

Abstract
The expression of certain HLA class I alleles, including HLA-B*27 and HLA-B*57, is associated with better control of human immunodeficiency virus type 1 (HIV-1) infection, but the mechanisms responsible are not fully understood. We sought evidence that pressure from the human restriction factor TRIM5α (hTRIM5α) could contribute to viral control. The hTRIM5α sensitivity of viruses from both HLA-B*57-positive (HLA-B*57+) and HLA-B*27+ patients who spontaneously controlled viral replication, but not viruses from viremic patients expressing these alleles, was significantly greater than that of viruses from patients not expressing these protective HLA-B alleles. Overall, a significant negative correlation between hTRIM5α sensitivity and viral load was observed. In HLA-B*57+ patients, the T242N mutation in the HLA-B*57-restricted TW10 CD8+ T lymphocyte (CTL) epitope was strongly associated with hTRIM5α sensitivity. In HLA-B*27+ controllers, hTRIM5α sensitivity was associated with a significant reduction in emergence of key CTL mutations. In several patients, viral evolution to avoid hTRIM5α sensitivity was observed but could be associated with reduced viral replicative capacity. Thus, in individuals expressing protective HLA-B alleles, the combined pressures exerted by CTL, hTRIM5α, and capsid structural constraints can prevent viral escape both by impeding the selection of necessary resistance/compensatory mutations and forcing the selection of escape mutations that increase hTRIM5α sensitivity or impair viral replicative capacity.
INTRODUCTION
The immune response mediated by cytotoxic CD8+ T lymphocytes (CTL) plays an important role in controlling viremia during human immunodeficiency virus type 1 (HIV-1) infection (1–5), which in turn influences the rate of loss of CD4+ T cells and the progression to AIDS (6, 7). It is clear, however, that the effectiveness of the immune response can be quite variable in different individuals, resulting in a clinical spectrum that ranges from individuals who maintain high viral loads and have rapid disease progression to those that are able to spontaneously control viral load to low levels in the absence of antiretroviral therapy.
The factors that determine the effectiveness of the CTL responses are beginning to be defined. The expression of certain HLA class I alleles, including HLA-B*57 and HLA-B*27, has been clearly shown to be associated with better control of HIV-1 infection, and these alleles are also overrepresented among patients who spontaneously control viral loads to low levels (8, 9). A number of explanations for this association have been proposed, including the ability of CTL from patients expressing these protective HLA alleles to target conserved viral proteins, such as the capsid protein (CA) (10–15), and induce the selection of viral CTL escape mutations in these structurally constrained proteins that carry a substantial fitness cost and are difficult to correct by the addition of compensatory mutations (16–23). Many other host and viral factors are potentially able to modify the efficacy of CD8+ T-cell responses. For example, both CD4+ and CD8+ T cells from different individuals can vary in their abilities to target viral antigens (12, 24–30) and to exercise effector functions (31–34). Similarly, differences in the replicative capacity of the transmitted virus (35, 36) and differences due to virus-specific sequence variation in epitopes targeted by CTL (17, 26) can modulate CD8 responses. Thus, the ability to control or not HIV-1 infection is likely to be multifactorial (35–37), and not all the factors involved may have been identified.
In this regard, we recently found that mutations in epitopes targeted by CTL could influence viral sensitivity to the human host restriction factor TRIM5α (hTRIM5α), suggesting that pressure from this restriction factor could also modulate the efficacy of CTL responses in some patients (38). TRIM5α interacts with the mature capsid lattice after its entry into target cells and directly promotes disassembly of the capsid structure, thereby preventing the completion of reverse transcription (39). In addition, the E3 ubiquitin ligase activity of TRIM5α is amplified following TRIM5α-capsid interactions, thereby stimulating a cascade of events that promote innate immune signaling and contribute directly to viral restriction by TRIM5α (40, 41). Restriction exerted by TRIM5α on retroviral replication varies according to the virus and to the host species (42). HIV-1 is strongly sensitive to restriction by Rhesus macaque TRIM5α (43), but viruses carrying CA sequences from laboratory-adapted HIV-1 strains and some clinical samples show only modest (less than 2-fold) sensitivity to hTRIM5α (44–48). However, we recently showed that viruses carrying CA sequences from two different clinical isolates were 4- to 7-fold more sensitive to hTRIM5α than the laboratory-adapted NL4-3 strain, and this sensitivity was explained by the presence of mutations known to promote escape from CTL responses directed against CA epitopes presented by HLA-B*27 and/or HLA-B*57 (38, 44). In this regard, it is noteworthy that studies in simian models have shown that differences in TRIM5α activity of this magnitude against infecting viruses can influence both viral replication and disease progression (49–52). Taken together, these findings are consistent with the possibility that hTRIM5α activity could reduce viral replication in patients expressing protective HLA alleles. We also found, however, that the impact of CA mutations on hTRIM5α sensitivity was context dependent, because a given mutation could produce a hTRIM5α-sensitive phenotype in viruses from one patient but have little or no impact on this parameter when introduced into a different viral isolate (38). Thus, it remained unclear how frequently either CTL-induced mutations or other polymorphisms might modify HIV-1 hTRIM5α sensitivity, and the potential impact of pressure from hTRIM5α on viral replication and evolution in vivo was not investigated.
To address these questions, we developed a recombinant virus assay to evaluate the impact of HIV-1 sensitivity to hTRIM5α in patients that did or did not express protective HLA-B alleles and to assess the possibility that selective pressure exerted by this restriction factor could influence viral evolution. The hTRIM5α sensitivity of viruses from patients expressing either HLA-B*57 or HLA-B*27 alleles and who spontaneously controlled virus replication was significantly higher than that of viruses from viremic patients, and a significant negative correlation was observed between hTRIM5α sensitivity and viral load. The presence of the classical T242N escape mutation in the HLA-B*57-restricted TW10 CTL epitope was strongly associated with increased hTRIM5α sensitivity, but T242N was not the only mutation capable of causing increased hTRIM5α sensitivity. Evidence consistent with viral evolution to avoid hTRIM5α sensitivity was observed, including the selection of the G248A mutation in association with T242N and the selection of alternative rare resistance mutations in the TW10 epitope that were deleterious for viral replication. Our data support the hypothesis that pressure mediated by hTRIM5α can influence viral escape from CTL responses and contribute to the improved control of viral replication observed in patients expressing protective HLA-B alleles.
MATERIALS AND METHODS
Study subjects.
Samples of plasma and peripheral blood mononuclear cells (PBMC) from HIV-1-infected patients were obtained from individuals participating in the ANRS CO21 CODEX cohort and from patients followed by the Service des Maladies Infectieuses et Tropicales, Hôpital Saint-Louis, Paris, France. In both cases, patients gave written informed consent and samples were used in accordance with protocols approved by the Comité de Protection des Personnes Ile-de-France VII and the Comité de Protection des Personnes Ile-de-France IV. Samples from 56 patients were evaluated. Clinical information is summarized in Table S1 in the supplemental material. Twenty-six samples were HLA-B*57 positive (HLA-B*57+), 15 were HLA-B*27 positive, and 16 did not express either of these HLA-B alleles. One subject (57C4; see Table S1 in the supplemental material) expressed both HLA-B*57 and HLA-B*27 alleles. Among the HLA-B*57-positive patients, 13 were classified as “controllers” (viral load [VL] < 500 HIV-1 RNA copies/ml for at least 10 years in the absence of antiretroviral therapy) and 13 were classified as “viremic” (VL > 500 RNA copies/ml). Among the HLA-B*27-positive patients, 6 were controllers and 9 were viremic.
Cell culture.
U373-X4 cells in which hTRIM5α activity had been inhibited by stable overexpression of untagged hTRIM5γ (U373-X4-TRIM5γ) and the corresponding control cell line that overexpresses β-galactosidase (U373-X4-LacZ) were established by transduction with pLenti6/V5-D-TOPO-based vectors using previously described techniques (38, 44). These cell lines were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum, 100 U/ml penicillin G, and 100 μg/ml streptomycin (complete medium) in the presence of 8 μg/ml blasticidin. The 293T cell line was maintained in complete medium.
Isolation of viral RNA and PCR.
All samples of plasma were stored at −80°C until use. For samples from patients with a VL of >500 RNA copies/ml, RNA was extracted from 140 μl of plasma using NucleoSpin RNA virus kits (Macherey-Nagel, Hoerd, Germany). For samples from patients with a VL of <500 RNA copies/ml, 2 to 5 ml of plasma was ultracentrifuged (160,000 × g; 30 min; 4°C) and RNA was isolated from the pellet using QIAamp viral RNA minikits (Qiagen, Valencia CA). Isolated viral RNA was immediately reverse transcribed and amplified. The RNA was used to amplify a viral sequence that spans the region coding CA and containing ≈100 bp upstream and downstream of this region. Two to four (VL > 500 copies/ml) or six (VL < 500 copies/ml) independent reverse transcription (RT)-PCRs were performed for samples from each patient using the SuperScript III one-step RT-PCR with the Platinum Taq High Fidelity system (Invitrogen, Carlsbad, NM) and 10 μM (each) primers: 5′-AARGATAGAKGTAAAAGACACCAAGGAAGC (forward) and 5′-TGTCCTTCCTTTCCACATTTCCAACA (reverse). RT-PCR parameters were (i) DNA synthesis for 30 min at 50°C, (ii) denaturation for 2 min at 90°C, (iii) 40 amplification cycles of 94°C for 30 s, 57°C for 30 s, 68°C for 70 s, and (iv) final extension for 10 min at 68°C. A nested PCR was then performed on the RT-PCR products obtained using the AccuPrime Pfx SuperMix kit (Invitrogen) and 10 μM (each) primers: 5′-AGATGTAAAAGACACCAAGGAAGCCTTAGA (forward) and 5′-CCTTCTTTGCCACAATTGAAACAYTTAA (reverse). PCR parameters were 1 cycle at 95°C for 5 min, 35 cycles at 95°C for 15 s, 57°C for 30 s, 68°C for 70 s, and 1 cycle at 68°C for 10 min. Finally, amplification products were purified using Nucleospin Extract II kits (Macherey-Nagel). All cDNAs were sequenced bidirectionally by the dideoxynucleotide method. Phylogenetic analysis confirmed that sequences from the patients were distinct from each other and from laboratory-adapted strains used in the laboratory (data not shown). For genotype/phenotype analysis and for constructing proviral plasmids expressing patient-derived sequences, the consensus sequence was used (see Table S2 in the supplemental material).
Production of recombinant viruses.
The pNL4-3-based vector pNL-SbfI-MscI, in which unique restriction sites were introduced in the N-terminal (SbfI) and C-terminal (MscI) sequences of CA, has been previously described (38). The CA sequence was removed from this plasmid by digestion with SbfI and MscI, and the linearized plasmid was ligated to a linker that was created by heating (95°C; 5 min) a mixture containing 0.5 μg each of oligonucleotides Linker-F (5′-GGACGCGTAGTGGGGGGACCTGG) and Linker-R (5′-CCAGGTCCCCCCACTACGCGTCCTGCA) in 20 μl of 1× oligonucleotide hybridization buffer (Invitrogen) and cooling the mixture to room temperature. The resulting circularized plasmid, pNL4-3-ΔCA-Δenv-lucR-RVA, which contains a unique MluI restriction site within the linker, was used to prepare a large stock of vector (Qiagen plasmid maxi kit), which was subsequently linearized by digestion with MluI.
