

Abstract
Vesicular stomatitis virus (VSV) is a promising oncolytic agent against various malignancies. Here, for the first time, we tested VSV in vitro and in vivo in a clinically relevant, immunocompetent mouse model of pancreatic ductal adenocarcinoma (PDA). Our system allows the study of virotherapy against PDA in the context of overexpression (80% of PDA patients) or no expression of human mucin 1 (MUC1), a major marker for poor prognosis in patients. In vitro, we tested three VSV recombinants, wild-type VSV, VSV-green fluorescent protein (VSV-GFP), and a safe oncolytic VSV-ΔM51-GFP, against five mouse PDA cell lines that either expressed human MUC1 or were MUC1 null. All viruses demonstrated significant oncolytic abilities independent of MUC1 expression, although VSV-ΔM51-GFP was somewhat less effective in two PDA cell lines. In vivo administration of VSV-ΔM51-GFP resulted in significant reduction of tumor growth for tested mouse PDA xenografts (+MUC1 or MUC1 null), and antitumor efficacy was further improved when the virus was combined with the chemotherapeutic drug gemcitabine. The antitumor effect was transient in all tested groups. The developed system can be used to study therapies involving various oncolytic viruses and chemotherapeutics, with the goal of inducing tumor-specific immunity while preventing premature virus clearance.
INTRODUCTION
Pancreatic cancer has the worst prognosis of all cancers and is estimated to be the fourth leading cause of cancer-related deaths in the United States (1). About 95% of pancreatic cancers are pancreatic ductal adenocarcinomas (PDAs), which are known to be highly invasive, with aggressive local growth and rapid metastases (2). To date, surgery remains the only potential cure for PDA. Other therapies, such as radiation therapy and chemotherapy, have shown little efficacy (3, 4). Thus, the development of new treatment strategies against PDA is of utmost importance.
PDA is generally driven by activating mutations in the KRAS proto-oncogene and is characterized by deregulation of several genes, including mucins (5, 6). In a tumor setting, the membrane-tethered glycoprotein mucin 1 (MUC1) becomes overexpressed and aberrantly glycosylated in more than 80% of human PDAs and in 100% of metastatic lesions (5). MUC1 plays an important role in the development and progression of PDA and other cancers and is a major marker for poor prognosis (7–11). Importantly, while the role of MUC1 in vesicular stomatitis virus (VSV) infection or oncolytic virus (OV) therapy has never been studied before, the O-linked carbohydrates of MUC1 purified from human breast milk can inhibit poxvirus (12), HIV (13, 14), and rotavirus (15), and MUC1 expression can block adeno-associated virus attachment (16).
OV therapy is an emerging therapeutic approach largely based on defects in the innate immunity of cancer cells or other abnormalities that increase cancer cell susceptibility to viral infection and virus-mediated death compared to healthy cells. VSV, a prototypic nonsegmented negative-strand RNA virus, has shown promising results against an array of cancers in preclinical studies (17) and is currently in a phase I clinical trial against hepatocellular carcinoma (trial NCT01628640). The undesirable natural neurotoxicity of wild-type (WT) VSV has been addressed by the generation of various VSV-based recombinants retaining their oncolytic activities but lacking neurotoxicity (17). One such oncolytic recombinant, VSV-ΔM51-GFP, has a deletion of the methionine at amino acid position 51 of the VSV matrix (M) protein, as well as a green fluorescent protein (GFP) open reading frame (ORF) inserted in position 5 of the viral genome (18). The ΔM51 mutation improves VSV oncoselectivity by preventing WT M protein's ability to shut down cellular gene expression (19–21). Therefore, VSV-ΔM51-GFP is unable to successfully replicate in healthy cells with intact type I interferon (IFN) responses. However, as many cancer cells are believed to have defective type I IFN signaling (22), they remain susceptible to VSV-ΔM51-GFP infection.
Our recent study analyzed several VSV recombinants in an array of human PDA cell lines in vitro (23, 24) and in xenografts in athymic mice (24). These studies demonstrated excellent abilities of VSV recombinants to infect and kill a majority of tested human PDAs and revealed that intact type I IFN signaling in some PDAs was responsible for their resistance to OV therapy (23). However, tumors in immunocompetent animals generate additional challenges for viruses, including the potential elimination of viruses before complete tumor killing can occur. Here, VSV was evaluated for the first time in an immunocompetent mouse PDA model. This system is based on xenografts of murine PDA cells originating from mice with spontaneous KRASG12D-driven PDAs (referred to as KC) either expressing human MUC1 (KCM cells) or MUC1 null (KCKO cells) (Fig. 1A) and thus allows for study of OV therapy in the context of MUC1 overexpression or lack of expression. This system can also be used to study combinational therapies involving chemotherapeutics or other combinational therapies. Therefore, we also examined VSV-ΔM51-GFP in combination with gemcitabine, the standard drug for treatment of pancreatic cancer.
Fig 1.

Mouse PDA cell lines used in this study. (A) KC mice producing KRASG12D-driven spontaneous PDAs (KC cells) were crossed with mice expressing human MUC1 (MUC1.Tg) or MUC1 null (MUC1KO) to generate the MUC1-positive KCM or MUC1-null KCKO cell lines, respectively. (B) MUC1 expression profile of PDA cell lines. For immunofluorescence (IF) analysis using confocal microscopy, cells were analyzed using HMFG2 antibody to detect the extracellular domain of human MUC1 and FITC-conjugated secondary antibody. Hoechst dye was used to stain for the nucleus, and wheat germ agglutinin (WGA) was used to stain the plasma membrane. For Western blot analysis, total cell lysates were separated by SDS-PAGE and then analyzed by Western blotting with HMFG2 antibody or CT2 antibody to detect the transmembrane domain of human MUC1. Western blotting using β-actin antibody was used as a loading control.
MATERIALS AND METHODS
Cell lines and culture.
The KC, KCM, and KCKO cell lines were generated from spontaneous PDA tumors in the corresponding mice (Fig. 1A). KC mice were generated on the C57BL/6 background by mating the P48-Cre mice with the LSL-KRASG12D mice (25). We generated the KC cell line (in which only mouse Muc1 is expressed) for this study using spontaneous PDA tumors from KC mice. The KCM and KCKO cells have been generated and characterized previously (7). The KCKO cells completely lack mouse Muc1 and human MUC1, while KCM cells express both mouse Muc1 and human MUC1. The murine cell line Panc02-Neo (transfected with neomycin empty vector) and Panc02-MUC1 (expressing full-length human MUC1) murine PDA cell line were a generous gift from Tony Hollingsworth (University of Nebraska) (26). In addition, 4T1 (murine mammary carcinoma; ATCC CRL-2539) and BHK-21 (Syrian golden hamster kidney fibroblasts; ATCC CCL-10) were used to grow VSV and/or as controls for viral replication. KCKO, KCM, KC, Panc02-MUC1, Panc02-Neo, and 4T1 cells were maintained in Dulbecco's modified Eagle's medium (DMEM; with 4.5 g/liter glucose, l-glutamine, and sodium pyruvate; Cellgro), and BHK-21 cells were maintained in modified Eagle's medium (MEM; Cellgro). MEM was also supplemented with 0.3% glucose (wt/vol). All cell growth media were supplemented with 9% fetal bovine serum (FBS; Gibco), 3.4 mM l-glutamine, 90 units (U) per ml penicillin, and 90 μg/ml streptomycin (Cellgro). Cells were kept in a 5% CO2 atmosphere at 37°C. The antibiotic G418 (30 mg/ml) was added to every other passage of Panc02-MUC1 and Panc02-Neo to select for cells maintaining the vector. For all experiments, cell lines were passaged no more than 10 times.
Immunofluorescence.
Cells were seeded in borosilicate glass chamber slides (Labtek catalog no. 155411) to be approximately 30% confluent in 24 h. Cells were washed with phosphate-buffered saline (PBS; Mediatech, Inc.) and then fixed with 3% paraformaldehyde (PFA) (Sigma-Aldrich) in distilled water (dH2O) for 15 min. Cells were permeabilized with a solution containing 20 mM HEPES (pH 7.5), 300 mM sucrose, 50 mM NaCl, 3 mM MgCl2, and 0.5% Triton X-100 on ice for 15 min, washed with PBS, and then blocked with 5% bovine serum albumin (BSA; Sigma-Aldrich) in PBS for 30 min, after which they were incubated with 1:100 HMFG2 antibody in 5% BSA at 4°C overnight. Cells were then incubated with secondary 1:100 anti-mouse-fluorescein isothiocyanate (FITC) antibody (catalog no. sc-2010; Santa Cruz Biotechnology, Inc.) in 5% BSA for 2 h at room temperature and then stained with 1 μM Hoechst and 1 mg/ml wheat germ agglutinin (WGA). Cells were washed with PBS and used for confocal imaging.
Western blotting.
Cellular lysates and Western blots were prepared as previously described (23). For MUC1, polyvinylidene difluoride (PVDF) membranes were incubated with 1:2,000 Armenian hamster monoclonal anti-human MUC1 cytoplasmic tail (CT2) (27), or 1:2,000 mouse HMFG2 monoclonal anti-human MUC1 (28) antibodies in Tris-buffered saline-Tween 20 (TBS-T) with 5% milk and 1% of 2% sodium azide. The HMFG2 antibody targets sparsely glycosylated variable-number tandem repeats (VNTR) within the human MUC1 extracellular domain. The CT2 antibody recognizes the last 17 amino acids (SSLSYNTPAVAATSANL) of the cytoplasmic tail (CT) of human MUC1 (29). Neither antibody allows for efficient detection of murine Muc1; the presence of murine Muc1 could not be confirmed. In addition, the following primary antibodies were used in TBS-T with 5% BSA and 1% of 2% sodium azide: 1:5,000 rabbit polyclonal anti-VSV antibodies (raised against VSV virion proteins), 1:1,000 rabbit anti-MX1/2/3 (catalog no. sc-5059; Santa Cruz Biotechnology, Inc.), 1:3,000 mouse anti-GFP (catalog no. 600-301-215; Rockland). Also used were the following antibodies from Cell Signaling (1:1,000): anti-STAT1 (catalog no. 9172), Stat1-P (catalog no. 9171), and IRF3-P (catalog no. 4947). The following horseradish peroxidase (HRP)-conjugated secondary antibodies in TBS-T with 5% milk antibodies were used: 1:4,000 goat antibody against Armenian hamster (Santa Cruz Biotechnology, Inc.; catalog no. sc-2443), and 1:4,000 goat anti-mouse and 1:2,000 goat anti-rabbit (catalog no. 115-035-003 and 111-035-003, respectively; Jackson-ImmunoResearch). The Amersham ECL Western Blotting Detection kit (catalog no. RPN2106; GE Healthcare) was used for detection. Membranes were reprobed with mouse anti-actin antibody (clone C4) to verify sample loading (30).
