

Abstract
Respiratory syncytial virus (RSV) is the leading cause of death due to a viral etiology in infants. RSV disease is characterized by epithelial desquamation, neutrophilic bronchiolitis and pneumonia, and obstructive pulmonary mucus. It has been shown that infection of BALB/cJ mice with RSV clinical isolate A2001/2-20 (2-20) results in a higher early viral load, greater airway necrosis, and higher levels of interleukin-13 (IL-13) and airway mucin expression than infection with RSV laboratory strain A2. We hypothesized that the fusion (F) protein of RSV 2-20 is a mucus-inducing viral factor. In vitro, the fusion activity of 2-20 F but not that of A2 F was enhanced by expression of RSV G. We generated a recombinant F-chimeric RSV by replacing the F gene of A2 with the F gene of 2-20, generating A2–2-20F. Similar to the results obtained with the parent 2-20 strain, infection of BALB/cJ mice with A2–2-20F resulted in a higher early viral load and higher levels of subsequent pulmonary mucin expression than infection with the A2 strain. A2–2-20F infection induced greater necrotic airway damage and neutrophil infiltration than A2 infection. We hypothesized that the neutrophil response to A2–2-20F infection is involved in mucin expression. Antibody-mediated depletion of neutrophils in RSV-infected mice resulted in lower tumor necrosis factor alpha levels, fewer IL-13-expressing CD4 T cells, and less airway mucin production in the lung. Our data are consistent with a model in which the F and attachment (G) glycoprotein functional interaction leads to enhanced fusion and F is a key factor in airway epithelium infection, pathogenesis, and subsequent airway mucin expression.
INTRODUCTION
Respiratory syncytial virus (RSV) is the most important cause of bronchiolitis and viral pneumonia in infants and the most common cause of hospitalization in infants younger than 1 year of age (1). RSV infects practically all children by the age of 2 years, causing approximately 400 infant deaths and up to 125,000 hospitalizations per year in the United States (2, 3). Worldwide, RSV is the leading cause of death due to a viral etiology in children younger than 1 year of age (3). RSV infection results in airway obstruction and lung inflammation, which lead to hyperresponsiveness involving leukocyte infiltration, mucus production, and epithelial damage (4). The virus targets the epithelial cells of the nasopharynx, trachea, and bronchi, resulting in the formation of thick mucus plugs mixed with cell debris, fibrin, and lymphocytes (5). An understanding of how RSV causes the epithelial damage and mucus production that lead to lung dysfunction may aid with the development of approaches to prevent severe respiratory illness.
Excess neutrophils are found in the airways of infants with acute RSV bronchiolitis as well as in the bronchoalveolar lavage (BAL) fluid of RSV-infected children (6–9). However, the precise role of neutrophils in RSV pathogenesis is unknown. Neutrophils recruited to airways cause epithelial damage through mechanisms such as the release of free oxygen radicals, neutrophil elastase, and proteolytic enzymes (10). In vitro, neutrophils can enhance RSV-induced epithelial damage and detachment of cells (11). It is not known whether this occurs in vivo. Furthermore, neutrophils are a major source of tumor necrosis factor alpha (TNF-α). Intratracheal administration of TNF-α in BALB/cJ mice induces mucus-associated proteins, such as gob5 and Muc5ac, as well as mucus expression in the airways (12).
We previously reported that infection of BALB/cJ mice with clinical strain RSV A2001/2-20 (2-20) results in greater disease severity and higher lung interleukin-13 (IL-13) levels than infection with laboratory strain A2 (13). Another laboratory RSV strain, line 19, also induces airway mucin expression in an IL-13-dependent manner, similar to RSV 2-20 (14, 15). RSV 2-20 is useful because it is a clinical isolate of known passage history (13). A chimeric virus containing the line 19 fusion (F) protein in the A2 genetic background indicated that the fusion protein of RSV plays a role in pulmonary mucus expression in the mouse model (16). Interestingly, the unique residues of the line 19 F protein do not occur in RSV clinical isolates, suggesting that line 19 may have acquired mutations during passage (A. L. Hotard and M. L. Moore, unpublished data).
We hypothesized that the F protein of 2-20 also plays a role in RSV-induced mucus production. In order to test this, we generated a chimeric virus strain containing the RSV 2-20 F gene in an RSV A2 backbone (referred to as strain A2–2-20F) and employed antibody depletion to further probe the role of neutrophils in RSV pathogenesis. Infection of BALB/cJ mice with chimeric virus strain A2–2-20F caused levels of early airway epithelium necrosis and neutrophil infiltration as well as pulmonary mucus expression higher than those caused by RSV A2. Neutrophil depletion caused diminished airway mucin expression and TNF-α production during 2-20 infection. Our results elucidate a novel pathway of RSV pathogenesis whereby the F protein results in airway cell debris and the ensuing robust neutrophil response mediates Th2 cell IL-13 expression and airway mucin expression. Mechanistic characterization identified a unique functional interaction between the RSV attachment (G) glycoprotein and F protein that may be important for RSV fusion and pathogenesis.
MATERIALS AND METHODS
Cells, viruses, and mice.
HEp-2 cells were obtained from the ATCC and propagated in minimal essential medium (MEM) supplemented with 10% fetal bovine serum (FBS) and penicillin G-streptomycin sulfate-amphotericin B solution (Mediatech, Manassas, VA). 293T cells were obtained from the ATCC and propagated in MEM supplemented with 10% FBS and penicillin G-streptomycin sulfate-amphotericin B solution. The A2 strain of RSV was provided by Stokes Peebles (Vanderbilt, Nashville, TN) and maintained by passage in HEp-2 cells. The 2-20 RSV strain was maintained by passage in HEp-2 cells (13). Viral stocks were propagated and titrated by plaque assay in HEp-2 cells as described previously (13). Female 6-week-old BALB/cJ mice were obtained from Jackson Laboratories (Bar Harbor, ME). All mice were maintained under specific-pathogen-free conditions. Mice were anesthetized by intramuscular injection of a ketamine-xylazine solution and infected intranasally (i.n.) with RSV stock or with mock-infected HEp-2 cell culture supernatant, as described previously (17). All animal procedures were conducted according to the guidelines of the Emory University Institutional Animal Care and Use Committee.
Generation and recovery of chimeric A2–2-20F RSV.
Total RNA was isolated from HEp-2 cells infected with RSV strain 2-20. cDNA was reverse transcribed as described previously using primer F-r (16). PCR amplicons were gel purified and sequenced. The 2-20 F cDNA sequence from three separate infections was compared to the published 2-20 F sequence and confirmed (13). The F cDNA was then PCR amplified using forward primer FStuI and reverse primer FSphI, which incorporated StuI and SphI restriction sites into the G-F intergenic region and F-M2 intergenic region, respectively, and added flanking EcoRI sites, as described previously (16). The 2-20 F cDNA was cloned into the EcoRI site of pGEM-9Zf (Promega, Madison WI) and then moved, using StuI and SphI, into a low-copy-number subclone (pLG4) harboring an XhoI/BamHI fragment of RSV antigenomic cDNA clone D46/6120 containing the RSV A2 G gene and the F gene flanked by StuI and SphI and the partial M2 sequence of RSV, thereby replacing the A2 F gene (16). The XhoI/BamHI fragment containing 2-20 F was then cloned into RSV antigenomic cDNA plasmid D46/6120, yielding the full-length recombinant A2–2-20F plasmid, in which the sequence of the F gene was confirmed.
BSRT7/5 cells, which constitutively express T7 polymerase (provided by Ursula J. Buchholz and Karl-Klaus Conzelmann), were transfected with A2–2-20F and four support plasmids which express RSV N, P, M2-1, and L proteins under the control of the T7 promoter (18, 19). Transfected cells were passaged until syncytia were observed, at which point they were scraped into the medium, snap-frozen, thawed, and used to infect HEp-2 cells (16). When the maximal cytopathic effect was observed in HEp-2 cells, serial dilutions of clarified supernatants were used to infect fresh HEp-2 cells, which were overlaid with agarose-medium. Single plaques were picked and amplified in HEp-2 cells to generate viral stocks. The F cDNA was PCR amplified from the recovered working A2–2-20F virus stock as described above, and the F cDNA sequence was confirmed. The stock was confirmed to be mycoplasma free through PCR-based testing (Venor GeM mycoplasma detection kit, PCR based; Sigma-Aldrich, St. Louis, MO).
Multistep virus growth curves.
Subconfluent HEp-2 cells in six-well dishes were infected in triplicate with RSV strain A2, 2-20, or A2–2-20F at a multiplicity of infection (MOI) of 1.0 in 250 μl. After 1 h of adsorption at room temperature on a rocking platform, the cells were washed with phosphate-buffered saline (PBS), and fresh medium was added. Supernatants were harvested from each well at 12 h, 24 h, 48 h, and 72 h, and RSV was titrated in duplicate by plaque assay on HEp-2 cells as described previously (13).
Quantification of lung viral load.
BALB/cJ mice were infected with 105 PFU of RSV, and lungs were harvested at the indicated time points. A BeadBeater apparatus (Biospec Products, Bartlesville, OK) was used to homogenize the lungs as previously described (13). Lung homogenates were serially diluted and used to inoculate subconfluent HEp-2 cells in 24-well dishes as described above and overlaid with MEM supplemented with 10% FBS, penicillin G-streptomycin sulfate-amphotericin B solution, and 0.75% methylcellulose (13). After 6 days, plaques were visualized by immunodetection as described previously (13).