To produce recombinant viruses carrying CA sequences from clinical isolates, 293T cells were grown in 6-well plates in 1 ml complete medium, and cultures at 70% confluence were transfected using the calcium phosphate method (53) with 125 μl of a suspension containing coprecipitated pNL4-3-ΔCA-Δenv-lucR-RVA vector (0.5 ng), cDNA obtained by amplification of the CA sequence from viral RNA (60 ng), and a vector permitting expression of the vesicular stomatitis virus (VSV) G protein (phCMV-G; 25 pg). Twenty-four hours after transfection, the medium was changed, and 48 h after transfection, supernatants were collected and centrifuged (1,000 × g; 5 min). hTRIM5α sensitivity of viruses produced in each transfection was evaluated in two independent experiments, once using freshly harvested culture supernatant, and once using an aliquot that had been frozen at −80°C.
Measurement of viral sensitivity to hTRIM5α and infectivity.
The techniques used to evaluate viral sensitivity to hTRIM5α have been previously described (38, 44, 54). Briefly, U373-X4 cells overexpressing β-galactosidase (U373-X4-LacZ) or hTRIM5γ (U373-X4-TRIM5γ) were incubated in the presence of 1,000 U/ml alpha interferon (IFN-α) (Sigma-Aldrich, St. Louis, MO) for 24 h to increase the expression of hTRIM5α and infected in triplicate in the presence of 2 μg/ml DEAE-dextran with serial 2-fold dilutions of culture supernatants from transfected cells. For viruses produced by recombination following transfection, 1/2, 1/4, and 1/8 dilutions of culture supernatant were used; for cloned recombinant viruses, aliquots of frozen culture supernatants containing nominally 20, 10, and 5 ng p24/ml were used. Luciferase activity was measured 40 h after infection as previously described. The slope (relative light units [RLU]/amount of virus) was calculated for each virus in each cell line. For all viral variants, RLU values were linearly related to the amount of virus used to infect the cells. For experiments evaluating cloned recombinant viruses, the actual p24 content of the virus preparations used to infect the cells was measured in each experiment, and these values were used to calculate slopes. The mutations introduced in CA were shown not to influence quantification of p24 (data not shown). To measure sensitivity to hTRIM5α, the results for the two cell lines were expressed as a ratio (slope for U373-X4-TRIM5γ/slope for U373-X4-LacZ). Reproducibility of the measurement of hTRIM5α sensitivity of viruses from the 56 subjects is shown in Fig. S1 in the supplemental material. No significant correlation was observed between RLU values of the recombinant viruses in U373-X4-TRIM5γ cells and their sensitivity to hTRIM5α (Spearman r = 0.17; P > 0.2). To evaluate viral replicative capacity, performed only for cloned recombinant viruses, the slope (RLU/p24) obtained in the target cells in which hTRIM5α activity had been inhibited by overexpression of hTRIM5γ (U373-X4-TRIM5γ cells) was used. To confirm the expression of hTRIM5α in the U373-X4-LacZ cells and its inhibition in the U373-X4-TRIM5γ cells, the infectivity of N-tropic and B-tropic murine leukemia viruses (N-MLV and B-MLV) in these cells was evaluated following IFN-α pretreatment, as previously described (38, 44), before and after completion of the studies evaluating clinical samples. In both cases, U373-X4-LacZ cells strongly inhibited the infectivity of N-MLV, whereas similar titers of N-MLV and B-MLV were observed in U373-X4-TRIM5γ cells (data not shown).
In preliminary experiments, we evaluated whether the production of recombinant viruses following transfection influenced the measurement of hTRIM5α sensitivity. To do so, we amplified, as described above, the CA sequence from the proviral plasmid used to generate hTRIM5α-sensitive viruses carrying the gag-protease sequence from clinical isolate NRC10 (38, 54) and compared the sensitivity of VSV-pseudotyped viruses produced by transfection with the proviral plasmid and those produced by cotransfecting the amplification product with the pNL4-3-ΔCA-Δenv-lucR-RVA vector. Sensitivities to hTRIM5α were very similar for these viruses (sensitivity to hTRIM5α: viruses from proviral plasmid, 10.8 ± 1.4; viruses from recombination after transfection, 10.7 ± 1.6, n = 5 independent experiments). For three samples, we also compared the results obtained using bulk amplification products and individual clonal sequences derived from the same amplification. As shown in Fig. S2 in the supplemental material, the hTRIM5α sensitivities of individual clones were generally quite similar to each other and similar to results obtained using bulk sequences, supporting the conclusion that results obtained for bulk sequences were representative the majority viral population present in the sample.
Generation of viral variants.
The CA sequence from one HLA-B*57+ patient (patient 57C3; see Tables S1 and S2 in the supplemental material) carried the Q244R and G248D mutations in the TW10 epitope rather than the more common T242N mutation. To permit evaluation of the impact of alternative CTL resistance mutations in this epitope on hTRIM5α sensitivity and replicative capacity of viruses carrying this CA sequence, the following strategy was used to construct viral variants. The CA cDNA was amplified as described above, and following an A-tailing reaction, the products were cloned into the pCR2.1-TOPO vector (Invitrogen). A clone carrying the consensus CA sequence from this patient was identified and digested with BamHI and XhoI restriction enzymes, and the segment encompassing the CA sequence was ligated into a pBluescript vector previously digested with the same enzymes, creating pB-C3. Two unique restriction sites were created in the N-terminal (SbfI) and C-terminal (MscI) sequences of CA by introducing silent mutations through sequential site-directed mutagenesis reactions (QuikChange site-directed mutagenesis kit; Stratagene) performed according to the manufacturer's instructions and using the oligonucleotides listed in Table 1. The resulting plasmid was digested with SbfI and MscI, and the fragment containing the CA sequence from this patient was used to replace the CA sequence of NL4-3 in the previously described pB-NL-SbfI-MscI plasmid (38), thereby creating pB-NL-C3-SbfI-MscI. This plasmid was then used to create variants in which CTL resistance mutations in the TW10 epitope were removed or added (Table 1; pB-NL-C3-N,-NR,-R, and -0) by site-directed mutagenesis using the oligonucleotides listed in Table 1. Finally, the BssHII-ClaI fragment from these pBluescript plasmids were cloned into pNL4-3-Δenv-lucR (55) previously digested with the same enzymes. All constructions were verified by sequencing the entire gag-protease region of the proviral plasmids. Viral stocks were produced as previously described (44).
Table 1.
Creation of viral variants by site-directed mutagenesisa
| Variant | Modification | Template plasmid | Forward primer |
|---|---|---|---|
| pB-C3-SbfI | Create SbfI site | pB-C3 | 5′CCCTATAGTACAGAACCTGCAGGGGCAAATGGTGCATCAG3′ |
| pB-C3-SbfI-MscI | Create MscI site | pB-C3-SbfI | 5′GTCAGGGAGTGGGGGGACCTGGCCATAAAGCAAGAG3′ |
| pB-NL-C3-R | D248G | pB-NL-C3-SbfI-MscI | 5′GTACCCTTCGGGAACAAATAGGTTGGATGACAAGTAATCCACC3′ |
| pB-NL-C3-0 | R244Q | pB-NL-C3-R | 5′CAGGAACTACTAGTACCCTTCAGGAACAAATAGGTTGGATGAC3′ |
| pB-NL-C3-NR | T242N | pB-NL-C3-R | 5′CATAGCAGGAACTACTAGTAACCTTCGGGAACAAATAGGTTGG3′ |
| pB-NL-C3-N | T242N | pB-NL-C3-0 | 5′CATAGCAGGAACTACTAGTAACCTTCAGGAACAAATAGGTTGG3′ |
The SbfI (CCTGCAGG) and MscI (TGGCCA) restriction sites or the codon modified by mutagenesis are underlined. The nucleotides modified in the target sequences are shown in bold. Only the forward primer is shown in the table; the reverse primer was the reverse complement of the forward primer.
ELISPOT assay.
IFN-γ secretion by HIV-specific CD8+ T cells was quantified ex vivo with an enzyme-linked immunosorbent spot assay (ELISPOT) as previously described (56) following incubation with peptides corresponding to the consensus sequence for the KK10 epitope (KRWIILGLNK), the TW10 epitope (TSTLQEQIGW), or variants of the TW10 epitope containing one or more resistance mutations. Peptides were synthesized by Neosystem Laboratories (Strasbourg, France) and used at a final concentration of 2 μg/ml. IFN-γ spot-forming cells (SFCs) were counted with a KS-ELISPOT system (Carl Zeiss Vision, Aalen, Germany) and expressed as SFCs/106 PBMC after the background of control unstimulated cells was subtracted. Wells were considered positive if they contained at least 50 SFCs/106 PBMC and exhibited at least twice the background level.
Databases and statistical analysis.
Virus subtype was determined using HIV BLAST (http://www.hiv.lanl.gov/). Results are presented as means ± standard errors of the means (SEM) unless otherwise indicated. Comparisons between 2 groups were performed using the Mann-Whitney test. Comparisons between 3 or more groups were performed using the Kruskal-Wallis test or by analysis of variance followed by Bonferroni's multiple comparisons test. Contingency tables were analyzed using the Fisher exact test.
Nucleotide sequence accession numbers.
The sequences determined in this study were deposited in GenBank under accession numbers KC966945 to KC967000.
RESULTS
Sensitivity to hTRIM5α of viruses from HLA-B*57+/HLA-B*27+ patients.
In a previous study, we found that for viruses from two individuals, mutations in CA associated with resistance to CTL targeting epitopes presented by HLA-B*57 and HLA-B*27 contributed to their increased sensitivity to hTRIM5α (38). To evaluate how frequently increased sensitivity to hTRIM5α was observed for viruses from these patients, we compared this parameter for viruses from 16 patients who did not express the HLA-B*57 or HLA-B*27 alleles associated with spontaneous control of HIV-1 replication (8) and from 40 patients who were HLA-B*57+ or HLA-B*27+ (see Table S1 in the supplemental material), including individuals whose viral load in the absence of antiretroviral therapy was <500 RNA copies/ml (HIV controllers) and those who were unable to control viral replication (viremic patients).
To perform these studies, we generated VSV-pseudotyped NL4-3-based recombinant viruses carrying CA sequences from plasma-derived viruses and expressing Renilla luciferase in the place of Nef. Recombinant viruses were collected, and their sensitivity to hTRIM5α was measured as previously described (38, 44), by determining the ratio of their infectivity in U373-X4-TRIM5γ cells, in which hTRIM5α activity has been blocked by overexpression of hTRIM5γ, and U373-X4-LacZ cells, which express physiological levels of hTRIM5α. Using this recombination-based approach allowed the measurement of hTRIM5α sensitivity in the majority population of circulating virions rather than in a few clones that might not be representative at the population level.
Circulating viruses from a substantial proportion of HIV-1-infected individuals demonstrated sensitivity to hTRIM5α (Fig. 1A). Viral sensitivity to hTRIM5α varied over a 6-fold range. Overall, sensitivity to hTRIM5α was significantly higher for viruses from HLA-B*57+ patients, but not HLA-B*27+ patients, than those from patients not expressing these protective HLA-B alleles (P < 0.05). Nevertheless, considerable overlap was observed, and viruses from patients expressing or not the protective HLA-B alleles could have low (less than 2-fold) or higher (up to 5- to 6-fold) sensitivity to hTRIM5α.
Fig 1.
