been characterized. Among the characterized YctJ-like enzymes, the N-substituted formamide deformylase NfdA (19), which catalyzes the hydrolysis of some specific N-substituted formamides, showed some homology with HylA (only 25% amino acid identity). HylA showed higher amino acid identity (46 to 51%) with several hypothetical YtcJ-like amidohydrolases encoded in the Sphingomonas wittichii RW1 genome (see Fig. S4 in the supplemental material): Swit_0715 (51%), Swit_3245 (50%), Swit_3246 (49%), Swit_3242 (49%), Swit_3243 (47%), and Swit_2059 (46%). Two additional RW1 hypothetical proteins (Swit_3759 and Swit_3767) show lower similarity (29 to 30% amino acid identity) to HylA (see Fig. S4 in the supplemental material). SignalP prediction suggested the presence of an N-terminal signal peptide in HylA with a cleavage site between position 24 and position 25. N-terminal amino acid sequencing confirmed that this signal peptide is missing in the purified HylA. Moreover, although the molecular mass of the mature protein deduced from the nucleotide sequence (59,747 Da) is slightly higher than the value estimated from SDS-PAGE (∼56 kDa) and the dimeric size measured in gel filtration (∼118 kDa), it is smaller than the unprocessed form of HylA (62,380 Da).
Fig 3.
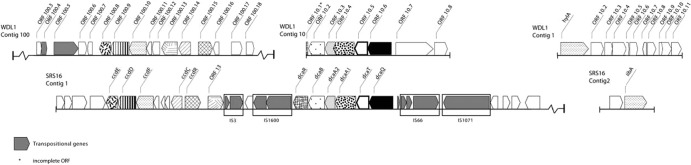
Organization of the genes involved in linuron and DCA metabolism in Variovorax sp. WDL1 compared to the corresponding genes in Variovorax sp. SRS16 (6). The arrows indicate the direction of transcription of the genes. Similar shaded patterns indicate isofunctional genes in WDL1 and SRS16. The following linuron/DCA catabolic functions correspond to the indicated genes (the coding genes in WDL1 and SRS16, respectively, are shown in parentheses): linuron hydrolase (hylA, libA), glutamine synthetase (orf10.6, dcaQ), glutamine amidotransferase (orf10.5, dcaT), chloroaniline dioxygenase large subunit (orf10.4, dcaA1), chloroaniline dioxygenase small subunit (orf10.3, dcaA2), chloroaniline dioxygenase reductase (orf10.2, dcaB), LysR family transcriptional regulator (orf10.1, dcaR), LysR family transcriptional regulator (orf100.15, ccdR), chlorocatechol 1,2-dioxygenase (orf100.14, ccdC), maleylacetate reductase (orf100.10, ccdF), chloromuconate cycloisomerase (orf100.9, ccdD), dienelactone hydrolase (orf100.8, ccdE), and the hypothetical Bug family protein (orf100.13, orf13). Further information about the different ORFs is shown in Table S1 in the supplemental material. The ORFs present on contig 100 in the genome of WDL1 upstream of ORF 100.3 and downstream of ORF 100.18 are represented in Table S1 in the supplemental material and the corresponding GenBank file (KC146404).
Expression of hylA in E. coli.
The linuron hydrolysis activity of HylA was confirmed by expression analysis in E. coli BL21(DE3)(pLys) carrying a vector containing hylA under the control of an IPTG-inducible promoter. The sequence encoding the N-terminal signal peptide was retained in this construct. In contrast to E. coli BL21(DE3)(pLys), for which no linuron degradation was observed, IPTG-induced cells of the E. coli containing pEXP5-CT_hylA showed complete linuron degradation in less than an hour (Fig. 4). Without IPTG induction, a significantly slower linuron degradation capacity was observed (10% after 1 h and 86% after 24 h) (Fig. 4). SDS-PAGE profiles showed two additional bands corresponding to the expected protein size of ∼56 kDa in the IPTG-supplemented cultures compared to the noninduced cultures (see Fig. S1B in the supplemental material), which probably correspond to the processed and unprocessed forms of the protein, with and without the signal peptide cleaved off, respectively.
Fig 4.