Histopathology.
Lungs were fixed in 10% neutral buffered formalin for 24 h and then transferred to 70% ethanol. Lungs were then embedded in paraffin blocks, and 5-μm-thick tissue sections were cut and then stained with hematoxylin-eosin (H&E) to examine histologic changes. A pathologist blinded to the groups scored the samples for severity of infiltrates of lymphocytes, neutrophils, macrophages, and eosinophils on a scale of from 0 to 4 for peribronchiolar, perivascular, interstitial, and alveolar spaces. Slides were scored for the presence or absence of bronchiolar exudates containing necrotic cell debris. Additional sections were also stained with periodic acid-Schiff (PAS) stain to quantify the amount of mucin expression in airways. PAS-stained slides were digitally scanned using a Mirax Midi microscope, and slides were analyzed for PAS-positive airways using HistoQuant software (3D Histotech, Budapest, Hungary), as we previously described (13). Each airway was outlined individually, and all airways in the section were included in the data.
Dual-functional split-reporter (DSP) fusion assay.
An assay using fused split-reporter proteins Renilla luciferase (RL) and green fluorescent protein (GFP), which recover activity when they reassociate due to cell fusion, allows quantification of viral protein-induced cell-cell fusion (20). The GFP reporter protein is made up of a small fragment of GFP fragment harboring a β sheet (21). Transfected cells expressing DSP1-7 or DSP8-12 were used (22). Plasmids expressing cDNA codons optimized for mammalian expression (GeneArt; Invitrogen, Carlsbad, CA) of RSV A2 F, A2 G, 2-20 F, and 2-20 G were cloned into pcDNA3.1(+) (Invitrogen), and the sequences were confirmed. 293T cells (90% confluent) were transfected with plasmids expressing A2 F, 2-20 F, A2 F and A2 G, 2-20 F and 2-20 G, or 2-20 F and A2 G plus DSP1-7. Additional wells were transfected with plasmids expressing DSP8-11. 293T cells were transfected with Lipofectamine 2000 (Invitrogen) and incubated in MEM with 10% FBS and 1% penicillin G-streptomycin sulfate-amphotericin B containing 250 nM RSV fusion inhibitor BMS-433771 (Alios Biopharma, San Francisco, CA) for 24 h at 37°C in 5% CO2. At 24 h posttransfection, cells were washed with 1 ml PBS and resuspended in 1 ml medium containing 1:1,000 EnduRen live cell substrate (Promega, Madison, WI). Cells expressing DSP1-7 as well as A2 F, 2-20 F, A2 F and A2 G, 2-20 F and 2-20 G, or 2-20 F and A2 G were mixed in an equal volume with cells expressing DSP8-11. One hundred microliters of each cell mixture was plated in a white 96-well plate, and RL activity was measured with a Top Count luminometer (PerkinElmer, Waltham, MA) at the indicated time points.
Western blotting of F and G levels in transfected 293T cells.
For immunoblotting, proteins were separated by SDS-PAGE, followed by transfer to a polyvinylidene difluoride membrane. After electroblotting, the membranes were probed using a SNAP i.d. system (Millipore, Billerica, MA). Briefly, the blot was saturated in 0.5% nonfat dry milk in Tris-buffered saline–Tween 20 (TBS-T). After blocking, the membrane was washed three times with TBS-T, followed by incubation with primary antibody against RSV F (palivizumab antibody, 1:1,0000; a gift from James Crowe, Vanderbilt, Nashville, TN) or RSV G (131-2G, 1:5,000; Millipore, Billerica, MA) for 10 min. Membranes were washed three times and incubated with horseradish peroxidase (HRP)-conjugated secondary antibodies (anti-mouse, 1:10,000; anti-human, 1:10,000; Sigma-Aldrich, St. Louis, MO) for 10 min. Signals were detected by chemiluminescence detection using an ECL Western blotting substrate reagent (Pierce Biology Protein Products, Rockford, IL).
Flow cytometry analysis of F and G surface levels in transfected 293T cells.
293T cells (90% confluent) were transfected with plasmids expressing A2 F, A2 G, 2-20 F, or 2-20 G in a pcDNA 3.1 vector and DSP1-7, as in the dual split-protein fusion assay. Cells were incubated for 36 h at 32°C to limit syncytium formation. Cells were harvested and washed in PBS containing 2% FBS and 0.1% NaN3. Cells were stained with palivizumab or anti-RSV G antibody (131-2G; Millipore) at a concentration of 1:100. Samples were incubated at 4°C in the dark for 2 h. Cells were then washed in 2 ml PBS containing 2% FBS and 0.1% NaN3 and centrifuged for 5 min at 456 × g. F samples were stained with anti-human phycoerythrin (PE; SouthernBiotech, Birmingham, AL) or human IgG1, K isotype control (SouthernBiotech). G samples were stained with anti-mouse allophycocyanin (APC; G; RMG1-1; Biolegend, San Diego, CA) or mouse IgG2a, K isotype control (eBM2a; eBioscience, San Diego, CA). Cells were analyzed using an LSR II flow cytometer (BD Biosciences, San Jose, CA). Data were analyzed using FlowJo software (Tree Star Inc., Ashland, OR).
Neutrophil depletion in BALB/cJ mice.
Seven- to 8-week-old BALB/cJ mice were depleted of neutrophils with 1 mg anti-Ly6G (1A8; Bio X Cell, West Lebanon, NH) given intraperitoneally (i.p.) 2 days prior to infection. Anti-Ly6G antibody (0.5 mg) was given to mice on days 0, 2, 4, and 6 postinfection. Control mice were treated with rat IgG2a isotype control (2A3; Bio X Cell) on days −2, 0, 2, 4, and 6 postinfection.
Flow cytometric analysis of blood cells.
Blood was collected by submandibular bleed on days 1 and 7 postinfection Peripheral blood mononuclear cells (PBMCs) were isolated using erythrocyte lysis buffer (0.15 M NH4Cl, 10 mM KHCO3, 0.1 mM EDTA) and counted with a hemocytometer. Cells were stained with anti-CD11b (M1/70; eBioscience) and anti-Gr-1 (RB6-8C5; BD Biosciences). A total of 5 × 105 cells were analyzed using an LSR II flow cytometer (BD Biosciences). The total number of neutrophils was calculated by multiplying the percentage of CD11b+ Gr-1+ cells by the total number of cells isolated. Data were analyzed using FlowJo software (Tree Star Inc.).
Flow cytometric analysis of lung mononuclear cells.
Mice were euthanized with sodium pentobarbital (8.5 mg/kg body weight) i.p., and lungs were harvested at 1 and 8 days postinfection. Cells were isolated using Ficoll and counted with a hemocytometer. Cells were stained with the following antibodies: anti-CD11b and anti-Gr-1 or anti-major histocompatibility complex class II (anti-MHC-II; M5/114.15.2; eBioscience), anti-CD11b (M1/70; eBioscience), and anti-Gr-1 (RB6-8C5; BD Biosciences). A total of 5 × 105 cells were analyzed using an LSR II flow cytometer (BD Biosciences). The total number of neutrophils was calculated by multiplying the percentage of CD11b+ Gr-1+ cells or the percentage of MHC-II-negative (MHC-II−) CD11b+ Ly6G-positive cells by the total number of cells isolated. The total number of macrophages was calculated by multiplying the percentage of MHC-II-positive (MHC-II+) CD11b+ Ly6G-negative cells by the total number of cells isolated. Cells were stained for IL-13 and gamma interferon (IFN-γ) using intracellular staining (ICS). Isolated cells were stimulated with phorbol myristate acetate (PMA)-ionomycin for 5 h and stained with antibodies to CD3 (145-2C11; BD Biosciences), CD4 (RM4-5; BD Biosciences), CD8 (53-6.7; BD Biosciences), IL-13 (ebio13A; eBioscience), and IFN-γ (XMG1.2; eBioscience). The total numbers of IFN-γ-expressing CD8+ T cells and IL-13-expressing CD4+ T cells in the lungs were determined by multiplying the percentage of lymphocytes (defined by forward and side scatter properties in flow cytometry) that were CD3+ CD8+ IFN-γ positive and CD3+ CD4+ IL-13 positive (IL-13+) by the total number of mononuclear cells isolated. Data were analyzed using FlowJo software (Tree Star Inc.).
Cytokine quantification.
Cytokines in lung homogenates were analyzed by Luminex bead analysis (Invitrogen) according to the manufacturer's protocol. Samples were centrifuged in a tabletop centrifuge at room temperature for 5 s, and 50 μl of sample was incubated with antibody-coated capture beads for 2 h at room temperature. After washing the beads three times, protein-specific biotinylated detector antibodies were added and incubated with the beads for 1 h. After removal of excess biotinylated antibodies, streptavidin conjugated to a fluorescent protein, R-phycoerythrin (streptavidin-RPE), was added, and the mixture was incubated for 30 min. After washing of unbound streptavidin-RPE, the beads were analyzed with a Luminex 200 machine (Austin, TX).
Generation of prefusion RSV fusion protein model.