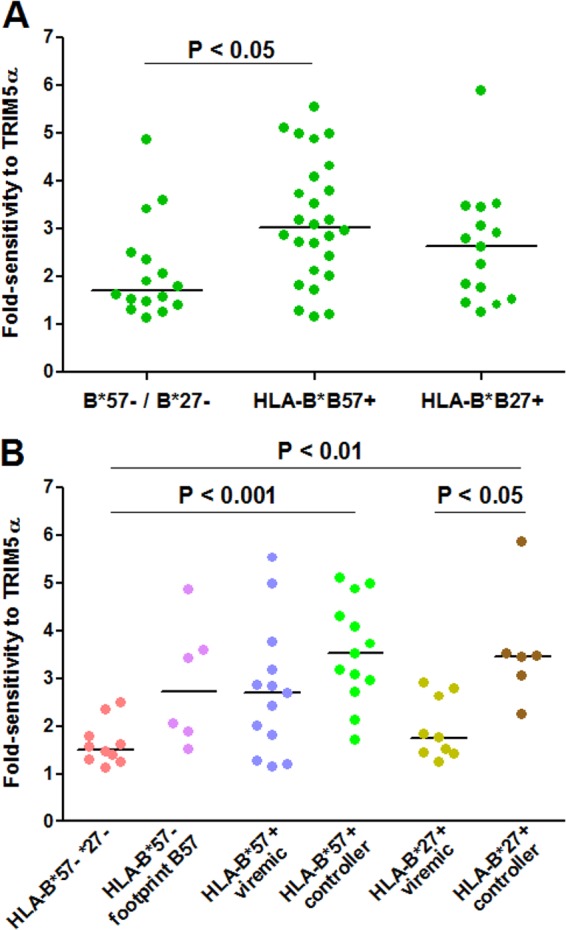
Sensitivity to hTRIM5α of recombinant viruses carrying CA sequences derived from plasma viruses. CA sequences of plasma viruses from HLA-B*57+ patients, HLA-B*27+, patients and HLA-B*57−/B*27− patients were amplified by RT-PCR, and amplification products were used to create recombinant viruses expressing Renilla luciferase in the place of Nef. Fold sensitivity to hTRIM5α was determined by measuring single-cycle infectivity using a luciferase-based assay after infection of U373-X4 cells in which hTRIM5α activity had been inhibited by stable overexpression of untagged hTRIM5γ (U373-X4-TRIM5γ) and in U373-X4 cells that express physiological levels of hTRIM5α (U373-X4-LacZ) and determining the ratio of these results. (A) Results for recombinant viruses from HLA-B*57+ (n = 26), HLA-B*27+ (n = 15), and HLA-B*57−/B*27− (n = 16) patients are compared. (B) Viruses from HLA-B*57− patients have been separated according to the presence or absence of 2 or more mutations associated with resistance to CTL targeting the 4 major epitopes in CA presented by HLA-B*57 (footprint B57). Viruses from HLA-B*57+ and HLA-B*27+ patients have been separated according to viral load in the absence of treatment (>500 copies/ml, viremic; <500 copies/ml, controller). Bars indicate medians. Statistical analysis: Mann-Whitney test (A) and Kruskal-Wallis test followed by Dunn's multiple comparison test (B).
Clinical/virological parameters associated with increased sensitivity to hTRIM5α.
Further evaluation identified several parameters that influenced viral sensitivity to hTRIM5α.
Footprint mutations.
It is known that the CA sequence of the viruses from HLA-B*57-negative patients can contain mutations associated with escape from CTL targeting epitopes presented by HLA-B*57. These mutations are thought to have been selected during prior passage of the virus in an HLA-B*57+ individual but to have persisted as “footprints” following transmission to an HLA-B*57− individual (57, 58). In our previous study, we found that for viruses from one HLA-B*57− patient, increased sensitivity to hTRIM5α resulted from the presence of such HLA-B*57 footprint mutations. To evaluate whether this was a common occurrence, we identified all HLA-B*57− patients whose viruses carried at least 2 of the following resistance/compensatory mutations selected in response to CTL targeting four immunodominant epitopes in CA presented by HLA-B*57 [epitope ISW9: A146P, I147L; epitope KF11: A163G, S165N; epitope TW10: H219Q, I223V, M228(I/L), T242N, G248A; epitope QW9: E312D]. Interestingly, viruses from all HLA-B*57− patients that had an index of sensitivity to hTRIM5α greater than 3 carried HLA-B*57 footprint mutations (Fig. 1B). Conversely, the hTRIM5α sensitivity of viruses from HLA-B*57−/HLA-B*27− patients without footprint mutations was generally low. Thus, HLA-B*57 selective pressure at any step of viral evolutionary history appeared to be a determinant of increased sensitivity to hTRIM5α.
HIV-1 subtype.
Most patients were infected with subtype B viruses (n = 33), but CRF02_AG (n = 17), A1 (n = 3), CRF01_AE (n = 2), and D (n = 1) subtypes were also identified (59, 60). The CA sequence of the two CRFs is derived from subtype A viruses, and therefore we compared the sensitivity to hTRIM5α of viruses carrying subtype A- and subtype B-derived CA sequences. For viruses from patients not expressing protective HLA-B alleles and that did not express HLA-B*57 footprint mutations, hTRIM5α sensitivity was low and not significantly different between the two subtypes (Fig. 2; P = 0.8 for subtypes A and B). In contrast, when viruses from HLA-B*57+ and HLA-B*27+ patients together with those carrying HLA-B*57 footprint mutations were compared, hTRIM5α sensitivity was significantly greater for viruses carrying a CA sequence from subtype B than for those carrying a CA sequence from subtype A (P < 0.001). Similarly, hTRIM5α sensitivity was significantly greater for subtype B viruses than subtype A viruses from viremic HLA-B*57+ and HLA-B*27+ patients (P < 0.01). These findings are consistent with the possibility that the subtype B capsid is more susceptible to becoming hTRIM5α sensitive. It should be recognized, however, that 16/18 of the controllers, whose viruses were more sensitive to hTRIM5α, were infected with subtype B viruses, and this may reflect the patient population from which the controller cohort was obtained. Further studies will be required to evaluate the impact of viral subtype on hTRIM5α sensitivity.
Fig 2.
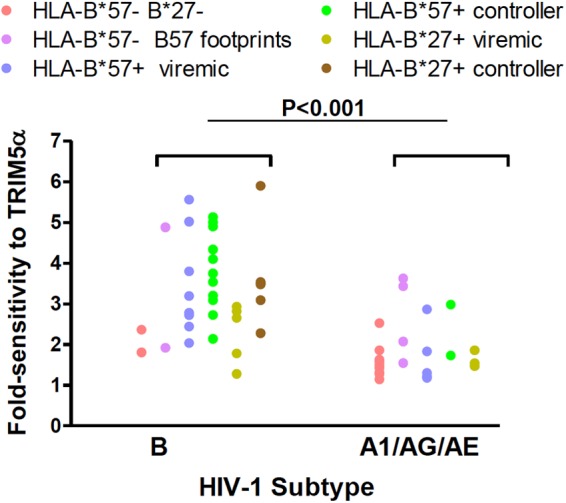
Comparison of sensitivity to hTRIM5α for viruses carrying subtype B and subtype A CA sequences. Viral fold sensitivity to hTRIM5α was measured as described in the Fig. 1 legend and is shown for subtype B viruses versus subtype A1 CRF02_AG and CRF01_AE viruses. Symbols are color coded according to the patient groups indicated in the legend and described in the Fig. 1 legend. Statistical analysis was performed using the Mann-Whitney test. This analysis was directed at identifying the effect of mutations selected by CTL targeting HLA-B*57- or HLA-B*27-restricted epitopes on hTRIM5α sensitivity according to virus subtype. Consequently, results for HLA-B*57−/B*27− patients whose viruses did not express B57 footprint mutations (red dots), which showed low hTRIM5α sensitivity regardless of virus subtype, were excluded from the statistical analysis.
Viral load.
The sensitivity to hTRIM5α of viruses from controllers (all of whom were either HLA-B*57+ or HLA-B*27+) was significantly greater than that of viruses from viremic patients (P < 0.001 using the Mann-Whitney test). The sensitivity to hTRIM5α of viruses from HLA-B*57+ patients who spontaneously controlled viral replication to <500 copies/ml, but not that of viruses from viremic patients expressing this HLA-B allele, was significantly greater than that of viruses without footprint mutations from patients not expressing these protective HLA-B alleles (P < 0.001; Fig. 1B). Similarly, the hTRIM5α sensitivity of viruses from HLA-B*27+ controllers was greater than that of viruses from patients without these protective HLA-B alleles (P < 0.01). These differences remained significant when analysis was restricted to subtype B viruses. To further evaluate the relationship between viral sensitivity to hTRIM5α and viral replication, we determined the correlation between hTRIM5α sensitivity and viral load for untreated patients evaluated in our study. As shown in Fig. 3, a significant negative correlation was observed between these two parameters (Pearson r = 0.49; P < 0.001). A significant correlation was also observed when the analysis was restricted to subtype B viruses (Pearson r = 0.38; P < 0.03; n = 33). These findings indicate that sensitivity to hTRIM5α had an impact on viral replication in these patients.
Fig 3.
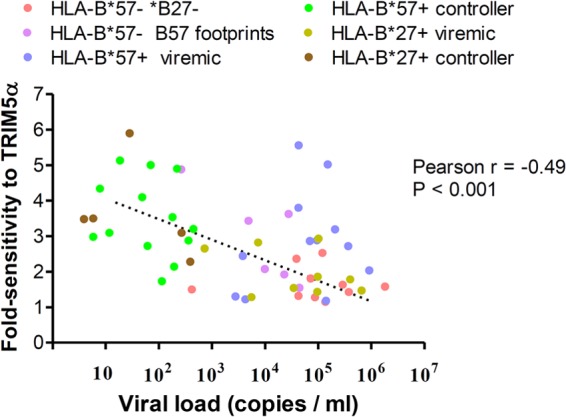
Correlation between viral sensitivity to hTRIM5α and viral load. Viral fold sensitivity to hTRIM5α was measured as described in the Fig. 1 legend and is plotted against plasma viral load measured at the time plasma used to amplify CA sequences was obtained. Analysis was restricted to patients not receiving antiretroviral therapy. Symbols are color coded according to the patient groups indicated in the figure and described in the Fig. 1 legend.
CTL resistance mutations influencing sensitivity to hTRIM5α in HLA-B*57+ patients.
Several genotypic features of viruses from HLA-B*57+ patients were identified that were associated with increased sensitivity to hTRIM5α.
Mutations associated with escape from CTL responses directed against the HLA-B*57-restricted epitope TW10.
In initial studies, we evaluated the relationship between viral sensitivity to hTRIM5α and the presence of CTL escape mutations in HLA-B*57-restricted epitopes. Consistent with prior studies, the T242N escape mutation in the immunodominant TW10 epitope was frequent in viruses from HLA-B*57+ patients (22/26 cases) (20, 61–64) and was also present in viruses from 2/6 HLA-B*57− patients carrying HLA-B*57 footprint mutations (38, 65). The hTRIM5α sensitivity of the viruses carrying the T242N escape mutation was significantly greater than that of viruses without the mutation (P < 0.01; Fig. 4A).
Fig 4.

Viral sensitivity to hTRIM5α and the CA genotype. Viral fold sensitivity to hTRIM5α was measured as described in the Fig. 1 legend and is shown for viruses expressing the indicated genotype at Gag amino acid 242 (A), viruses expressing the indicated genotype at Gag amino acid 146 (B), viruses expressing the indicated genotypes at Gag amino acids 242 and 248 (C), and viruses expressing the indicated number of compensatory mutations [H219Q, I223V, M228(I/L)] in the cyclophilin A binding loop (D). Symbols are color coded according to the patient groups indicated in the legend and described in the Fig. 1 legend. Bars indicate medians. Statistical analysis was performed using the Mann-Whitney test (panels A and B) or analysis of variance followed by Bonferroni's multiple comparison test (panels C and D). Note that these analyses were directed at identifying mutations selected by CTL targeting HLA-B57-restricted epitopes that modify hTRIM5α sensitivity. Consequently, results for HLA-B57− patients whose viruses did not express B57 footprint mutations (red dots) were excluded from the statistical analyses.
