

Abstract
It has been proposed that herpes simplex virus 1 with VP22 deleted requires secondary mutation of VHS for viability. Here we show that a replication-competent Δ22 virus constructed by homologous recombination maintains a wild-type (Wt) VHS gene and has no other gross mutations. By contrast, Δ22 viruses recovered from a bacterial artificial chromosome contain multiple amino acid changes within a conserved region of VHS. Hence, the mode of virus rescue influences the acquisition of secondary mutations.
TEXT
The herpes simplex virus 1 (HSV-1) virion host shutoff protein (VHS), encoded by gene UL41, is a tegument protein that is released into the cytoplasm of the infected cell, where it degrades both host and viral mRNA via an endoribonucleolytic activity, resulting in translational arrest (1–5). Although it has been believed for some time that VHS does not discriminate between cellular and viral mRNAs, a recent publication has suggested that it exhibits differential degradation of some classes of viral transcripts during infection (6). Nonetheless, it has been postulated that VHS must be downregulated in the infected cell to allow late virus proteins to be optimally expressed. Viruses with the major tegument protein VP16 deleted show unrestrained levels of translational arrest, a feature that can be rescued by additionally mutating the UL41 gene (7, 8). Furthermore, VP16 interacts directly with VHS (9), suggesting that VP16 may downregulate the activity of newly synthesized VHS late in infection by sequestering it. A second major tegument protein, VP22, is an additional binding partner of VP16 (10). Previous attempts to isolate a VP22 knockout virus by using a bacterial artificial chromosome (BAC) indicated that VP22 null viruses could be rescued only when gross secondary mutations in the UL41 gene had been acquired, thereby abrogating VHS activity (11). This led the authors to conclude that VHS is lethal for the virus in the absence of VP22. Other studies on a BAC-recovered Δ22 virus have also identified a spontaneous secondary frameshift mutation resulting in a truncated VHS (12, 13).
We have previously characterized an HSV-1 Δ22 virus which had been constructed by classical homologous recombination in the background of the strain 17 (s17) genome (14). This virus was originally recovered from a VP22 complementing cell line, but when it was found to replicate with wild-type (Wt) kinetics in noncomplementing Vero cells, it was subsequently propagated on Vero cells (14). In Vero cells Δ22 virus plaque size was reduced by only 50% compared to Wt size (Fig. 1A and B), suggesting that, in these cells at least, the virus was not substantially attenuated. Considering the results described above, we sequenced the Δ22 UL41 gene from DNA amplified by PCR from our current Δ22 virus stock and found that the UL41 gene was intact and had a sequence identical to that of the reference s17 sequence (GenBank no. JN555585.1; s17REF). To assess if other secondary mutations had been introduced to the virus, we carried out next-generation sequencing of the full Δ22 genome, as has been described previously (15). Sequence data sets were parsed through QUASR (16) and aligned against s17REF using BWA (17). The aligned read data were processed using SAMTools (18), and consensus sequences were generated. Single-nucleotide polymorphism (SNP) and indel differences between the Δ22 consensus sequence and s17REF were determined using BaseByBase (http://athena.bioc.uvic.ca/). This sequencing revealed no major changes in the Δ22 genome, with only 29 nucleotide changes compared to s17REF (0.02%), 14 of which resulted in coding changes, and one nucleotide deletion (Table 1). While our replication-competent Δ22 virus could have acquired viability by incorporating alternative secondary mutations such as in the VHS promoter or the UL13 kinase, deletion of which has been shown to result in a VHS null virus (19), no such mutations were apparent. Notably, we identified a single nucleotide polymorphism in the Δ22 UL41 gene, encoding a V to A change at residue 271, that had not been found by PCR sequencing. This change was present in around half of the genomic population, implying that the two variants were coreplicating (see Fig. 4B).
Fig 1.