Structural models are based on the recently reported coordinates of the RSV F protein in its prefusion conformation (Protein Data Bank [PDB] accession number 4JHW) (23). The UCSF chimera (24) and MacPyMol programs were used for structure analysis and presentation.
Statistical analyses.
Unless otherwise indicated, groups were compared by one-way analysis of variance (ANOVA) and the Tukey multiple-comparison test (P < 0.05). Values below the limit of detection were assigned a value of half the limit of detection, as shown in the figures.
RESULTS
RSV A2–2-20F replication in human cells and viral load in BALB/cJ mice.
RSV strain 2-20 infection causes airway mucin expression in BALB/cJ mice (13). The fusion (F) protein of the mucus-inducing RSV strain line 19 was shown to be a factor in airway mucin expression induced by RSV infection in BALB/cJ mice (16). We hypothesized that the 2-20 F protein may similarly be a mucin-inducing factor in RSV infection. We generated a chimeric RSV strain that contains the 2-20 gene in an RSV A2 genetic background (RSV A2–2-20F). We first compared the in vitro growth of RSV A2–2-20F to that of RSV A2 and RSV 2-20. In HEp-2 cells, RSV A2–2-20F grew to lower titers (P < 0.05, ANOVA) than its parent strains at 48 h postinfection, and there were no significant differences between strains at any other time points (Fig. 1A). BALB/cJ mice are semipermissive for RSV replication. We previously showed that RSV 2-20 exhibits a higher viral load on day 1 postinfection and a lower peak viral load than RSV A2 (13). The viral loads of RSV strains A2, 2-20, and A2–2-20F were compared over a time course. BALB/cJ mice were infected with 105 PFU of each strain. RSV A2–2-20F and 2-20 had significantly higher viral loads than A2 on day 1 postinfection, although these titers were near the limit of detection of the plaque assay. RSV A2 had a significantly higher viral load on days 4 and 6 postinfection (Fig. 1B), and A2 grew to a higher peak titer in vivo. However, early viral load differences may play a role in RSV pathogenesis in BALB/cJ mice. The similar viral loads of 2-20 and A2–2-20F show that the 2-20 F gene is a determinant of the viral load.
Fig 1.
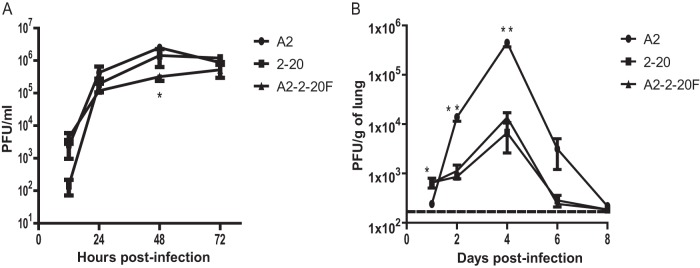
In vitro growth and in vivo viral load of RSV strains A2, 2-20, and A2–2-20F. (A) Infectious yield in supernatants of HEp-2 cells infected at an MOI of 1.0 with RSV A2, 2-20, or A2–2-20F. Error bars represent SEMs of two replicates. Data are means ± SEMs. *, the infectious yield of A2 was significantly higher than that of A2–2-20F at 48 h postinfection (P < 0.05, ANOVA). The results shown represent those from three experiments with similar data. (B) BALB/cJ mice were infected with 5 × 105 PFU of A2, 2-20, or A2–2-20F (n = 5/group). Lungs were harvested on the indicated days postinfection, and infectious RSV was titrated by plaque assay. Data are means ± SEMs. *, at day 1 postinfection, values for 2-20 and A2–2-20F were significantly higher than those for A2 (P < 0.05, ANOVA); **, at day 4 postinfection, the value for A2 was significantly higher than the values for 2-20 and A2–2-20F (P < 0.05, ANOVA). Dashed line, limit of detection. The results shown represent those for three experiments with similar data.
RSV A2–2-20F causes early lung lesions in BALB/cJ mice.
Lung injury is common in severe RSV infection, shown by loss of lung epithelial cells, edema, hemorrhage, and alveolar inflammation (25). Lung damage is thought to be mediated by both direct effects of the virus and the immune response (26). We previously reported that RSV 2-20 infects the airway epithelium at 1 day postinfection in mice (13). We sought to determine if early viral load correlates with early lung lesion development in RSV A2–2-20F-infected mice. At day 1 postinfection, mice infected with RSV A2–2-20F exhibited increased interstitial pneumonia involving increased thickening of alveolar walls associated with inflammatory cell infiltrates compared to mock-infected and RSV A2-infected mice (Fig. 2A and B). RSV 2-20-infected mice also had significantly more interstitial pneumonia than mock-infected mice (Fig. 2A and B). There was no difference in the interstitial pneumonia scores between groups on day 4, 6, or 8 postinfection (data not shown). The airways of RSV 2-20- and A2–2-20F-infected mice contained more necrotic cell debris than mock- and A2-infected mice on day 1 postinfection (Fig. 2C). One of 10 mock-infected mice and 1 of 10 A2-infected mice exhibited airway necrotic cell debris. In contrast, necrotic cell debris was observed in 6 of 10 mice infected with RSV 2-20 and 8 of 10 mice infected with RSV A2–2-20F. Epithelial cell debris and BAL fluid cells in the airways of infected mice reportedly contribute to the pathogenesis of RSV (23). Our data show that A2–2-20F-infected mice exhibit lung epithelial damage and inflammation on day 1 postinfection.
Fig 2.

RSV A2–2-20F infection results in early lung lesions in BALB/cJ mice. BALB/cJ mice were mock infected or infected with 105 PFU of the indicated strain. Lungs were harvested on day 1 postinfection, stained with H&E, and analyzed for histologic changes. (A) Interstitial pneumonia (IP) score for each group. *, significant difference from mock-infected animals (P < 0.01, ANOVA); **, significant difference from mock- and A2-infected animals (P < 0.05, ANOVA). (B) Representative airways indicating interstitial pneumonia in mock-, A2-, 2-20-, and A2–2-20F-infected mice. (C) Representative airways containing cell debris in mock-, A2-, 2-20-, and A2–2-20F-infected mice. The results shown represent those from two experiments with similar data.
RSV A2–2-20F infection causes airway mucin expression.
Pulmonary mucus expression is a hallmark of severe RSV disease in infants (27). RSV 2-20 infection of BALB/cJ mice triggers higher airway mucin expression than infection with RSV A2 (13). We used the chimeric virus RSV A2–2-20F to determine whether the 2-20 F protein is responsible for this phenotype. Periodic acid-Schiff (PAS) staining was used to examine the mucin distribution in the airways of RSV-infected mice. Mice were infected with 105 PFU of RSV, and lungs were harvested on day 8 postinfection. Both 2-20- and A2–2-20F-infected mice exhibited significantly greater PAS positivity than mock- and A2-infected mice (Fig. 3). Greater than 5% of the airways in 2-20- and A2–2-20F-infected mice had approximately 10% mucin positivity, whereas approximately 1% of the airways in A2-infected mice had such mucin positivity (Fig. 3). These results show that both 2-20 infection and A2–2-20F infection result in more airway mucin expression in the airways of BALB/cJ mice than A2 infection, suggesting that the 2-20 F protein is a mucus-inducing factor in RSV infection.
Fig 3.

2-20 F is a mucogenic factor in RSV infection. BALB/cJ mice were mock infected (n = 3) or infected with 105 PFU of A2 (n = 5), 2-20 (n = 5), or A2–2-20F (n = 5), and lungs were harvested on day 8 postinfection. The PAS-positive area was determined for each airway. (A) Results for >200 individual airways are shown per group. Each symbol represents one airway. †, significant difference compared to mock-infected animals (P < 0.001); *, significant difference compared to A2-infected animals (P < 0.05, ANOVA); $, significant difference compared to A2-infected animals (P < 0.001, ANOVA). (B) The same percent PAS positivity data as in panel A plotted against the size of all airways. The results are representative of those from two experiments with similar results.
Fusion activity.
We sought to determine the underlying mechanisms of 2-20 F-protein-mediated pathogenesis. We hypothesized that the 2-20 F protein has greater fusion activity than the A2 F protein. To quantify cell-to-cell fusion, we used a bioluminescence reporter-based content mixing assay (20). 293T cells were transfected with A2 F, 2-20 F, A2 F and A2 G, 2-20 F and 2-20 G, or 2-20 F and A2 G plus DSP1-7 and mixed with 293T cells transfected with DSP8-11. An RSV fusion inhibitor, BMS-433771, was used during transfection to eliminate effector cell-to-effector cell fusion. This inhibitor was removed prior to cell mixing to allow effector cell-to-target cell fusion. Transfected cells expressed total cell (Fig. 4A and B) and surface (Fig. 4C and D) levels of F and G proteins equivalently. Contrary to our hypothesis, A2 F and 2-20 F had equal fusion activity (Fig. 4E and F). It has been shown that coexpression of the RSV attachment (G) glycoprotein promotes fusion (28). We therefore also tested whether RSV A2 G and/or 2-20 G can enhance fusion. Both G proteins increased the fusion activity of 2-20 F but not that of A2 F. RSV F and G glycoproteins interact to form a complex on the surface of infected cells, suggesting that G plays a role in RSV fusion (29). We identified a specific, functional interaction between 2-20 F and RSV G.
Fig 4.