Because CTL escape mutations accumulate in parallel in several HLA-B*57-restricted CA epitopes, it was important to determine if the T242N mutation contributed directly to the increased hTRIM5α sensitivity or whether this statistical difference reflected the appearance of T242N in association with other CTL escape mutations. Two lines of evidence indicated that the T242N mutation was indeed directly responsible. First, no significant correlations were observed between hTRIM5α sensitivity and the presence of common CTL escape mutations in the ISW9, KF11, or QW9 epitopes, as is illustrated for the 146P mutation associated with escape from CTL targeting the HLA-B*57-restricted ISW9 epitope (Fig. 4B). Second, for viruses from two HLA-B*57+ patients, we have previously shown that reversion of the T242N mutation led to a significant reduction in hTRIM5α sensitivity (38). Thus, T242N, the most frequently observed escape mutation occurring in CA of viruses from HLA-B*57+ patients, appeared to be a major determinant of hTRIM5α sensitivity.
In subtype B viruses from HLA-B*57+ patients, the T242N mutation is often accompanied by the G248A mutation in the TW10 epitope (20, 61, 62). Following transmission of a virus from an HLA-B*57+ individual to an HLA-B*57− individual, the T242N mutation can revert, leaving the G248A mutation as a footprint (62, 65), a genotype that was observed in viruses from 2/6 HLA-B*57− patients in our study whose viruses carried HLA-B*57 footprint mutations (Fig. 4C). Although G248A has been classified both as a CTL escape mutation (62) and a compensatory mutation (61), the forces that drive the selection of G248A have not been well defined. In patients infected with subtype B viruses, the recognition by CTL of peptides containing T242N alone and T242N and G248A was found to be quite similar (62), and G248A alone was less likely than T242N alone to provide escape from T-cell clones targeting TW10 (25). The replicative capacity of viruses expressing T242N alone and T242N and G248A have also been found to be similar, suggesting that G248A has little effect on the replicative defect resulting from T242N (61). Another possible function of the G248A mutation could be the reduction of viral sensitivity to hTRIM5α caused by the T242N mutation. Consistent with this possibility, we observed that the sensitivity to hTRIM5α was significantly lower for viruses carrying the T242N and G248A mutations than for viruses with only the T242N mutation (P < 0.05; Fig. 4C). This finding remained significant when analysis was restricted to subtype B viruses (P < 0.01).
Viruses from HLA-B*57+ patients with the T242N mutation also accumulate mutations in the CypA-binding loop of capsid, and these mutations can partially correct the replicative defect that results from T242N (19, 61, 62, 65). These mutations were also observed in the HLA-B*57+ patients studied here [H219Q, n = 3; I223V, n = 7; M228(I/L), n = 5]. No obvious relationship was observed between hTRIM5α sensitivity and the presence of individual mutations, specific combinations of these mutations, or the total number of these mutations present (Fig. 4D and data not shown), suggesting that CypA-binding loop mutations were not a major determinant of hTRIM5α sensitivity. However, viruses from only 4 HLA-B*57+ patients had more than one of these mutations, and further studies are required to draw definite conclusions. In this context, we previously found that reverting mutations in the CypA-binding loop did not modify hTRIM5α sensitivity of viruses carrying the T242N mutation (38).
High sensitivity to hTRIM5α is associated with low frequency of CTL escape mutations in viruses from HLA-B*27+ controllers.
A highly immunodominant HLA-B*27-restricted CTL epitope is present in CA spanning Gag amino acids 263 to 272. Resistance to CTL recognizing this KK10 epitope commonly starts with the selection of the L268M mutation. Although this mutation has little fitness cost, it does not impair peptide binding to HLA-B*27 and often does not provide strong CTL escape in clonotypic assays (21, 22, 25, 66, 67). Much stronger CTL escape is provided by the R264K mutation. Because this mutation strikingly impairs viral replicative capacity, its emergence requires the simultaneous presence of one or more compensatory mutations, including S173(A/T), N252(S/H), and E260D, which can correct these defects (22, 38, 67–69). We showed previously that one reason the R264K mutation impacts viral fitness is that it can strongly increase sensitivity to hTRIM5α (38). The requirement for multiple mutations to provide CTL escape without compromising viral replicative capacity is thought to explain the often late emergence of escape to the KK10 epitope (70, 71).
Unlike what was observed for HLA-B*57+ patients, the higher hTRIM5α sensitivity of viruses from HLA-B*27+ patients that spontaneously controlled viral replication was not explained by the selection of CTL resistance mutations in the HLA-B*27-restricted KK10 epitope. The frequency of selection of the L268M mutation was similar in viremic patients and controllers (Fig. 5; 5/9 and 3/6, respectively, P = 1.0). Interestingly, viruses from 5/9 viremic patients had additional CTL resistance mutations in the KK10 epitope (R264K, n = 4; K263R, n = 1), whereas none of the viruses from controllers carried these mutations (P = 0.04 using Fisher's exact test). The absence of additional KK10 resistance mutations in controllers could not be explained by weak pressure from CTL, since IFN-γ ELISPOT responses by the patient's PBMC to the wild-type KK10 peptide, studied for 4 patients, were generally robust (3,998, 5,212, 5,358, and 398 spot-forming cells/106 PBMC). Consistent with prior studies, the R264K mutation in the viremic patients was always accompanied by the compensatory mutations S173(A/T) or E260D. In addition, subtype B viruses from viremic patients, but not those from controllers, were more likely to carry N252(S/H) mutations, irrespective of the presence of other KK10 resistance or compensatory mutations (P = 0.02; S252 is the consensus sequence for subtype D and CRF02_AG viruses). No single mutation or combination of mutations were identified in viruses from HLA-B*27+ patients whose presence was significantly correlated with increased hTRIM5α sensitivity (data not shown). Taken together, these findings suggest that in HLA-B*27+ controllers, viral replication is constrained through a combination of strong CTL pressure and viral sensitivity to hTRIM5α. The conjunction of these two pressures may reduce viral replication below the threshold necessary to select the multiple mutations required for escape, allowing the patients to maintain controller status.
Fig 5.

Capsid sequence of viruses from HLA-B*27+ patients. Shown are the subtype and partial amino acid sequence of CA for viruses from HLA-B*27+ patients whose viral load in the absence of treatment was >500 RNA copies/ml (viremic) or <500 copies/ml (controller). The HLA-B*27-restricted KK10 epitope (amino acids 263 to 272) and the position of common compensatory mutations (amino acids 173, 252, and 260) have been highlighted. Note that viruses from patient 57C4, who was both HLA-B*27+ and HLA-B*57+, had the T242N mutation in the HLA-B*57-restricted TW10 epitope, which is likely to have contributed to their increased hTRIM5α sensitivity.
Viral sensitivity to hTRIM5α and selection of rare CTL resistance mutations.
Viruses from one of the HLA-B*57+ controller patients (patient 57C3; see Table S1 in the supplemental material) carried the unusual Q244R and G248D mutations in theTW10 epitope, mutations that have previously been observed exclusively in patients with spontaneous control of HIV replication (20, 72–74). Viruses from this patient had low sensitivity to hTRIM5α, consistent with the absence of the T242N mutation. The appearance of these rare mutations is thought to foster host control of viral replication, because they can carry an increased fitness cost relative to the T242N mutation and can continue to be targeted by CTL responses (20, 74). The selective pressure that leads to the emergence of these seemingly deleterious mutations remains undefined, and we evaluated this question for the patient in our series.
A possible explanation for the emergence of such rare mutations in the TW10 epitope is that the usual T242N mutation would not provide escape from the patient's CTL response. This was not the case for our patient. We observed positive IFN-γ ELISPOT responses by the patient's PBMC to the wild-type TW10 peptide but not to peptides containing the T242N mutation or T242N and G248A (Fig. 6A). In contrast, IFN-γ ELISPOT responses were stronger against the peptide carrying the Q244R and G248D mutations expressed by the patient's virus than those to the wild-type peptide. This seemingly paradoxical finding may indicate that acquisition of the Q244R and G248D mutations initially conferred escape to a preexisting CTL response directed at the wild-type sequence, but the patient subsequently mounted an efficient secondary response targeting the mutated epitope. This phenomenon has been previously described for responses to viruses carrying only the G248D mutation. The G248D mutation can provide escape from CTL targeting the wild-type sequence (25), but following its introduction, the secondary CTL response recognizes the mutant epitope better than the wild-type sequence (20).
Fig 6.
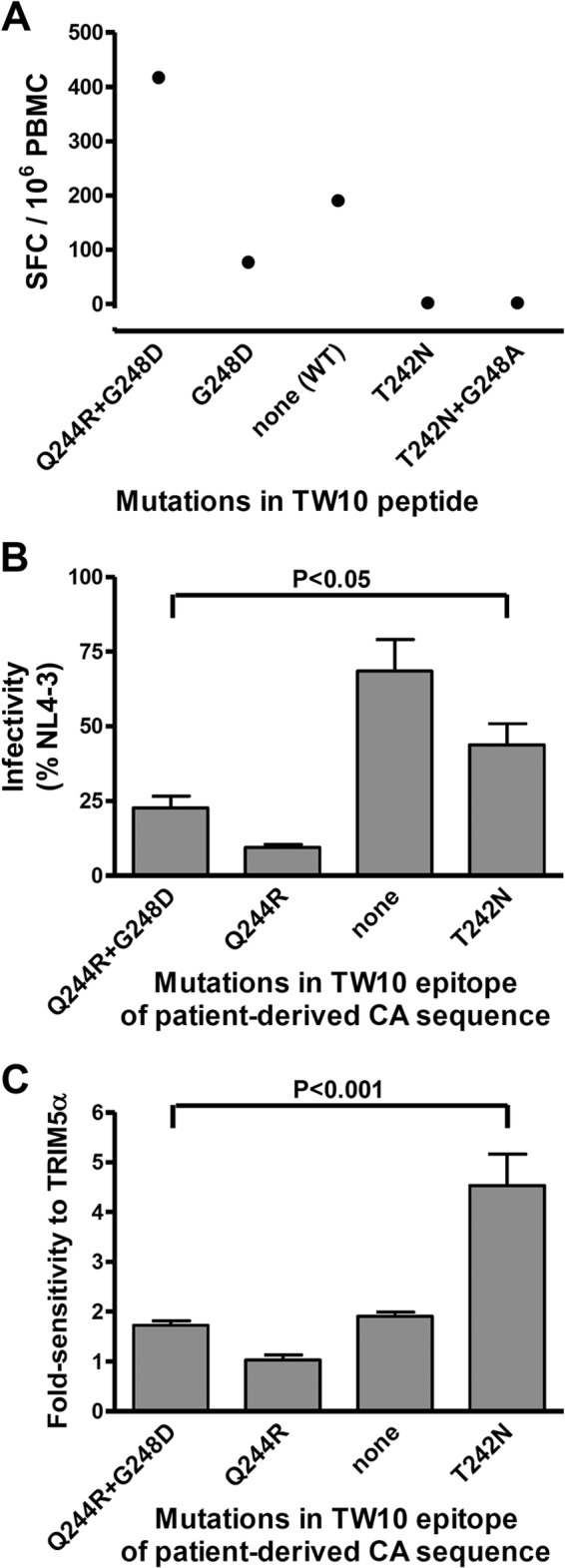
Effect of mutations in the TW10 epitope on CTL recognition, viral infectivity, and sensitivity to hTRIM5α. (A) PBMC from patient 57C3, who spontaneously controlled viral replication, were incubated with synthetic peptides having the consensus sequence of the TW10 epitope [TSTLQEQIGW, none (WT)] or peptides containing the indicated resistance mutations, and numbers of spot-forming cells (SFC) were measured using an IFN-γ-ELISPOT assay. (B) Recombinant viruses expressing Renilla luciferase in the place of Nef and carrying the CA sequence of viruses from the same patient (Q244R+G248D) and variants in which the indicated resistance mutations in the TW10 epitope had been added or removed were created. Single-cycle infectivity was measured using a luciferase-based assay after infection of U373-X4 cells in which hTRIM5α activity had been inhibited by stable overexpression of untagged hTRIM5γ (U373-X4-TRIM5γ). Results are expressed as a percentage of those obtained for a similar virus expressing the CA sequence of NL4-3. (C) The fold sensitivity to hTRIM5α of these viruses was also measured as described in the Fig. 1 legend. Results in panels B and C are the means ± SEM from 4 experiments performed using viruses obtained from two independent transfections. Statistical analysis was performed by analysis of variance followed by Bonferroni's multiple comparison test.