HSV-1 lacking VP22 fails to package VHS. (A) Fifty PFU of s17 (Wt) and Δ22 viruses were plated on Vero cells and fixed and stained with crystal violet 4 days later. (B) The relative areas of Wt and Δ22 plaques from 3 separate experiments was measured using Image J software. Values are shown as means ± standard errors of the means of 10 plaques. (C) HeLa cells treated with 5 μg/ml actinomycin D were infected with s17 (Wt), Δvhs, or Δ22 viruses or left uninfected (M). Five hours later the cells were metabolically labeled with 50 μCi/ml [35S]methionine for 1 h, and total lysates were analyzed by SDS-PAGE and autoradiography. (D) Gradient-purified virions from s17 (Wt) and Δ22 viruses were analyzed by SDS-PAGE, followed by Coomassie blue staining (left) or Western blotting (right) with antibodies against the major capsid protein VP5 and the tegument proteins VP22, VP16, and VHS.
Table 1.
Coding changes in the genome of our s17 Δ22 virus
| s17REF locationa | ORFb | Gene productc | Coding change(s) in the Δ22 virus |
|---|---|---|---|
| 9984 | UL1/UL2 | gL/uracil DNA glycosylase | R216H/A34T |
| 26671 | UL12 | DNase | R73H |
| 26863 | UL12 | DNase | C9Y |
| 41209 | UL20 | Membrane protein | A94V |
| 47095 | UL23 | TK | R237C |
| 56878 | UL28 | DNA packaging | A428V |
| 60783 | UL29 | ssDNA binding protein | E424D |
| 68223 | UL32 | DNA packaging | A314T |
| 93835 | UL42 | DNA pol subunit | A242T |
| 109553 | UL52 | Helicase/primase | A169T |
| 115555 | UL55 | Nuclear matrix protein | Deletion of 1 nt |
| 134619 | US2 | Unknown | V105M |
| 140564 | US7 | Glycoprotein I | T259M |
| 140771 | US7 | Glycoprotein I | M328K |
| 143184 | US8A | Nucleolar phosphoprotein | C146Y |
Nucleotide numbers refer to the s17REF sequence (GenBank no. JN555585.1, deposited May 2012).
ORF, open reading frame.
gL, glycoprotein L; TK, thymidine kinase; ssDNA, single-stranded DNA; pol, polymerase.
Fig 4.

Rescue of an HSV-1 s17 Δ22 BAC produces multiple coding changes affecting amino acids in conserved box III of VHS. (A) Line drawing of the HSV-1 VHS gene open reading frame denoting the 4 conserved boxes (I to IV) and the VP16 binding domain (9, 27). (B) Amino acid variations identified in the original Δ22 virus, Δ22 viruses derived from a s17 BAC, the revertant of Δ22 (Δ22Rev), and strain F. Sequences have been compared to s17REF.
To investigate if VHS was functional in our Δ22 virions, we infected HeLa cells at a multiplicity of infection of 20 in the presence of 5 μg/ml actinomycin D to inhibit mRNA transcription, metabolically labeled cells 5 h later with 50 μCi/ml [35S]methionine for 1 h, and analyzed cell lysates by SDS-PAGE and autoradiography. As expected, in actinomycin D-treated cells Wt virus reduced the overall level of incorporated label compared to that in uninfected cells, indicating a shutoff of total protein synthesis (Fig. 1C, Wt), while a Δvhs virus had no effect on the level of protein synthesis (Fig. 1C, Δvhs). Interestingly, the Δ22 virus also failed to shut off protein synthesis, suggesting that no functional VHS was associated with the tegument of this virus. However, Western blotting of extracellular virus particles purified on Ficoll gradients and equalized by the major capsid protein VP5 revealed that this lack of activity was likely a consequence of greatly reduced VHS content in Δ22 virions compared to Wt virions (Fig. 1D), in spite of equal packaging of VP16 (14).
