

Abstract
On entry into the nucleus, herpes simplex virus 1 (HSV-1) DNA localizes to nuclear bodies known as ND10. Gene repression imposed by ND10 is released by a viral protein, ICP0, via degradation of the ND10 constituents promyelocytic leukemia protein (PML) and Sp100 and the subsequent dispersal of ND10 bodies. In order to understand the dynamic interaction between ICP0 and ND10, we carried out deletion mapping to identify the domains of ICP0 responsible for its association with ND10. Here, we report the following. (i) An ND10 entry signal (ND10-ES), located between residues 245 and 474, is required for ICP0 to penetrate and fuse with ND10. ICP0 lacking ND10-ES adheres to the surface of ND10 but fails to enter. (ii) In the absence of ND10-ES, the E3 ubiquitin ligase of ICP0 facilitates the transient adhesion of the truncated ICP0 to the ND10 surface, whereas the presence of ND10-ES in ICP0 renders ND10 fusion regardless of the E3 ligase activity. (iii) The C terminus of ICP0 is required for retention of ICP0 in ND10 but plays no role in the recruitment process. (iv) The adverse effects of an inactive RING domain on viral replication are partially reversed by deleting either ND10-ES or the C-terminal retention domain, suggesting that additional ICP0 functions require the release of ICP0 from ND10. Based on these results, we conclude that association of ICP0 and ND10 is a dynamic process, in which three sequential steps—adhesion, fusion, and retention—are adopted to stabilize the interaction. A faithful execution of these steps defines the ultimate productivity of the virus.
INTRODUCTION
Following herpes simplex virus 1 (HSV-1) entry into cells, the nucleocapsid is transported to the nuclear pore, where it releases the viral DNA into the nucleus. Sensing the foreign invasion, the infected cells attempt to immediately silence viral DNA by intrinsic immune responses, which mobilize existing restriction factors before the induction of additional antiviral molecules (1). At least two multiprotein complexes, the REST/CoREST/HDAC complex and nuclear domain 10 (ND10), have been described as part of the intrinsic defenses that inhibit initial HSV-1 viral DNA expression (2–7).
ND10s, also known as promyelocytic leukemia protein (PML) nuclear bodies or PML oncogenic domains, are dynamic nuclear structures that contain the constant constituents PML and Sp100 and numerous transient components, including gene regulatory proteins such as Daxx, CBP, p53, Rb, etc. (8–10). The dynamic complexity of ND10 postulates the importance of ND10s in different aspects of cell life. Indeed, ND10s function in many cellular regulatory processes, including cell cycle regulation, apoptosis, DNA repair, cell senescence, and also antiviral defense (11–14).
The antiviral effects of ND10s were initially proposed based on the observation that exposure of cells to interferon (IFN) increases the number and size of ND10 bodies as well as the total amounts of PML and Sp100 (15, 16). Knockout mice without PML are prone to infections, while fibroblasts from PML−/− mice fail to mount antiviral responses following exposure to IFN (17, 18). The participation of ND10s in IFN-induced viral inhibition, which requires protein synthesis of IFN-responsive factors, is regarded as part of the innate immune response. More recent studies indicate that ND10s are also part of intrinsic antiviral defenses that use cellular histones and their associated repressors to silence viral DNA. Mounting evidence suggests that ND10s also serve as sites for epigenetic regulation of foreign DNAs. For example, incoming DNA, from viral infection or DNA transfection, is located in the vicinity of ND10s and leads to enlarged ND10s (19–21). ND10 components—Daxx and ATRX—are found to regulate histone assembly on minigenes introduced into the cell (22). Moreover, many chromatin-remodeling proteins such as CoREST and CLOCK are recruited to ND10s upon infection (23, 24). Given the fact that ND10 participates in multiple antiviral pathways, it is considered one of the most important cell antiviral mechanisms and a key target regulated by many different viruses (11).
For HSV-1, an α protein designated infected cell protein 0 (ICP0) is critical for viral countermeasures mounted against cell intrinsic defenses (3, 6, 25, 26). ICP0 gene is essential for viral replication in low-multiplicity infection of cultured cells but is dispensable at high multiplicity (25), which indicates that ICP0 functions through saturating cellular factors. ICP0 is a multifunctional viral protein targeting diverse cellular pathways. One well-characterized function of ICP0 is the E3 ubiquitin ligase activity located in its RING finger domain (27, 28). Upon HSV-1 infection, ICP0 is responsible for the proteasomal degradation of many cellular proteins, including PML and Sp100 (29, 30), some of the centromeric proteins (CENPs) that are important for centromere architecture (31, 32), DNA-PK (DNA-dependent protein kinase) involved in DNA repair pathway (33), and the IFN-inducible protein IFI16 that is responsible for IFN induction triggered by foreign DNA (34). In addition to functioning through the E3 ligase activity, ICP0 also interacts with numerous cellular proteins to regulate cell homeostatic status during infection. As exemplified in Fig. 1A, frame 3, ICP0 interacts with CoREST and disrupts the CoREST-HDAC interaction (3). ICP0 binds to USP7 (ubiquitin-specific protease 7) to regulate the ubiquitin-proteasome pathway (35, 36). It also interacts with RNF8 (37), cyclin D3 (38, 39), and EF-1δ (40), tuning various cell pathways such as DNA repair, cell cycle, and protein translation. A fundamental question in HSV-host interaction is how ICP0 achieves multitasking and coordinates its multiple functions to ensure a robust HSV-1 infection.
Fig 1.

Schematic diagrams of ICP0 gene structure and ICP0 mutants used in this study. (A) Line 1, HSV-1 genome, with the repeated sequences shown in patterned boxes. Line 2, position of the two copies of ICP0 in the terminal and internal repeats and expanded illustration of ICP0 gene, with three exons and two introns. Frame 3, ICP0 cDNA used in constructing recombinant viruses. The positions of some known functional domains are illustrated: RING finger domain (white R in black oval) (27, 28); nuclear localization sequence (NLS) (51); SUMO interaction motif (SIM) (56); USP7 interaction domain (USP7) (44); EF-1δ interaction domain (EF-1δ) (40); ICP0 dimerization domain (Dimer) (44); and amino acids important for interactions between ICP0 and cellular partners (thin vertical rectangles). T67 is important for binding to RNF8 (37), D199 is involved in binding to cyclin D3 (38, 39), and D671/E673 are responsible for CoREST interaction (46). Line 4, amino acid positioning in ICP0 cDNA. (B1 to B3) ICP0 truncations constructed and studied in this paper. The RING finger mutant of C116G/C156A substitutions (RFm) is illustrated as a black oval with a white X. All constructs are tagged with mCherry (stars) at their N termini. The amino acid inclusions or deletions are demonstrated in the parentheses following the virus names.
During infection, ICP0 undergoes dynamic trafficking. Newly synthesized ICP0 is first localized to ND10 nuclear bodies (41). The next event observed is the degradation of ND10 proteins PML and Sp100 (29, 30). Between 3 and 5 h after infection, PML essentially disappears, leading to the dispersal of ND10, and subsequently ICP0 diffuses throughout the nucleus (41, 42). Later on, ICP0 disappears from the nucleus and accumulates in the cytoplasm (43). The spatial-temporal trafficking of ICP0 likely plays an important role in coordinating the interactions between ICP0 and its multiple targets. In order to delineate ICP0 functions, we chose to investigate the first trafficking event—interaction with ND10. We sought to apply reverse genetics to identify the domains of ICP0 that determine the interaction of ICP0 with ND10. Previously, Meredith et al. have identified the C terminus of ICP0 as essential for the localization of ICP0 in ND10 (44). In this report, we show that ICP0 contains three domains that define adhesion, fusion, and retention of ICP0 in ND10 bodies. In this context, the C terminus of ICP0 functions as an anchor for retention rather than recruitment of ICP0 to the ND10 structures.
MATERIALS AND METHODS
Cells.
Immortalized human embryonic lung (HEL) fibroblasts received originally from Thomas E. Shenk (Princeton University) and human osteosarcoma U2OS cells from ATCC were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and in McCoy's 5A medium (Invitrogen) supplemented with 10% serum, respectively.
Construction of recombinant viruses.