Another possible explanation for the emergence of these mutations is that their inhibitory effect on viral replicative capacity could be less than that produced by T242N. To test this possibility, we created a recombinant NL4-3-based virus carrying the CA sequence from the patient's virus along with variants in which resistance mutations in the TW10 epitope were removed or added and evaluated the single-cycle infectivity of these viruses in target cells in which hTRIM5α activity had been blocked by overexpression of hTRIM5γ. Viruses expressing the patient's CA sequence, which includes the Q244R and G248D mutations in the TW10 epitope, had an infectivity in cells not expressing hTRIM5α that was <25% that of NL4-3, and removing the G248D mutation further impaired infectivity (Fig. 6B). Mutations in the TW10 epitope made a strong contribution to the low infectivity of this virus, because removing both of these mutations improved infectivity to 69% of that of NL4-3. Importantly, the infectivity of viruses carrying the T242N mutation was significantly better than that of the patient's viruses expressing the Q244R and G248D mutations. Thus, the Q244R and G248D mutations were selected over T242N despite their greater negative impact on replicative capacity.
Since neither the efficiency of CTL resistance nor a protective effect on viral replicative capacity could explain the selection of Q244R and G248D over the T242N mutation, we considered the possibility that avoidance of sensitivity to hTRIM5α might be a driving force. Consistent with this possibility, viruses carrying the CA sequence from this patient had low sensitivity to hTRIM5α (Fig. 6C), and removing the G248D mutation or Q244R and G248D had no significant effect on this parameter, suggesting that the original virus had intrinsically low sensitivity to hTRIM5α. Inserting the T242N mutation in place of Q244R and G248D, however, led to a significant increase in hTRIM5α sensitivity, approaching values seen for the most sensitive viruses from other patients in this series. Thus, the selection of rare capsid mutations that carry a substantial fitness cost may be explained by the dual constraint of escaping an active CTL response while maintaining low sensitivity to hTRIM5α.
DISCUSSION
Among all the genetic determinants studied, the expression of HLA-B*57 and HLA-B*27 is among the parameters that are most strongly associated with a better outcome in HIV infection (8). The findings in this study suggest that pressure exerted by hTRIM5α helps explain this unusually strong protective effect. We found that the hTRIM5α sensitivity of viruses from both HLA-B*57+ and HLA-B*27+ patients who spontaneously controlled viral replication was significantly greater than that of viruses from patients not expressing these protective HLA-B alleles and observed a significant negative correlation between hTRIM5α sensitivity and viral load for the 56 HIV-infected individuals evaluated. Interestingly, the mechanisms responsible for protection differed for patients expressing HLA-B*57 and HLA-B*27 alleles. Viruses from HLA-B27+ controllers were intrinsically more sensitive to hTRIM5α, presumably reflecting the context-dependent effects of polymorphisms expressed by these viruses. Multiple simultaneous mutations are needed to escape CTL pressure in HLA-B*27+ individuals, and additional mutations are likely required to reduce hTRIM5α sensitivity. Given the low levels of viral replication in these controllers, the probability of acquiring all these mutations simultaneously is low, and the virus remains effectively blocked by the combination of pressure from CTLs and TRIM5α. In contrast, for HLA-B*57+ patients, the CTL escape mutation T242N in the immunodominant TW10 epitope conferred increased hTRIM5α susceptibility to subtype B viruses and reduced the replicative capacity of the escaped viruses in hTRIM5α-expressing target cells. Viruses from some HLA-B*57+ HIV controllers introduce rare escape mutations in this epitope, but these mutations, which allow the virus to retain low hTRIM5α sensitivity, can carry a substantial fitness cost. Thus, escape from CTL pressure in individuals expressing protective HLA-B alleles may fail either because the virus is unable to select necessary resistance/compensatory mutations or must settle for suboptimal escape mutations that increase hTRIM5α sensitivity or impair viral replicative capacity.
Viruses from both HLA-B*27+ and HLA-B*57+ patients who controlled viral load to low levels had greater hTRIM5α sensitivity than those from viremic patients. In this context, two additional findings should be emphasized. First, although the T242N mutation was strongly linked to increased hTRIM5α sensitivity in viruses from HLA-B*57+ patients, our findings confirm that additional CA polymorphisms can produce this phenotype in patients expressing other alleles. Second, we found that the selective pressure exerted by hTRIM5α has the potential to impair viral replicative capacity in the absence of emergence of hTRIM5α-sensitive viruses. Miura et al. (20) previously showed that viruses from HLA-B*57+ patients who can control viral replication, but not those from viremic patients, can forego the T242N mutation and select alternative escape mutations in the TW10 epitope. These rare mutations can have several liabilities. Importantly, they often carry a substantial fitness cost (20). In addition, although mutations such as G248D do provide escape from CTL targeting the wild-type epitope, they are susceptible to targeting by secondary CTL responses directed at the mutant epitope. The combined impairment of viral replicative capacity and persisting CTL pressure is thought to promote control of viral replication in these patients. Our results explain the persistence of mutants with apparently deleterious properties by indicating that avoidance of hTRIM5α sensitivity may be the dominant pressure contributing to their emergence. For the patient studied by us, selection of the T242N mutation, rather than the Q244R and G248D mutations, would have resulted in excellent escape from CTL recognizing the TW10 epitope and would have impaired replicative capacity to a much smaller extent. However, the T242N mutation would have rendered the virus quite sensitive to hTRIM5α, and this may have favored the emergence of the alternative rare mutations. Similarly, avoidance of sensitivity to hTRIM5α could explain why viruses from some HLA-B*57+ patients do not develop resistance mutations in key epitopes despite the presence of strong CTL responses directed against these epitopes (25, 61, 75). Thus, pressure from hTRIM5α can influence viral replication even in cases where viral sensitivity to hTRIM5α, per se, is not observed.
The replicative capacity of viruses from HLA-B*27+ and HLA-B*57+ controllers was generally 3- to 5-fold higher in target cells not expressing hTRIM5α than in target cells expressing hTRIM5α. Although greater viral sensitivity to TRIM5α may be required to have a strong impact on cross-species transmission (76), studies in some simian models have shown that only modest changes in sensitivity to TRIM5α can influence both transmission and disease progression (49–52). For example, simian immunodeficiency virus SIVmac251 has been highly adapted for replication in Rhesus monkeys, and replication of this virus is inhibited by only 3- to 4-fold in cells expressing the most active allelic variants of Macaca mulatta TRIM5α and inhibited only 2-fold in cells expressing the less active allelic variants (51). Despite these small differences in TRIM5α activity, infected animals expressing the more active allelic variants were found to have significantly lower viral loads at set-point, lower depletion of central memory CD4 T cells, and a lower rate of progression to AIDS than animals expressing the less-active allelic variants. Similarly, 2- to 3-fold changes in viral replicative capacity due to drug resistance mutations or sequence variation in pol are associated with significant differences in viral load and CD4+ T cell counts (77–79). Thus, relatively small differences in viral replication, including those due to differences in TRIM5α sensitivity, appear to be clinically relevant in infected hosts.
For viruses infecting HLA-B*57+ individuals, maintaining low sensitivity to hTRIM5α does not necessarily require avoidance of the T242N mutation. We observed that some viruses carrying the T242N mutation expressed low hTRIM5α sensitivity, indicating that other polymorphisms in the CA also influence this phenotype. One such mutation identified by us is G248A in the TW10 epitope. This mutation is almost always found in association with T242N in viruses from HLA-B*57+ patients but can occur alone (20, 62, 73, 74, 80, 81). Unlike T242N, G248A alone is often not an effective escape variant for patients infected with subtype B viruses (19, 25), and its presence has little impact on CTL escape provided by T242N (62). These findings suggest that the beneficial effects of G248A in reducing hTRIM5α sensitivity and improving viral replicative capacity (38, 82) may contribute to its selection. In viruses that express both mutations, the order of appearance is usually not known, although several examples of G248A preceding T242N have been described (62, 73, 80, 81), consistent with the possibility that G248A may facilitate the subsequent emergence of T242N in some patients. Additional sites, including both intrasubtype and subtype-specific polymorphisms, also influenced hTRIM5α sensitivity, emphasizing the context dependence of this phenomenon. Further work will be required to comprehensively define the set of residues influencing hTRIM5α sensitivity. In this regard, Rahm et al. (83) recently identified 10 amino acids in CA under positive selection, including residues involved in CTL escape, and found that some of the minor variants showed increased sensitivity to hTRIM5α.
Finally, the extent that hTRIM5α is able to exert pressure on viral replication is likely to differ between individuals. Variation in hTRIM5α expression has been described (84), possibly reflecting polymorphisms in transcription factor-binding sites (85). In addition, overall hTRIM5α activity can be influenced by its induction by IFN-α (44, 86, 87) and the relative expression of hTRIM5α and the inhibitory hTRIM5 splicing variants (gamma, delta, iota) (88). Furthermore, several hTRIM5α allelic variants have been identified with impaired activity, including those carrying the H43Y and G249D polymorphisms, although both the effect of these polymorphisms on TRIM5α activity and the impact of expression of these variants on disease progression remain controversial (44, 83, 89–97). Interestingly, strain-specific differences in CA can also influence viral sensitivity to different TRIM5α allelic variants (44, 83). The occurrence of rare CA mutations with clearly reduced replicative capacity but low hTRIM5α sensitivity may be preferentially selected in individuals with particularly strong hTRIM5α-restricting activity. Conversely, expression of low hTRIM5α activity could explain why some patients with hTRIM5α-sensitive viruses were unable to control viral replication. Studies evaluating hTRIM5α mRNA levels in PBMC ex vivo have not found correlations with viral load in infected patients (84, 98), but further studies directly measuring hTRIM5α activity against autologous viruses in target cells from individual patients will be required to fully evaluate the variability of this parameter and its impact on replicative capacity and the evolutionary trajectory of the virus.
In conclusion, this study indicates that viral sensitivity to hTRIM5α can have an impact on the development of CTL resistance mutations in key HLA-B*57- and HLA-B*27-restricted epitopes. In order to develop resistance to CA epitopes, the virus has to adapt to a minimum of three simultaneous selective pressures mediated by CTLs, hTRIM5α, and structural CA constraints (82, 99). The evolution of the virus may follow different paths depending on the relative strengths of these three constraints, but with an outcome that will invariably tend to reduce viral replication, whether it is unable to introduce necessary resistance/compensatory mutations and remains sensitive to CTL pressure, escapes CTL pressure by introducing mutations that increase sensitivity to hTRIM5α, or escapes using alternative pathways that do not increase hTRIM5α sensitivity but carry an increased fitness cost and/or fail to provide optimal CTL escape. Taken together, these findings support the idea that pressure from hTRIM5α contributes to the control of viral replication observed in patients expressing protective HLA-B haplotypes.
Supplementary Material
ACKNOWLEDGMENTS
We thank the physicians who cared for the patients and the individuals who participated in this study for their cooperation.
This work was supported by grants from the Agence Nationale de Recherches sur le Sida et les Hépatites Virales (ANRS) and Sidaction. C.G. and E.B. were recipients of fellowships from the Assistance Publique—Hôpitaux de Paris and ANRS, respectively.
Footnotes
Published ahead of print 17 July 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01313-13.