Western blotting of infected-cell lysates produced in Vero, HeLa, or BHK cells indicated that VHS was barely detectable in Δ22-infected cells (Fig. 2A), suggesting that the low virion content of VHS was a reflection of a low steady-state level of the protein. There are several possibilities for this low level of VHS in Δ22-infected cells, including reduced stability of the VHS protein, reduced transcription of the VHS gene, and reduced translation of the VHS mRNA. Of the noncoding changes found in the Δ22 genome, none were located in the VHS transcription unit, and hence there were no obvious secondary mutations that could affect VHS translation. However, other studies have shown that VHS protein levels but not mRNA levels are increased in transient transfection by coexpression of both VP16 and VP22, suggesting that VP22 itself may play a specific role in the accumulation of VHS protein in the cell (20). While a similar low level of cellular VHS has been reported in other Δ22 virus infections (21, 22), these infections have also been shown to exhibit a functional shutoff of protein synthesis (21). Likewise in our own virus, metabolic labeling of infected HeLa cells 8 and 16 h after infection revealed that overall protein synthesis was reduced in Δ22-infected cells compared to Wt or Δvhs infections at the later time (Fig. 2B), suggesting that, in spite of its low concentration, VHS expressed from this virus is functional and may lack regulation late in infection.
Fig 2.
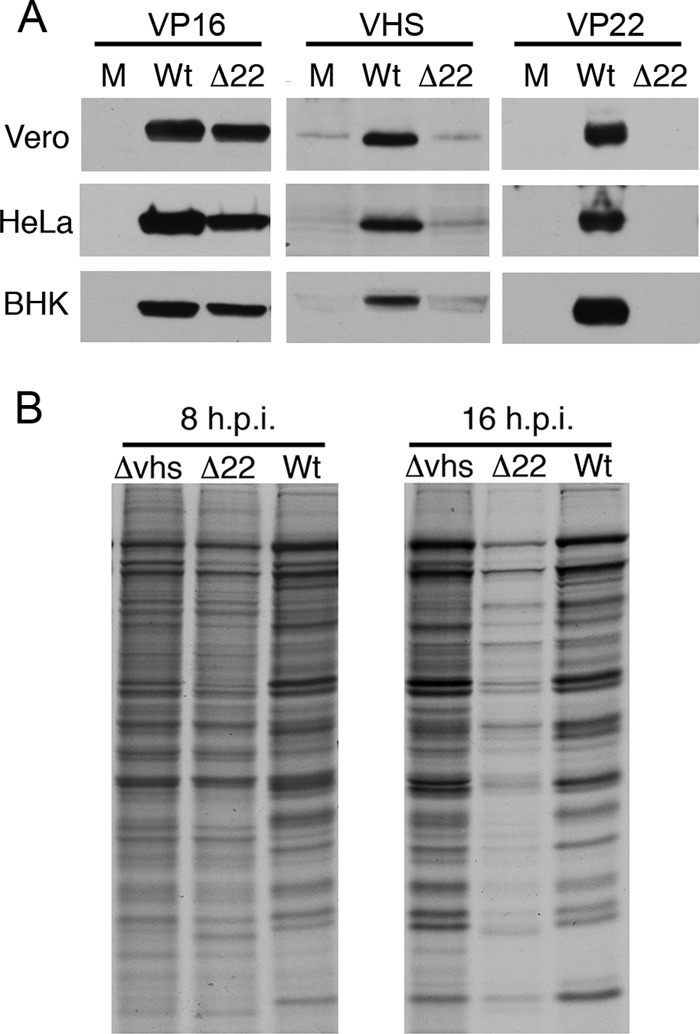
Relative protein shutoff in Δ22-infected cells. (A) Vero, HeLa, and BHK cells were infected with s17 (Wt) or Δ22 virus at a multiplicity of infection of 5 or left uninfected (M). After 16 h, total lysates were harvested and analyzed by SDS-PAGE and Western blotting for VP16, VHS, and VP22. (B) HeLa cells infected with s17 (Wt), Δvhs, or Δ22 virus at a multiplicity of infection of 5 were labeled at 8 or 16 h postinfection (h.p.i.) with 50 μCi/ml [35S]methionine for 30 min, and total lysates were analyzed by SDS-PAGE and autoradiography.