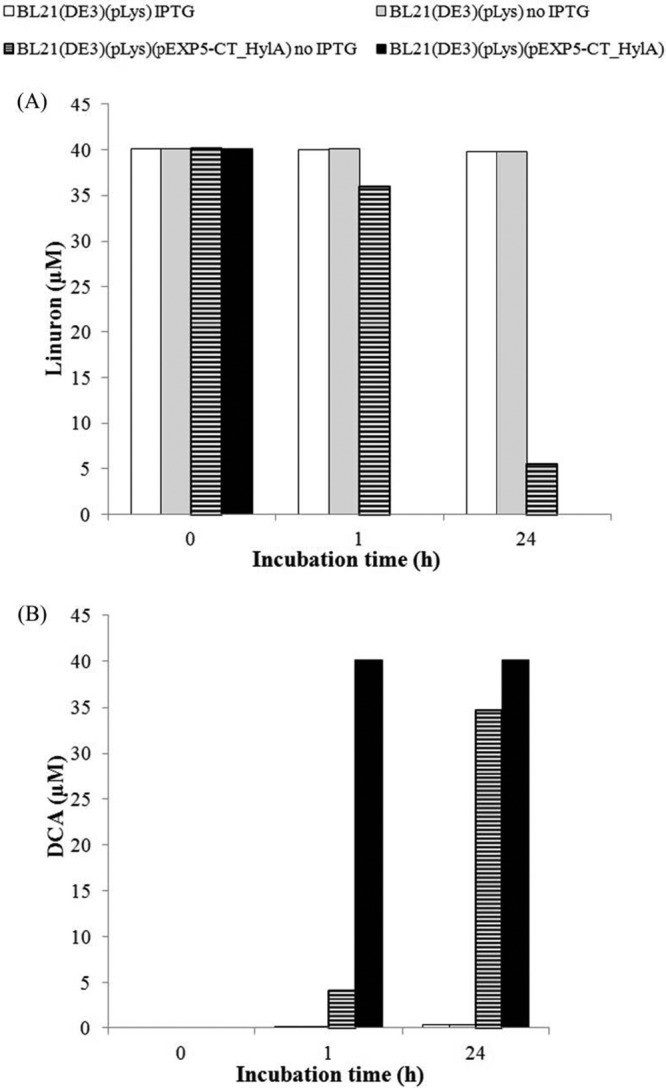
Linuron degradation by E. coli expressing recombinant HylA. Activity of BL21(DE3)(pLys) control cells was compared with that of cells carrying recombinant vector pEXP-CT_HylA. Linuron was added (final concentration of 40 μM) to both IPTG-induced and noninduced cultures 5 h after addition of IPTG (1 mM). Linuron degradation (A) and DCA production (B) were measured by HPLC after 1 h and after 24 h of incubation.
Presence of hylA in other linuron-degrading bacteria.
Different linuron-degrading strains (Variovorax sp. strains SRS16, PBL-E5, PBL-H6, and PBS-H4 and Hydrogenophaga sp. strain PBL-H3) were examined by PCR for the presence of hylA. In addition to strain WDL1, hylA was detected only in strain PBS-H4, the only strain that does not contain libA (6). The hylA amplicon recovered from PBS-H4 showed 99.7% nucleotide identity with hylA of strain WDL1, with a different amino acid sequence at three positions. Spontaneous mutants of strain WDL1 that had lost the capability to degrade linuron after growth on a medium without linuron did not show amplification with the hylA-specific primers (data not shown).