Viruses.
Recombinant wild-type VSV (VSV-rWT, Indiana serotype) (31) and VSV-ΔM51-GFP (18) were kindly provided by Jack Rose (Yale University), and VSV-GFP (32) was kindly provided by Asit Pattnaik (University of Nebraska). VSV stocks were prepared using BHK-21 cells infected at a multiplicity of infection (MOI) of 0.005 CIU (cell infectious units) and incubated at 37°C in MEM-based medium containing 5% FBS. Virus-containing medium was collected at 24 h postinfection (p.i.) and centrifuged at 3,000 × g for 10 min at room temperature to remove large cellular debris. Virus was purified by the method of Kalvodova et al. (33), with slight modifications. In brief, clarified supernatants were underlaid with 5 ml 20% (wt/vol) sucrose in HEN buffer (10 mM HEPES [pH 7.4], 1 mM EDTA, 100 mM NaCl) and centrifuged at 28,000 rpm for 3.5 h at 4°C in a Beckman SW32 Ti rotor. The resulting viral pellet was resuspended in HEPES buffered saline (HBS) (pH 7.5) (21 mM HEPES, 140 mM NaCl, 45 mM KCl, 0.75 mM Na2HPO4, 0.1% [wt/vol] dextrose) at 4°C overnight and then centrifuged in a 7.5 to 27.5% continuous gradient of Optiprep (Axis Shield) in HBS at 26.5 × 103 rpm for 30 min at 4°C using a Beckman SW40 Ti rotor. The virus-containing band was collected from the gradient, diluted with ET buffer (1 mM Tris-HCl, pH 7.5, 1 mM EDTA), pelleted by centrifugation at 27,000 rpm for 1.5 h at 4°C using a Beckman SW40 Ti rotor, and then resuspended in PBS.
Cell viability assay.
Cells (in triplicate) were seeded in 96-well plates so that they reached approximately 80% confluence at 24 h and then were infected with VSV at an MOI of 0.001, 0.1, or 10 CIU/cell (based on titration on KCKO cells) or mock infected in growth medium without FBS. Virus-containing medium was aspirated 1 h p.i. and replaced with growth medium containing 5% FBS. Virus replication was measured by GFP fluorescence readings approximately every 12 h p.i. for 5 days (CytoFluor Series 4000, with excitation filter of 450/20 nm, emission filter of 530/25 nm, and gain of 63; Perseptive Biosystems). Cell viability was analyzed 120 h p.i. by a 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) cell viability assay (Biotium).
Type I interferon sensitivity.
Cells were seeded in 24-well plates so that they reached approximately 80% confluence at 24 h. Cells were mock treated or treated with 5,000, 15,000, or 30,000 U/ml human alpha interferon (IFN-α) (catalog no. 407294; Calbiochem) in growth medium containing 5% FBS. Twenty-four hours posttreatment, the cells were infected with serial dilutions of VSV-ΔM51-GFP, and infectious foci were counted at 12 h p.i. by fluorescence microscopy. Treatments and infections were performed in duplicate.
Plaque reduction neutralization test.
BHK-21 cells were seeded in 96-well plates to reach confluence in 24 h. Mouse serum was first diluted 1:20 to 1:40,960 for analysis. VSV-ΔM51-GFP stock diluted 1:32,000 (a dilution determined to produce approximately 50 infectious foci per well) was incubated with the serum dilutions for 1 h at 37°C. Serum/virus dilutions were then used to infect cells for 1 h at 37°C, with rocking every 10 min. Serum/virus dilutions were removed and the cells overlaid with growth media with 5% FBS and 1% Bacto agar. Infectious foci were counted by fluorescence microscopy at 16 h p.i. Antibody dilution titers were calculated as the inverse of the serum dilution resulting in one-half of the number of foci obtained with VSV-ΔM51-GFP alone. All serum samples were tested in triplicate.
Detecting antibodies generated against KCM cells.
KCM cells were seeded in 96-well plates to reach confluence in 24 h. Cells were fixed and permeabilized as described above. Cells were blocked with 5% BSA in PBS for 20 min at room temperature and then incubated with mouse serum dilutions as prepared for the plaque reduction neutralization assay, but without incubating with virus, overnight at 4°C. Cells were washed with PBS and then incubated with peroxidase-conjugated goat anti-mouse IgG antibodies (1:300; Jackson ImmunoResearch) for 1 h. For detection, cells were washed with PBS and then incubated with o-phenylenediamine (OPD; Thermo Scientific) for 15 min. OPD was inactivated by addition of 2.5 M sulfuric acid. Optical density was read at 490 nm. All serum samples were tested in triplicate.
MUC1.Tg mice.
Mice were handled and maintained under veterinary supervision in accordance with the University of North Carolina at Charlotte Institutional Animal Care and Use Committee (IACUC) approved protocol. All experiments were conducted using MUC1.Tg mice (Fig. 1A). Previously generated and characterized MUC1.Tg mice (inbred CS7BI/6 background) express human MUC1 under its own promoter and in a tissue-specific manner, and these mice exhibit T and B cell tolerance when immunized with human MUC1 antigen, making it a relevant model to study (34). For genotypic confirmation, DNA from MUC1.Tg mice was isolated from tail clippings when mice were 11 to 17 days old and analyzed by PCR. Primers used for identification of MUC1-positive MUC1.Tg mice were 5′-CTTGCCAGCCATAGCACCAAG-3′ and 5′-CTCCACGTCGTGGACATTGATG-3′. Genotype was confirmed by the presence of a 340-bp amplification product seen on 1% agarose gels (35).
In vivo treatment of tumors with VSV-ΔM51-GFP.
All cell lines used in animal experiments were negative for an extended panel of pathogens as tested by Charles River Laboratories. For the short-term in vivo efficacy study, 16- to 18-week-old male MUC1.Tg mice (n = 29) were injected in the right flank with 1 × 106 KCM in 100 μl of PBS. Mice were palpated for tumor formation starting on day 5 post-tumor injection (p.t.i.) and then randomly divided into 5 groups: PBS, VSV-ΔM51-GFP, UV-killed VSV-ΔM51-GFP, VSV-ΔM51-GFP + gemcitabine, and gemcitabine alone (n = 6 per group, n = 5 for UV-killed VSV-ΔM51-GFP). At 5 days p.t.i., mice were treated once with a single intraperitoneal (i.p.) injection of either 50 μl PBS or gemcitabine (50 mg/kg of body weight) dissolved in 50 μl PBS. On days 7, 9, and 11 p.t.i., the PBS and gemcitabine groups received intratumoral (i.t.) administration of either 50 μl PBS or 1 × 108 CIU in 50 μl PBS (based on BHK-21 titer) of infectious VSV-ΔM51-GFP or UV-killed VSV-ΔM51-GFP. The same amounts of particles for infectious VSV-ΔM51-GFP or UV-killed VSV-ΔM51-GFP were used (based on virus titration prior to UV-mediated inactivation of the killed virus). Tumor size was monitored by caliper measurements every day until day 12 and every other day afterward. Body weight was measured once weekly. Tumor volume was calculated according to the following formula: volume in mm3 = [length in cm × (width in cm)2]/2. Mice were sacrificed 18 days p.t.i., at which time the animals showed no clinical signs indicating severe morbidity. To conduct a survival study using VSV-ΔM51-GFP against KCKO and KCM tumors, 8- to 11-week-old MUC1.Tg male mice were subcutaneously injected in the flank with either 1 × 106 KCM or KCKO cell lines in 100 μl of PBS (n = 8 each). Mice were palpated for tumor formation starting at 5 days p.t.i. and then were randomly divided into 2 groups per cell line (n = 4 per group). One group per cell line served as a control and received i.t. administration of 50 μl PBS on days 8, 10, and 12 p.t.i. The other group received i.t. administration of VSV-ΔM51-GFP on days 8, 10, and 12 p.t.i. with an initial dose of 7.2 × 107 CIU in 50 μl PBS (based on BHK-21 titer) followed by two doses of 4.3 × 107 CIU in 50 μl PBS. Tumor size was monitored by caliper measurements every other day, and body weight was measured once weekly. Tumor volume was calculated according to the following formula: volume in mm3 = [length in cm × (width in cm)2]/2. Mice were sacrificed when the length or width of the tumor reached 1.5 cm, the tumors became ulcerated, or the mice presented with clinical signs indicating severe morbidity. Data were analyzed using GraphPad software and are expressed as means ± standard errors of the means (SEM). Comparison of groups was done by two-way analysis of variance (ANOVA) only when the groups had the same number of animals (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Analysis of tumor samples.
Tumors were isolated at sacrifice and sectioned for analysis. One section was used to make tissue homogenate to check for the presence of viral RNA or infectious virus. Homogenized tumor sections were prepared in DMEM using a tissue homogenizer and then centrifuged at 13,000 rpm for 10 min at 4°C to remove large cellular debris. RNA was extracted from the supernatant using the Quick-RNA MiniPrep Kit (Zymo Research). RNA was reverse transcribed using random hexamers and SmartScribe reverse transcriptase (Clontech). The resulting cDNA was PCR amplified using the primers VG31, 5′-CCCAATCCATTCATCATGAGTTCC-3′, and VG32, 5′-CACTTCATAGTGACGCGTAAACAG-3′, which bind part of the intergenic region on either side of the VSV M gene. PCR was conducted for 35 or 40 cycles with an annealing temperature of 55°C. PCR products were electrophoresed on a 2% agarose gel stained with ethidium bromide and visualized using a GelDoc-It imager (UVP Imaging, Upland, CA). A second tumor section was used to make tissue lysates for Western blot analysis. Western blot detection was performed with tumor lysates, using 1:5,000 rabbit polyclonal anti-VSV antibodies (raised against VSV virions), 1:1,000 anti-VSV N antibodies, or 1:1,000 anti-VSV G antibodies. A third tumor section was formalin fixed and paraffin embedded and analyzed by hematoxylin and eosin (H&E) staining and by immunohistochemistry (IHC). IHC was performed to look for the presence of VSV N and G proteins (antibody dilution, 1:50).