For N-terminal deletions of ICP0, forward primers 5′-GCAGGATCCGCAGACTACGTACCGCCCGC-3′, 5′-GCAGGATCCGCGGTCTCGGGGGGGAGC-3′, and 5′-GCAGGATCCGGGCCCTCCCGCGGCGCCCC-3′ were paired with reverse primer for PCR amplification of ICP0 truncations to construct recombinant viruses RHG105, RHG106, and RHG107. For C-terminal deletions, reverse primers 5′-GCAGTCGACTTACCCGGGCCCACCCTGGCCGCG-3′ and 5′-GCAGTCGACTTAGGGCAGGCAGTCCCCCGTG-3′ were paired with forward primer for PCR amplification of ICP0 truncations to construct recombinant viruses RHG104 and RHG103. For RHG110 and RHG118, plasmids containing full-length ICP0 or ICP0 84 to 775 were digested with SnaBI and SalI, whereas PCR products amplified with primers 5′-GGGCCCTCCCGCGGCGCCGCCCCCTC-3′ and 5′-TATTGTTTTCCCTCGTCCCG-3′ were digested with SalI. The blunt end generated by SnaBI was ligated to amino acid 475. Additional RING finger mutations C116G/C156A in recombinant viruses RHG120, RHG108, RHG109, RHG117, and RHG119 were constructed as described elsewhere (45). The PCR fragment amplified with primers 5′-GCAGAATTCCCCGCCCCGGATGTCTGGGTG-3′ and 5′-GCAGTCGACTTACAGGTACCGCGGGGCGAAC-3′ was digested with XhoI plus KpnI to swap for the wild-type RING finger. The mCherry gene was PCR amplified with primers 5′-ATGGTGAGCAAGGGCGAGGAGG-3′ and 5′-GCAGGATCCCTTGTACAGCTCGTCCATG-3′ and ligated in frame to the N termini of the above ICP0 deletions.
The above-described series of mCherry-ICP0 deletions with their flanking sequences were cloned into the KO5 plasmid as previously described (46). The series of KO5 constructs were then electroporated into the RR1 strain containing ICP0-null bacterial artificial chromosome (BAC) (46). BAC DNAs extracted from positive colonies were transfected into U2OS cells to generate recombinant viruses. All viruses were plaque purified at least three times on U2OS cells. Viral DNAs were isolated and verified for the existence of ICP0 mutations in both terminal and internal repeats.
Confocal microscopy.
HEL cells grown on four-well glass slides (Thermo Fisher Scientific) were exposed to 10 PFU/cell of recombinant viruses for 1 h. Inocula were then removed, and cells were incubated in growth medium. At the time points indicated in Results, the cells were fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and blocked with phosphate-buffered saline (PBS) containing 5% horse serum and 1% bovine serum albumin. The cells were then allowed to react with primary antibodies at 4°C overnight, rinsed, and subjected to reaction with fluorescein isothiocyanate (FITC)-conjugated goat anti-rabbit (Sigma) plus Texas Red-conjugated goat anti-mouse (Invitrogen) secondary antibodies or Alexa 488-conjugated goat anti-mouse plus Alexa 594-conjugated goat anti-rabbit secondary antibodies (Invitrogen). Images were taken with a Zeiss confocal microscope. Three-dimensional (3D) images were constructed by Volocity 3D image analysis software.
Western blotting.
HEL cells were mock infected or infected with 10 PFU/cell of recombinant viruses for 1 h. Inocula were then removed, and cells were incubated in DMEM supplied with 10% newborn calf serum. At the hours indicated, the cells were harvested, washed, and lysed in radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% NP-40, 0.25% sodium deoxycholate, 1 mM phenylmethylsulfonyl fluoride). The total cell lysate was sonicated, electrophoretically separated on SDS-PAGE, and then transferred to polyvinylidene difluoride membrane. The membrane was blocked with 1× TBST (20 mM Tris, pH 7.5, 150 mM NaCl, 0.5% Tween 20) containing 5% nonfat dry milk and probed with primary antibodies as indicated in Results. The membrane was then incubated with horseradish peroxidase-conjugated goat anti-mouse or goat anti-rabbit secondary antibody (Sigma) and visualized with ECL Western Blotting Detection Reagent (GE Healthcare).
Antibodies.
Polyclonal anti-ICP0 exon 2 antibody was a generous gift from Bernard Roizman (University of Chicago), polyclonal and monoclonal anti-PML antibodies were purchased from Santa Cruz Biotechnology, Inc., polyclonal anti-actin antibody was purchased from Rockland Immunochemicals Inc., and polyclonal and monoclonal anti-mCherry antibodies were purchased from Clontech.
One-step growth curve.
HEL cells were infected with viruses at 0.1 or 2 PFU/cell for 1 h. Inocula were then removed, and cells were incubated in DMEM supplied with 10% newborn calf serum. At the time points indicated in Results, cells were harvested and resuspended in milk. To titrate viral yields, cells harvested at different time points were sonicated, diluted, and plated on U2OS cells. After 2 h of incubation, the inocula were removed and the cells were cultured in McCoy's 5A medium supplemented with 10% newborn calf serum and 0.1% human gamma globulin.
qPCR.
HEL cells were infected with viruses at 0.1 or 2 PFU/cell for 1 h. Inocula were then removed, and cells were incubated in DMEM supplied with 10% newborn calf serum. At 2 and 24 h postinfection, total DNA was isolated from infected cells by phenol-chloroform extraction and subjected to quantitative PCR (qPCR) assay using ABsolute qPCR SYBR green ROX mix (Thermo Scientific) and a Stratagene Mx3005P Thermocycler. Primers 5′-GCAGCTAGCATGGCGACTGACATTGATATG-3′ and 5′-GCAGAATTCCTAAAACAGGGAGTTGCAATA-3′ targeting the ICP27 gene were used for viral DNA quantitation. The QuantumRNA Universal 18S internal standard purchased from Invitrogen Life Technologies was used as an endogenous control to normalize the DNA amount. The DNA fold increase from 2 hours postinfection (hpi) to 24 hpi was calculated by comparative ΔΔCT analysis.
RESULTS
Construction of viruses with specific deletions or point mutations in ICP0.
In order to define the sequence requirements for the association of ICP0 with ND10, we performed systematic deletion mapping of ICP0 in the context of HSV-1 infection. As shown schematically in Fig. 1, we generated a recombinant virus, RHG101, in which a cDNA copy of ICP0 (47, 48) was tagged with mCherry and cloned to replace the ICP0 gene in the HSV-1 genome, using the BAC technique as described elsewhere (46, 49). Then, a series of recombinant viruses carrying deletions or mutations in ICP0 were constructed in the same fashion. The ICP0 mutant viruses (Fig. 1) were grouped into C-terminal truncations (Fig. 1B1), N-terminal truncations (Fig. 1B2), and internal truncations (Fig. 1B3). The localization of mutant ICP0s was determined with the aid of confocal microscopy.
Fig 2.