2-20 F is more fusogenic than A2 F when coexpressed with RSV G protein. (A and B) Total cell steady-state levels of RSV F (A) and RSV G (B) proteins expressed in 293T cells after transfection (GAPDH, glyceraldehyde-3-phosphate dehydrogenase). Blots were probed with palivizumab and HRP-conjugated anti-human secondary antibodies or anti-RSV G and HRP-conjugated antimouse secondary antibodies. The results are representative of those from three experiments. Numbers to the right of the gels are molecular sizes (in kilodaltons). (C and D) Transfected cells were stained with palivizumab and anti-human PE or anti-RSV G and anti-mouse APC to quantify F (C) and G (D) surface levels. Dashed lines, isotype control-stained samples. The results are representative of those from 3 experiments. (E and F) DSP-expressing cells transfected with A2 F, 2-20 F, A2 F and A2 G, 2-20 F and 2-20 G, or 2-20 F and A2 G were mixed in equal amounts. Luciferase activity was measured at the indicated time points. The results are representative of those from three experiments (4 to 5 wells per group) with similar results.
RSV A2–2-20F infection results in lung neutrophil recruitment in BALB/cJ mice.
Neutrophils are present in the lungs of children with severe RSV infection (9, 30). Additionally, neutrophils are among the first cells to be recruited to the site of infection and have been shown to contribute to cell damage (11, 31). H&E staining revealed neutrophil infiltration in the lungs of A2–2-20F-infected mice (data not shown). We used flow cytometry to determine neutrophil levels in the lungs of infected BALB/cJ mice on day 1 postinfection and found significantly higher levels in the lungs of A2–2-20F-infected BALB/cJ mice than in those of mock- and A2-infected mice (Fig. 5). Thus, the 2-20 F protein contributes to pulmonary neutrophil recruitment. 2-20-infected mice showed an intermediate phenotype with higher neutrophil levels than in A2-infected mice but slightly less than in A2–2-20F-infected mice (Fig. 5). There are many sequence differences between the 2-20 and A2 genomes (GenBank accession numbers M74568 and JX069798, respectively), but the A2–2-20F recombinant narrows the candidate pool to residues within the F protein. A2–2-20F permits the analysis of the 2-20 F protein specifically.
Fig 5.
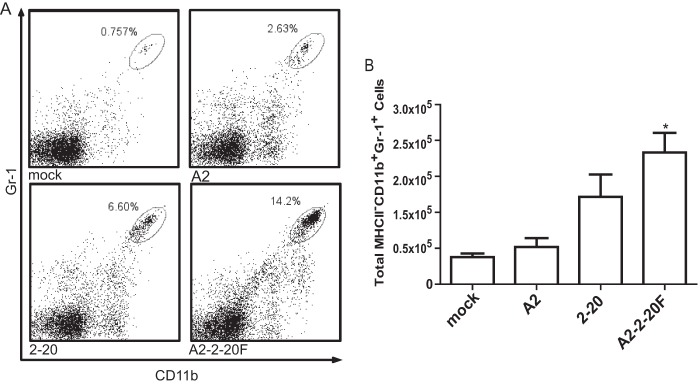
A2–2-20F causes neutrophil infiltration in the lungs of BALB/cJ mice. Mice were mock infected or infected with the indicated RSV strain. Lungs were harvested at day 1 postinfection, and cells were isolated and stained for CD11b and Gr-1. (A) Representative flow plots from the indicated group; (B) quantification from three replicates. *, significant differences between mock- and A2-infected mice (P < 0.05, ANOVA).
Neutrophil depletion in RSV-infected BALB/cJ mice.
Anti-Ly6G clone monoclonal antibody 1A8 has been shown to be specific for neutrophils (32). BALB/cJ mice were treated with anti-Ly6G in order to investigate the role of neutrophils in the context of RSV-induced airway mucin expression. Lung and peripheral blood mononuclear cells were isolated from mock- and RSV-infected mice that were treated with anti-Ly6G or IgG2a isotype control antibody. Administration of this anti-Ly6G antibody in the context of RSV infection resulted in depletion of neutrophils for the duration of infection in both the blood and lungs of BALB/cJ mice (Fig. 6). Mock-infected mice had greater numbers of neutrophils in the lung on day 1 than day 7 postinfection, indicating that the mock preparation does induce an early neutrophil response in the lung (Fig. 6G and H). The macrophage population in anti-Ly6G-treated mice was also investigated. The levels of MHC-II+ Gr-1+ CD11b+ cells were not significantly affected by antibody treatment (Fig. 7). Eosinophils do not express Ly6G and therefore are not affected by 1A8 antibody treatment (32).
Fig 6.

Anti-Ly6G treatment results in depletion of neutrophils in the blood and lungs of RSV-infected mice. BALB/cJ mice were treated with IgG2a control antibody or anti-Ly6G antibody. These mice were mock infected or infected with 1 × 106 PFU RSV 2-20. Blood was taken at days 1 and 7 postinfection. Lungs were harvested on days 1 and 8 postinfection. Cells were isolated by use of a Ficoll gradient and stained for CD11b and Gr-1. (A) Representative flow plots of PBMCs from the indicated group on day 1 postinfection. (B) Representative flow plots of PBMCs from the indicated groups on day 7 postinfection. (C) Quantification of PBMCs on day 1. Results are representative of those from three replicate experiments. (D) Quantification of PBMCs on day 7. Results are representative of those from three replicate experiments. (E) Representative flow plots of lung cells from the indicated group on day 1 postinfection. (F) Representative flow plots of lung cells from the indicated group on day 8 postinfection. (G) Quantification of lung cells on day 1. Results are representative of those from three replicate experiments. (H) Quantification of lung cells on day 8. Results are representative of those from three replicate experiments. Bars, significant differences (P < 0.05, ANOVA).
Fig 7.

1A8 administration does not affect macrophage numbers in mock- or RSV-infected mice. BALB/cJ mice were treated with IgG2a isotype control antibody or anti-Ly6G antibody. These mice were mock infected (n = 3 or 5 per group) or infected with 1 × 106 PFU of 2-20 (n = 3 or 5). Lungs were harvested at day 6 postinfection Cells were isolated by the use of a Ficoll gradient and stained for MHC-II and CD11b. Data were analyzed using FlowJo software. (A) Representative flow plots gated for MHC-II+ CD11b+ cells; (B) quantification of three replicates.
To assess the contribution of neutrophils to RSV clearance, we examined the viral loads in neutrophil-depleted and isotype control-treated BALB/cJ mice. Though neutrophil-depleted mice had a slightly higher viral load on days 4, 6, and 8 postinfection, this difference never reached significance (Fig. 8). These results show that neutrophils do not contribute to viral clearance in this RSV infection model.
Fig 8.
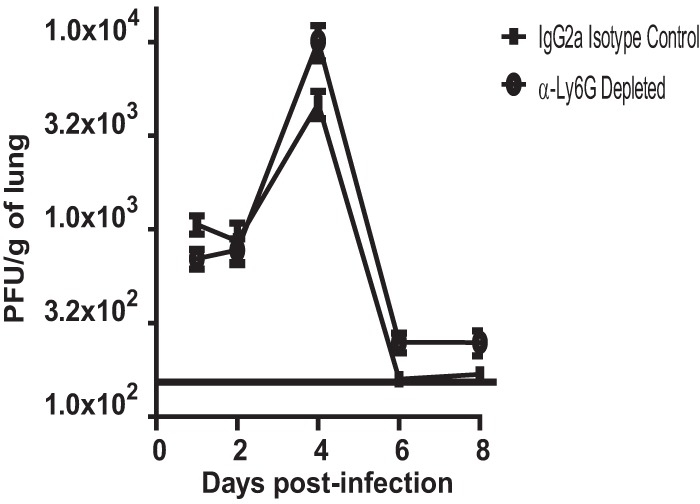
Viral load after neutrophil depletion. BALB/cJ mice were treated with IgG2a isotype control antibody or anti-Ly6G antibody. BALB/cJ mice were mock infected (n = 5) or infected with 1 × 106 PFU of 2-20 (n = 5). Lungs were harvested on the indicated days, and infectious RSV was titrated by an immunodetection plaque assay. Solid line, limit of detection. Data represent the results of three replicate experiments.
Neutrophil depletion during RSV infection modulates airway mucin expression.
Neutrophils have been shown to be associated with lung mucus in models such as those for chronic obstructive pulmonary disease and asthma (23, 24). We hypothesized that neutrophils also modulate mucin expression in the lungs of RSV-infected mice. To examine this, lungs were harvested on day 8 postinfection from mock- or RSV-infected mice treated with anti-Ly6G or isotype control antibody and stained with PAS. Neutrophil depletion in RSV-infected mice resulted in significantly more PAS-positive airways than in RSV-infected isotype control-treated mice (Fig. 9). These data indicate that neutrophils play a role in airway mucin expression during pathogenic RSV 2-20 infection.
Fig 9.