Taken together, these data suggest that our Δ22 virus expresses a Wt VHS protein that, despite its low abundance, functions in protein shutoff and does not undergo gross mutation during passaging in noncomplementing cells. There are three differences between the study described in reference 11 and our own work (14). First, our VP22 knockout virus was produced in s17, not strain F. Second, our virus was rescued in a complementing cell line, not Vero cells. Lastly, our s17 Δ22 virus was generated by homologous recombination, not from a BAC. Any combination of these differences may impact the outcome of virus rescue. The strain F VHS gene has 2 amino acid changes compared to s17 according to the published sequence (GenBank no. GU734771.1) and our own sequencing data (Fig. 4B), which may reflect different relative activities of the two proteins. However, comparison of the VHS activities associated with F and s17 virions showed that the two were equivalent (Fig. 3A, +Act D) and exhibited similar protein synthesis profiles in an ongoing infection (Fig. 3B, −Act D).
Fig 3.

Relative shutoff of protein synthesis in s17- and strain F-infected HeLa cells. HeLa cells treated with 5 μg/ml actinomycin D (Act D) (A) or left untreated (B) were infected with s17 or strain F or left uninfected (M). Cells were metabolically labeled at the indicated times with 50 μCi/ml [35S]methionine for 1 h (A) or 30 min (B), and total lysates were analyzed by SDS-PAGE and autoradiography.
To assess if the outcome for UL41 would be different if the s17 Δ22 virus was generated by BAC recombination, we replaced the VP22-encoding gene, UL49, in an infectious s17 BAC (23) with the kanamycin resistance gene aphA1 using Red-mediated recombination as described by others (24). The aphA1 cassette was PCR amplified from plasmid pEPkan-S using primers bearing 40-bp extensions homologous to the UL49 flanking region (Table 2), and the product was electroporated into Escherichia coli strain GS1783 harboring the s17 BAC. Kanamycin-resistant colonies were analyzed by PCR and restriction fragment length polymorphism and reconstituted by cotransfecting BHK cells with the recombinant BAC and a Cre recombinase-encoding plasmid (pGS403) to remove the BAC backbone. Under these conditions, although the UL41 sequence of the input BAC was confirmed as Wt, sequencing of nine UL41 clones generated by PCR cloning of rescued virus identified multiple coding changes in each of them (Fig. 4B, Δ22Bac). Many of the changes affected amino acids that clustered in box III of VHS, a conserved region that has been shown by others to contain residues essential for VHS activity (25, 26).
Table 2.
Sequences of PCR primers
| Primer | Sequencea (5′–3′) |
|---|---|
| Forward | TAATTGTCCG CGCATCCGAC CCTAGCGTGT TCGTGGAACC AGGATGACGACGATAAGTAGGG |
| Reverse | GAACCCCTGT TGGTGCTTTA TTGTCTGGGT ACGGAAGTTT ggttccacga acacgctagg gtcggatgcg cggacaatta CAACCAATTAACCAATTCTGATTAG |
Sequences annealing to plasmid pEPkan-S are underlined; duplicated reverse complementary sequences are in lowercase.
We conclude that our success in generating a Δ22 virus with Wt VHS was influenced by the mode of Δ22 virus rescue and that direct transfection of infectious DNA lacking the VP22 gene into cells where no VP22 is present leads to pressure to inactivate VHS at the first stage of virus rescue. In this respect, it is of interest that we found it highly problematic to generate a revertant virus of our original Δ22 virus, a process that also involves transfection of genomic DNA lacking the VP22 gene, and when UL41 in our Δ22Rev was sequenced, it was found to contain a single amino acid change (R257C) that was not present in the originating Δ22 genome (Fig. 4B, Δ22Rev). Hence, this study demonstrates that if the right conditions are used it is possible to generate and propagate an HSV-1 VP22 knockout virus that maintains an intact VHS sequence and, importantly, that VHS is not inherently lethal for virus replication in the absence of VP22.