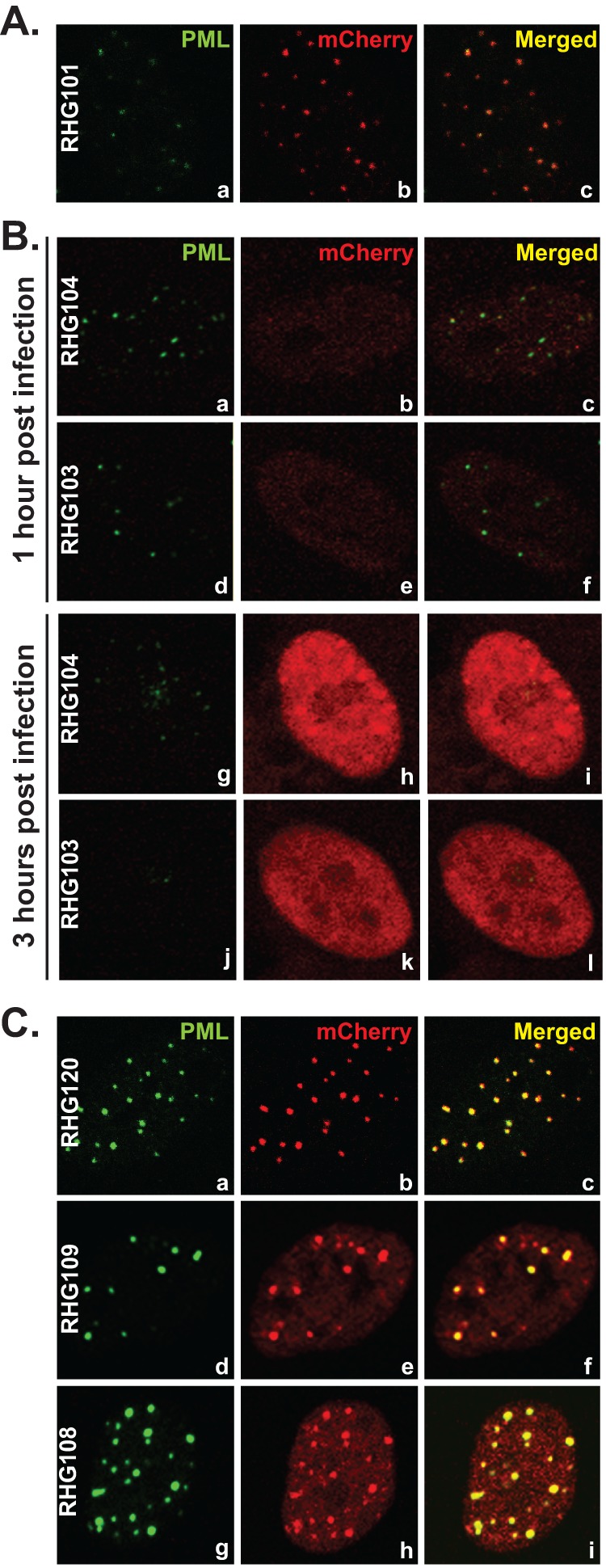
Localization of ICP0 C-terminal truncations. (A) HEL cells were infected with RHG101 at 10 PFU/cell. At 3 h postinfection, cells were fixed and stained with polyclonal anti-PML and monoclonal anti-mCherry antibodies. Representative pictures of the infected cells were taken with a Zeiss 780 confocal microscope. (B) HEL cells were infected with RHG103 or RHG104 at 10 PFU/cell. At 1 or 3 h postinfection, cells were stained and photographed as described for panel A. (C) HEL cells were infected with RHG120, RHG108, or RHG109 at 10 PFU/cell. At 3 h postinfection, cells were stained and photographed as described for panel A.
Fig 3.
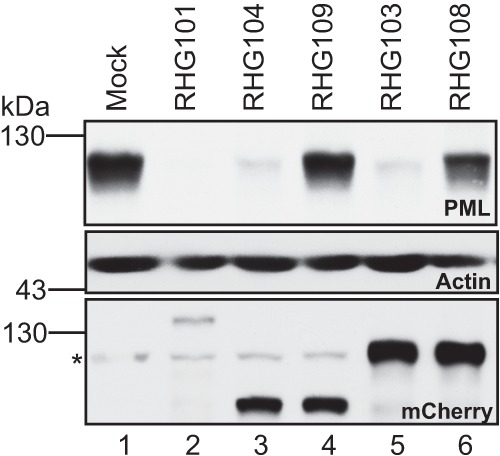
PML degradation by ICP0 C-terminal truncations. HEL cells were either mock infected or infected with 10 PFU/cell of mutant viruses as indicated. At 8 h postinfection, total cell lysates were separated on denaturing polyacrylamide gel and blotted for proteins as indicated in each gel panel. The asterisk (*) points to a nonspecific cellular protein that cross-reacts with the polyclonal anti-mCherry antibody. EZ-Run prestained Rec protein ladder from Fisher was used as a marker.
The C terminus of ICP0 is required for retention but not for recruitment of ICP0 to ND10.
Meredith et al. reported that a deletion of the C-terminal residues 594 to 775 led to a diffused nuclear distribution of ICP0 and a mild decrease in viral replication. They concluded that the C-terminal domain is required for ICP0 localization to ND10 nuclear bodies (44). Consistent with these observations, we also found that ICP0 containing C-terminal deletions in recombinant viruses RHG103 and RHG104 were dispersed throughout the nucleus at early times postinfection (Fig. 2B). Panels a through c of Fig. 2A show that full-length ICP0 with an N-terminal mCherry-tag was localized to ND10, just like wild-type ICP0 or ICP0 tagged at the C terminus reported elsewhere (41, 45). At 1 h postinfection, ND10 nuclear bodies stained with PML antibody were generally intact in cells infected by RHG103 and RHG104, whereas the barely detectable ICP0 truncations were clearly diffused throughout the nucleus (Fig. 2B, panels a through f). At 3 h postinfection, the ND10 structures were barely visible or had disappeared altogether (Fig. 2B, panels g and j). The ICP0 truncations, despite the elevated level, were still diffused throughout the nucleus (Fig. 2B, panels h and k). To determine whether the disappearance of ND10s in Fig. 2B was the consequence of PML degradation but not of PML disaggregation, we measured the amounts of PML by Western blot analyses. As shown in Fig. 3, at 8 h after infection, PMLs in HEL cells infected with viruses RHG103 and RHG104 were degraded to the same extent as in cells infected with full-length ICP0 (Fig. 3, top panel, lanes 5, 3, and 2, respectively). The full-length ICP0 had the lowest steady-state level (Fig. 3, bottom panel, lane 2), consistent with the quick turnover of wild-type ICP0 reported elsewhere (45). Anti-actin antibody was used as a loading control.
Two hypotheses could explain the observation that PML was degraded in the apparent absence of accumulation of ICP0 at ND10 bodies. The first hypothesis postulates that PML cycles in and out of ND10 and can be degraded during its sojourn outside ND10. Therefore, C-terminal truncations meet and degrade PML without being localized to ND10. The alternative hypothesis is that ICP0 enters ND10 and degrades PML inside ND10. In this scenario, the C terminus of ICP0 acts as a retention anchor in ND10 bodies. The deletion of the C terminus shortens its sojourn in ND10 bodies. To differentiate between the two hypotheses, we constructed the mutant viruses RHG108 and RHG109, in which the ICP0s are truncated at the C terminus and which at the same time contain the C116A and C156G substitutions in the RING finger domain. ICP0 with substitutions of C116G and C156A completely abolishes the E3 ligase activity and fails to degrade PML (46, 50). The full-length ICP0 protein carrying the C116G/C156A substitutions is sequestered in ND10 bodies due to its failure in completing the E3 enzymatic reaction (46). The mCherry tag at the N terminus did not change the sequestration of C116G/C156A mutant in ND10 (Fig. 2C, panels a to c). However, ICP0s carrying the RING finger substitutions and lacking the C-terminal domains encoded by RHG108 and RHG109 entered into and were sequestered in the ND10 bodies, as shown in Fig. 2C, panels d to i. These results indicate that (i) the absence of the C-terminal domain did not impair the recruitment of ICP0 to ND10 and (ii) upon entry into ND10, the additional RING finger mutation sequestered the enzymatic reaction in a transition state and blocked mutant ICP0 from cycling out of ND10s. Based on these observations, we conclude that the rapid dispersal of ICP0 throughout the nucleus—a characteristic of ICP0 encoded by RHG103 and RHG104 (Fig. 2B, panels b, e, h, and k)—is the result of a lack of ICP0 retention. Therefore, the function of ICP0 C-terminal residues 689 to 775 is to serve as an anchor for retention rather than recruitment of ICP0 to ND10 bodies.
ICP0 with deletions encompassing amino acids 245 to 474 is recruited to ND10 surface but fails to enter.
The nuclear localization sequence (NLS) of ICP0 has been mapped to residues between 475 and 549 (51). In order to determine the sequence required for recruiting ICP0 to ND10, we systematically truncated ICP0 from the N terminus but left NLS intact (Fig. 1B2). Cells infected with recombinant viruses RHG105, RHG106, and RHG107 containing ICP0 with N-terminal truncations are shown in Fig. 4. Compared to the wild-type ICP0 (Fig. 2A, panels a to c), deletion of the first 83 or 241 amino acids did not affect the ability of the truncations to localize at ND10 (Fig. 4A and B, panels a to f). However, further deletion of residues 242 to 474 led to a complete dispersal of ICP0, at both 1 h and 3 h postinfection (Fig. 4A and B, panels g to i). As expected, the ability of ICP0 to degrade PML correlated with the presence of the RING finger domain located between amino acids 84 and 241 (Fig. 4C). ICP0 undergoes proteasome-dependent degradation and proteasome-independent proteolytic cleavage during HSV-1 infection (45). The smaller bands on the mCherry blot (Fig. 4C) are likely the degradation intermediates.
Fig 4.