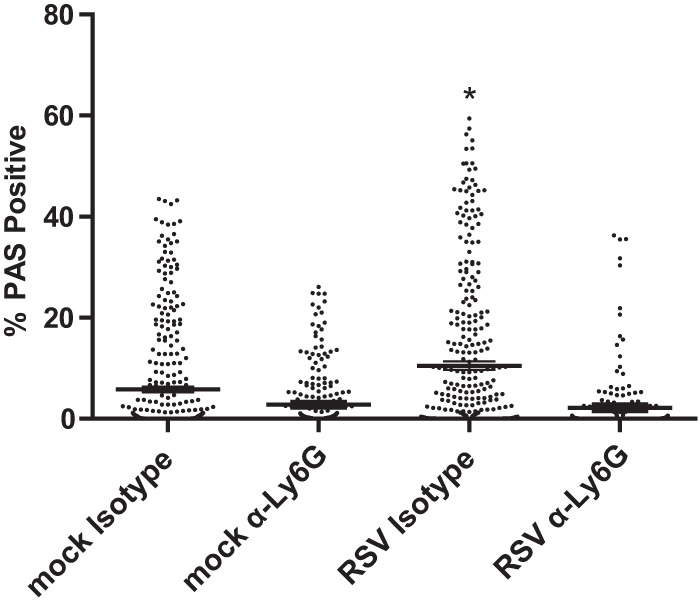
Neutrophil depletion decreased mucin production. BALB/c mice were treated with IgG2a isotype control antibody or anti-Ly6G antibody. These mice were mock infected (n = 5 per group) or infected with 1 × 106 PFU of 2-20 (n = 5). Lungs were harvested at 8 days postinfection and processed for PAS staining. Data show the percentage of the area that was PAS positive for each airway; >200 individual airways are shown per group. Each symbol represents one airway. *, significant differences from other groups (P < 0.05, ANOVA). Data represents the results from two replicate experiments.
Neutrophil depletion in the setting of RSV infection decreases lung TNF-α levels.
TNF-α is a cytokine that has been linked to mucus production. Intratracheal administration of TNF-α to BALB/cJ mice was shown to induce mucus production (12). Because activated neutrophils release TNF-α, we postulated that depletion of neutrophils would result in less TNF-α in the lungs (33). Indeed, RSV-infected mice treated with anti-Ly6G antibody had significantly decreased TNF-α levels in the lungs on day 1 postinfection (Fig. 10). This decrease coincided with a decrease in neutrophil levels in the lungs due to antibody depletion.
Fig 10.
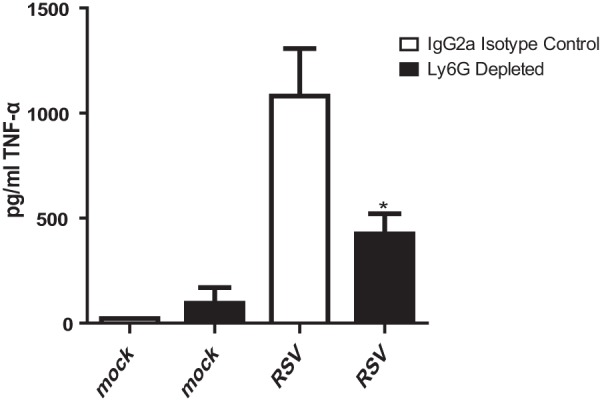
Neutrophil depletion results in decreased TNF-α levels during RSV infection. BALB/c mice were treated with IgG2a isotype control antibody or anti-Ly6G antibody. These mice were mock infected (n = 5) or infected with 1 × 106 PFU of 2-20 (n = 5). Lungs were harvested at day 1 postinfection to measure TNF-α levels. *, significant differences compared to RSV-infected isotype control antibody-treated mice (P < 0.001, ANOVA). Data represent those from four replicate experiments.
Neutrophil depletion during RSV infection results in fewer pulmonary IL-13+ CD4 T cells in depleted mice than in control mice.
IL-13 is a key factor that mediates mucus production in the lungs (15, 34, 35). This cytokine is necessary for both RSV line 19- and 2-20-induced PAS positivity (13, 16). We previously showed that RSV 2-20 infection results in greater IL-13 production than RSV A2 infection (13). Here, we sought to determine a cell source of IL-13 in the RSV infection model and define the effect of neutrophil depletion on IL-13 production. IL-13-producing CD4 T cells were quantified on day 6 postinfection. Neutrophil depletion did not affect the number of lung CD4+ T cells or CD8+ T cells (data not shown) but resulted in fewer IL-13+ CD4 T cells than in isotype control-treated mice (Fig. 11). These CD4+ T cells are likely an important source of IL-13 that causes mucin expression during RSV infection. Our data show that neutrophils modulate CD4 T cell IL-13 expression in RSV infection.
Fig 11.

Neutrophil depletion reduces IL-13-producing CD4+ T cells in lungs of RSV-infected BALB/cJ mice. BALB/cJ mice were injected i.p. with anti-Ly6G antibody or an IgG2a isotype control. These mice were mock infected (n = 3) or infected with 1 × 106 PFU of RSV 2-20 (n = 3). Lungs were harvested on day 6 postinfection, and cells were isolated and stained for CD3, CD4, and IL-13. (A) Representative flow plots gated for MHC-II+ CD11b+ cells; (B) results for three experiments combined.
DISCUSSION
Using a genetically controlled chimeric virus approach, we have shown that the F protein of RSV 2-20 increases the pathogenicity of the laboratory strain A2, as evidenced by greater interstitial pneumonia, increased mucin levels, and more necrotic cell debris in the airways. An increased early viral load correlated with airway necrotic cell debris. Additionally, infection with RSV A2–2-20F resulted in greater neutrophil infiltration into the lungs of BALB/cJ mice than mock and A2 infection. Our data show that neutrophils play an important immunomodulatory role in RSV infection. Depletion of neutrophils had no effect on viral load but resulted in less airway mucin expression, lower lung TNF-α levels, and fewer IL-13-producing CD4+ T cells compared to the findings for isotype control antibody-treated animals during RSV infection. Therefore, neutrophils contribute to pulmonary mucus expression and RSV pathogenesis, potentially through modulation of TNF-α expression and IL-13 expression by CD4+ T cells.
It was previously shown that the fusion protein of RSV strain line 19 is a factor that plays a role in pulmonary mucin expression in the setting of RSV infection (16). BALB/cJ mouse infection with chimeric A2-line 19F resulted in a peak viral load higher than that achieved with infection with the parent viruses A2 and line 19 (16). The unique amino acids in RSV line 19 F, however, are not found in any clinical isolates of RSV (Hotard and Moore, unpublished). The passage history of the line 19 strain is unclear and included many passages through human embryonic diploid lung (MRC-5) cells (36). On the other hand, 2-20 is a clinical isolate with a defined low number of passages (13). Unlike A2-line 19F, A2–2-20F had a viral load pattern similar to that of parent virus 2-20. This suggests that although infection of BALB/cJ mice with both line 19 and 2-20 induces pulmonary mucin expression, the underlying mechanisms leading to its phenotype are distinct. Our data show that both 2-20 and A2–2-20F infection results in early (day 1) lung debris in the airway epithelium of BALB/cJ mice. We hypothesize that this early damage affects the subsequent immune response and results in mucus production. We previously showed that RSV 2-20 viral antigen is detectable in the airway epithelium on day 1 postinfection (13), while at 4 days postinfection viral antigen is localized to the alveolar region (37). Our data are consistent with a model in which 2-20 F renders the mouse airway epithelium susceptible to infection. Infection of the airway epithelium results in necrosis, neutrophil recruitment, and subsequent mucin expression.
Although RSV F can trigger fusion without G, its cognate attachment protein, our data suggest that G and F interaction promotes fusion. Activation of most Paramyxovirinae subfamily F proteins involves interaction with the attachment protein (38). In an in vitro fusion assay, 2-20 F was more fusogenic than A2 F when it was coexpressed with either A2 G or homotypic 2-20 G protein. These data suggest that RSV G boosts 2-20 F fusion activity, but the precise mechanism is unknown. The F and G proteins likely interact to form a complex on the surface of infected cells (29). Hydrophobic residues located in the lower half of the head of the measles virus (MeV) F protein were shown to be important for fusion and interactions with the MeV attachment protein (39). We hypothesize that this increased fusion activity due to 2-20 F affects subsequent RSV pathogenesis. In previous studies, infection of both primary and immortalized human airway epithelial cells did not result in obvious syncytium formation (40, 41). Cell-to-cell fusion, however, was observed in recent studies performed in well-differentiated pediatric bronchial epithelial cells (WD-PECs) cultured at an air-liquid interface (42). Interestingly, more syncytia were observed in WD-PECs infected with an RSV clinical isolate than in WD-PECs infected with the RSV A2 strain (42). Cell-to-cell fusion was also seen in WD-PECs after infection with Sendai virus (SeV) (43). It has been shown that specific residues in the fusion protein of SeV modulate its virulence by influencing both the spread of the virus and the severity of inflammation (44).
There are 15 amino acid differences between the structures of the RSV A2 and 2-20 fusion proteins (Fig. 12). Residues 4, 8, 16, 20, and 25 are not present in the protein because they are part of the signal peptide that directs the F0 precursor to the endoplasmic reticulum. Residue 124 is present in the short, 27-residue peptide between the furin cleavage sites. Of the remaining residues, mutations at positions 519 and 524 are in immediate proximity to the membrane and may influence interaction with lipids, which reportedly affects the bioactivity of viral fusion proteins (45). For instance, a membrane proximal glycine residue of the influenza HA2 fusion protein is required for viral fusion and infectivity (46). Residue 384 (Fig. 12) lies in a region where the head and stalk domains come together, which was previously shown to be a determinant for prefusion MeV F protein stability and fusion activity (48). When all other residues were highlighted in a structural model of the RSV F protein in its prefusion conformation, we noted a concentration of changes in a microdomain mapping closely to the top of the prefusion F trimer (residues 63, 66, and 76). These residues may contribute to metastability (48). In future studies, we will explore whether residues contribute to controlling the thermodynamic stability of the prefusion RSV F trimer and/or affect its interaction with the RSV G protein.