Statistical analysis software.
All statistical analyses were performed using GraphPad Prism, version 5.0c for Mac OS X (GraphPad Software, San Diego, CA).
RESULTS
Susceptibility of murine PDA cells to VSV.
The immunocompetent model of PDA described in this study can be used with various mouse PDA cell lines either expressing human MUC1 or MUC1 null. VSV has never been tested (in vitro or in vivo) against any mouse PDA cells before. Our previous study with human PDA cells in athymic nude mice showed very good correlation between the oncolytic efficacy of VSV-ΔM51-GFP in vitro and in vivo (24). Therefore, we wanted to test first in vitro if VSV can infect and kill such PDA cell lines and whether oncolytic efficacy of VSV would be negatively affected by MUC1 overexpression in PDA cells. The first set of cell lines, KC, KCKO, and KCM (Fig. 1A), originate from spontaneous PDAs expressing or lacking the human (MUC1) and/or mouse (Muc1) mucin 1 gene (35, 36). KC cells express murine Muc1 while KCM cells express both murine Muc1 and human MUC1, and KCKO cells lack mucin 1 expression from either species. MUC1 expression in KC, KCKO, and KCM cells may not be the only difference between these cell lines, as an accumulation of additional mutations is likely during spontaneous PDA formation. Therefore, in addition to these cell lines, we also tested two isogenic cell lines, Panc02-Neo and Panc02-MUC1, which should differ only in their human MUC1 expression profile and were previously characterized in detail (26, 37). The MUC1 expression phenotypes of these five cell lines were confirmed by Western blotting and immunofluorescence (Fig. 1B) as well as flow cytometry (data not shown).
The major focus of our study was the recombinant VSV-ΔM51-GFP retaining its oncolytic abilities without the neurotoxicity associated with WT-like VSV (38, 39). In addition, the insertion of a GFP gene at position 5 of the VSV-ΔM51-GFP genome allows for monitoring of virus replication and spread based on VSV-driven GFP expression (18). To examine if the ΔM51 or GFP insertion would have any effect on the oncolytic abilities of VSV-ΔM51-GFP, our initial in vitro experiments included the VSV-GFP recombinant (similar to VSV-ΔM51-GFP but encoding WT M) and VSV-rWT (lacking either modification) for comparison. To analyze the ability of viruses to infect and kill the described mouse PDA cell lines in vitro, the cells were mock infected or infected at increasing MOIs: 0.001, 0.1, or 10 CIU/cell (MOI values were calculated based on the titration of viruses on KCKO cells, so the same amounts of infectious particles were added to each cell line). Virus replication was monitored by GFP fluorescence readings (for VSV-ΔM51-GFP and VSV-GFP) taken approximately every 12 h. Cell viability was determined using an MTT assay performed at 120 h p.i. As shown in Fig. 2, both VSV-rWT and VSV-GFP killed all cells by 120 h p.i. VSV-GFP behaved similarly to VSV-rWT, which is in agreement with previous reports indicating that insertion of the GFP gene into VSV-rWT produced a virus with similar oncolytic ability (40–42).
Fig 2.
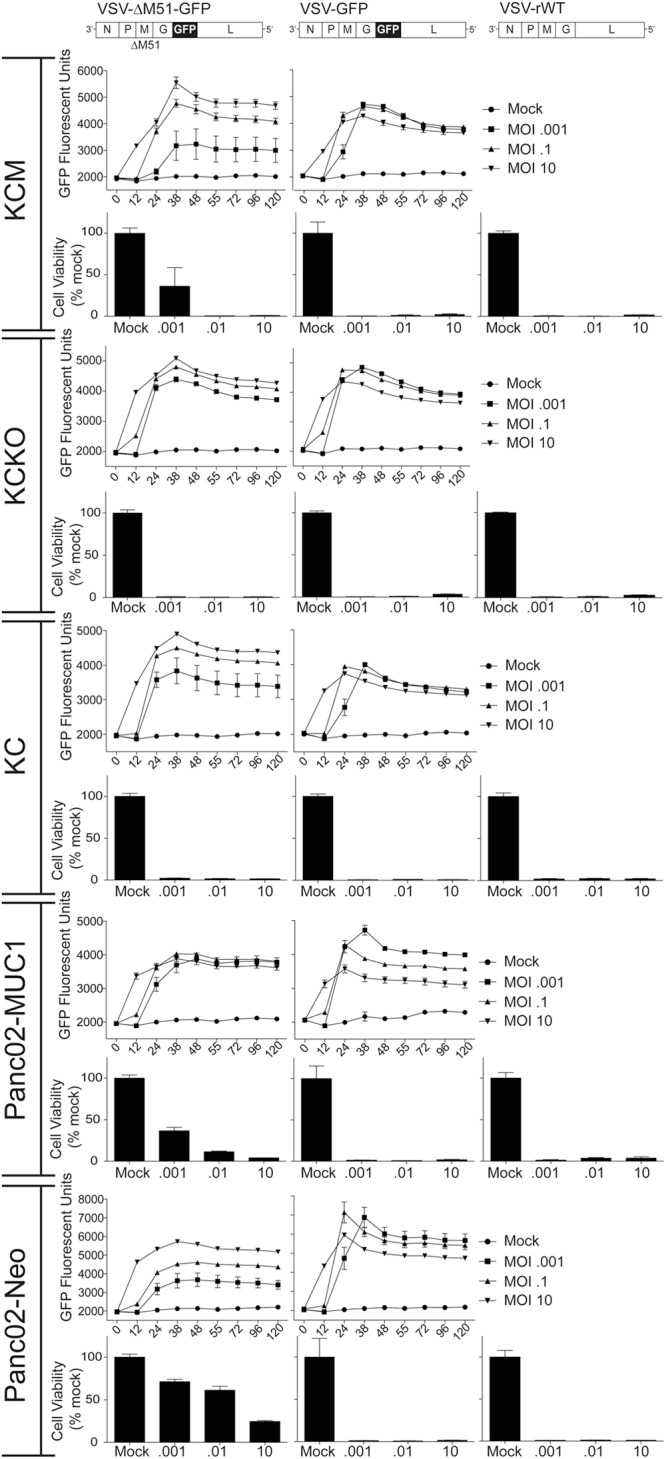
Mouse PDA cell viability following infection with VSV recombinants. Cells were mock infected or infected with viruses at an MOI of 0.001, 0.1, or 10.0 CIU/cell based on their titration on KCKO cells. VSV-ΔM51-GFP and VSV-GFP replication-driven GFP expression was measured by CytoFluor GFP fluorescence readings at the indicated time points. Cell viability was analyzed 120 h p.i. by an MTT cell viability assay and is expressed as a percentage of mock-treated cells. All MTT assays were done in triplicate, and the data represent the means ± SEM.
Both GFP expression and cell viability assay indicated that VSV-ΔM51-GFP was less effective in KCM and Panc02-MUC1 at an MOI of 0.001 and in Panc02-Neo at all tested MOIs (especially at MOIs of 0.001 and 1). This result could be explained by differences in the infectivity of this virus on these mouse PDA cell lines. To test it, the titer of VSV-ΔM51-GFP stock was determined on all five cell lines, and the oncolytic abilities of VSV-ΔM51-GFP correlated with its abilities to initiate infection in the tested cell lines (Fig. 3A). We hypothesized that if murine PDA cells differ only in their initial susceptibility to infection, then virus replication would be different after the lower-MOI infection but similar when all cells are infected at a higher MOI. To test this hypothesis, multistep (MOI of 0.001 CIU/cell) and one-step (MOI of 5 CIU/cell) growth kinetics of virus replication were examined (Fig. 3B). As predicted, VSV-ΔM51-GFP was somewhat attenuated in KCM, Panc02-MUC1, and Panc02-Neo when cells were infected at the cell type-specific MOI of 0.001, but it replicated very similarly in all tested cell lines when they were infected at the cell type-specific MOI of 5 (Fig. 3B).
Fig 3.
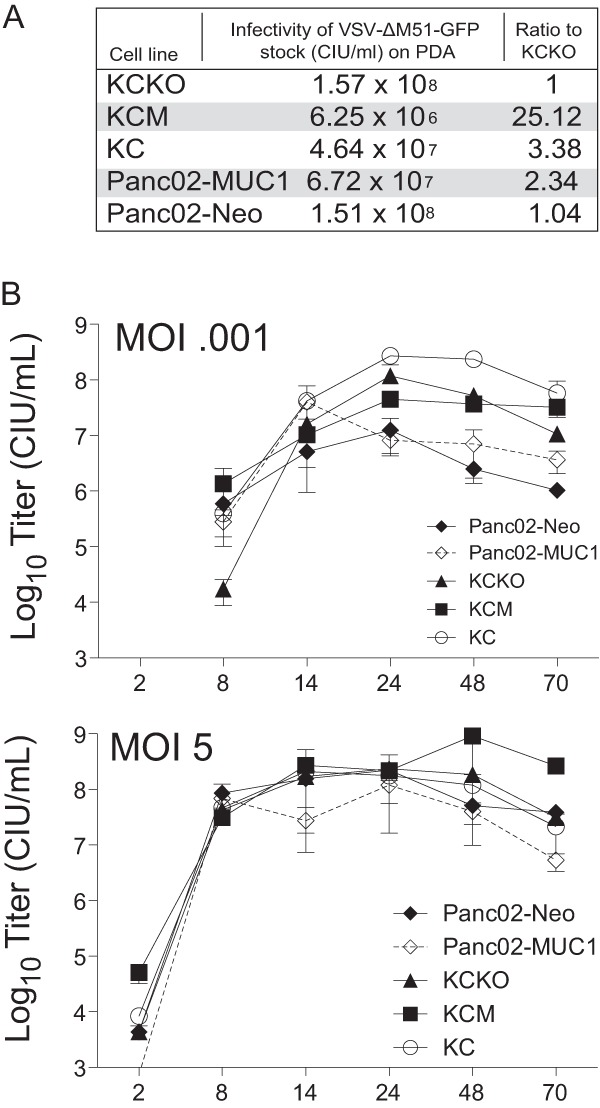
Infectivity and growth kinetics of VSV-ΔM51-GFP in mouse PDA cells. (A) Serial dilutions of VSV-ΔM51-GFP were used to infect PDA cell lines to calculate relative infectivity of this virus on tested cell lines. (B) For growth kinetics, cells were infected with VSV-ΔM51-GFP at a cell line-specific MOI of 0.001 or 5.0. At the times indicated, supernatant was collected, and virus titers were determined by plaque assay on BHK-21 cells. All infections were done in triplicate, and the data represent means ± SEM.