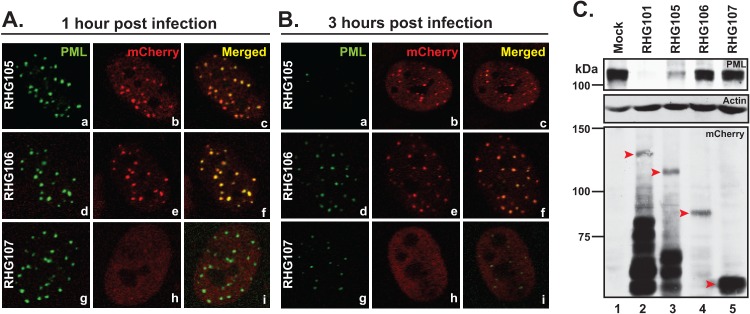
Localization and ability to degrade PML of ICP0 N-terminal truncations. (A and B) HEL cells were infected with 10 PFU/cell of recombinant viruses as indicated. At 1 h (A) or 3 h (B) postinfection, cells were stained and photographed as described above. (C) HEL cells were either mock infected or infected with 10 PFU/cell of mutant viruses as indicated. At 8 h postinfection, total cell lysates were separated and blotted for proteins as indicated. Red arrowheads indicate the full-length or truncated ICP0s. The Precision Plus protein standards system from Bio-Rad was used as a marker.
Although both RHG106 and RHG107 caused little degradation of PML (Fig. 4C, lanes 4 and 5), there was a dramatic change in the ICP0 distribution, from a distinct ND10 localization in RHG106-infected cells (Fig. 4A and B, panels d to f) to a complete diffusion throughout the nucleus in cells infected by RHG107 (Fig. 4A and B, panels g to i). As discussed above, upon interacting with its substrates in ND10, ICP0 with inactive E3 ubiquitin ligase is sequestered and unable to release itself from the enzymatic transition state. The fact that neither RHG106 nor RHG107 contained the RING finger domain but only RHG106 showed a sequestration of ICP0 truncation at ND10 led us to conclude that residues 242 to 474 are responsible for recruiting ICP0 into ND10. Without the domain encompassing residues 242 to 474, ICP0 cannot be recruited to meet with its substrates residing within the ND10 bodies. Thus, enzymatic sequestration does not occur.
To better define the domain within residues 242 to 474, we constructed a recombinant virus RHG110, in which the codon 244 of ICP0 was fused in frame to codon 475. The deletion of residues 245 to 474 brought the RING finger domain in close proximity to the nuclear localization sequence (Fig. 1B3). Immunofluorescence studies revealed that in approximately 30% of the RHG110-infected cells, the ICP0 encoded by RHG110 aggregated in dot structures that were juxtaposed to rather than coincided with the ND10 bodies (Fig. 5A, panel c, arrowheads). The juxtaposition of the internal truncation and ND10 was unmistakably clear in the 3D image reconstructed through z-sectioning (Fig. 5B, panel a). Under the same imaging conditions, RHG120, whose ICP0 contains C116G/C156A substitutions and is sequestered in ND10, showed the perfect colocalization (Fig. 5B, panel b). We also examined ICP0 in prototype strain HSV-1(F), and wild-type ICP0 was perfectly colocalized with ND10 under the same imaging conditions (Fig. 5B, panel c). To exclude potential artifacts, the experiment was validated by staining duplicated slides with monoclonal anti-PML plus polyclonal anti-mCherry antibodies or with polyclonal anti-PML plus monoclonal anti-mCherry antibodies. In both sets of experiments, ICP0 was found juxtaposed to ND10 in one-third of the RHG110-infected cells (Fig. 6A, panels a to c and g to i), indicating that the domain of residues 245 to 474 in fact contains a sequence necessary for ICP0 to penetrate and comingle with the components of the ND10 bodies. We define this domain as the ND10 entry signal (ND10-ES). ICP0 without ND10-ES docks at the ND10 surface but does not enter the ND10 nuclear bodies.
Fig 5.

Docking of ICP0 at ND10 in the absence of ND10-ES. (A and B) HEL cells were infected with RHG110 at 10 PFU/cell for 3 h. The infected cells were fixed and stained with polyclonal anti-PML and monoclonal anti-mCherry antibodies. (A) Representative images taken with a Zeiss 780 confocal microscope. Arrowheads point to the juxtaposition between ND10 and the ICP0 lacking ND10-ES. (B) Three-dimensional images of cells infected with RHG110, RHG120, or HSV-1(F). (C) HEL cells were mock infected or infected with 10 PFU/cell of recombinant viruses RHG101 or RHG110. At 8 h postinfection, total cell lysates were separated and blotted for proteins as indicated. (D) HEL cells were infected with RHG110 at 20 PFU/cell for 1.5 and 3 h. The infected cells were stained as described above. About 400 cells were tabulated for ICP0 subcellular distribution and categorized as cytoplasmic, juxtaposed to ND10, or dispersed throughout the nucleus. The percentage of each ICP0 distribution was then plotted with Microsoft Excel.
Fig 6.

Involvement of the ubiquitination-proteasome pathway in recruiting ICP0 to ND10. (A) Duplicated HEL cells were infected with RHG110 or RHG117 at 10 PFU/cell for 3 h. The infected cells were fixed and stained with either monoclonal anti-PML plus polyclonal anti-mCherry antibodies (a to f) or polyclonal anti-PML plus monoclonal anti-mCherry antibodies (g to l). Representative pictures of the infected cells were taken with a Zeiss 780 confocal microscope. (B) HEL cells were pretreated with 10 μM MG132 for 1 h before being infected with RHG110 or RHG117 at 10 PFU/cell for 3 h in the presence of MG132. The cells were then stained and photographed as described above. (C) HEL cells were infected with RHG118 and RHG119 at 10 PFU/cell for 3 h. The cells were stained and photographed as described above.
In the remaining 70% of RHG110-infected cells, ICP0 lacking the ND10-ES domain was diffused throughout the nucleus with an elevated expression level at early times postinfection (Fig. 5A, panels d to f). Quantitative assessment of RHG110-infected cells further revealed that prolonged infection time caused a decrease in the number of cells containing juxtaposed ICP0. In Fig. 5D, HEL cells infected with RHG110 at 20 PFU/cell were tabulated. At both 1.5 and 3 h postinfection, about one-half of the 400 cells counted in this study contained ICP0 at a detectable level. Within these infected cells, a decrease from 28% at 1.5 hpi to 17% at 3 hpi was observed for cells containing juxtaposed ICP0 (Fig. 5D). Further incubation by RHG110 at this multiplicity of infection showed extensive cytoplasmic localization of ICP0 (data not shown). These results suggest that the juxtaposition of ICP0 to the ND10 bodies is transient and can be observed only in a fraction of the infected cells shortly after the initiation of ICP0 accumulation under current sensitivity for detection.
ICP0 without ND10-ES in RHG110-infected cells did not degrade PML (Fig. 5C, lane 3) either because the domain of entry signal was required for the E3 ubiquitin ligase activity or because there was no contact between ICP0 adhering to the ND10 surface and PML in the interior of ND10 bodies.
The active RING finger domain stabilizes the adhesion of ICP0 at the ND10 surface in the absence of ND10-ES.
The striking differences in the subcellular distribution of mutant ICP0s in cells infected by RHG107 (Fig. 4A and B, panels g to i) and RHG110 (Fig. 5) indicated that not only is the ND10-ES within residues 245 to 474 important but the upstream sequence, including the RING finger, is also involved in recruiting ICP0 to ND10. When both N terminus and ND10-ES domains were deleted in RHG107, ICP0 completely lost its interaction with ND10, being dispersed throughout the nucleus with no aggregation observed (Fig. 4A and B, panels g to i). To determine whether the RING finger domain is involved in recruiting ICP0 to ND10, we constructed mutant virus RHG117 (Fig. 1B3), in which ICP0 is truncated at ND10-ES and at the same time carries the C116G and C156A substitutions as described above. In cells infected with RHG117, the mutant ICP0 was evenly distributed throughout the nucleus early in infection, an observation verified by reciprocal antibody staining (Fig. 6A, panels d to f and j to l). Cell tabulation showed that over 99% of cells infected by RHG117 contained dispersed ICP0, consistent with the results in cells infected by recombinant virus RHG107 (Fig. 4A and B, panels g to i). Therefore, a functional RING domain is necessary for ICP0 to adhere at the ND10 surface.