DISCUSSION
This paper reports on the identification of a novel enzyme, designated HylA, that hydrolyzes the phenylurea herbicide linuron to DCA in Variovorax sp. WDL1. The novel amidohydrolase differs from other previously reported and characterized linuron hydrolases, i.e., PuhA identified in Arthrobacter globiformis D47 (20), the PuhA orthologue PuhB in Mycobacterium brisbanense strain JK1 (21) and LibA in Variovorax sp. SRS16 (6). First, they belong to different protein families. HylA belongs to the YctJ-like family within the metal-dependent amidohydrolase superfamily. PuhA and PuhB are members of a separate family within this superfamily, while LibA belongs to the amidase signature (AS) family (6). Second, HylA shows different enzyme kinetics toward linuron. LibA, PuhA, and PuhB show a 2-fold lower Km (∼7 to 8 μM) than HylA (14.97 ± 0.81 μM), indicating that the former have a somewhat higher affinity for linuron. Conversely, PuhA and PuhB demonstrated lower turnover numbers (4.25 ± 0.22 s−1 and 4.3 ± 0.13 s−1, respectively) than that of HylA (1.55 × 103 ± 0.04 × 103 s−1). No kcat data are currently available for LibA. LibA shows optimal linuron hydrolysis activity in a lower temperature range (22 to 30°C) than that of HylA. Maximum linuron hydrolysis by PuhA and PuhB takes place between 30°C and 35°C, which is comparable to the temperature range for HylA (see Fig. S3 in the supplemental material). Third, the phenylurea herbicide substrate range of HylA differs from those of LibA, PuhA, and PuhB. The substrate range of HylA is limited compared to those of PuhA and PuhB, which show activity toward a wide range of N-methoxy-N-methyl as well as N,N-dimethyl phenylurea herbicides, but its activity is less restricted than the one of LibA, which among all tested phenylurea herbicides hydrolyzes only linuron. Furthermore, in contrast to the monomeric linuron hydrolase LibA (6), HylA is a dimer in its active form. Other amidohydrolases have been shown to have a dimeric structure in solution (22, 23), and only the dimer is active in some cases (17). Finally, in contrast to LibA, PuhA, and PuhB, HylA contains an N-terminal signal sequence for secretion across the cytoplasmic membrane. This signal sequence was indeed absent in the mature enzyme isolated from whole cells. Apparently, the enzyme is exported to the periplasm to exert its function and HylA-mediated linuron hydrolysis is a periplasmic process in WDL1 (24).
Some observations indicate that HylA is responsible for the initial hydrolysis in the linuron degradation pathway in Variovorax sp. WDL1. The hylA gene is missing in spontaneous mutants of WDL1 that lack linuron hydrolysis activity but retain the capacity to degrade DCA. The appearance of mutants defective in xenobiotic degradation on nonselective medium is a common observation in organic xenobiotic-degrading bacteria (25). Strain WDL1 is able to degrade linuron to mineral products and to degrade DCA. As such, hylA is expected to compose a complete linuron mineralization pathway together with a multicomponent dioxygenase and a chlorocatechol cleavage pathway in WDL1. In the draft genome sequence of WDL1, both a gene cluster that accommodates putative proteins of a multicomponent chloroaniline dioxygenase (consisting of ORF 10.6 to ORF 10.1) and a gene cluster that encodes putative proteins linked to a modified chlorocatechol ortho-cleavage pathway (ORF 100.8 to ORF 100.10, ORF 100.14, and ORF 100.15) were identified (Fig. 3; see Table S1 in the supplemental material). ORF 10.1 to ORF 10.6 can be linked to the multicomponent aniline dioxygenase components that were identified by differential SDS-PAGE analysis by Breugelmans et al. (8) as being increasingly expressed when strain WDL1 was grown on linuron. In contrast to hylA, the translation products of these two gene clusters show high amino acid similarity with the corresponding proteins in SRS16 (see Table S1 in the supplemental material). Both gene clusters also show an organization highly similar to those in SRS16 (Fig. 3), with minor differences in the chlorocatechol catabolic gene cluster. In strain SRS16, chlorocatechol degradation appears to be encoded by a gene cluster (ccdRC orf10 orf9 orf8 ccdFDE) whose organization differs substantially from those encoding modified ortho-cleavage pathways in other organisms (6). Similar to SRS16, three ORFs that cannot directly be linked to chlorocatechol degradation (ORFs 100.11 to 100.13) are located within the gene cluster (Fig. 3; see Table S1). However, only ORF 100.11 is a putative orthologue of the corresponding ORF_9 in SRS16 (82% amino acid identity; see Table S1). The deduced translation product of ORF 100.13 shows significant amino acid similarity (43% amino acid identity) to ORF 13, a hypothetical transporter component (periplasmic solute-binding protein) encoded by a gene flanking the chlorocatechol cleavage gene cluster in SRS16 (Fig. 3; see Table S1). Thus, as previously suggested for Variovorax sp. SRS16, these observations suggest a similar patchwork assembly of catabolic gene modules in Variovorax sp. WDL1 that allows the strain to use the xenobiotic compound linuron as the sole source of carbon and energy, but instead of libA in strain SRS16, hylA completes the linuron degradation pathway in WDL1. PCR analyses of all linuron-degrading strains in our collection show that they contain either libA or hylA but never both. This might indicate that linuron hydrolysis genes were acquired by horizontal gene transfer and that the evolvement of a linuron catabolic pathway in Variovorax can involve different nonrelated isofunctional linuron hydrolytic enzymes. This can be explained by independent acquisition of the corresponding gene modules and hence divergent evolution.