Type I IFN signaling in mouse PDA cell lines.
The observed attenuation of VSV-ΔM51-GFP (but not VSV-GFP) in KCM, Panc02-MUC1, and Panc02-Neo (at least at some tested MOIs) suggested that the M51 deletion in the M protein, rather than GFP insertion, was responsible for this attenuation. Because WT M protein prevents a robust innate antiviral response (the M51 deletion in M allows for nuclear export and translation of cellular mRNA), we hypothesized that intact (or residual) type I IFN signaling may play a role in the reduced susceptibility of KCM, Panc02-MUC1, and Panc02-Neo to VSV-ΔM51-GFP. This information is very important in predicting VSV success in vivo, as our recent analysis of a panel of human PDA cell lines showed that responsiveness to IFN-α treatment and the expression of the IFN-stimulated gene (ISG) MxA could be predictive of resistance to VSV-ΔM51-GFP in vitro as well as in vivo (23, 24). To test this hypothesis, all mouse PDA cell lines were tested for their IFN responsiveness. Cells were mock treated or treated with three different concentrations of human IFN-α and analyzed for virus infectivity after the treatment. As controls, we used IFN-α-responsive BHK-21 and nonresponsive 4T1 cells. Unlike BHK-21 cells (which showed a 1,000-fold decrease in VSV-ΔM51-GFP infectivity following IFN treatment), none of the tested mouse PDA cell lines mounted robust antiviral responses following IFN-α treatment (Fig. 4A). In addition, all cells were infected with VSV-ΔM51-GFP and cell lysates were collected at 4, 12, and 24 h p.i. Analysis of cell lysates indicated that the expression levels of Mx1, the murine version of human MxA, were similar for all cell lines and did not increase upon infection (Fig. 4B). However, while all the cell lines were able to sense virus infection (as determined by phosphorylation of IRF3), phosphorylation of STAT1 was observed at 12 and 24 h p.i. in Panc02-MUC1 and Panc02-Neo only. Together, our results indicate that while none of the tested mouse PDA cell lines have a robust type I IFN response, Panc02-MUC1 and Panc02-Neo demonstrated a limited antiviral response, which may explain their reduced susceptibility to VSV-ΔM51-GFP. A possible mechanism of VSV-ΔM51-GFP attenuation in KCM cells will be discussed below.
Fig 4.
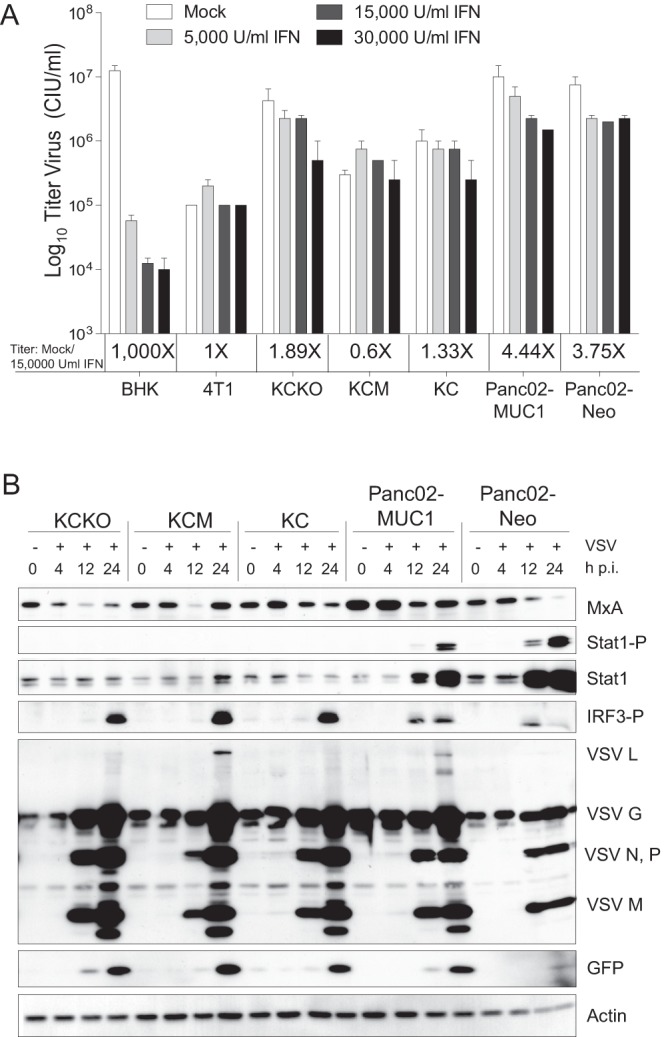
Type I interferon status of mouse PDA cell lines. (A) Monolayer cultures of PDA cells and control 4T1 and BHK-21 cell lines were mock treated or treated with 5,000, 15,000, or 30,000 U/ml human IFN-α in growth medium with 5% FBS. Twenty-four hours posttreatment, the cells were infected with serial dilution of VSV-ΔM51-GFP, and the infectious foci were counted 12 h p.i. by fluorescence microscopy to calculate the virus titer. Treatments and infections were performed in duplicate; the data represent the means ± SEM. (B) Expression of cellular antiviral proteins and VSV-ΔM51-GFP proteins during infection. Monolayer cultures of cells were infected with VSV-ΔM51-GFP at an MOIBHK-21 of 10.0. Total cell lysates were collected at the indicated time points. Lysates were separated by SDS-PAGE and then analyzed by Western blotting with antibodies to detect the indicated proteins. Western blotting using β-actin antibody was used as a loading control.
Efficacy of VSV-ΔM51-GFP in vivo in immunocompetent Muc1.Tg mice.
Based on previous studies that demonstrated unacceptable neurotoxicity of VSV-rWT and VSV-GFP, we decided to conduct our in vivo experiments with VSV-ΔM51-GFP (43–45). We focused our initial experiment on mice bearing KCM tumors, given that MUC1-expressing tumors are more clinically relevant and more challenging. Previous comparative studies of KCM and KCKO cell lines in vitro and in vivo demonstrated that KCM cells display a much more aggressive phenotype, evidenced by an increase in invasiveness of KCM cells, an increase in proliferation, and deregulation of the mitogen-activated protein kinase (MAPK) pathway (7). The efficacy of VSV-ΔM51-GFP was compared to that of gemcitabine, the most common chemotherapeutic used against pancreatic cancer (46). In addition, the efficacy of OV therapy alone was also compared to combinational therapy (“chemovirotherapy”) using both VSV-ΔM51-GFP and gemcitabine.
Subcutaneous injections of KCM cells were used to establish tumors in the flank of MUC1.Tg mice, which express human MUC1 under its own promoter and in a tissue-specific manner (34, 47). The MUC1.Tg mice exhibit T and B cell tolerance when immunized with human MUC1 antigen, making it a relevant model to study (34).
On day 5, when tumors were palpable, mice were treated i.p. with a single dose of gemcitabine (50 mg/kg in PBS) or PBS control. Depending on the group, tumors were injected i.t. with VSV-ΔM51-GFP, UV-killed VSV-ΔM51-GFP, or PBS on days 7, 9, and 11 (Fig. 5A). UV-killed virus was used as a control to determine whether viral replication was required for antitumor effects of VSV-ΔM51-GFP and if the presence of viral components alone without virus replication would affect tumor progression. Mice were monitored for signs of distress, and tumor size was measured daily for the first 8 days and then every other day afterward. Mice were sacrificed 18 days p.t.i., at which time the animals showed no clinical signs that would indicate severe morbidity. KCM tumors injected with PBS as a control continued to grow at a steady rate. In agreement with previously published data demonstrating resistance of KCM tumors to gemcitabine, tumor growth with gemcitabine alone was comparable to that observed with PBS treatment (48). Treatment with VSV-ΔM51-GFP alone and VSV-ΔM51-GFP plus gemcitabine showed a statistically significant reduction in tumor burden beginning on day 12 compared to PBS treatment (Fig. 5A). This significance was maintained until day 18, at which point the mice were sacrificed. The greatest therapeutic effect was seen in the combinational therapy of VSV-ΔM51-GFP plus gemcitabine, which showed a significant reduction in tumor burden compared to the use of virus alone at day 18 (Fig. 5A). Surprisingly, KCM tumors injected with UV-killed virus grew larger than the PBS control, with a significantly increased tumor burden on days 16 and 18. It is unclear why UV-killed virus would result in enhanced tumor growth, and we are planning to address this question in future studies.
Fig 5.
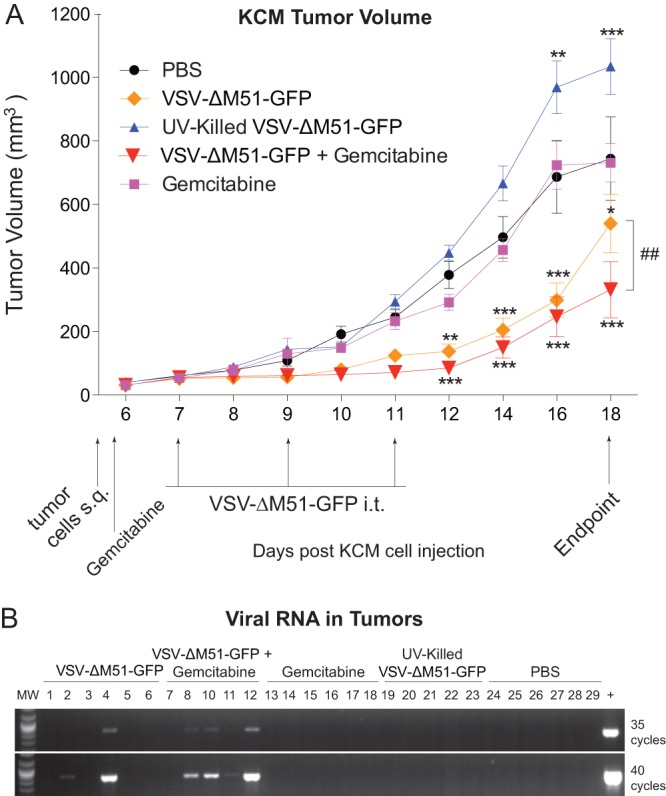
In vivo short-term efficacy of VSV-ΔM51-GFP against KCM tumors. (A) MUC1.Tg male mice, 16 to 18 weeks old, were subcutaneously (s.q.) injected with KCM cells in the right flank (n = 30). Tumors were established by day 5, and the mice were randomly divided into 5 groups (n = 6 per group). On day 5, mice were administered one dose of gemcitabine or PBS i.p. On day 7, treatments began with groups being administered 108 CIU VSV-ΔM51-GFP, UV-killed VSV-ΔM51-GFP, or PBS three times, on days 7, 9, and 11. Tumor size was monitored by caliper measurements, and tumor weight was calculated according to the standard ellipsoid formula: weight in grams = (length in cm × width2)/2. Mice were sacrificed at day 18 p.t.i. Bonferroni post hoc tests compared all groups to PBS and VSV-ΔM51-GFP to VSV-ΔM51-GFP + gemcitabine (*, P < 0.05; **, P < 0.01; ***, P < 0.001; ##, P < 0.01). (B) At the endpoint (as indicated in panel A), tumor sections from each group were homogenized, and RNA was extracted from the supernatant. RNA was reverse transcribed, and the resulting cDNA was PCR amplified using the primers that bind part of the intergenic region on either side of the VSV M gene. PCR was conducted for 35 or 40 cycles, and samples were electrophoresed on a 2% agarose gel stained with ethidium bromide.