Fig 12.
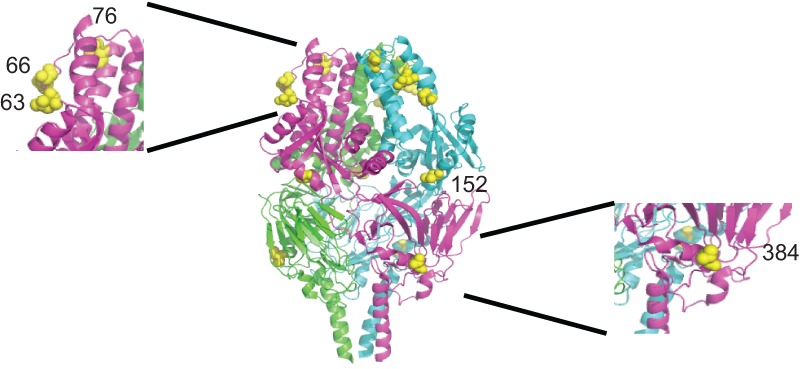
Side-view ribbon representation of the prefusion RSV F-protein trimer (PDB accession number 4JHW), colored by monomer. Red spheres highlight sequence differences between RSV strains A2 and 2-20 in one monomer. (Insets) Closeup views of areas with differing residues; a first set maps toward the top of the prefusion F structure (residues 63, 66, and 76), and residue 384 toward the lateral face of prefusion F. Residue 152 is also labeled.
The relationship between viral load and disease in RSV pathogenesis is not defined. A previous study indicated that higher viral loads in infants are a predictor of greater disease severity (49, 50). Other studies, however, have shown no correlation between viral titers and disease severity (51, 52). In the mouse model of 2-20 and A2–2-20F infection, pathogenesis correlated with the early viral load and not the peak viral load. The cell tropism of clinical RSV strains may play a role in their differential pathogenesis. RSV has been shown to infect cells in both the bronchial and bronchiolar epithelia and alveolar cells in the lungs of RSV-infected children and mice (53, 54). RSV 2-20 viral antigen is localized to the airway epithelium at greater levels than A2 antigen at day 1 postinfection (13). A2 infection also resulted in less necrotic cell debris and a lower viral load at this early time point. Peak viral load, although correlated with disease severity in some cases, may not be the most important factor in RSV airway pathogenesis. Early viral load may be responsible for driving disease that is not dependent on later peak viral loads (55). We hypothesize that localization of the RSV load to the airways, rather than peak viral load per se, drives airway pathogenesis. We speculate that elevated fusion activity may permit infection of the mouse airway epithelium. Host entry factors specific for airway and alveolar cells may impact airway penetration as well. RSV F has been shown to interact with nucleolin (56). However, we have not identified factors that specifically interact with 2-20 F in a cell type-dependent manner.
Previous studies reveal that neutrophils are prevalent bronchial infiltrates in human RSV infection (7, 9, 30). The neutrophil response occurs prior to the RSV-induced CD8+ T cell response in infants (57). Depletion of neutrophils during influenza virus infection is mice results in higher virus titers in the lung and elevated mortality (58, 59). In the present study, however, neutrophil depletion did not affect the viral load in RSV-infected mice. CD8+ T cells have been shown to be important for RSV clearance in mice (60). We did not see any differences in CD8+ T cell numbers in neutrophil-depleted mice (data not shown) but found that neutrophils affect the CD4+ T cell IL-13 expression and TNF-α expression. Neutrophils have been linked to mucus plugging in severe RSV infection and asthma (6, 8, 23). Previous studies have shown that dysregulated or excessive neutrophil responses in the airways may contribute to disease during severe influenza virus infections (61). Low-, intermediate-, or high-virulence influenza virus strains differ in their ability to recruit neutrophils to airways. In severe influenza virus infection, neutrophils had a beneficial role in ultimately limiting disease (62). Neutrophils may be playing a similar damaging role in RSV infection. In lung bacterial infections, bacteria induce neutrophil sequestration and eventual damage (63). When neutrophils were depleted in BALB/cJ mice during RSV infection, we found few mucin-positive cells in the airways. TNF-α expression was higher in RSV-infected, neutrophil-depleted mice than nondepleted, infected mice. Neutrophils are a source of TNF-α in the lungs (64), which in turn facilitates the recruitment of neutrophils during airway inflammation and has been implicated in stimulation of mucus factor MUC5AC expression (14, 65).
Increased concentrations of TNF-α and IL-13 have been measured in the BAL fluid of asthmatic patients following allergen stimulation (66). In a model of allergic asthma, IL-13 instillation results in TNF-α expression by neutrophils (67) and is necessary for mucus production (68). It has been shown that RSV infection promotes a Th2-type inflammatory response in the lung, subsequently inducing a Th2-like effector phenotype in regulatory T cells and increasing susceptibility to asthma (69). BALB/cJ mouse infection with mucus-inducing RSV strains line 19 and 2-20 results in lung IL-13 expression (13, 14, 16). Neutrophils are seen in BAL fluid samples from severe asthmatics but not from mild or moderate asthmatics (70). In a model of allergic asthma, IL-13 was shown to activate neutrophils (67, 71). It was also found that IL-13 administration induced substantial airway neutrophilia (72). In neutrophil-depleted, RSV-infected BALB/cJ mice, there was a drop in IL-13+ CD4 T cells compared to their levels in isotype-treated, RSV-infected mice. These results indicate that although IL-13 has been shown to act on neutrophils, neutrophils also modulate the IL-13 cellular immune response. Further elucidation of this pathway involving IL-13 and TNF-α may lead to therapies to combat RSV infection-induced pulmonary mucus.
Neutrophils have long been associated with bronchiolitis. For the first time, we analyzed the role of neutrophils in a mouse model of RSV bronchiolitis. We have identified a novel pathway wherein the F protein affects the early immune response to RSV, including the neutrophil response, and subsequently influences downstream cytokine and mucus expression. Our data suggest that the early viral load and damage influenced by the activity of the RSV F protein are related to the neutrophil and mucus responses.
ACKNOWLEDGMENTS
This work was supported by the following grants: NIH 1R01AI087798 and NIH 1U19AI095227.
We thank the Emory Children's Pediatric Research Center flow cytometry core and immunology core, which are supported by Children's Healthcare of Atlanta (CHOA). We thank Naoyuki Kondo and Zene Matsuda for the DSP1-7 and DSP8-11 plasmids and Ursula Buchholz and Karl-Klaus Conzelmann for the BSRT7/5 cells. We thank James Crowe, Jr., for palivizumab. We also thank Carla Shoffeitt for histology technical assistance.