The identification of HylA as an alternative linuron hydrolase used by linuron-degrading Variovorax strains poses questions about its ecological function in environmental linuron degradation. In the past, ecological studies showed that the dynamics of linuron-degrading activity in the environment as a response to linuron application (i.e., in linuron-treated agricultural soil and on-farm biopurification systems), is not always explained by the abundance of libA (26). We are currently investigating whether hylA provides an alternative explanation in those case studies, whether organisms containing hylA or libA coexist or compete in linuron-treated and -contaminated environments, and whether still other linuron hydrolysis gene functions exist.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by project OT10/03 of KU Leuven, EU project BIOTREAT (EU grant no. 266039), and the research project MIRESOWA funded by Danish Council for Strategic Research grant 2104-08-001.
We thank J. Desair and M. Ghequire for help and advice in protein purification and B. Horemans, P. Albers, V. Dunon, K. Simoens, and P. J. Vaysse for advice and technical assistance.
Footnotes
Published ahead of print 28 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/AEM.01478-13.
REFERENCES
- 1.Satsuma K. 2010. Mineralisation of the herbicide linuron by Variovorax sp strain RA8 isolated from Japanese river sediment using an ecosystem model (microcosm). Pest Manag. Sci. 66:847–852 [DOI] [PubMed] [Google Scholar]
- 2.Sorensen SR, Rasmussen J, Jacobsen CS, Jacobsen OS, Juhler RK, Aamand J. 2005. Elucidating the key member of a linuron-mineralizing bacterial community by PCR and reverse transcription-PCR denaturing gradient gel electrophoresis 16S rRNA gene fingerprinting and cultivation. Appl. Environ. Microbiol. 71:4144–4148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Breugelmans P, D'Huys PJ, De Mot R, Springael D. 2007. Characterization of novel linuron-mineralizing bacterial consortia enriched from long-term linuron-treated agricultural soils. FEMS Microbiol. Ecol. 62:374–385 [DOI] [PubMed] [Google Scholar]
- 4.Dejonghe W, Berteloot E, Goris J, Boon N, Crul K, Maertens S, Hofte M, De Vos P, Verstraete W, Top EM. 2003. Synergistic degradation of linuron by a bacterial consortium and isolation of a single linuron-degrading Variovorax strain. Appl. Environ. Microbiol. 69:1532–1541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wallnofer P. 1969. Decomposition of urea herbicides by Bacillus sphaericus, isolated from soil. Weed Res. 9:333–339 [Google Scholar]
- 6.Bers K, Leroy B, Breugelmans P, Albers P, Lavigne R, Sorensen SR, Aamand J, De Mot R, Wattiez R, Springael D. 2011. A novel hydrolase identified by genomic-proteomic analysis of phenylurea herbicide mineralization by Variovorax sp. strain SRS16. Appl. Environ. Microbiol. 77:8754–8764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pease HL. 1962. Separation and colorimetric determination of monuron and diuron residues. J. Agric. Food Chem. 10:279–281 [Google Scholar]
- 8.Breugelmans P, Leroy B, Bers K, Dejonghe W, Wattiez R, De Mot R, Springael D. 2010. Proteomic study of linuron and 3,4-dichloroaniline degradation by Variovorax sp. WDL1: evidence for the involvement of an aniline dioxygenase-related multicomponent protein. Res. Microbiol. 161:208–218 [DOI] [PubMed] [Google Scholar]