The importance of an active RING finger implicated a potential involvement of the ubiquitination-proteasome pathway in the ND10 recruitment of ICP0. To test this hypothesis, we conducted RHG110 infection in the presence of the proteasome inhibitor MG132. As shown in Fig. 6B, panels a to c, inhibition of the proteasome pathway dispersed ICP0 lacking ND10-ES in all RHG110-infected cells. In contrast, addition of MG132 had no effects on the subcellular distribution of ICP0 lacking ND10-ES but containing C116G/C156A mutations (Fig. 6B, panels d to f). The fact that MG132 treatment alters the distribution of ICP0 truncation indicates that the ND10 juxtaposition of ICP0 lacking ND10-ES is not simply the result of aggregation of misfolded proteins.
Based on the studies described above, we conclude that in the absence of ND10-ES, the ubiquitin-proteasome pathway is required to stabilize the transient adhesion of ICP0 at the ND10 surface. Either inactivation of the ICP0 RING finger or inhibition of proteasome causes ICP0 lacking ND10-ES to lose its ability to aggregate at the ND10 surface and results in the failure to recruit the truncated ICP0 to ND10. In the presence of ND10-ES, ICP0 entered ND10 regardless of the E3 ligase activity (Fig. 2C and 4A and B, panels a to f), suggesting that the ND10-ES of ICP0 is a dominant driving force for recruitment of ICP0 into ND10 bodies. A transient adhesion process of ICP0 at the ND10 surface can be captured by fluorescence staining only when ND10-ES is deleted (Fig. 5A and B).
The first 83 amino acids do not play a role in ICP0 recruitment to ND10.
As shown in Fig. 4A and B, panels a to c, we found that deletion of the first 83 amino acids of ICP0 in RHG105 had no effects on ICP0 localization at ND10. In order to determine whether the dominant entry force of ND10-ES obscured any potential domains residing within residues 1 to 83, we constructed recombinant virus RHG118, in which both N-terminal residues 1 to 83 and the ND10-ES domain were deleted (Fig. 1B3). As shown in Fig. 6C, panels a to c, ICP0 was juxtaposed to the ND10 bodies, exhibiting the same distribution pattern as observed in RHG110-infected cells (Fig. 6A, panels a to c and g to i). Consistent with this observation, additional point mutations of C116G and C156A in the RHG119 virus caused dispersal of ICP0 (Fig. 6C, panels d to f), just like what the mutations did to RHG117 virus (Fig. 6A, panels d to f and j to l). These results and the results from RHG105 (Fig. 4A and B, panels a to c) led to a conclusion that the first 83 amino acids are dispensable in the recruitment of ICP0 to ND10 bodies.
Furthermore, RHG110, RHG117, RHG118, and RHG119 are recombinant viruses independently developed by the BAC system. The consistent results from comparison of RHG110 to RHG118 and of RHG117 to RHG119 validated our conclusion that ND10-ES is required for ICP0 to comingle with ND10 components whereas a functional RING finger stabilizes the adhesion of ICP0 at the ND10 surface in the absence of ND10-ES.
Failure of releasing ICP0 from ND10 severely blocks viral replication.
To further investigate the effects of ND10 fusion and ND10 retention on viral replication, we performed one-step growth curve analysis for mutant viruses RHG101, RHG120, RHG110, RHG117, RHG103, and RHG108 (Fig. 7A). In cultures exposed to 0.1 PFU/cell (Fig. 7A), the replication of RHG101 appeared to be comparable to that of the wild-type HSV-1(F), as we reported elsewhere (3), suggesting that the insertion of the mCherry tag at the N terminus of ICP0 and the disruption of thymidine kinase gene by BAC did not affect viral replication in cultured cells (solid line with diamond). Consistent with the previous report (44), deletion of the C terminus of ICP0 in RHG103 had a <10-fold decrease in viral yields (Fig. 7A, solid line with triangle). For RHG108, the C156G/C156A substitutions in addition to the C-terminal deletion further impaired viral replication (dashed line with triangle), causing an ∼100-fold decrease compared to RHG101. The deletion of ND10-ES in recombinant virus RHG110 rendered a larger delay in the first 24 h, generating a >100-fold reduction in viral yields compared to RHG101 (solid line with circle). The delays in replication of RHG103, RHG108, and RHG110 were overcome between 24 to 48 h, with viral titers rising to levels comparable to those of RHG101 at 48 h postinfection. Additional C116G/C156A mutations in recombinant virus RHG117 further decreased viral yields to about 1,000-fold at 24 h, and the delay was sustained at 48 h (dashed line with circle). Interestingly, RHG120, which carries the C116G/C156A substitutions in full-length ICP0, generated the least viral growth within the first 24 h (dashed line with diamond). At 24 h postinfection, RHG120 produced 10-fold fewer infectious particles than RHG117, which carries the same mutations but is truncated at ND10-ES, and 100-fold fewer than RHG108, which carries the same mutations but is truncated at the retention sequence. To confirm these results, we performed quantitative PCR to directly measure DNA replication. As shown in Fig. 7C, within 24 h after infection, the viral DNA fold increase in cells infected with 0.1 PFU/cell of RHG120 was about 5 times less than that of RHG117 and was 45 times less than that of RHG108, suggesting that RHG120 had the least DNA replication. Figure 7D shows the relative cycle numbers of input viral DNAs normalized to cellular 18S rDNA. The lesser ΔCT value of RHG120, which reflects the presence of more viral DNA at initial infection, is likely due to a higher particle-to-PFU ratio in RHG120 replication. All recombinant viral stocks studied here are generated in U2OS cells under the same culture conditions. The higher DNA-to-PFU ratio again reflects the severe defect of RHG120 replication, even in U2OS cells.
Fig 7.
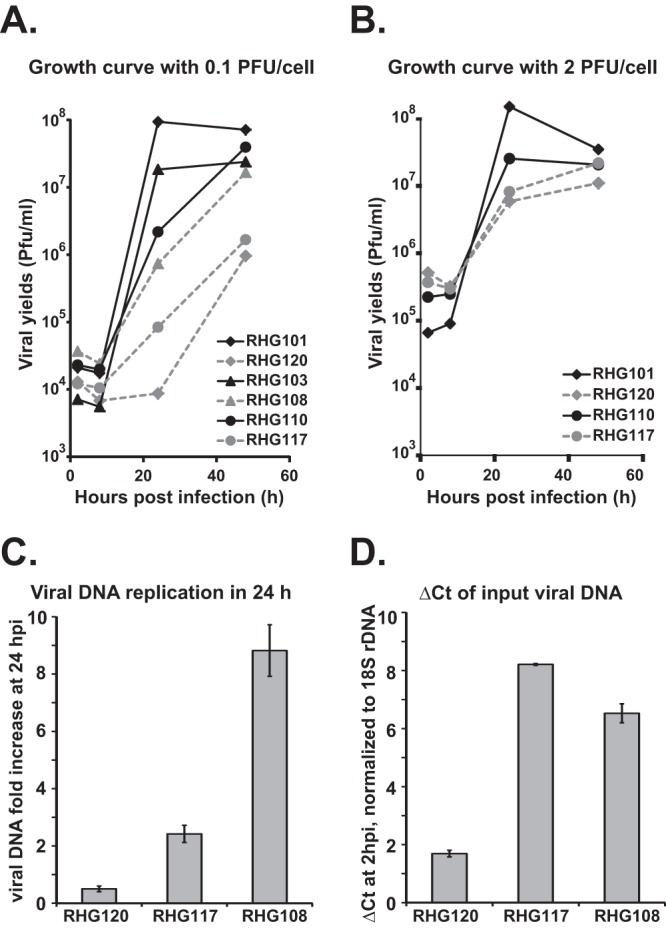
Viral replication of mutant viruses defective in ND10 retention or ND10 fusion. (A and B) One-step growth curves for the mutant viruses indicated. HEL cells were infected with the indicated viruses at either 0.1 PFU/cell (A) or 2 PFU/cell (B). At 2, 8, 24, and 48 hours postinfection, cells were harvested and lysed for titration. The viral yields were plotted against infection times using Microsoft Excel. (C and D) HEL cells were infected with the indicated viruses at 0.1 PFU/cell. At 2 and 24 h postinfection, total DNAs were extracted and comparative qPCR was performed using primers targeting ICP27 or 18S rDNA. Viral DNA fold increases of the 24-h samples relative to the 2-h samples were plotted by the ΔΔCT method (C). The cycle numbers for 2-h viral DNA normalized against 18S rDNA (ΔCT) were plotted (D).