Footnotes
Published ahead of print 10 July 2013
REFERENCES
- 1. Leader S, Kohlhase K. 2003. Recent trends in severe respiratory syncytial virus (RSV) among US infants, 1997 to 2000. J. Pediatr. 143:S127–S132 [DOI] [PubMed] [Google Scholar]
- 2. Shay DK, Holman RC, Newman RD, Liu LL, Stout JW, Anderson LJ. 1999. Bronchiolitis-associated hospitalizations among US children, 1980-1996. JAMA 282:1440–1446 [DOI] [PubMed] [Google Scholar]
- 3. Thompson WW, Shay DK, Weintraub E, Brammer L, Cox N, Anderson LJ, Fukuda K. 2003. Mortality associated with influenza and respiratory syncytial virus in the United States. JAMA 289:179–186 [DOI] [PubMed] [Google Scholar]
- 4. Nakanishi A, Morita S, Iwashita H, Sagiya Y, Ashida Y, Shirafuji H, Fujisawa Y, Nishimura O, Fujino M. 2001. Role of gob-5 in mucus overproduction and airway hyperresponsiveness in asthma. Proc. Natl. Acad. Sci. U. S. A. 98:5175–5180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Aherne W, Bird T, Court SD, Gardner PS, McQuillin J. 1970. Pathological changes in virus infections of the lower respiratory tract in children. J. Clin. Pathol. 23:7–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bataki EL, Evans GS, Everard ML. 2005. Respiratory syncytial virus and neutrophil activation. Clin. Exp. Immunol. 140:470–477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Everard ML, Swarbrick A, Wrightham M, McIntyre J, Dunkley C, James PD, Sewell HF, Milner AD. 1994. Analysis of cells obtained by bronchial lavage of infants with respiratory syncytial virus infection. Arch. Dis. Child. 71:428–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. McNamara PS, Flanagan BF, Baldwin LM, Newland P, Hart CA, Smyth RL. 2004. Interleukin 9 production in the lungs of infants with severe respiratory syncytial virus bronchiolitis. Lancet 363:1031–1037 [DOI] [PubMed] [Google Scholar]
- 9. Smith PK, Wang SZ, Dowling KD, Forsyth KD. 2001. Leucocyte populations in respiratory syncytial virus-induced bronchiolitis. J. Paediatr. Child Health 37:146–151 [DOI] [PubMed] [Google Scholar]
- 10. Miyamoto M, Prause O, Sjostrand M, Laan M, Lotvall J, Linden A. 2003. Endogenous IL-17 as a mediator of neutrophil recruitment caused by endotoxin exposure in mouse airways. J. Immunol. 170:4665–4672 [DOI] [PubMed] [Google Scholar]
- 11. Wang SZ, Smith PK, Lovejoy M, Bowden JJ, Alpers JH, Forsyth KD. 1998. The apoptosis of neutrophils is accelerated in respiratory syncytial virus (RSV)-induced bronchiolitis. Clin. Exp. Immunol. 114:49–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Busse PJ, Zhang TF, Srivastava K, Lin BP, Schofield B, Sealfon SC, Li XM. 2005. Chronic exposure to TNF-alpha increases airway mucus gene expression in vivo. J. Allergy Clin. Immunol. 116:1256–1263 [DOI] [PubMed] [Google Scholar]
- 13. Stokes KL, Chi MH, Sakamoto K, Newcomb DC, Currier MG, Huckabee MM, Lee S, Goleniewska K, Pretto C, Williams JV, Hotard A, Sherrill TP, Peebles RS, Jr, Moore ML. 2011. Differential pathogenesis of respiratory syncytial virus clinical isolates in BALB/c mice. J. Virol. 85:5782–5793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lukacs NW, Strieter RM, Chensue SW, Widmer M, Kunkel SL. 1995. TNF-alpha mediates recruitment of neutrophils and eosinophils during airway inflammation. J. Immunol. 154:5411–5417 [PubMed] [Google Scholar]
- 15. Tekkanat KK, Maassab HF, Cho DS, Lai JJ, John A, Berlin A, Kaplan MH, Lukacs NW. 2001. IL-13-induced airway hyperreactivity during respiratory syncytial virus infection is STAT6 dependent. J. Immunol. 166:3542–3548 [DOI] [PubMed] [Google Scholar]
- 16. Moore ML, Chi MH, Luongo C, Lukacs NW, Polosukhin VV, Huckabee MM, Newcomb DC, Buchholz UJ, Crowe JE, Jr, Goleniewska K, Williams JV, Collins PL, Peebles RS., Jr 2009. A chimeric A2 strain of respiratory syncytial virus (RSV) with the fusion protein of RSV strain line 19 exhibits enhanced viral load, mucus, and airway dysfunction. J. Virol. 83:4185–4194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Graham BS, Perkins MD, Wright PF, Karzon DT. 1988. Primary respiratory syncytial virus infection in mice. J. Med. Virol. 26:153–162 [DOI] [PubMed] [Google Scholar]
- 18. Buchholz UJ, Granzow H, Schuldt K, Whitehead SS, Murphy BR, Collins PL. 2000. Chimeric bovine respiratory syncytial virus with glycoprotein gene substitutions from human respiratory syncytial virus (HRSV): effects on host range and evaluation as a live-attenuated HRSV vaccine. J. Virol. 74:1187–1199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Collins PL, Hill MG, Camargo E, Grosfeld H, Chanock RM, Murphy BR. 1995. Production of infectious human respiratory syncytial virus from cloned cDNA confirms an essential role for the transcription elongation factor from the 5′ proximal open reading frame of the M2 mRNA in gene expression and provides a capability for vaccine development. Proc. Natl. Acad. Sci. U. S. A. 92:11563–11567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kondo N, Miyauchi K, Meng F, Iwamoto A, Matsuda Z. 2010. Conformational changes of the HIV-1 envelope protein during membrane fusion are inhibited by the replacement of its membrane-spanning domain. J. Biol. Chem. 285:14681–14688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cabantous S, Terwilliger TC, Waldo GS. 2005. Protein tagging and detection with engineered self-assembling fragments of green fluorescent protein. Nat. Biotechnol. 23:102–107 [DOI] [PubMed] [Google Scholar]
- 22. Ishikawa H, Meng F, Kondo N, Iwamoto A, Matsuda Z. 2012. Generation of a dual-functional split-reporter protein for monitoring membrane fusion using self-associating split GFP. Protein Eng. Des. Sel. 25:813–820 [DOI] [PubMed] [Google Scholar]
- 23. McLellan JS, Chen M, Leung S, Graepel KW, Du X, Yang Y, Zhou T, Baxa U, Yasuda E, Beaumont T, Kumar A, Modjarrad K, Zheng Z, Zhao M, Xia N, Kwong PD, Graham BS. 2013. Structure of RSV fusion glycoprotein trimer bound to a prefusion-specific neutralizing antibody. Science 340:1113–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 25:1605–1612 [DOI] [PubMed] [Google Scholar]
- 25. Bruder D, Srikiatkhachorn A, Enelow RI. 2006. Cellular immunity and lung injury in respiratory virus infection. Viral Immunol. 19:147–155 [DOI] [PubMed] [Google Scholar]
- 26. Moore ML, Peebles RS., Jr 2006. Respiratory syncytial virus disease mechanisms implicated by human, animal model, and in vitro data facilitate vaccine strategies and new therapeutics. Pharmacol. Ther. 112:405–424 [DOI] [PubMed] [Google Scholar]
- 27. Johnson JE, Gonzales RA, Olson SJ, Wright PF, Graham BS. 2007. The histopathology of fatal untreated human respiratory syncytial virus infection. Mod. Pathol. 20:108–119 [DOI] [PubMed] [Google Scholar]
- 28. Rawling J, Garcia-Barreno B, Melero JA. 2008. Insertion of the two cleavage sites of the respiratory syncytial virus fusion protein in Sendai virus fusion protein leads to enhanced cell-cell fusion and a decreased dependency on the HN attachment protein for activity. J. Virol. 82:5986–5998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Low KW, Tan T, Ng K, Tan BH, Sugrue RJ. 2008. The RSV F and G glycoproteins interact to form a complex on the surface of infected cells. Biochem. Biophys. Res. Commun. 366:308–313 [DOI] [PubMed] [Google Scholar]
- 30. Heidema J, Lukens MV, van Maren WW, van Dijk ME, Otten HG, van Vught AJ, van der Werff DB, van Gestel SJ, Semple MG, Smyth RL, Kimpen JL, van Bleek GM. 2007. CD8+ T cell responses in bronchoalveolar lavage fluid and peripheral blood mononuclear cells of infants with severe primary respiratory syncytial virus infections. J. Immunol. 179:8410–8417 [DOI] [PubMed] [Google Scholar]
- 31. Yu KL, Wang XA, Civiello RL, Trehan AK, Pearce BC, Yin Z, Combrink KD, Gulgeze HB, Zhang Y, Kadow KF, Cianci CW, Clarke J, Genovesi EV, Medina I, Lamb L, Wyde PR, Krystal M, Meanwell NA. 2006. Respiratory syncytial virus fusion inhibitors. Part 3. Water-soluble benzimidazol-2-one derivatives with antiviral activity in vivo. Bioorg. Med. Chem. Lett. 16:1115–1122 [DOI] [PubMed] [Google Scholar]
- 32. Daley JM, Thomay AA, Connolly MD, Reichner JS, Albina JE. 2008. Use of Ly6G-specific monoclonal antibody to deplete neutrophils in mice. J. Leukoc. Biol. 83:64–70 [DOI] [PubMed] [Google Scholar]
- 33. Foley SC, Hamid Q. 2007. Images in allergy and immunology: neutrophils in asthma. J. Allergy Clin. Immunol. 119:1282–1286 [DOI] [PubMed] [Google Scholar]
- 34. Tanabe T, Fujimoto K, Yasuo M, Tsushima K, Yoshida K, Ise H, Yamaya M. 2008. Modulation of mucus production by interleukin-13 receptor alpha2 in the human airway epithelium. Clin. Exp. Allergy 38:122–134 [DOI] [PubMed] [Google Scholar]
- 35. Wills-Karp M, Luyimbazi J, Xu X, Schofield B, Neben TY, Karp CL, Donaldson DD. 1998. Interleukin-13: central mediator of allergic asthma. Science 282:2258–2261 [DOI] [PubMed] [Google Scholar]
- 36. Herlocher ML, Ewasyshyn M, Sambhara S, Gharaee-Kermani M, Cho D, Lai J, Klein M, Maassab HF. 1999. Immunological properties of plaque purified strains of live attenuated respiratory syncytial virus (RSV) for human vaccine. Vaccine 17:172–181 [DOI] [PubMed] [Google Scholar]