ACKNOWLEDGMENTS
We thank David Leib, Klaus Osterrieder, and Greg Smith for providing the HSV-1 strain 17 BAC, plasmid pEPkan-S, and competent GS1783 bacteria, respectively. We also thank Stacey Efstathiou for help with constructing the s17 Δ22BAC, Duncan Wilson for the VHS antibody, and Roger Everett for the Δvhs virus. We thank Tony Brooks (UCL Genomics) for Illumina sequencing and acknowledge the infrastructure support provided by the MRC Centre for Molecular Medical Virology and the use of the UCL Legion High Performance Computing Facility and associated support services in the completion of this work.
This work was funded by Medical Research Council grants G0601605 & G0700814. J.B. receives funding from the National Institute for Health Research University College London Hospitals Biomedical Research Centre. L.H. is a BBSRC-funded CASE student.
Footnotes
Published ahead of print 17 July 2013
REFERENCES
- 1. Elgadi MM, Hayes CE, Smiley JR. 1999. The herpes simplex virus vhs protein induces endoribonucleolytic cleavage of target RNAs in cell extracts. J. Virol. 73:7153–7164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Smibert CA, Johnson DC, Smiley JR. 1992. Identification and characterization of the virion-induced host shutoff product of herpes simplex virus gene UL41. J. Gen. Virol. 73:467–470 [DOI] [PubMed] [Google Scholar]
- 3. Everly DN, Jr, Read GS. 1997. Mutational analysis of the virion host shutoff gene (UL41) of herpes simplex virus (HSV): characterization of HSV type 1 (HSV-1)/HSV-2 chimeras. J. Virol. 71:7157–7166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Read GS, Karr BM, Knight K. 1993. Isolation of a herpes simplex virus type 1 mutant with a deletion in the virion host shutoff gene and identification of multiple forms of the vhs (UL41) polypeptide. J. Virol. 67:7149–7160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Fenwick ML, Everett RD. 1990. Inactivation of the shutoff gene (Ul41) of herpes-simplex virus type-1 and type-2. J. Gen. Virol. 71:2961–2967 [DOI] [PubMed] [Google Scholar]
- 6. Taddeo B, Zhang WR, Roizman B. 2013. The herpes simplex virus host shutoff RNase degrades cellular and viral mRNAs made before infection but not viral mRNA made after infection. J. Virol. 87:4516–4522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lam Q, Smibert CA, Koop KE, Lavery C, Capone JP, Weinheimer SP, Smiley JR. 1996. Herpes simplex virus VP16 rescues viral mRNA from destruction by the virion host shutoff function. EMBO J. 15:2575–2581 [PMC free article] [PubMed] [Google Scholar]
- 8. Mossman KL, Sherburne R, Lavery C, Duncan J, Smiley JR. 2000. Evidence that herpes simplex virus VP16 is required for viral egress downstream of the initial envelopment event. J. Virol. 74:6287–6299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smibert CA, Popova B, Xiao P, Capone JP, Smiley JR. 1994. Herpes simplex virus VP16 forms a complex with the virion host shutoff protein vhs. J. Virol. 68:2339–2346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Elliott G, Mouzakitis G, O'Hare P. 1995. VP16 interacts via its activation domain with VP22, a tegument protein of herpes simplex virus, and is relocated to a novel macromolecular assembly in coexpressing cells. J. Virol. 69:7932–7941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sciortino MT, Taddeo B, Giuffre-Cuculletto M, Medici MA, Mastino A, Roizman B. 2007. Replication-competent herpes simplex virus 1 isolates selected from cells transfected with a bacterial artificial chromosome DNA lacking only the UL49 gene vary with respect to the defect in the UL41 gene encoding host shutoff RNase. J. Virol. 