Reduced tumor sizes in the groups containing infectious (but not inactivated) VSV-ΔM51-GFP could be attributed to continued virus infection, replication, and oncolytic action. Therefore, we hypothesized that if the virus was still replicating at the endpoint within the tumor, then VSV infectious particles, proteins, or RNA could be detectable. Tumor lysates were analyzed for the presence of infectious VSV-ΔM51-GFP particles (using plaque assay on BHK-21 cells) and VSV RNA via RNA isolation, cDNA synthesis, and PCR using VSV RNA-specific primers. In addition, a section of each tumor was sliced and histologically stained for the presence of VSV proteins using antibodies against VSV G and N proteins. In addition, another tumor section was used to isolate total protein that was analyzed by Western blotting for antibodies against different VSV proteins or GFP. Interestingly, despite the differences in the tumor volumes in treatment groups, we were unable to detect VSV infectious particles by plaque assay or VSV proteins by either IHC or Western blotting (data not shown). Only when total RNA from tumor lysates was analyzed by reverse transcription (RT)-PCR (35 or 40 cycles of PCR), 2 of the 6 VSV-ΔM51-GFP-treated mice and 4 of the 6 VSV-ΔM51-GFP- plus gemcitabine-treated mice showed evidence of viral material in the tumors (Fig. 5B). No viral RNA was detected in UV-killed VSV-ΔM51-GFP-treated mice, suggesting that in the tumors treated with infectious virus, VSV replication took place at least at some point during treatment. Importantly, the products of RT-PCR, as shown in Fig. 5B, were sequenced, and all viral products retained the M51 deletion (data not shown).
While live VSV-ΔM51-GFP significantly reduced the KCM tumor burden up to 18 days following subcutaneous injections of cancer cells, OV therapy (alone or in combination with gemcitabine) did not abolish tumor growth (Fig. 5A). Therefore, we conducted a survival study to determine whether VSV-ΔM51-GFP treatment could result in a sustained antitumor effect (Fig. 6). In this experiment, in addition to the more aggressive KCM-based model (MUC1 overexpression), we included KCKO-based tumors (MUC1 null). First, subcutaneous injections of KCM or KCKO cells were used to establish tumors in the flanks of MUC1.Tg mice. When tumors were palpable, mice were treated i.t. with PBS as a control or with VSV-ΔM51-GFP every other day for three treatments (days 8, 10, and 12) (Fig. 6). Mice were monitored for signs of distress, and tumor size was measured every other day. Mice were sacrificed when tumor length reached 1.5 cm, tumors became ulcerated, or animals presented clinical signs indicative of morbidity. The results confirm that KCM is a more aggressive form of PDA, with tumor growth greatly exceeding that of KCKO. VSV-ΔM51-GFP treatment of both cell lines temporarily delayed tumor growth compared to the control mice (Fig. 6). For KCM, all control mice were sacrificed by day 20, with all animals being sacrificed by day 26. For KCKO, there was a significant decrease in tumor burden starting at day 12, which lasted through day 32, when significance could no longer be determined due to the need to sacrifice the control animals needed for comparison. However, by day 34 control tumors and those treated with VSV-ΔM51-GFP reached similar sizes. Regardless of treatment, most KCKO tumors never grew as large as the KCM tumors. This observation is in agreement with previous studies in which mice bearing KCKO tumors present a less-challenging, more-stable form of PDA disease as the cells grow at a lower rate than the more aggressive KCM cells (7).
Fig 6.
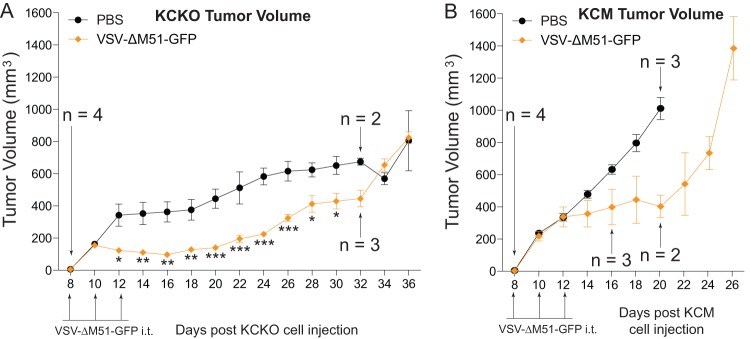
In vivo long-term efficacy of VSV-ΔM51-GFP against KCM and KCKO tumors. Male MUC1.Tg mice, 8 to 11 weeks old, were subcutaneously injected with KCM or KCKO cells in the right flank (n = 8 per group). Tumors were established by day 8, and then mice were randomly divided into 2 groups (n = 4 per group) per cell line. Mice were administered PBS or VSV-ΔM51-GFP i.t. on days 8, 10, and 12. Tumor size was monitored by caliper measurements, and tumor weight was calculated according to the standard ellipsoid formula: grams = (length in cm × width2)/2. Mice were sacrificed when the length of the tumor reached 1.5 cm, tumors became ulcerated, or mice presented clinical signs indicating morbidity. Comparison of groups was done using a 2-way ANOVA only at the time points at which the groups had the same population (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
Humoral immune response in immunocompetent MUC1.Tg mice at the endpoint.
While our in vitro and short-term in vivo results show promising oncolytic abilities of VSV-ΔM51-GFP against both KCM and KCKO cells, there is a clear indication that this treatment is not having a long-term, sustained anticancer effect. The design of our experiment did not allow us to look at early time points to assess intratumoral virus replication, oncolysis, and innate immune resources against VSV. However, premature inactivation of virus by the humoral immune response in an immunocompetent subject could negatively affect the efficacy of OV therapy. To determine the production of neutralizing antibodies against VSV-ΔM51-GFP, serum samples were examined from the KCM experiments in which all mice were sacrificed 18 days following KCM cell injection (Fig. 5A). Serial dilutions of mouse sera were first incubated with a known amount of VSV-ΔM51-GFP. The serum/VSV incubation was then used to infect BHK-21 cells for plaque assay analysis. In the VSV-ΔM51-GFP alone and VSV-ΔM51-GFP plus gemcitabine groups, it is evident that a strong humoral immune response was mounted against the virus (Fig. 7A). Importantly, much lower antibody levels were detected in sera from mice treated with UV-killed VSV-ΔM51-GFP, indicating that active virus replication was necessary for a robust humoral response.
Fig 7.
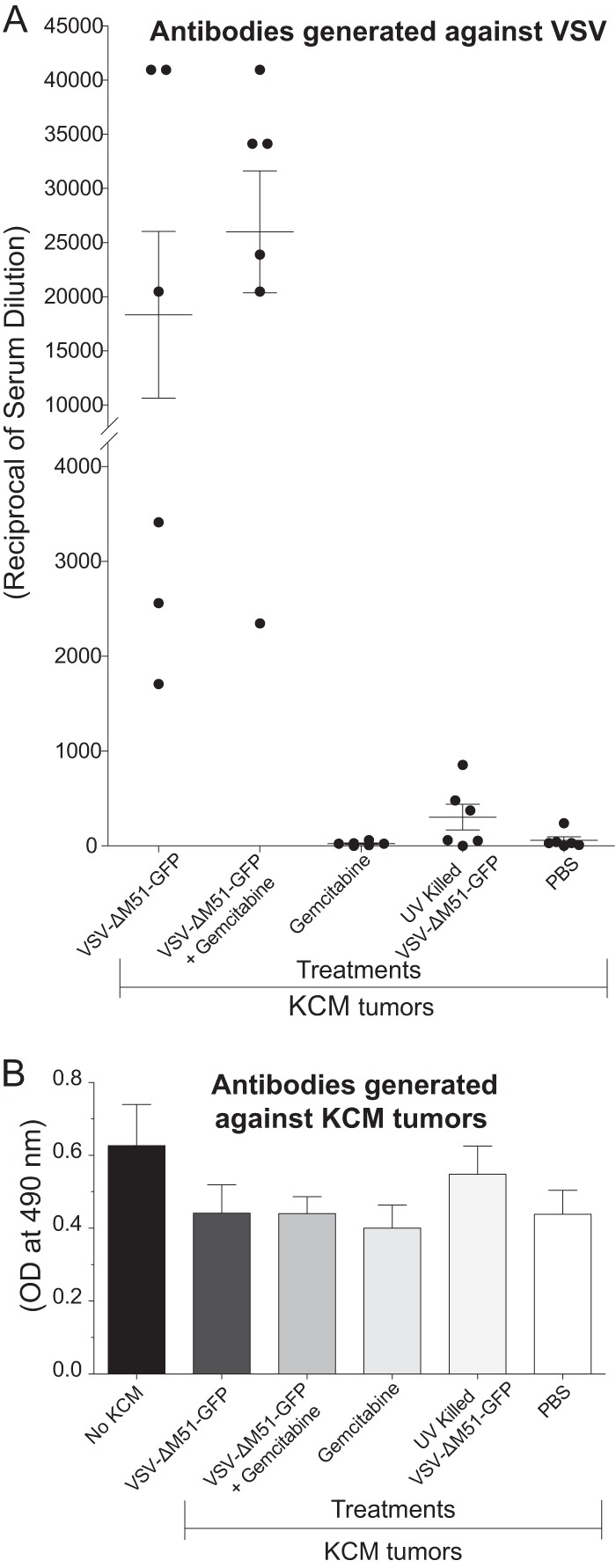
Humoral response against virus and cancer cells. (A) VSV-neutralizing antibody detection in mouse serum. Serum dilutions from all animals whose results are shown in Fig. 5 were incubated together with VSV-ΔM51-GFP and then added to BHK-21 cells. Infectious foci were counted by fluorescence microscopy at 16 h p.i. Antibody titers were determined to be the inverse of the dilution with one-half the number of plaques obtained with VSV-ΔM51-GFP alone. All serum samples were tested in triplicate. (B) KCM cell-specific antibody detection in serum. KCM cells were fixed, permeabilized, blocked in BSA, and incubated with dilutions of mouse sera. Detection was with HRP-conjugated secondary antibodies and the OPD substrate, and OD reading was done at 490 nm. All serum samples were repeated in triplicate, and the data represent means ± SEM.