REFERENCES
- 1. Jin X, Bauer DE, Tuttleton SE, Lewin S, Gettie A, Blanchard J, Irwin CE, Safrit JT, Mittler J, Weinberger L, Kostrikis LG, Zhang L, Perelson AS, Ho DD. 1999. Dramatic rise in plasma viremia after CD8(+) T cell depletion in simian immunodeficiency virus-infected macaques. J. Exp. Med. 189:991–998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Koup RA, Safrit JT, Cao Y, Andrews CA, McLeod G, Borkowsky W, Farthing C, Ho DD. 1994. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J. Virol. 68:4650–4655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, Racz P, Tenner-Racz K, Dalesandro M, Scallon BJ, Ghrayeb J, Forman MA, Montefiori DC, Rieber EP, Letvin NL, Reimann KA. 1999. Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283:857–860 [DOI] [PubMed] [Google Scholar]
- 4. Goonetilleke N, Liu MK, Salazar-Gonzalez JF, Ferrari G, Giorgi E, Ganusov VV, Keele BF, Learn GH, Turnbull EL, Salazar MG, Weinhold KJ, Moore S, Letvin N, Haynes BF, Cohen MS, Hraber P, Bhattacharya T, Borrow P, Perelson AS, Hahn BH, Shaw GM, Korber BT, McMichael AJ. 2009. The first T cell response to transmitted/founder virus contributes to the control of acute viremia in HIV-1 infection. J. Exp. Med. 206:1253–1272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mudd PA, Martins MA, Ericsen AJ, Tully DC, Power KA, Bean AT, Piaskowski SM, Duan L, Seese A, Gladden AD, Weisgrau KL, Furlott JR, Kim YI, Veloso de Santana MG, Rakasz E, Capuano S, III, Wilson NA, Bonaldo MC, Galler R, Allison DB, Piatak M, Jr, Haase AT, Lifson JD, Allen TM, Watkins DI. 2012. Vaccine-induced CD8+ T cells control AIDS virus replication. Nature 491:129–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mellors JW, Rinaldo CR, Jr, Gupta P, White RM, Todd JA, Kingsley LA. 1996. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science 272:1167–1170 [DOI] [PubMed] [Google Scholar]
- 7. Streeck H, Jolin JS, Qi Y, Yassine-Diab B, Johnson RC, Kwon DS, Addo MM, Brumme C, Routy JP, Little S, Jessen HK, Kelleher AD, Hecht FM, Sekaly RP, Rosenberg ES, Walker BD, Carrington M, Altfeld M. 2009. Human immunodeficiency virus type 1-specific CD8+ T-cell responses during primary infection are major determinants of the viral set point and loss of CD4+ T cells. J. Virol. 83:7641–7648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carrington M, Walker BD. 2012. Immunogenetics of spontaneous control of HIV. Annu. Rev. Med. 63:131–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Goulder PJ, Walker BD. 2012. HIV and HLA class I: an evolving relationship. Immunity 37:426–440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dinges WL, Richardt J, Friedrich D, Jalbert E, Liu Y, Stevens CE, Maenza J, Collier AC, Geraghty DE, Smith J, Moodie Z, Mullins JI, McElrath MJ, Horton H. 2010. Virus-specific CD8+ T-cell responses better define HIV disease progression than HLA genotype. J. Virol. 84:4461–4468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Geldmacher C, Currier JR, Herrmann E, Haule A, Kuta E, McCutchan F, Njovu L, Geis S, Hoffmann O, Maboko L, Williamson C, Birx D, Meyerhans A, Cox J, Hoelscher M. 2007. CD8 T-cell recognition of multiple epitopes within specific Gag regions is associated with maintenance of a low steady-state viremia in human immunodeficiency virus type 1-seropositive patients. J. Virol. 81:2440–2448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kiepiela P, Ngumbela K, Thobakgale C, Ramduth D, Honeyborne I, Moodley E, Reddy S, de Pierres C, Mncube Z, Mkhwanazi N, Bishop K, van der Stok M, Nair K, Khan N, Crawford H, Payne R, Leslie A, Prado J, Prendergast A, Frater J, McCarthy N, Brander C, Learn GH, Nickle D, Rousseau C, Coovadia H, Mullins JI, Heckerman D, Walker BD, Goulder P. 2007. CD8+ T-cell responses to different HIV proteins have discordant associations with viral load. Nat. Med. 13:46–53 [DOI] [PubMed] [Google Scholar]
- 13. Streeck H, Lichterfeld M, Alter G, Meier A, Teigen N, Yassine-Diab B, Sidhu HK, Little S, Kelleher A, Routy JP, Rosenberg ES, Sekaly RP, Walker BD, Altfeld M. 2007. Recognition of a defined region within p24 Gag by CD8+ T cells during primary human immunodeficiency virus type 1 infection in individuals expressing protective HLA class I alleles. J. Virol. 81:7725–7731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Novitsky V, Gilbert P, Peter T, McLane MF, Gaolekwe S, Rybak N, Thior I, Ndung'u T, Marlink R, Lee TH, Essex M. 2003. Association between virus-specific T-cell responses and plasma viral load in human immunodeficiency virus type 1 subtype C infection. J. Virol. 77:882–890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zuniga R, Lucchetti A, Galvan P, Sanchez S, Sanchez C, Hernandez A, Sanchez H, Frahm N, Linde CH, Hewitt HS, Hildebrand W, Altfeld M, Allen TM, Walker BD, Korber BT, Leitner T, Sanchez J, Brander C. 2006. Relative dominance of Gag p24-specific cytotoxic T lymphocytes is associated with human immunodeficiency virus control. J. Virol. 80:3122–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boutwell CL, Rowley CF, Essex M. 2009. Reduced viral replication capacity of human immunodeficiency virus type 1 subtype C caused by cytotoxic-T-lymphocyte escape mutations in HLA-B57 epitopes of capsid protein. J. Virol. 83:2460–2468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Crawford H, Lumm W, Leslie A, Schaefer M, Boeras D, Prado JG, Tang J, Farmer P, Ndung'u T, Lakhi S, Gilmour J, Goepfert P, Walker BD, Kaslow R, Mulenga J, Allen S, Goulder PJ, Hunter E. 2009. Evolution of HLA-B*5703 HIV-1 escape mutations in HLA-B*5703-positive individuals and their transmission recipients. J. Exp. Med. 206:909–921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Crawford H, Prado JG, Leslie A, Hue S, Honeyborne I, Reddy S, van der Stok M, Mncube Z, Brander C, Rousseau C, Mullins JI, Kaslow R, Goepfert P, Allen S, Hunter E, Mulenga J, Kiepiela P, Walker BD, Goulder PJ. 2007. Compensatory mutation partially restores fitness and delays reversion of escape mutation within the immunodominant HLA-B*5703-restricted Gag epitope in chronic human immunodeficiency virus type 1 infection. J. Virol. 81:8346–8351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Martinez-Picado J, Prado JG, Fry EE, Pfafferott K, Leslie A, Chetty S, Thobakgale C, Honeyborne I, Crawford H, Matthews P, Pillay T, Rousseau C, Mullins JI, Brander C, Walker BD, Stuart DI, Kiepiela P, Goulder P. 2006. Fitness cost of escape mutations in p24 Gag in association with control of human immunodeficiency virus type 1. J. Virol. 80:3617–3623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Miura T, Brockman MA, Schneidewind A, Lobritz M, Pereyra F, Rathod A, Block BL, Brumme ZL, Brumme CJ, Baker B, Rothchild AC, Li B, Trocha A, Cutrell E, Frahm N, Brander C, Toth I, Arts EJ, Allen TM, Walker BD. 2009. HLA-B57/B*5801 human immunodeficiency virus type 1 elite controllers select for rare Gag variants associated with reduced viral replication capacity and strong cytotoxic T-lymphocyte [corrected] recognition. J. Virol. 83:2743–2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schneidewind A, Brockman MA, Sidney J, Wang YE, Chen H, Suscovich TJ, Li B, Adam RI, Allgaier RL, Mothe BR, Kuntzen T, Oniangue-Ndza C, Trocha A, Yu XG, Brander C, Sette A, Walker BD, Allen TM. 2008. Structural and functional constraints limit options for cytotoxic T-lymphocyte escape in the immunodominant HLA-B27-restricted epitope in human immunodeficiency virus type 1 capsid. J. Virol. 82:5594–5605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schneidewind A, Brockman MA, Yang R, Adam RI, Li B, Le Gall S, Rinaldo CR, Craggs SL, Allgaier RL, Power KA, Kuntzen T, Tung CS, LaBute MX, Mueller SM, Harrer T, McMichael AJ, Goulder PJ, Aiken C, Brander C, Kelleher AD, Allen TM. 2007. Escape from the dominant HLA-B27-restricted cytotoxic T-lymphocyte response in Gag is associated with a dramatic reduction in human immunodeficiency virus type 1 replication. J. Virol. 81:12382–12393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wright JK, Naidoo VL, Brumme ZL, Prince JL, Claiborne DT, Goulder PJ, Brockman MA, Hunter E, Ndung'u T. 2012. Impact of HLA-B*81-associated mutations in HIV-1 Gag on viral replication capacity. J. Virol. 86:3193–3199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Almeida JR, Price DA, Papagno L, Arkoub ZA, Sauce D, Bornstein E, Asher TE, Samri A, Schnuriger A, Theodorou I, Costagliola D, Rouzioux C, Agut H, Marcelin AG, Douek D, Autran B, Appay V. 2007. Superior control of HIV-1 replication by CD8+ T cells is reflected by their avidity, polyfunctionality, and clonal turnover. J. Exp. Med. 204:2473–2485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen H, Ndhlovu ZM, Liu D, Porter LC, Fang JW, Darko S, Brockman MA, Miura T, Brumme ZL, Schneidewind A, Piechocka-Trocha A, Cesa KT, Sela J, Cung TD, Toth I, Pereyra F, Yu XG, Douek DC, Kaufmann DE, Allen TM, Walker BD. 2012. TCR clonotypes modulate the protective effect of HLA class I molecules in HIV-1 infection. Nat. Immunol. 13:691–700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dahirel V, Shekhar K, Pereyra F, Miura T, Artyomov M, Talsania S, Allen TM, Altfeld M, Carrington M, Irvine DJ, Walker BD, Chakraborty AK. 2011. Coordinate linkage of HIV evolution reveals regions of immunological vulnerability. Proc. Natl. Acad. Sci. U. S. A. 108:11530–11535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Perez CL, Larsen MV, Gustafsson R, Norstrom MM, Atlas A, Nixon DF, Nielsen M, Lund O, Karlsson AC. 2008. Broadly immunogenic HLA class I supertype-restricted elite CTL epitopes recognized in a diverse population infected with different HIV-1 subtypes. J. Immunol. 180:5092–5100 [DOI] [PubMed] [Google Scholar]
- 28. Berger CT, Frahm N, Price DA, Mothe B, Ghebremichael M, Hartman KL, Henry LM, Brenchley JM, Ruff LE, Venturi V, Pereyra F, Sidney J, Sette A, Douek DC, Walker BD, Kaufmann DE, Brander C. 2011. High-functional-avidity cytotoxic T lymphocyte responses to HLA-B-restricted Gag-derived epitopes associated with relative HIV control. J. Virol. 85:9334–9345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Betts MR, Nason MC, West SM, De Rosa SC, Migueles SA, Abraham J, Lederman MM, Benito JM, Goepfert PA, Connors M, Roederer M, Koup RA. 2006. HIV nonprogressors preferentially maintain highly functional HIV-specific CD8+ T cells. Blood 107:4781–4789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Vingert B, Benati D, Lambotte O, de Truchis P, Slama L, Jeannin P, Galperin M, Perez-Patrigeon S, Boufassa F, Kwok WW, Lemaitre F, Delfraissy JF, Theze J, Chakrabarti LA. 2012. HIV controllers maintain a population of highly efficient Th1 effector cells in contrast to patients treated in the long term. J. Virol. 86:10661–10674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Day CL, Kaufmann DE, Kiepiela P, Brown JA, Moodley ES, Reddy S, Mackey EW, Miller JD, Leslie AJ, DePierres C, Mncube Z, Duraiswamy J, Zhu B, Eichbaum Q, Altfeld M, Wherry EJ, Coovadia HM, Goulder PJ, Klenerman P, Ahmed R, Freeman GJ, Walker BD. 2006. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature 443:350–354 [DOI] [PubMed] [Google Scholar]