The above results suggest that RING domain mutations C116G/C156A had detrimental effects on HSV-1 replication, which can be explained by two potential mechanisms: (i) ICP0 carrying an inactive RING finger is unable to degrade cellular restrictive proteins, including PML; and (ii) mutated ICP0 is completely sequestered at ND10 and unable to carry out additional functions designated to occur outside ND10. Our observations that among the viruses RHG120, RHG117, and RHG108, all of which contain the C116G/C156A mutations and are incapable of PML degradation, the ones that are defective in ND10 recruitment (RHG117 [Fig. 6A]) or retention (RHG108 [Fig. 2C]) replicated better support the second scenario. Theoretically, protein sequestration can be overcome by saturating the sequestering factor(s). To test whether we can eliminate the difference in RHG117 and RHG120 replications by accelerating ICP0 expression, we performed growth curve analysis with cultures exposed to 2 PFU/cell of viruses. We found that with the increased multiplicity of infection the difference between RHG120 and RHG117 replications was greatly reduced (Fig. 7B), suggesting that the elements restricting RHG120 replication can be saturated by increasing the multiplicity of infection.
Compared to viruses containing the wild-type RING finger (RHG101, RHG103, and RHG110), replications of counterpart viruses lacking the RING finger (RHG120, RHG108, and RHG117) had less replication at both 0.1 and 2 PFU/cell, suggesting that the E3 ligase activity is the most important function of ICP0. However, additional functions of ICP0 occurring outside the ND10 structures also greatly affect the overall outcome of HSV-1 infection. Defect in ICP0 recruitment led to complete diffusion of ICP0 throughout the nucleus (Fig. 6A, panels d to f), whereas defect in ICP0 retention caused partial diffusion of ICP0 (Fig. 2C, panels g to i). In both cases, the incomplete ND10 sequestration partially reversed the adverse effects of the mutant RING finger on viral replication, suggesting that functions in addition to the E3 ligase activity rely heavily on the ability of ICP0 to be freed from the ND10 restraint.
DISCUSSION
The objective of this study is to define the interaction between ICP0 and ND10 nuclear bodies and the biological outcome of this interaction. We conducted deletion mapping in the context of HSV-1 infection, which allows us to study the ICP0-ND10 interaction with biological relevance and to dissect the dynamic process in infected cells. Truncations and mutations of ICP0 blocked some of the dynamic interactions in their transitional phases and revealed three consecutive steps in ICP0-ND10 association that are otherwise too transient to be observed. These steps are adhesion, fusion, and retention, as shown in Fig. 8A.
Fig 8.

ICP0-ND10 interaction is a sequential process of adhesion, fusion, and retention. (A) Wild-type ICP0 adheres to the surface of ND10. This is a transient process that quickly leads to the fusion of ICP0 to ND10 bodies. PML and Sp100 are degraded, while ICP0 is retained at ND10. ND10 is subsequently dispersed. (B) ICP0 lacking the retention sequence adheres to the surface of ND10 and also rapidly fuses with ND10. The lack of retention sequence leads to accelerated cycling of ICP0 in and out of ND10. PML can be degraded without retention. (C) ICP0 lacking the entry signal adheres to the surface of ND10. The RING domain of ICP0 interacts with an unknown surface component(s) of ND10 in a proteasome-dependent manner. This interaction is unstable, and ICP0 is released without degrading PML.
The consecutive steps of adhesion and fusion occur so rapidly that only the fused ICP0, or ICP0-ND10 colocalization, has been reported in the literature thus far (41). We identified the ND10-ES domain located between residues 245 and 474 that is responsible for the adhered ICP0 comingling with ND10 components. Deletion of ND10-ES blocks the fusion of ICP0 with ND10, which prolongs the duration of adhesion so that ICP0 juxtaposed to ND10 is observed (Fig. 8C). The affinity of ICP0 lacking ND10-ES to the ND10 surface is not very strong. Only a fraction of mutant ICP0 actually decorates the surface of ND10 bodies, whereas prolonged infection leads to less ICP0 juxtaposing to the ND10 surface. This suggests that the truncated ICP0 excluded from entering ND10 lingers at the surface only briefly and is disseminated quickly afterwards. Since the infection process is not synchronized from cell to cell, about one-third of the infected cells are captured with juxtaposed ICP0 under current experimental conditions and sensitivity.
An active RING domain is essential for the adhesion of ICP0 lacking ND10-ES at the ND10 surface, whereas MG132 treatment also abolishes the association, suggesting that some part of the proteasome pathway is required for the transient adhesion. The fact that MG132 treatment completely disaggregates the internal truncation indicates that adhesion of ICP0 to the ND10 surface is not simply the result of protein misfolding. The truncated ICP0 also undergoes normal nuclear import at early infection and translocates to the cytoplasm in late infection just like the wild-type ICP0, suggesting that in the absence of ND10-ES certain parts of ICP0 maintain regular function to sustain the cellular trafficking. Cuchet-Lourenco et al. recently reported a direct interaction between ICP0 241 to 388 and PML isoform I in a yeast 2-hybrid system (52), which coincides with ND10-ES described in our study. However, in the infection context they reported ICP0 interacting with ectopic PML I only in cells depleted of all endogenous PML isoforms but not in regular cell cultures (52), which implies that in the absence of other PML isoforms the biochemical and functional properties of ectopic PML I have likely changed. Therefore, additional studies are needed to verify whether PML I interacts with ICP0 under natural conditions and whether PML I is the cellular factor important for ICP0 for entry into ND10.
Everett et al. has previously reported the juxtaposition of ICP4 and PML in the live imaging of HSV-1-infected cells (53), suggesting a potential juxtaposition of viral DNA to ND10 early in infection. Our results clearly indicated a transient state in which ICP0 also attaches to the surface of ND10. The relative interactions among viral DNA, ICP4, and ICP0 at the surface of ND10 are not clear at this point. The involvement of the proteasome pathway in stabilizing the surface interaction of ICP0 lacking ND10-ES certainly raises interesting questions, such as what might be ubiquitinated or degraded at the ND10 surface and the significance of modifying these surface proteins in the interaction between viral DNA and ND10 nuclear bodies.
At first glance, ICP0 mutants lacking the C termini appear as if they do not localize to ND10. In fact, the C-terminal third of ICP0 has been described as a sequence required for ND10 localization (44, 52). In our study, we found that ICP0 mutants containing C-terminal truncation as well as the C116G/C156A substitutions are capable of being recruited to and sequestered at ND10. We postulate that the RING finger mutation traps ICP0 truncation in the enzymatic transition state and blocks it from cycling out of the ND10 bodies. Interestingly, Cuchet-Lourenco et al. also demonstrated that ectopic PML isoform I partially restored the association of a C-terminal truncation at ND10 when all endogenous PML isoforms were knocked down (52). The fact that interaction between the endogenous PML I and C-terminal truncation in a normal infection, if it exists, is not enough to retain the truncation at ND10 (Fig. 2B and reference 44) implies that the phenomenon described by Cuchet-Lourenco et al. is likely the result of sequestration of an enzymatic transition state caused by an impeded E3 reaction, the consequence of a changed biochemical property of ectopic PML I in the absence of other PML isoforms.
Therefore, we conclude that there is a dynamic equilibrium between ICP0 contained in the ND10 bodies and that in the nucleoplasm. The function of the C terminus is to retain ICP0 at ND10 for a certain duration, likely to engage in additional action(s) other than the PML degradation. Loss of the C terminus abrogates that function and accelerates the cycling of ICP0 in and out of the ND10 bodies, as shown in Fig. 8B. Our observation that a shortened sojourn of ICP0 inside ND10 does not substantially change the rate of PML degradation suggests that the dynamic cycling of ICP0 in and out of ND10 does not affect substrate recognition and binding of ICP0 when it comes to PML.
As shown in Fig. 1A, the retention domain coincides with the CoREST interaction motif (46). We have previously reported that CoREST is recruited to ND10 during infection (23), whereas CoREST-ICP0 interaction is critical for dislodging HDAC-1 and -2 from the REST/CoREST/HDAC repressor complex (2, 46). It will be worthy of investigation to find out whether ICP0 finds and interacts with CoREST inside the ND10 nuclear bodies for derepression. The ND10 retention domain also overlaps the ICP0 dimerization domain located between amino acids 633 and 775 (44). Previous reports have demonstrated that ICP0 dimerization alters the cellular distribution of ectopically expressed ICP0 in transfected cells (54, 55). It needs to be verified whether the dimerization exists in infected cells and whether it plays a role in ICP0-ND10 association.