81:10924–10932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Duffy C, Lavail JH, Tauscher AN, Wills EG, Blaho JA, Baines JD. 2006. Characterization of a UL49-null mutant: VP22 of herpes simplex virus type 1 facilitates viral spread in cultured cells and the mouse cornea. J. Virol. 80:8664–8675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mbong EF, Woodley L, Dunkerley E, Schrimpf JE, Morrison LA, Duffy C. 2012. Deletion of the herpes simplex virus 1 UL49 gene results in mRNA and protein translation defects that are complemented by secondary mutations in UL41. J. Virol. 86:12351–12361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Elliott G, Hafezi W, Whiteley A, Bernard E. 2005. Deletion of the herpes simplex virus VP22-encoding gene (UL49) alters the expression, localization, and virion incorporation of ICP0. J. Virol. 79:9735–9745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Milne I, Bayer M, Cardle L, Shaw P, Stephen G, Wright F, Marshall D. 2010. Tablet—next generation sequence assembly visualization. Bioinformatics 26:401–402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Watson SJ, Welkers MR, Depledge DP, Coulter E, Breuer JM, de Jong MD, Kellam P. 2013. Viral population analysis and minority-variant detection using short read next-generation sequencing. Philos. Trans. R. Soc. Lond. B Biol. Sci. 368:20120205. 10.1098/rstb.2012.0205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Overton H, McMillan D, Hope L, Wong-Kai-In P. 1994. Production of host shutoff-defective mutants of herpes simplex virus type 1 by inactivation of the UL13 gene. Virology 202:97–106 [DOI] [PubMed] [Google Scholar]
- 20. Taddeo B, Sciortino MT, Zhang W, Roizman B. 2007. Interaction of herpes simplex virus RNase with VP16 and VP22 is required for the accumulation of the protein but not for accumulation of mRNA. Proc. Natl. Acad. Sci. U. S. A. 104:12163–12168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Duffy C, Mbong EF, Baines JD. 2009. VP22 of herpes simplex virus 1 promotes protein synthesis at late times in infection and accumulation of a subset of viral mRNAs at early times in infection. J. Virol. 83:1009–1017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Everly DN, Jr, Read GS. 1999. Site-directed mutagenesis of the virion host shutoff gene (UL41) of herpes simplex virus (HSV): analysis of functional differences between HSV type 1 (HSV-1) and HSV-2 alleles. J. Virol. 73:9117–9129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gierasch WW, Zimmerman DL, Ward SL, Vanheyningen TK, Romine JD, Leib DA. 2006. Construction and characterization of bacterial artificial chromosomes containing HSV-1 strains 17 and KOS. J. Virol. Methods 135:197–206 [DOI] [PubMed] [Google Scholar]
- 24. Tischer BK, Kaufer BB, Sommer M, Wussow F, Arvin AM, Osterrieder N. 2007. A self-excisable infectious bacterial artificial chromosome clone of varicella-zoster virus allows analysis of the essential tegument protein encoded by ORF9. J. Virol. 81:13200–13208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jones FE, Smibert CA, Smiley JR. 1995. Mutational analysis of the herpes simplex virus virion host shutoff protein: evidence that vhs functions in the absence of other viral proteins. J. Virol. 69:4863–4871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Kwong AD, Frenkel N. 1989. The herpes simplex virus virion host shutoff function. J. Virol. 63:4834–4839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Berthomme H, Jacquemont B, Epstein A. 1993. The pseudorabies virus host-shutoff homolog gene: nucleotide sequence and comparison with alphaherpesvirus protein counterparts. Virology 193:1028–1032 [DOI] [PubMed] [Google Scholar]