The absence of a sustained antitumor response suggests that no effective humoral immune responses were mounted against the tumors following OV treatment. To test whether VSV-ΔM51-GFP treatment induced humoral responses against KCM tumors, mouse sera were analyzed for antibodies directed against KCM cells (Fig. 7B). KCM cells were fixed and permeabilized and then incubated with serial dilution of mouse sera from treated animals to detect KCM-specific antibodies. Results indicate that there was no significant difference in humoral response against KCM cells in any of the treatments compared to sera from mice that were never injected with KCM (Fig. 7B).
DISCUSSION
In the present study, for the first time, we tested VSV against mouse PDA cells in vitro and in vivo and in a clinically relevant, immunocompetent mouse model of PDA. VSV has never been tested in any immunocompetent model of pancreatic cancer. We have utilized a system that allows the study of oncolytic viruses (VSV or other OVs) in the context of MUC1 overexpression, as seen in approximately 80% of PDA patients, or no expression (49, 50). Our data show that VSV can infect and kill all tested mouse PDA cell lines in vitro and cause transient reduction of KCM and KCKO tumors in vivo.
Our study was focused on five mouse PDA cell lines overexpressing MUC1 or MUC1 null, which could all be used in this immunocompetent model of PDA. Although our main focus was on the performance of VSV in vivo, OV therapy success is generally dependent on the abilities of the virus to infect and kill cells, and our previous study with human PDA cells in athymic nude mice showed very good correlation between the oncolytic efficacy of VSV-ΔM51-GFP in vitro and in vivo (24). Therefore, in addition to in vivo experiments, we conducted a series of in vitro experiments to determine the abilities of VSV to infect, replicate, and kill these five mouse PDA cell lines, as well as to examine cellular characteristics of these cell lines important for their susceptibility to OV therapy.
We were particularly interested in determining the ability of VSV to kill cancer cells overexpressing MUC1. MUC1 is overexpressed and aberrantly glycosylated in more than 80% of human PDAs and in 100% of metastatic lesions (5). It not only plays an important role in development and progression of PDA and other cancers but also is a major marker of poor prognosis, and its expression often confers resistance of cancer cells to chemotherapeutics (7, 8, 11). In general, our data suggest that VSV is tolerant to the expression of MUC1, at least in the tested PDA cell lines. The tolerance of VSV to MUC1 is a significant result. Although the role of MUC1 in oncolytic therapy has never been studied before, the O-linked carbohydrates of MUC1 purified from human breast milk can inhibit poxvirus (12), HIV (13, 14), and rotavirus (15), and MUC1 expression can block adeno-associated virus attachment (16). Although VSV-ΔM51-GFP was slightly less infectious in MUC1-expressing KCM cells than in MUC1-null KCKO cells, the opposite could be seen in Panc02-based cell lines (Fig. 2 and 3). Moreover, as VSV-rWT and VSV-GFP were not attenuated at all in KCM or Panc02-MUC1, our data suggest that VSV can successfully infect PDA cells regardless of their MUC1 expression status.
We observed some variations in the susceptibility of different cell lines specifically to VSV-ΔM51-GFP (but not VSV-rWT or VSV-GFP) infection and virus-induced oncolysis. Specifically, VSV-ΔM51-GFP had reduced oncolytic ability in KCM, Panc02-MUC1, and Panc02Neo. The M51 deletion improves VSV oncoselectivity by preventing WT M protein's ability to shut down cellular gene expression (19–21). Therefore, VSV-ΔM51-GFP is unable to successfully replicate in healthy cells with intact type I IFN responses. Our recent analysis of a panel of human PDA cell lines showed that responsiveness to IFN-α treatment and the expression of the IFN-stimulated genes could be predictive of resistance to VSV-ΔM51-GFP (23, 24). When cell lines were treated with type I IFN for 24 h prior to infection, some reduction, though modest, in virus infectivity was seen in Panc02-MUC1 and Panc02-Neo. Also, all cell lines were able to sense virus infection (as determined by phosphorylation of IRF3); however, where KCKO, KC, and KCM cell lines showed IRF3 phosphorylation at 24 h p.i., Panc02-MUC1 and Panc02Neo cells showed IRF3 phosphorylation as early as 12 h p.i. Additionally, only Panc02-MUC1 and Panc02Neo cells showed any detectable phosphorylation of STAT1 following VSV infection (indicative of type I IFN signaling). Together, our results indicate that while none of the tested mouse PDA cell lines have robust type I IFN responses, Panc02-MUC1 and Panc02-Neo have limited antiviral signaling, which may explain their reduced susceptibility specifically to VSV-ΔM51-GFP.
At present, we cannot determine a mechanism for the somewhat low in vitro susceptibility of KCM cells to VSV-ΔM51-GFP compared to that of KCKO cells. However, it should be noted that MUC1 expression in KC, KCKO, and KCM might not be the only difference between these cell lines, as an accumulation of additional mutations is likely during spontaneous PDA formation. Some of these mutations could confer resistance of KCM cells to VSV-ΔM51-GFP (but not VSV-rWT or VSV-GFP) independently of MUC1 or type I IFN signaling.
We focused on KCM cells for our in vivo experiments because xenografts of KCM lead to more aggressive tumors than KCKO or KC and because most human PDA patients exhibit MUC1 overexpression (5). Our in vivo data demonstrated that VSV-ΔM51-GFP significantly reduced the tumor burden in mice with subcutaneous KCM xenografts after 18 days. However, this effect was not sustained when mice were monitored for survival. Furthermore, a similar result was shown even for KCKO-derived tumors, which exhibit much slower tumor growth than do KCM-derived tumors (7).
Our previous in vivo study with human PDA cells in athymic nude mice showed that cell lines that were susceptible to VSV-ΔM51-GFP-mediated oncolysis in vitro also showed a sustained antitumor response in vivo (24). Similarly, many other immunocompromised models using cell lines susceptible to VSV in vitro demonstrated a sustained oncolytic effect in vivo, including subcutaneous G62 glioblastoma tumors (51), SMT-91-01 rhabdoid tumors (52), and KU-7 orthotopic bladder tumors (53). Here, however, VSV-ΔM51-GFP demonstrated very good oncolytic activities against KCM and especially KCKO cells in vitro, but no long-term, sustained anticancer effect in vivo. The major difference between these studies is possible premature clearance of virus by the adaptive immune response in immunocompetent mice that could negatively affect the efficacy of OV therapy. Mice treated with “live” replicative virus (but not UV-killed virus) developed a robust antibody response directed at VSV-ΔM51-GFP. Previous studies also showed an immune response to VSV with an M mutation (VSV-rM51R-M), with antibody titers being comparable to those seen against WT VSV (38). In the context of oncolytic therapy, one study showed a transient reduction of multiple myeloma and a significant antibody response generated against virus (54). Additionally, transient reduction of melanoma tumor burden and robust antibody response against VSV was seen even in a model where VSV was capable of only a single-cycle replication (55).
In addition to a humoral response to VSV (which peaks a week after infection), a negative role for innate immune responses to VSV has been demonstrated, with neutrophils (peak response at 36 h p.i.) (43) and natural killer (NK) cells (peak response, 3 to 4 days p.i.) (56) responsible for early-stage virus clearance (42, 56–58). A limitation of our study is that our experimental design did not allow for analysis of early time points, so we cannot rule out that these innate immune responses contributed to a limited efficacy of VSV-ΔM51-GFP in vivo.
Several approaches to inhibit host responses to VSV have been reported. Neutrophil and NK cell depletion as well as inhibitors of NK cell-activating chemokines greatly increased VSV spread and animal survival (42, 57, 58). Cell-based delivery seeks to use infected “Trojan horse” carrier cells, i.e., T cells that home to the tumor and deliver VSV without initial immune detection (59, 60). Additional methods to hide virus employ DNA aptamer technology (61) or pegylation of VSV (62) to shield the virus from neutralizing antibodies. Even more, a putative cotherapeutic like cyclophosphamide (CPA) was shown to suppress an immune response to virus (63), but one oncolytic study with mesothelioma showed reduced therapeutic efficacy of the VSV and CPA cotherapy (64).
In addition to premature immune clearance of virus, limited penetration and spread within the tumor mass were noted in several previous studies using VSV and other OVs, and this may also contribute to the limited antitumor effect of VSV in this study. While we did not conduct a time course analysis of VSV infection within tumors in mice, we were unable to detect VSV infectious particles or VSV proteins at the endpoint. Only when total RNA from tumor lysates was analyzed by RT-PCR was viral RNA detected in 2 of the 6 VSV-ΔM51-GFP-treated mice and 4 of the 6 VSV-ΔM51-GFP plus gemcitabine-treated mice (no viral RNA was detected in UV-killed VSV-ΔM51-GFP-treated mice). These results suggest possible limited replication and spread of the virus within tumor (in addition to immune clearance of VSV). Future studies will focus on the analysis of VSV replication following various VSV-ΔM51-GFP doses and administration protocols to determine the contributions of various factors to the limited antitumor effect of VSV in this study. Several approaches could be employed to address such limitations. For example, using VSV encoding suicide genes like herpesvirus thymidine kinase (41) or cytosine deaminase (65, 66) enhanced the bystander effect and increased tumor regression compared to parental virus treatment. Penetration and spread within the tumor mass also were shown to be improved by incorporation of fusogenic proteins from Newcastle disease virus (67, 68) or simian parainfluenza virus (69) and resulted in enhanced tumor killing and prolonged survival compared to parental virus. Also, prolonged tumor exposure to virus and increased survival were reported when tumor vasculature was targeted, either by blocking arterial flow with starch microspheres (70) or reducing angiogenesis (71). Future experiments will test whether these approaches can improve tumor reduction and prolong survival as well as allow us to assess intratumoral replication, virus spread, and direct oncolysis at earlier time points, which we were unable to address in this study.