By homologous analysis, Boutell et al. identified seven SUMO interaction motif-like sequences (SLS) in ICP0. ICP0 was demonstrated to interact with SUMO 2/3 but not SUMO 1 in yeast 2-hybrid analysis. Among the most conserved SLSs, mutations in SLS4 completely disrupted the interaction of ICP0 with SUMO 2/3 in vitro, whereas mutations in SLS5 and SLS7 had no effect (56). SLS4 is located in the middle of ND10-ES. The involvement of SUMO interaction in the ND10 entry of ICP0 remains an important question to be answered.
In wild-type HSV-1 infection, ICP0 is released from ND10 after the ND10 dispersal. Our results suggest that this release is essential for viral replication. The RING finger mutation of RHG120, of which the ND10 sequestration of ICP0 is more thorough than those with defects in either recruitment or retention, has the most detrimental effect on overall viral production. The release of ICP0 indicated that additional functions carried out in the nucleus or in the cytoplasm are also important for a full-blown infection.
ND10 and its organizer PML have been a hot spot for viral oncogenic studies (57–61). As a tumor suppressor gene, PML deficiency and subsequent ND10 loss have been observed in many cancer types, including viral-infection-related cancers. For example, PML is downregulated in cervical carcinogenesis caused by human papillomavirus (58) and gastric carcinoma associated with Epstein-Barr virus (EBV) (61). HSV infection in a healthy individual establishes latency and is sporadically reactivated to cause recurrent skin lesions. HSV has not been considered a tumorigenic virus until recently, partly due to the extensive PML degradation caused by HSV infection. A few studies have been conducted to assess the cancer risk for HSV patients, and mild increases of oral cancer have been linked to HSV infection (62, 63). Unlike the nuclear antigen EBNA1 of EBV, which is constantly expressed in latent EBV infection, ICP0 expression cannot be detected during the latent infection of HSV. However, frequent HSV reactivation, especially in immunosuppressed patients, may increase the chance of constant PML deficiency and potentially the risk of cancer.
ACKNOWLEDGMENTS
These studies were supported by National Cancer Institute grant CA78766, awarded to Bernard Roizman at University of Chicago, and by the Wayne State University Startup fund, awarded to Haidong Gu.
We thank Anura Shrivastava for her participation in viral titration. We thank the MICR Core facility at Wayne State University for technical support on confocal imaging.
Footnotes
Published ahead of print 17 July 2013
REFERENCES
- 1. Bieniasz PD. 2004. Intrinsic immunity: a front-line defense against viral attack. Nat. Immunol. 5:1109–1115 [DOI] [PubMed] [Google Scholar]
- 2. Gu H, Liang Y, Mandel G, Roizman B. 2005. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc. Natl. Acad. Sci. U. S. A. 102:7571–7576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gu H, Roizman B. 2007. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc. Natl. Acad. Sci. U. S. A. 104:17134–17139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ferenczy MW, Ranayhossaini DJ, Deluca NA. 2011. Activities of ICP0 involved in the reversal of silencing of quiescent herpes simplex virus 1. J. Virol. 85:4993–5002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. 2009. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat. Med. 15:1312–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lukashchuk V, Everett RD. 2010. Regulation of ICP0-null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx. J. Virol. 84:4026–4040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Glass M, Everett RD. 2013. Components of PML Nuclear Bodies (ND10) act cooperatively to repress herpesvirus infection. J. Virol. 87:2174–2185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bernardi R, Pandolfi PP. 2003. Role of PML and the PML-nuclear body in the control of programmed cell death. Oncogene 22:9048–9057 [DOI] [PubMed] [Google Scholar]
- 9. Pearson M, Pelicci PG. 2001. PML interaction with p53 and its role in apoptosis and replicative senescence. Oncogene 20:7250–7256 [DOI] [PubMed] [Google Scholar]
- 10. Mallette FA, Goumard S, Gaumont-Leclerc MF, Moiseeva O, Ferbeyre G. 2004. Human fibroblasts require the Rb family of tumor suppressors, but not p53, for PML-induced senescence. Oncogene 23:91–99 [DOI] [PubMed] [Google Scholar]
- 11. Tavalai N, Stamminger T. 2008. New insights into the role of the subnuclear structure ND10 for viral infection. Biochim. Biophys. Acta 1783:2207–2221 [DOI] [PubMed] [Google Scholar]
- 12. Bernardi R, Papa A, Pandolfi PP. 2008. Regulation of apoptosis by PML and the PML-NBs. Oncogene 27:6299–6312 [DOI] [PubMed] [Google Scholar]
- 13. Bernardi R, Pandolfi PP. 2007. Structure, dynamics and functions of promyelocytic leukaemia nuclear bodies. Nat. Rev. Mol. Cell Biol. 8:1006–1016 [DOI] [PubMed] [Google Scholar]
- 14. Geoffroy MC, Chelbi-Alix MK. 2011. Role of promyelocytic leukemia protein in host antiviral defense. J. Interferon Cytokine Res. 31:145–158 [DOI] [PubMed] [Google Scholar]
- 15. Chelbi-Alix MK, Pelicano L, Quignon F, Koken MH, Venturini L, Stadler M, Pavlovic J, Degos L, de Thé H. 1995. Induction of the PML protein by interferons in normal and APL cells. Leukemia 9:2027–2033 [PubMed] [Google Scholar]
- 16. Stadler M, Chelbi-Alix MK, Koken MH, Venturini L, Lee C, Saïb A, Quignon F, Pelicano L, Guillemin MC, Schindler C, de Thé H. 1995. Transcriptional induction of the PML growth suppressor gene by interferons is mediated through an ISRE and a GAS element. Oncogene 11:2565–2573 [PubMed] [Google Scholar]
- 17. Regad T, Saib A, Lallemand-Breitenbach V, Pandolfi PP, de Thé H, Chelbi-Alix MK. 2001. PML mediates the interferon-induced antiviral state against a complex retrovirus via its association with the viral transactivator. EMBO J. 20:3495–3505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chee AV, Lopez P, Pandolfi PP, Roizman B. 2003. Promyelocytic leukemia protein mediates interferon-based anti-herpes simplex virus 1 effects. J. Virol. 77:7101–7105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Maul GG, Ishov AM, Everett RD. 1996. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology 217:67–75 [DOI] [PubMed] [Google Scholar]
- 20. Everett RD, Murray J, Orr A, Preston CM. 2007. Herpes simplex virus type 1 genomes are associated with ND10 nuclear substructures in quiescently infected human fibroblasts. J. Virol. 81:10991–11004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kalamvoki M, Roizman B. 2008. Nuclear retention of ICP0 in cells exposed to HDAC inhibitor or transfected with DNA before infection with herpes simplex virus 1. Proc. Natl. Acad. Sci. U. S. A. 105:20488–20493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Newhart A, Rafalska-Metcalf IU, Yang T, Negorev DG, Janicki SM. 2012. Single cell analysis of Daxx and ATRX-dependent transcriptional repression. J. Cell Sci. 125:5489. 10.1242/jcs.110148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gu H, Roizman B. 2009. Engagement of the lysine-specific demethylase/HDAC1/CoREST/REST complex by herpes simplex virus 1. J. Virol. 83:4376–4385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kalamvoki M, Roizman B. 2010. Circadian CLOCK histone acetyl transferase localizes at ND10 nuclear bodies and enables herpes simplex virus gene expression. Proc. Natl. Acad. Sci. U. S. A. 107:17721–17726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Roizman B, Knipe DM, Whitley RJ. 2007. Herpes simplex viruses, p 2501–2601 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 26. Lilley CE, Chaurushiya MS, Boutell C, Everett RD, Weitzman MD. 2011. The intrinsic antiviral defense to incoming HSV-1 genomes includes specific DNA repair proteins and is counteracted by the viral protein ICP0. PLoS Pathog. 7:e1002084. 10.1371/journal.ppat.1002084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Boutell C, Sadis S, Everett RD. 2002. Herpes simplex virus type 1 immediate-early protein ICP0 and its isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 76:841–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hagglund R, Van Sant C, Lopez P, Roizman B. 2002. Herpes simplex virus 1-infected cell protein 0 contains two E3 ubiquitin ligase sites specific for different E2 ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. U. S. A. 99:631–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Everett RD, Freemont P, Saitoh H, Dasso M, Orr A, Kathoria M, Parkinson J. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 72:6581–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chelbi-Alix MK, de The H. 1999. Herpes virus induced proteasome-dependent degradation of the nuclear bodies-associated PML and Sp100 proteins. Oncogene 18:935–941 [DOI] [PubMed] [Google Scholar]