- 9.Parret AHA, Schoofs G, Proost P, De Mot R. 2003. Plant lectin-like bacteriocin from a rhizosphere-colonizing Pseudomonas isolate. J. Bacteriol. 185:897–908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Besemer J, Borodovsky M. 2005. GeneMark: web software for gene finding in prokaryotes, eukaryotes and viruses. Nucleic Acids Res. 33:W451–W454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 12.Petersen TN, Brunak S, von Heijne G, Nielsen H. 2011. SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat. Methods 8:785–786 [DOI] [PubMed] [Google Scholar]
- 13.Drummond A, Ashton B, Cheung M, Heled J, Kearse M, Moir R, Stones-Havas S, Thierer T, Wilson A. 2011. Geneious, version 5.6.3. Geneious, Auckland, New Zealand [Google Scholar]
- 14.Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52:696–704 [DOI] [PubMed] [Google Scholar]
- 15.Goujon M, McWilliam H, Li W, Valentin F, Squizzato S, Paern J, Lopez R. 2010. A new bioinformatics analysis tools framework at EMBL-EBI. Nucleic Acids Res. 38:W695–W699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Han JI, Choi HK, Lee SW, Orwin PM, Kim J, Laroe SL, Kim TG, O'Neil J, Leadbetter JR, Lee SY, Hur CG, Spain JC, Ovchinnikova G, Goodwin L, Han C. 2011. Complete genome sequence of the metabolically versatile plant growth-promoting endophyte Variovorax paradoxus S110. J. Bacteriol. 193:1183–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Werner AK, Sparkes IA, Romeis T, Witte C-P. 2008. Identification, biochemical characterization, and subcellular localization of allantoate amidohydrolases from Arabidopsis and soybean. Plant Physiol. 146:418–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Duarte M, Ferreira da Silva F, Lünsdorf H, Junca H, Gales L, Pieper DH, Nunes OC. 2011. Gulosibacter molinativorax ON4T molinate hydrolase, a novel cobalt-dependent amidohydrolase. J. Bacteriol. 193:5810–5815 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Fukatsu H, Hashimoto Y, Goda M, Higashibata H, Kobayashi M. 2004. Amine-synthesizing enzyme N-substituted formamide deformylase: screening, purification, characterization, and gene cloning. Proc. Natl. Acad. Sci. U. S. A. 101:13726–13731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turnbull GA, Ousley M, Walker A, Shaw E, Morgan JAW. 2001. Degradation of substituted phenylurea herbicides by Arthrobacter globiformis strain D47 and characterization of a plasmid-associated hydrolase gene, puhA. Appl. Environ. Microbiol. 67:2270–2275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Khurana JL, Jackson CJ, Scott C, Pandey G, Horne I, Russell RJ, Herlt A, Easton CJ, Oakeshott JG. 2009. Characterization of the phenylurea hydrolases A and B: founding members of a novel amidohydrolase subgroup. Biochem. J. 418:431–441 [DOI] [PubMed] [Google Scholar]
- 22.Ho Y-Y, Hsieh H-C, Huang C-Y. 2011. Biochemical characterization of allantoinase from Escherichia coli BL21. Protein J. 30:384–394 [DOI] [PubMed] [Google Scholar]
- 23.Wang WC, Hsu WH, Chien FT, Chen CY. 2001. Crystal structure and site-directed mutagenesis studies of N-carbamoyl-D-amino-acid amidohydrolase from Agrobacterium radiobacter reveals a homotetramer and insight into a catalytic cleft. J. Mol. Biol. 306:251–261 [DOI] [PubMed] [Google Scholar]
- 24.von Heijne G. 1990. The signal peptide. J. Membr. Biol. 115:195–201 [DOI] [PubMed] [Google Scholar]
- 25.Top EM, Springael D. 2003. The role of mobile genetic elements in bacterial adaptation to xenobiotic organic compounds. Curr. Opin. Biotechnol. 14:262–269 [DOI] [PubMed] [Google Scholar]
- 26.Bers K, Sniegowski K, De Mot R, Springael D. 2012. Dynamics of the linuron hydrolase libA gene pool size in response to linuron application and environmental perturbations in agricultural soil and on-farm biopurification systems. Appl. Environ. Microbiol. 78:2783–2789 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.