- 32. Hersperger AR, Pereyra F, Nason M, Demers K, Sheth P, Shin LY, Kovacs CM, Rodriguez B, Sieg SF, Teixeira-Johnson L, Gudonis D, Goepfert PA, Lederman MM, Frank I, Makedonas G, Kaul R, Walker BD, Betts MR. 2010. Perforin expression directly ex vivo by HIV-specific CD8 T-cells is a correlate of HIV elite control. PLoS Pathog. 6:e1000917. 10.1371/journal.ppat.1000917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Migueles SA, Laborico AC, Shupert WL, Sabbaghian MS, Rabin R, Hallahan CW, Van Baarle D, Kostense S, Miedema F, McLaughlin M, Ehler L, Metcalf J, Liu S, Connors M. 2002. HIV-specific CD8+ T cell proliferation is coupled to perforin expression and is maintained in nonprogressors. Nat. Immunol. 3:1061–1068 [DOI] [PubMed] [Google Scholar]
- 34. Migueles SA, Osborne CM, Royce C, Compton AA, Joshi RP, Weeks KA, Rood JE, Berkley AM, Sacha JB, Cogliano-Shutta NA, Lloyd M, Roby G, Kwan R, McLaughlin M, Stallings S, Rehm C, O'Shea MA, Mican J, Packard BZ, Komoriya A, Palmer S, Wiegand AP, Maldarelli F, Coffin JM, Mellors JW, Hallahan CW, Follman DA, Connors M. 2008. Lytic granule loading of CD8+ T cells is required for HIV-infected cell elimination associated with immune control. Immunity 29:1009–1021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Deeks SG, Walker BD. 2007. Human immunodeficiency virus controllers: mechanisms of durable virus control in the absence of antiretroviral therapy. Immunity 27:406–416 [DOI] [PubMed] [Google Scholar]
- 36. Theze J, Chakrabarti LA, Vingert B, Porichis F, Kaufmann DE. 2011. HIV controllers: a multifactorial phenotype of spontaneous viral suppression. Clin. Immunol. 141:15–30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Blankson JN. 2010. Control of HIV-1 replication in elite suppressors. Discov. Med. 9:261–266 [PubMed] [Google Scholar]
- 38. Battivelli E, Migraine J, Lecossier D, Yeni P, Clavel F, Hance AJ. 2011. Gag cytotoxic T lymphocyte escape mutations can increase sensitivity of HIV-1 to human TRIM5alpha, linking intrinsic and acquired immunity. J. Virol. 85:11846–11854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, Sodroski J. 2006. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc. Natl. Acad. Sci. U. S. A. 103:5514–5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim J, Tipper C, Sodroski J. 2011. Role of the TRIM5α RING domain E3 ubiquitin ligase activity in capsid disassembly, reverse transcription blockade and restriction of simian immunodeficiency virus. J. Virol. 85:8116–8132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Pertel T, Hausmann S, Morger D, Zuger S, Guerra J, Lascano J, Reinhard C, Santoni FA, Uchil PD, Chatel L, Bisiaux A, Albert ML, Strambio-De-Castillia C, Mothes W, Pizzato M, Grutter MG, Luban J. 2011. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472:361–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Johnson WE, Sawyer SL. 2009. Molecular evolution of the antiretroviral TRIM5 gene. Immunogenetics 61:163–176 [DOI] [PubMed] [Google Scholar]
- 43. Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. 2004. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427:848–853 [DOI] [PubMed] [Google Scholar]
- 44. Battivelli E, Lecossier D, Matsuoka S, Migraine J, Clavel F, Hance AJ. 2010. Strain-specific differences in the impact of human TRIM5alpha, different TRIM5alpha alleles, and the inhibition of capsid-cyclophilin A interactions on the infectivity of HIV-1. J. Virol. 84:11010–11019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hatziioannou T, Perez-Caballero D, Cowan S, Bieniasz PD. 2005. Cyclophilin interactions with incoming human immunodeficiency virus type 1 capsids with opposing effects on infectivity in human cells. J. Virol. 79:176–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Keckesova Z, Ylinen LM, Towers GJ. 2006. Cyclophilin A renders human immunodeficiency virus type 1 sensitive to Old World monkey but not human TRIM5 alpha antiviral activity. J. Virol. 80:4683–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sokolskaja E, Berthoux L, Luban J. 2006. Cyclophilin A and TRIM5alpha independently regulate human immunodeficiency virus type 1 infectivity in human cells. J. Virol. 80:2855–2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Stremlau M, Song B, Javanbakht H, Perron M, Sodroski J. 2006. Cyclophilin A: an auxiliary but not necessary cofactor for TRIM5alpha restriction of HIV-1. Virology 351:112–120 [DOI] [PubMed] [Google Scholar]
- 49. Letvin NL, Rao SS, Montefiori DC, Seaman MS, Sun Y, Lim SY, Yeh WW, Asmal M, Gelman RS, Shen L, Whitney JB, Seoighe C, Lacerda M, Keating S, Norris PJ, Hudgens MG, Gilbert PB, Buzby AP, Mach LV, Zhang J, Balachandran H, Shaw GM, Schmidt SD, Todd JP, Dodson A, Mascola JR, Nabel GJ. 2011. Immune and genetic correlates of vaccine protection against mucosal infection by SIV in monkeys. Sci. Transl. Med. 3:81ra36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lim SY, Chan T, Gelman RS, Whitney JB, O'Brien KL, Barouch DH, Goldstein DB, Haynes BF, Letvin NL. 2010. Contributions of Mamu-A*01 status and TRIM5 allele expression, but not CCL3L copy number variation, to the control of SIVmac251 replication in Indian-origin rhesus monkeys. PLoS Genet. 6:e1000997. 10.1371/journal.pgen.1000997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lim SY, Rogers T, Chan T, Whitney JB, Kim J, Sodroski J, Letvin NL. 2010. TRIM5alpha modulates immunodeficiency virus control in rhesus monkeys. PLoS Pathog. 6:e1000738. 10.1371/journal.ppat.1000738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Rogers TF, Lim SY, Sundsvold TJ, Chan T, Hsu A, Letvin NL. 2010. Variability in a dominant block to SIV early reverse transcription in rhesus monkey cells predicts in vivo viral replication and time to death. Virol. J. 7:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Jordan M, Wurm F. 2004. Transfection of adherent and suspended cells by calcium phosphate. Methods 33:136–143 [DOI] [PubMed] [Google Scholar]
- 54. Matsuoka S, Dam E, Lecossier D, Clavel F, Hance AJ. 2009. Modulation of HIV-1 infectivity and cyclophilin A-dependence by Gag sequence and target cell type. Retrovirology 6:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nora T, Bouchonnet F, Labrosse B, Charpentier C, Mammano F, Clavel F, Hance AJ. 2008. Functional diversity of HIV-1 envelope proteins expressed by contemporaneous plasma viruses. Retrovirology 5:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lacabaratz-Porret C, Urrutia A, Doisne JM, Goujard C, Deveau C, Dalod M, Meyer L, Rouzioux C, Delfraissy JF, Venet A, Sinet M. 2003. Impact of antiretroviral therapy and changes in virus load on human immunodeficiency virus (HIV)-specific T cell responses in primary HIV infection. J. Infect. Dis. 187:748–757 [DOI] [PubMed] [Google Scholar]
- 57. Matthews PC, Leslie AJ, Katzourakis A, Crawford H, Payne R, Prendergast A, Power K, Kelleher AD, Klenerman P, Carlson J, Heckerman D, Ndung'u T, Walker BD, Allen TM, Pybus OG, Goulder PJ. 2009. HLA footprints on human immunodeficiency virus type 1 are associated with interclade polymorphisms and intraclade phylogenetic clustering. J. Virol. 83:4605–4615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. McMichael A, Klenerman P. 2002. HIV/AIDS. HLA leaves its footprints on HIV. Science 296:1410–1411 [DOI] [PubMed] [Google Scholar]
- 59. Descamps D, Chaix ML, Montes B, Pakianather S, Charpentier C, Storto A, Barin F, Dos Santos G, Krivine A, Delaugerre C, Izopet J, Marcelin AG, Maillard A, Morand-Joubert L, Pallier C, Plantier JC, Tamalet C, Cottalorda J, Desbois D, Calvez V, Brun-Vezinet F, Masquelier B, Costagliola D. 2010. Increasing prevalence of transmitted drug resistance mutations and non-B subtype circulation in antiretroviral-naive chronically HIV-infected patients from 2001 to 2006/2007 in France. J. Antimicrob. Chemother. 65:2620–2627 [DOI] [PubMed] [Google Scholar]
- 60. Fleury H, Recordon-Pinson P, Caumont A, Faure M, Roques P, Plantier JC, Couturier E, Dormont D, Masquelier B, Simon F. 2003. HIV type 1 diversity in France, 1999–2001: molecular characterization of non-B HIV type 1 subtypes and potential impact on susceptibility to antiretroviral drugs. AIDS Res. Hum. Retroviruses 19:41–47 [DOI] [PubMed] [Google Scholar]
- 61. Brockman MA, Schneidewind A, Lahaie M, Schmidt A, Miura T, Desouza I, Ryvkin F, Derdeyn CA, Allen S, Hunter E, Mulenga J, Goepfert PA, Walker BD, Allen TM. 2007. Escape and compensation from early HLA-B57-mediated cytotoxic T-lymphocyte pressure on human immunodeficiency virus type 1 Gag alter capsid interactions with cyclophilin A. J. Virol. 81:12608–12618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Leslie AJ, Pfafferott KJ, Chetty P, Draenert R, Addo MM, Feeney M, Tang Y, Holmes EC, Allen T, Prado JG, Altfeld M, Brander C, Dixon C, Ramduth D, Jeena P, Thomas SA, St. John A, Roach TA, Kupfer B, Luzzi G, Edwards A, Taylor G, Lyall H, Tudor-Williams G, Novelli V, Martinez-Picado J, Kiepiela P, Walker BD, Goulder PJ. 2004. HIV evolution: CTL escape mutation and reversion after transmission. Nat. Med. 10:282–289 [DOI] [PubMed] [Google Scholar]
- 63. Altfeld M, Addo MM, Rosenberg ES, Hecht FM, Lee PK, Vogel M, Yu XG, Draenert R, Johnston MN, Strick D, Allen TM, Feeney ME, Kahn JO, Sekaly RP, Levy JA, Rockstroh JK, Goulder PJ, Walker BD. 2003. Influence of HLA-B57 on clinical presentation and viral control during acute HIV-1 infection. AIDS 17:2581–2591 [DOI] [PubMed] [Google Scholar]