- 31. Gross S, Catez F, Masumoto H, Lomonte P. 2012. Centromere architecture breakdown induced by the viral E3 ubiquitin ligase ICP0 protein of herpes simplex virus type 1. PLoS One 7:e44227. 10.1371/journal.pone.0044227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Everett RD, Earnshaw WC, Findlay J, Lomonte P. 1999. Specific destruction of kinetochore protein CENP-C and disruption of cell division by herpes simplex virus immediate-early protein Vmw110. EMBO J. 18:1526–1538 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lees-Miller SP, Long MC, Kilvert MA, Lam V, Rice SA, Spencer CA. 1996. Attenuation of DNA-dependent protein kinase activity and its catalytic subunit by the herpes simplex virus type 1 transactivator ICP0. J. Virol. 70:7471–7477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Orzalli MH, DeLuca NA, Knipe DM. 2012. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. U. S. A. 109:E3008–E3017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Boutell C, Canning M, Orr A, Everett RD. 2005. Reciprocal activities between herpes simplex virus type 1 regulatory protein ICP0, a ubiquitin E3 ligase, and ubiquitin-specific protease USP7. J. Virol. 79:12342–12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Daubeuf S, Singh D, Tan Y, Liu H, Federoff HJ, Bowers WJ, Tolba K. 2009. HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood 113:3264–3275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Chaurushiya MS, Lilley CE, Aslanian A, Meisenhelder J, Scott DC, Landry S, Ticau S, Boutell C, Yates JR, Schulman BA, Hunter T, Weitzman MD. 2012. Viral E3 ubiquitin ligase-mediated degradation of a cellular E3: viral mimicry of a cellular phosphorylation mark targets the RNF8 FHA domain. Mol. Cell 46:79–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Van Sant C, Kawaguchi Y, Roizman B. 1999. A single amino acid substitution in the cyclin D binding domain of the infected cell protein no. 0 abrogates the neuroinvasiveness of herpes simplex virus without affecting its ability to replicate. Proc. Natl. Acad. Sci. U. S. A. 96:8184–8189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kawaguchi Y, Van Sant C, Roizman B. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J. Virol. 71:7328–7336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kawaguchi Y, Bruni R, Roizman B. 1997. Interaction of herpes simplex virus 1 alpha regulatory protein ICP0 with elongation factor 1delta: ICP0 affects translational machinery. J. Virol. 71:1019–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maul GG, Everett RD. 1994. The nuclear location of PML, a cellular member of the C3HC4 zinc-binding domain protein family, is rearranged during herpes simplex virus infection by the C3HC4 viral protein ICP0. J. Gen. Virol. 75:1223–1233 [DOI] [PubMed] [Google Scholar]
- 42. Maul GG, Guldner HH, Spivack JG. 1993. Modification of discrete nuclear domains induced by herpes simplex virus type 1 immediate early gene 1 product (ICP0). J. Gen. Virol. 74:2679–2690 [DOI] [PubMed] [Google Scholar]
- 43. Everett RD, Maul GG. 1994. HSV-1 IE protein Vmw110 causes redistribution of PML. EMBO J. 13:5062–5069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Meredith M, Orr A, Elliott M, Everett RD. 1995. Separation of sequence requirements for HSV-1 Vmw110 multimerisation and interaction with a 135-kDa cellular protein. Virology 209:174–187 [DOI] [PubMed] [Google Scholar]
- 45. Gu H, Poon AP, Roizman B. 2009. During its nuclear phase the multifunctional regulatory protein ICP0 undergoes proteolytic cleavage characteristic of polyproteins. Proc. Natl. Acad. Sci. U. S. A. 106:19132–19137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Gu H, Roizman B. 2009. The two functions of herpes simplex virus 1 ICP0, inhibition of silencing by the CoREST/REST/HDAC complex and degradation of PML, are executed in tandem. J. Virol. 83:181–187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Zhu XX, Chen JX, Silverstein S. 1991. Isolation and characterization of a functional cDNA encoding ICP0 from herpes simplex virus type 1. J. Virol. 65:957–960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lopez P, Van Sant C, Roizman B. 2001. Requirements for the nuclear-cytoplasmic translocation of infected-cell protein 0 of herpes simplex virus 1. J. Virol. 75:3832–3840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Horsburgh BC, Hubinette MM, Tufaro F. 1999. Genetic manipulation of herpes simplex virus using bacterial artificial chromosomes. Methods Enzymol. 306:337–352 [DOI] [PubMed] [Google Scholar]
- 50. Lium EK, Silverstein S. 1997. Mutational analysis of the herpes simplex virus type 1 ICP0 C3HC4 zinc ring finger reveals a requirement for ICP0 in the expression of the essential alpha27 gene. J. Virol. 71:8602–8614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mullen MA, Ciufo DM, Hayward GS. 1994. Mapping of intracellular localization domains and evidence for colocalization interactions between the IE110 and IE175 nuclear transactivator proteins of herpes simplex virus. J. Virol. 68:3250–3266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cuchet-Lourenco D, Vanni E, Glass M, Orr A, Everett RD. 2012. Herpes simplex virus 1 ubiquitin ligase ICP0 interacts with PML isoform I and induces its SUMO-independent degradation. J. Virol. 86:11209–11222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Everett RD, Sourvinos G, Orr A. 2003. Recruitment of herpes simplex virus type 1 transcriptional regulatory protein ICP4 into foci juxtaposed to ND10 in live, infected cells. J. Virol. 77:3680–3689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lium EK, Panagiotidis CA, Wen X, Silverstein SJ. 1998. The NH2 terminus of the herpes simplex virus type 1 regulatory protein ICP0 contains a promoter-specific transcription activation domain. J. Virol. 72:7785–7795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ciufo DM, Mullen MA, Hayward GS. 1994. Identification of a dimerization domain in the C-terminal segment of the IE110 transactivator protein from herpes simplex virus. J. Virol. 68:3267–3282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Boutell C, Cuchet-Lourenco D, Vanni E, Orr A, Glass M, McFarlane S, Everett RD. 2011. A viral ubiquitin ligase has substrate preferential SUMO targeted ubiquitin ligase activity that counteracts intrinsic antiviral defence. PLoS Pathog. 7:e1002245. 10.1371/journal.ppat.1002245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Frappier L. 2012. Contributions of Epstein-Barr nuclear antigen 1 (EBNA1) to cell immortalization and survival. Viruses 4:1537–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Singh N, Sobti RC, Suri V, Nijhawan R, Sharma S, Das BC, Bharadwaj M, Hussain S. 2013. Downregulation of tumor suppressor gene PML in uterine cervical carcinogenesis: impact of human papillomavirus infection (HPV). Gynecol. Oncol. 128:420–426 [DOI] [PubMed] [Google Scholar]
- 59. Chung YL, Wu ML. 2013. Promyelocytic leukemia protein links DNA damage response and repair to hepatitis B virus related hepatocarcinogenesis. J. Pathol. 10.1002/path.4195 [DOI] [PubMed] [Google Scholar]
- 60. Rabellino A, Scaglioni PP. 2013. PML degradation: multiple ways to eliminate PML. Front. Oncol. 3:60. 10.3389/fonc.2013.00060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sivachandran N, Dawson CW, Young LS, Liu FF, Middeldorp J, Frappier L. 2012. Contributions of the Epstein-Barr virus EBNA1 protein to gastric carcinoma. J. Virol. 86:60–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Sheu JJ, Keller JJ, Lin HC. 2012. Increased risk of cancer after Bell's palsy: a 5-year follow-up study. J. Neurooncol. 110:215–220 [DOI] [PubMed] [Google Scholar]
- 63. Jalouli J, Jalouli MM, Sapkota D, Ibrahim SO, Larsson PA, Sand L. 2012. Human papilloma virus, herpes simplex virus and Epstein-Barr virus in oral squamous cell carcinoma from eight different countries. Anticancer Res. 32:571–580 [PubMed] [Google Scholar]