Importantly, in this study, no humoral immune responses against tumor cells could be detected in any treatment group. This is consistent with the previous results that indicate tolerance of MUC1.Tg mice toward MUC1-expressing cells (34). It should be noted that a lack of antibody production against the tumor does not rule out that other antitumor immune responses were generated. Virus-induced long-term antitumor responses include increase in inflammatory cytokines, dendritic cell migration to lymph nodes, NK cell activation, and antitumor cytotoxic T lymphocytes (CTLs) (72). Specifically for VSV, increased infiltration of CTLs generated against virus and tumor epitopes, increased infiltration of B cells, interleukin-28 (IL-28) induction to promote NK cell activation, and downregulation of regulatory T cells were reported (73–80). As our study was limited only to antitumor antibody analysis (no detectable response), we cannot rule out activation of those other antitumor immune responses. However, even if such responses were generated, they were not potent enough to induce long-term antitumor effects.
Future studies will look at VSV's ability to shift the immune response toward cancer cells. VSV engineered to express a relevant tumor-associated antigen (TAA) such as MUC1 could be created. Studies with other tumor types utilized recombinant VSVs encoding TAAs like human melanoma-associated antigen (Ag) dopachrome tautomerase (73) or chicken ovalbumin (75, 81) demonstrated improved antitumor efficacy of VSV associated with the ability to generate increased numbers of TAA-specific T cells (75, 82). Also, other VSV recombinants expressing immune system-modulating cytokines or cancer suppressor proteins could be tested and compared to VSV-ΔM51-GFP (17). Interestingly, the outcome of treatment may be dependent on the amount of virus used. At lower concentration, for an oncolytic VSV recombinant, antitumor effects were improved compared to higher concentrations (83), suggesting that the increased viral presence biases the immune response against viral antigens rather than tumor cell antigens. However, recently it was shown with a VSV vaccine vector that robust replication of VSV could be required for efficient adaptive immune responses against non-VSV antigen (84). Therefore, in our future experiments we will study dose-dependent efficacy of VSV-ΔM51-GFP against PDA tumors VSV.
Additional strategies seek to use combinational therapies to improve oncolysis. A recombinant virus expressing a sodium iodide symporter, when coupled with iodine-131 radiation therapy, resulted in an enhanced oncolytic effect in radiation-sensitive tumors (85). Finally, OV therapy has been tested in combination with chemotherapeutics like obatoclax (86) or EM20-25 (87) (inhibitors of BCL-2) or doxorubin (intercalates DNA) (88), with all showing enhanced oncolysis compared to VSV monotherapy. When we tested a combinational treatment of VSV-ΔM51-GFP and a commonly used PDA chemotherapeutic, gemcitabine, significant improvement was observed compared to use of VSV-ΔM51-GFP alone. In addition to its role as a chemotherapeutic, gemcitabine was shown to deplete B cells (89). While the antibody response was similar in the VSV-ΔM51-GFP alone and VSV-ΔM51-GFP plus gemcitabine groups at the endpoint, it is possible that gemcitabine contributed to the prolonged tumor reduction in this group. It should also be noted that gemcitabine had no effect on VSV-ΔM51-GFP replication in vitro (data not shown). Future experiments will study the potential of VSV with gemcitabine (or other drugs) using additional concentrations and treatment schedules.
Overall, the immunocompetent murine system described here is a clinically relevant model of PDA to study oncolytic virotherapy against PDA tumors (MUC1 positive or null) using OVs as a monotherapy or in combination with other treatments.
ACKNOWLEDGMENTS
We are grateful to Jack Rose (Yale University) for the VSV-rWT and VSV-ΔM51-GFP viruses, Asit K. Pattnaik (University of Nebraska) for the VSV-GFP virus, Tony Hollingsworth (University of Nebraska) for Panc02-Neo and Panc02-MUC1 cell lines, and Sandra Gendler (Mayo Clinic Arizona) for the MUC1.Tg mice. We thank David Gray, Natascha Moestl, and Priyanka Grover for technical assistance. We also acknowledge the Research Histology Core/Imaging Core at The Cannon Research Center, Carolinas Medical Center, Charlotte, NC, for providing histology support for this project.
This work was supported by NIH grant 1R15CA167517-01 (to V.Z.G.) from the National Cancer Institute.
Footnotes
Published ahead of print 17 July 2013
REFERENCES
- 1. Tarver T. 2012. Cancer facts & figures 2012. American Cancer Society (ACS). J. Consum. Health Internet 16:366–367 [Google Scholar]
- 2. Stathis A, Moore MJ. 2010. Advanced pancreatic carcinoma: current treatment and future challenges. Nat. Rev. Clin. Oncol. 7:163–172 [DOI] [PubMed] [Google Scholar]
- 3. Farrow B, Albo D, Berger DH. 2008. The role of the tumor microenvironment in the progression of pancreatic cancer. J. Surg. Res. 149:319–328 [DOI] [PubMed] [Google Scholar]
- 4. Lindsay TH, Jonas BM, Sevcik MA, Kubota K, Halvorson KG, Ghilardi JR, Kuskowski MA, Stelow EB, Mukherjee P, Gendler SJ, Wong GY, Mantyh PW. 2005. Pancreatic cancer pain and its correlation with changes in tumor vasculature, macrophage infiltration, neuronal innervation, body weight and disease progression. Pain 119:233–246 [DOI] [PubMed] [Google Scholar]
- 5. Jonckheere N, Skrypek S, Van Seuningen I. 2010. Mucins and pancreatic cancer. Cancers 2:1794–1812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Reichert M, Rustgi AK. 2011. Pancreatic ductal cells in development, regeneration, and neoplasia. J. Clin. Invest. 121:4572–4578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Besmer DM, Curry JM, Roy LD, Tinder TL, Sahraei M, Schettini JL, Hwang SI, Lee YY, Gendler SJ, Mukherjee P. 2011. Pancreatic ductal adenocarcinoma (PDA) mice lacking Mucin 1 have a profound defect in tumor growth and metastasis. Cancer Res. 71:4432–4442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Horm TM, Schroeder JA. 2013. MUC1 and metastatic cancer: expression, function and therapeutic targeting. Cell Adh. Migr. 7:187–198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kalra AV, Campbell RB. 2007. Mucin impedes cytotoxic effect of 5-FU against growth of human pancreatic cancer cells: overcoming cellular barriers for therapeutic gain. Br. J. Cancer 97:910–918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kalra AV, Campbell RB. 2009. Mucin overexpression limits the effectiveness of 5-FU by reducing intracellular drug uptake and antineoplastic drug effects in pancreatic tumours. Eur. J. Cancer. 45:164–173 [DOI] [PubMed] [Google Scholar]
- 11. Roy LD, Sahraei M, Subramani DB, Besmer D, Nath S, Tinder TL, Bajaj E, Shanmugam K, Lee YY, Hwang SI, Gendler SJ, Mukherjee P. 2011. MUC1 enhances invasiveness of pancreatic cancer cells by inducing epithelial to mesenchymal transition. Oncogene 30:1449–1459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Habte HH, Kotwal GJ, Lotz ZE, Tyler MG, Abrahams M, Rodriques J, Kahn D, Mall AS. 2007. Antiviral activity of purified human breast milk mucin. Neonatology 92:96–104 [DOI] [PubMed] [Google Scholar]
- 13. Habte HH, de Beer C, Lotz ZE, Tyler MG, Kahn D, Mall AS. 2008. Inhibition of human immunodeficiency virus type 1 activity by purified human breast milk mucin (MUC1) in an inhibition assay. Neonatology 93:162–170 [DOI] [PubMed] [Google Scholar]
- 14. Saeland E, de Jong MA, Nabatov AA, Kalay H, Geijtenbeek TB, van Kooyk Y. 2009. MUC1 in human milk blocks transmission of human immunodeficiency virus from dendritic cells to T cells. Mol. Immunol. 46:2309–2316 [DOI] [PubMed] [Google Scholar]
- 15. Kvistgaard AS, Pallesen LT, Arias CF, Lopez S, Petersen TE, Heegaard CW, Rasmussen JT. 2004. Inhibitory effects of human and bovine milk constituents on rotavirus infections. J. Dairy Sci. 87:4088–4096 [DOI] [PubMed] [Google Scholar]
- 16. Walters RW, Pilewski JM, Chiorini JA, Zabner J. 2002. Secreted and transmembrane mucins inhibit gene transfer with AAV4 more efficiently than AAV5. J. Biol. Chem. 277:23709–23713 [DOI] [PubMed] [Google Scholar]
- 17. Hastie E, Grdzelishvili V. 2012. Vesicular stomatitis virus as a flexible platform for oncolytic virotherapy against cancer. J. Gen. Virol. 93:2529–2545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wollmann G, Rogulin V, Simon I, Rose JK, van den Pol AN. 2010. Some attenuated variants of vesicular stomatitis virus show enhanced oncolytic activity against human glioblastoma cells relative to normal brain cells. J. Virol. 84:1563–1573 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ahmed M, McKenzie MO, Puckett S, Hojnacki M, Poliquin L, Lyles DS. 2003. Ability of the matrix protein of vesicular stomatitis virus to suppress beta interferon gene expression is genetically correlated with the inhibition of host RNA and protein synthesis. J. Virol. 77:4646–4657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kopecky SA, Willingham MC, Lyles DS. 2001. Matrix protein and another viral component contribute to induction of apoptosis in cells infected with vesicular stomatitis virus. J. Virol. 75:12169–12181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stojdl DF, Lichty BD, ten Oever BR, Paterson JM, Power AT, Knowles S, Marius R, Reynard J, Poliquin L, Atkins H, Brown EG, Durbin RK, Durbin JE, Hiscott J, Bell JC. 2003. VSV strains with defects in their ability to shutdown innate immunity are potent systemic anti-cancer agents. Cancer Cell 4:263–275 [DOI] [PubMed] [Google Scholar]