- 64. Altfeld M, Kalife ET, Qi Y, Streeck H, Lichterfeld M, Johnston MN, Burgett N, Swartz ME, Yang A, Alter G, Yu XG, Meier A, Rockstroh JK, Allen TM, Jessen H, Rosenberg ES, Carrington M, Walker BD. 2006. HLA alleles associated with delayed progression to AIDS contribute strongly to the initial CD8(+) T cell response against HIV-1. PLoS Med. 3:e403. 10.1371/journal.pmed.0030403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Schneidewind A, Tang Y, Brockman MA, Ryland EG, Dunkley-Thompson J, Steel-Duncan JC, St. John MA, Conrad JA, Kalams SA, Noel F, Allen TM, Christie CD, Feeney ME. 2009. Maternal transmission of human immunodeficiency virus escape mutations subverts HLA-B57 immunodominance but facilitates viral control in the haploidentical infant. J. Virol. 83:8616–8627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ladell K, Hashimoto M, Iglesias MC, Wilmann PG, McLaren JE, Gras S, Chikata T, Kuse N, Fastenackels S, Gostick E, Bridgeman JS, Venturi V, Arkoub ZA, Agut H, van Bockel DJ, Almeida JR, Douek DC, Meyer L, Venet A, Takiguchi M, Rossjohn J, Price DA, Appay V. 2013. A molecular basis for the control of preimmune escape variants by HIV-specific CD8(+) T cells. Immunity 38:425–436 [DOI] [PubMed] [Google Scholar]
- 67. Kelleher AD, Long C, Holmes EC, Allen RL, Wilson J, Conlon C, Workman C, Shaunak S, Olson K, Goulder P, Brander C, Ogg G, Sullivan JS, Dyer W, Jones I, McMichael AJ, Rowland-Jones S, Phillips RE. 2001. Clustered mutations in HIV-1 Gag are consistently required for escape from HLA-B27-restricted cytotoxic T lymphocyte responses. J. Exp. Med. 193:375–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Schneidewind A, Brumme ZL, Brumme CJ, Power KA, Reyor LL, O'Sullivan K, Gladden A, Hempel U, Kuntzen T, Wang YE, Oniangue-Ndza C, Jessen H, Markowitz M, Rosenberg ES, Sekaly RP, Kelleher AD, Walker BD, Allen TM. 2009. Transmission and long-term stability of compensated CD8 escape mutations. J. Virol. 83:3993–3997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Streeck H, Li B, Poon AF, Schneidewind A, Gladden AD, Power KA, Daskalakis D, Bazner S, Zuniga R, Brander C, Rosenberg ES, Frost SD, Altfeld M, Allen TM. 2008. Immune-driven recombination and loss of control after HIV superinfection. J. Exp. Med. 205:1789–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Feeney ME, Tang Y, Roosevelt KA, Leslie AJ, McIntosh K, Karthas N, Walker BD, Goulder PJ. 2004. Immune escape precedes breakthrough human immunodeficiency virus type 1 viremia and broadening of the cytotoxic T-lymphocyte response in an HLA-B27-positive long-term-nonprogressing child. J. Virol. 78:8927–8930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Goulder PJ, Phillips RE, Colbert RA, McAdam S, Ogg G, Nowak MA, Giangrande P, Luzzi G, Morgan B, Edwards A, McMichael AJ, Rowland-Jones S. 1997. Late escape from an immunodominant cytotoxic T-lymphocyte response associated with progression to AIDS. Nat. Med. 3:212–217 [DOI] [PubMed] [Google Scholar]
- 72. Bailey JR, Williams TM, Siliciano RF, Blankson JN. 2006. Maintenance of viral suppression in HIV-1-infected HLA-B*57+ elite suppressors despite CTL escape mutations. J. Exp. Med. 203:1357–1369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Durand CM, O'Connell KA, Apuzzo LG, Langan SJ, Imteyaz H, Ahonkhai AA, Ceccato CM, Williams TM, Margolick JB, Blankson JN. 2010. HIV-1 Gag evolution in recently infected human leukocyte antigen-B*57 patients with low-level viremia. AIDS 24:2405–2408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. O'Connell KA, Brennan TP, Bailey JR, Ray SC, Siliciano RF, Blankson JN. 2010. Control of HIV-1 in elite suppressors despite ongoing replication and evolution in plasma virus. J. Virol. 84:7018–7028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Migueles SA, Laborico AC, Imamichi H, Shupert WL, Royce C, McLaughlin M, Ehler L, Metcalf J, Liu S, Hallahan CW, Connors M. 2003. The differential ability of HLA B*5701+ long-term nonprogressors and progressors to restrict human immunodeficiency virus replication is not caused by loss of recognition of autologous viral Gag sequences. J. Virol. 77:6889–6898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Kirmaier A, Wu F, Newman RM, Hall LR, Morgan JS, O'Connor S, Marx PA, Meythaler M, Goldstein S, Buckler-White A, Kaur A, Hirsch VM, Johnson WE. 2010. TRIM5 suppresses cross-species transmission of a primate immunodeficiency virus and selects for emergence of resistant variants in the new species. PLoS Biol. 8:e1000462. 10.1371/journal.pbio.1000462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Barbour JD, Hecht FM, Wrin T, Segal MR, Ramstead CA, Liegler TJ, Busch MP, Petropoulos CJ, Hellmann NS, Kahn JO, Grant RM. 2004. Higher CD4+ T cell counts associated with low viral pol replication capacity among treatment-naive adults in early HIV-1 infection. J. Infect. Dis. 190:251–256 [DOI] [PubMed] [Google Scholar]
- 78. Deeks SG, Wrin T, Liegler T, Hoh R, Hayden M, Barbour JD, Hellmann NS, Petropoulos CJ, McCune JM, Hellerstein MK, Grant RM. 2001. Virologic and immunologic consequences of discontinuing combination antiretroviral-drug therapy in HIV-infected patients with detectable viremia. N. Engl. J. Med. 344:472–480 [DOI] [PubMed] [Google Scholar]
- 79. Nijhuis M, Deeks S, Boucher C. 2001. Implications of antiretroviral resistance on viral fitness. Curr. Opin. Infect. Dis. 14:23–28 [DOI] [PubMed] [Google Scholar]
- 80. Norstrom MM, Buggert M, Tauriainen J, Hartogensis W, Prosperi MC, Wallet MA, Hecht FM, Salemi M, Karlsson AC. 2012. Combination of immune and viral factors distinguishes low-risk versus high-risk HIV-1 disease progression in HLA-B*5701 subjects. J. Virol. 86:9802–9816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Feeney ME, Tang Y, Pfafferott K, Roosevelt KA, Draenert R, Trocha A, Yu XG, Verrill C, Allen T, Moore C, Mallal S, Burchett S, McIntosh K, Pelton SI, St. John MA, Hazra R, Klenerman P, Altfeld M, Walker BD, Goulder PJ. 2005. HIV-1 viral escape in infancy followed by emergence of a variant-specific CTL response. J. Immunol. 174:7524–7530 [DOI] [PubMed] [Google Scholar]
- 82. von Schwedler UK, Stray KM, Garrus JE, Sundquist WI. 2003. Functional surfaces of the human immunodeficiency virus type 1 capsid protein. J. Virol. 77:5439–5450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rahm N, Gfeller D, Snoeck J, Martinez R, McLaren PJ, Ortiz M, Ciuffi A, Telenti A. 2013. Susceptibility and adaptation to human TRIM5alpha alleles at positive selected sites in HIV-1 capsid. Virology 441:162–170 [DOI] [PubMed] [Google Scholar]
- 84. Sewram S, Singh R, Kormuth E, Werner L, Mlisana K, Karim SS, Ndung'u T. 2009. Human TRIM5alpha expression levels and reduced susceptibility to HIV-1 infection. J. Infect. Dis. 199:1657–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cagliani R, Fumagalli M, Biasin M, Piacentini L, Riva S, Pozzoli U, Bonaglia MC, Bresolin N, Clerici M, Sironi M. 2010. Long-term balancing selection maintains trans-specific polymorphisms in the human TRIM5 gene. Hum. Genet. 128:577–588 [DOI] [PubMed] [Google Scholar]
- 86. Carthagena L, Parise MC, Ringeard M, Chelbi-Alix MK, Hazan U, Nisole S. 2008. Implication of TRIM alpha and TRIMCyp in interferon-induced anti-retroviral restriction activities. Retrovirology 5:59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Sakuma R, Mael AA, Ikeda Y. 2007. Alpha interferon enhances TRIM5alpha-mediated antiviral activities in human and rhesus monkey cells. J. Virol. 81:10201–10206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Battivelli E, Migraine J, Lecossier D, Matsuoka S, Perez-Bercoff D, Saragosti S, Clavel F, Hance AJ. 2011. Modulation of TRIM5alpha activity in human cells by alternatively spliced TRIM5 isoforms. J. Virol. 85:7828–7835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Goldschmidt V, Bleiber G, May M, Martinez R, Ortiz M, Telenti A. 2006. Role of common human TRIM5alpha variants in HIV-1 disease progression. Retrovirology 3:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Javanbakht H, An P, Gold B, Petersen DC, O'Huigin C, Nelson GW, O'Brien SJ, Kirk GD, Detels R, Buchbinder S, Donfield S, Shulenin S, Song B, Perron MJ, Stremlau M, Sodroski J, Dean M, Winkler C. 2006. Effects of human TRIM5alpha polymorphisms on antiretroviral function and susceptibility to human immunodeficiency virus infection. Virology 354:15–27 [DOI] [PubMed] [Google Scholar]
- 91. Liu FL, Qiu YQ, Li H, Kuang YQ, Tang X, Cao G, Tang NL, Zheng YT. 2011. An HIV-1 resistance polymorphism in TRIM5alpha gene among Chinese intravenous drug users. J. Acquir. Immune Defic. Syndr. 56:306–311 [DOI] [PubMed] [Google Scholar]
- 92. Nakajima T, Nakayama EE, Kaur G, Terunuma H, Mimaya JI, Ohtani H, Mehra N, Shioda T, Kimura A. 2009. Impact of novel TRIM5alpha variants, Gly110Arg and G176del, on the anti-HIV-1 activity and the susceptibility to HIV-1 infection. AIDS 23:2091–2100 [DOI] [PubMed] [Google Scholar]
- 93. Nakayama EE, Carpentier W, Costagliola D, Shioda T, Iwamoto A, Debre P, Yoshimura K, Autran B, Matsushita S, Theodorou I. 2007. Wild type and H43Y variant of human TRIM5alpha show similar anti-human immunodeficiency virus type 1 activity both in vivo and in vitro. Immunogenetics 59:511–515 [DOI] [PubMed] [Google Scholar]
- 94. Price H, Lacap P, Tuff J, Wachihi C, Kimani J, Ball TB, Luo M, Plummer FA. 2010. A TRIM5alpha exon 2 polymorphism is associated with protection from HIV-1 infection in the Pumwani sex worker cohort. AIDS 24:1813–1821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Sawyer SL, Wu LI, Akey JM, Emerman M, Malik HS. 2006. High-frequency persistence of an impaired allele of the retroviral defense gene TRIM5alpha in humans. Curr. Biol. 16:95–100 [DOI] [PubMed] [Google Scholar]
- 96. Speelmon EC, Livingston-Rosanoff D, Li SS, Vu Q, Bui J, Geraghty DE, Zhao LP, McElrath MJ. 2006. Genetic association of the antiviral restriction factor TRIM5alpha with human immunodeficiency virus type 1 infection. J. Virol. 80:2463–2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. van Manen D, Rits MA, Beugeling C, van Dort K, Schuitemaker H, Kootstra NA. 2008. The effect of Trim5 polymorphisms on the clinical course of HIV-1 infection. PLoS Pathog. 4:e18. 10.1371/journal.ppat.0040018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Saez-Cirion A, Hamimi C, Bergamaschi A, David A, Versmisse P, Melard A, Boufassa F, Barre-Sinoussi F, Lambotte O, Rouzioux C, Pancino G. 2011. Restriction of HIV-1 replication in macrophages and CD4+ T cells from HIV controllers. Blood 118:955–964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Watts JM, Dang KK, Gorelick RJ, Leonard CW, Bess JW, Jr, Swanstrom R, Burch CL, Weeks KM. 2009. Architecture and secondary structure of an entire HIV-1 RNA genome. Nature 460:711–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.