- 37. Anderson JJ, Norden J, Saunders D, Toms GL, Scott R. 1990. Analysis of the local and systemic immune responses induced in BALB/c mice by experimental respiratory syncytial virus infection. J. Gen. Virol. 71(Pt 7):1561–1570 [DOI] [PubMed] [Google Scholar]
- 38. Waning DL, Russell CJ, Jardetzky TS, Lamb RA. 2004. Activation of a paramyxovirus fusion protein is modulated by inside-out signaling from the cytoplasmic tail. Proc. Natl. Acad. Sci. U. S. A. 101:9217–9222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Apte-Sengupta S, Negi S, Leonard VH, Oezguen N, Navaratnarajah CK, Braun W, Cattaneo R. 2012. Base of the measles virus fusion trimer head receives the signal that triggers membrane fusion. J. Biol. Chem. 287:33026–33035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Zhang L, Peeples ME, Boucher RC, Collins PL, Pickles RJ. 2002. Respiratory syncytial virus infection of human airway epithelial cells is polarized, specific to ciliated cells, and without obvious cytopathology. J. Virol. 76:5654–5666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang L, Collins PL, Lamb RA, Pickles RJ. 2011. Comparison of differing cytopathic effects in human airway epithelium of parainfluenza virus 5 (W3A), parainfluenza virus type 3, and respiratory syncytial virus. Virology 421:67–77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Villenave R, Thavagnanam S, Sarlang S, Parker J, Douglas I, Skibinski G, Heaney LG, McKaigue JP, Coyle PV, Shields MD, Power UF. 2012. In vitro modeling of respiratory syncytial virus infection of pediatric bronchial epithelium, the primary target of infection in vivo. Proc. Natl. Acad. Sci. U. S. A. 109:5040–5045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Villenave R, Touzelet O, Thavagnanam S, Sarlang S, Parker J, Skibinski G, Heaney LG, McKaigue JP, Coyle PV, Shields MD, Power UF. 2010. Cytopathogenesis of Sendai virus in well-differentiated primary pediatric bronchial epithelial cells. J. Virol. 84:11718–11728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Luque LE, Bridges OA, Mason JN, Boyd KL, Portner A, Russell CJ. 2010. Residues in the heptad repeat A region of the fusion protein modulate the virulence of Sendai virus in mice. J. Virol. 84:810–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Smith EC, Culler MR, Hellman LM, Fried MG, Creamer TP, Dutch RE. 2012. Beyond anchoring: the expanding role of the Hendra virus fusion protein transmembrane domain in protein folding, stability, and function. J. Virol. 86:3003–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Walker JA, Kawaoka Y. 1993. Importance of conserved amino acids at the cleavage site of the haemagglutinin of a virulent avian influenza A virus. J. Gen. Virol. 74(Pt 2):311–314 [DOI] [PubMed] [Google Scholar]
- 47. Lee JK, Prussia A, Snyder JP, Plemper RK. 2007. Reversible inhibition of the fusion activity of measles virus F protein by an engineered intersubunit disulfide bridge. J. Virol. 81:8821–8826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. DeVincenzo JP, El Saleeby CM, Bush AJ. 2005. Respiratory syncytial virus load predicts disease severity in previously healthy infants. J. Infect. Dis. 191:1861–1868 [DOI] [PubMed] [Google Scholar]
- 50. Houben ML, Coenjaerts FE, Rossen JW, Belderbos ME, Hofland RW, Kimpen JL, Bont L. 2010. Disease severity and viral load are correlated in infants with primary respiratory syncytial virus infection in the community. J. Med. Virol. 82:1266–1271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wright PF, Gruber WC, Peters M, Reed G, Zhu Y, Robinson F, Coleman-Dockery S, Graham BS. 2002. Illness severity, viral shedding, and antibody responses in infants hospitalized with bronchiolitis caused by respiratory syncytial virus. J. Infect. Dis. 185:1011–1018 [DOI] [PubMed] [Google Scholar]
- 52. Legg JP, Hussain IR, Warner JA, Johnston SL, Warner JO. 2003. Type 1 and type 2 cytokine imbalance in acute respiratory syncytial virus bronchiolitis. Am. J. Respir. Crit. Care Med. 168:633–639 [DOI] [PubMed] [Google Scholar]
- 53. Taylor G, Stott EJ, Hughes M, Collins AP. 1984. Respiratory syncytial virus infection in mice. Infect. Immun. 43:649–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stott EJ, Taylor G. 1985. Respiratory syncytial virus. Brief review. Arch. Virol. 84:1–52 [DOI] [PubMed] [Google Scholar]
- 55. DeVincenzo JP. 2005. Factors predicting childhood respiratory syncytial virus severity: what they indicate about pathogenesis. Pediatr. Infect. Dis. J. 24:S177–S183 [DOI] [PubMed] [Google Scholar]
- 56. Tayyari F, Marchant D, Moraes TJ, Duan W, Mastrangelo P, Hegele RG. 2011. Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat. Med. 17:1132–1135 [DOI] [PubMed] [Google Scholar]
- 57. Lukens MV, van de Pol AC, Coenjaerts FE, Jansen NJ, Kamp VM, Kimpen JL, Rossen JW, Ulfman LH, Tacke CE, Viveen MC, Koenderman L, Wolfs TF, van Bleek GM. 2010. A systemic neutrophil response precedes robust CD8(+) T-cell activation during natural respiratory syncytial virus infection in infants. J. Virol. 84:2374–2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Fujisawa H. 2008. Neutrophils play an essential role in cooperation with antibody in both protection against and recovery from pulmonary infection with influenza virus in mice. J. Virol. 82:2772–2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Tate MD, Brooks AG, Reading PC. 2008. The role of neutrophils in the upper and lower respiratory tract during influenza virus infection of mice. Respir. Res. 9:57. 10.1186/1465-9921-9-57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Cannon MJ, Openshaw PJ, Askonas BA. 1988. Cytotoxic T cells clear virus but augment lung pathology in mice infected with respiratory syncytial virus. J. Exp. Med. 168:1163–1168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Sakai S, Kawamata H, Mantani N, Kogure T, Shimada Y, Terasawa K, Sakai T, Imanishi N, Ochiai H. 2000. Therapeutic effect of anti-macrophage inflammatory protein 2 antibody on influenza virus-induced pneumonia in mice. J. Virol. 74:2472–2476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Tate MD, Ioannidis LJ, Croker B, Brown LE, Brooks AG, Reading PC. 2011. The role of neutrophils during mild and severe influenza virus infections of mice. PLoS One 6:e17618. 10.1371/journal.pone.0017618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Craig A, Mai J, Cai S, Jeyaseelan S. 2009. Neutrophil recruitment to the lungs during bacterial pneumonia. Infect. Immun. 77:568–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Xing Z, Jordana M, Kirpalani H, Driscoll KE, Schall TJ, Gauldie J. 1994. Cytokine expression by neutrophils and macrophages in vivo: endotoxin induces tumor necrosis factor-alpha, macrophage inflammatory protein-2, interleukin-1 beta, and interleukin-6 but not RANTES or transforming growth factor-beta 1 mRNA expression in acute lung inflammation. Am. J. Respir. Cell Mol. Biol. 10:148–153 [DOI] [PubMed] [Google Scholar]
- 65. Smirnova MG, Birchall JP, Pearson JP. 2000. TNF-alpha in the regulation of MUC5AC secretion: some aspects of cytokine-induced mucin hypersecretion on the in vitro model. Cytokine 12:1732–1736 [DOI] [PubMed] [Google Scholar]
- 66. Luttmann W, Matthiesen T, Matthys H, Virchow JC., Jr 1999. Synergistic effects of interleukin-4 or interleukin-13 and tumor necrosis factor-alpha on eosinophil activation in vitro. Am. J. Respir. Cell Mol. Biol. 20:474–480 [DOI] [PubMed] [Google Scholar]
- 67. Shim JJ, Dabbagh K, Ueki IF, Dao-Pick T, Burgel PR, Takeyama K, Tam DC, Nadel JA. 2001. IL-13 induces mucin production by stimulating epidermal growth factor receptors and by activating neutrophils. Am. J. Physiol. Lung Cell. Mol. Physiol. 280:L134–L140 [DOI] [PubMed] [Google Scholar]
- 68. Walter DM, McIntire JJ, Berry G, McKenzie AN, Donaldson DD, DeKruyff RH, Umetsu DT. 2001. Critical role for IL-13 in the development of allergen-induced airway hyperreactivity. J. Immunol. 167:4668–4675 [DOI] [PubMed] [Google Scholar]
- 69. Krishnamoorthy N, Khare A, Oriss TB, Raundhal M, Morse C, Yarlagadda M, Wenzel SE, Moore ML, Peebles RS, Jr, Ray A, Ray P. 2012. Early infection with respiratory syncytial virus impairs regulatory T cell function and increases susceptibility to allergic asthma. Nat. Med. 18:1525–1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Wenzel SE, Szefler SJ, Leung DY, Sloan SI, Rex MD, Martin RJ. 1997. Bronchoscopic evaluation of severe asthma. Persistent inflammation associated with high dose glucocorticoids. Am. J. Respir. Crit. Care Med. 156:737–743 [DOI] [PubMed] [Google Scholar]
- 71. Zaitsu M, Hamasaki Y, Matsuo M, Kukita A, Tsuji K, Miyazaki M, Hayasaki R, Muro E, Yamamoto S, Kobayashi I, Ichimaru T, Kohashi O, Miyazaki S. 2000. New induction of leukotriene A(4) hydrolase by interleukin-4 and interleukin-13 in human polymorphonuclear leukocytes. Blood 96:601–609 [PubMed] [Google Scholar]
- 72. Pope SM, Brandt EB, Mishra A, Hogan SP, Zimmermann N, Matthaei KI, Foster PS, Rothenberg ME. 2001. IL-13 induces eosinophil recruitment into the lung by an IL-5- and eotaxin-dependent mechanism. J. Allergy Clin. Immunol. 108:594–601 [DOI] [PubMed] [Google Scholar]