- 22. Obuchi M, Fernandez M, Barber GN. 2003. Development of recombinant vesicular stomatitis viruses that exploit defects in host defense to augment specific oncolytic activity. J. Virol. 77:8843–8856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moerdyk-Schauwecker M, Shah NR, Murphy AM, Hastie E, Mukherjee P, Grdzelishvili VZ. 2013. Resistance of pancreatic cancer cells to oncolytic vesicular stomatitis virus: role of type I interferon signaling. Virology 436:221–234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Murphy AM, Besmer DM, Moerdyk-Schauwecker M, Moestl N, Ornelles DA, Mukherjee P, Grdzelishvili VZ. 2012. Vesicular stomatitis virus as an oncolytic agent against pancreatic ductal adenocarcinoma. J. Virol. 86:3073–3087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hingorani SR, Petricoin EF, Maitra A, Rajapakse V, King C, Jacobetz MA, Ross S, Conrads TP, Veenstra TD, Hitt BA, Kawaguchi Y, Johann D, Liotta LA, Crawford HC, Putt ME, Jacks T, Wright CV, Hruban RH, Lowy AM, Tuveson DA. 2003. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell 4:437–450 [DOI] [PubMed] [Google Scholar]
- 26. Morikane K, Tempero RM, Sivinski CL, Nomoto M, Van Lith ML, Muto T, Hollingsworth MA. 1999. Organ-specific pancreatic tumor growth properties and tumor immunity. Cancer Immunol. Immunother. 47:287–296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schroeder JA, Thompson MC, Gardner MM, Gendler SJ. 2001. Transgenic MUC1 interacts with epidermal growth factor receptor and correlates with mitogen-activated protein kinase activation in the mouse mammary gland. J. Biol. Chem. 276:13057–13064 [DOI] [PubMed] [Google Scholar]
- 28. Taylor-Papadimitriou J, Peterson JA, Arklie J, Burchell J, Ceriani RL, Bodmer WF. 1981. Monoclonal antibodies to epithelium-specific components of the human milk fat globule membrane: production and reaction with cells in culture. Int. J. Cancer 28:17–21 [DOI] [PubMed] [Google Scholar]
- 29. Croce MV, Isla-Larrain M, Remes-Lenicov F, Colussi AG, Lacunza E, Kim KC, Gendler SJ, Segal-Eiras A. 2006. MUC1 cytoplasmic tail detection using CT33 polyclonal and CT2 monoclonal antibodies in breast and colorectal tissue. Histol. Histopathol. 21:849–855 [DOI] [PubMed] [Google Scholar]
- 30. Moyer SA, Baker SC, Lessard JL. 1986. Tubulin: a factor necessary for the synthesis of both Sendai virus and vesicular stomatitis virus RNAs. Proc. Natl. Acad. Sci. U. S. A. 83:5405–5409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lawson ND, Stillman EA, Whitt MA, Rose JK. 1995. Recombinant vesicular stomatitis viruses from DNA. Proc. Natl. Acad. Sci. U. S. A. 92:4477–4481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Das SC, Nayak D, Zhou Y, Pattnaik AK. 2006. Visualization of intracellular transport of vesicular stomatitis virus nucleocapsids in living cells. J. Virol. 80:6368–6377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kalvodova L, Sampaio JL, Cordo S, Ejsing CS, Shevchenko A, Simons K. 2009. The lipidomes of vesicular stomatitis virus, Semliki Forest virus, and the host plasma membrane analyzed by quantitative shotgun mass spectrometry. J. Virol. 83:7996–8003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Rowse GJ, Tempero RM, VanLith ML, Hollingsworth MA, Gendler SJ. 1998. Tolerance and immunity to MUC1 in a human MUC1 transgenic murine model. Cancer Res. 58:315–321 [PubMed] [Google Scholar]
- 35. Tinder TL, Subramani DB, Basu GD, Bradley JM, Schettini J, Million A, Skaar T, Mukherjee P. 2008. MUC1 enhances tumor progression and contributes toward immunosuppression in a mouse model of spontaneous pancreatic adenocarcinoma. J. Immunol. 181:3116–3125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Spicer AP, Rowse GJ, Lidner TK, Gendler SJ. 1995. Delayed mammary tumor progression in Muc-1 null mice. J. Biol. Chem. 270:30093–30101 [DOI] [PubMed] [Google Scholar]
- 37. Corbett TH, Roberts BJ, Leopold WR, Peckham JC, Wilkoff LJ, Griswold DP, Jr, Schabel FM., Jr 1984. Induction and chemotherapeutic response of two transplantable ductal adenocarcinomas of the pancreas in C57BL/6 mice. Cancer Res. 44:717–726 [PubMed] [Google Scholar]
- 38. Ahmed M, Marino TR, Puckett S, Kock ND, Lyles DS. 2008. Immune response in the absence of neurovirulence in mice infected with M protein mutant vesicular stomatitis virus. J. Virol. 82:9273–9277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Johnson JE, Nasar F, Coleman JW, Price RE, Javadian A, Draper K, Lee M, Reilly PA, Clarke DK, Hendry RM, Udem SA. 2007. Neurovirulence properties of recombinant vesicular stomatitis virus vectors in non-human primates. Virology 360:36–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Dalton KP, Rose JK. 2001. Vesicular stomatitis virus glycoprotein containing the entire green fluorescent protein on its cytoplasmic domain is incorporated efficiently into virus particles. Virology 279:414–421 [DOI] [PubMed] [Google Scholar]
- 41. Fernandez M, Porosnicu M, Markovic D, Barber GN. 2002. Genetically engineered vesicular stomatitis virus in gene therapy: application for treatment of malignant disease. J. Virol. 76:895–904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wu L, Huang TG, Meseck M, Altomonte J, Ebert O, Shinozaki K, García-Sastre A, Fallon J, Mandeli J, Woo SL. 2008. rVSV(M Delta 51)-M3 is an effective and safe oncolytic virus for cancer therapy. Hum. Gene Ther. 19:635–647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Bi Z, Barna M, Komatsu T, Reiss CS. 1995. Vesicular stomatitis virus infection of the central nervous system activates both innate and acquired immunity. J. Virol. 69:6466–6472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Reiss CS, Plakhov IV, Komatsu T. 1998. Viral replication in olfactory receptor neurons and entry into the olfactory bulb and brain. Ann. N. Y. Acad. Sci. 855:751–761 [DOI] [PubMed] [Google Scholar]
- 45. van den Pol A, Dalton K, Rose J. 2002. Relative neurotropism of a recombinant rhabdovirus expressing a green fluorescent envelope glycoprotein. J. Virol. 76:1309–1327 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ansari D, Tingstedt B, Andersson R. 2013. Pancreatic cancer—cost for overtreatment with gemcitabine. Acta Oncol. 52:1146–1151 [DOI] [PubMed] [Google Scholar]
- 47. Kovarik A, Peat N, Wilson D, Gendler SJ, Taylor-Papadimitriou J. 1993. Analysis of the tissue-specific promoter of the MUC1 gene. J. Biol. Chem. 268:9917–9926 [PubMed] [Google Scholar]
- 48. Mukherjee P, Basu GD, Tinder TL, Subramani DB, Bradley JM, Arefayene M, Skaar T, De Petris G. 2009. Progression of pancreatic adenocarcinoma is significantly impeded with a combination of vaccine and COX-2 inhibition. J. Immunol. 182:216–224 [PMC free article] [PubMed] [Google Scholar]
- 49. Jonckheere N, Van Seuningen I. 2010. The membrane-bound mucins: from cell signalling to transcriptional regulation and expression in epithelial cancers. Biochimie 92:1–11 [DOI] [PubMed] [Google Scholar]
- 50. Kim GE, Bae HI, Park HU, Kuan SF, Crawley SC, Ho JJ, Kim YS. 2002. Aberrant expression of MUC5AC and MUC6 gastric mucins and sialyl Tn antigen in intraepithelial neoplasms of the pancreas. Gastroenterology 123:1052–1060 [DOI] [PubMed] [Google Scholar]
- 51. Muik A, Dold C, Geiß Y, Volk A, Werbizki M, Dietrich U, von Laer D. 2012. Semireplication-competent vesicular stomatitis virus as a novel platform for oncolytic virotherapy. J. Mol. Med. (Berl.) 90:959–970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wu Y, Lun X, Zhou H, Wang L, Sun B, Bell JC, Barrett JW, McFadden G, Biegel JA, Senger DL, Forsyth PA. 2008. Oncolytic efficacy of recombinant vesicular stomatitis virus and myxoma virus in experimental models of rhabdoid tumors. Clin. Cancer Res. 14:1218–1227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hadaschik BA, Zhang K, So AI, Fazli L, Jia W, Bell JC, Gleave ME, Rennie PS. 2008. Oncolytic vesicular stomatitis viruses are potent agents for intravesical treatment of high-risk bladder cancer. Cancer Res. 68:4506–4510 [DOI] [PubMed] [Google Scholar]
- 54. Naik S, Nace R, Barber GN, Russell SJ. 2012. Potent systemic therapy of multiple myeloma utilizing oncolytic vesicular stomatitis virus coding for interferon-beta. Cancer Gene Ther. 19:443–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Galivo F, Diaz RM, Wongthida P, Thompson J, Kottke T, Barber G, Melcher A, Vile R. 2010. Single-cycle viral gene expression, rather than progressive replication and oncolysis, is required for VSV therapy of B16 melanoma. Gene Ther. 17:158–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chen N, Restivo A, Reiss CS. 2001. Leukotrienes play protective roles early during experimental VSV encephalitis. J. Neuroimmunol. 120:94–102 [DOI] [PubMed] [Google Scholar]
- 57. Altomonte J, Wu L, Chen L, Meseck M, Ebert O, García-Sastre A, Fallon J, Woo SL. 2008. Exponential enhancement of oncolytic vesicular stomatitis virus potency by vector-mediated suppression of inflammatory responses in vivo. Mol. Ther. 16:146–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Altomonte J, Wu L, Meseck M, Chen L, Ebert O, Garcia-Sastre A, Fallon J, Mandeli J, Woo SL. 2009. Enhanced oncolytic potency of vesicular stomatitis virus through vector-mediated inhibition of NK and NKT cells. Cancer Gene Ther. 16:266–278 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kottke T, Diaz RM, Kaluza K, Pulido J, Galivo F, Wongthida P, Thompson J, Willmon C, Barber GN, Chester J, Selby P, Strome S, Harrington K, Melcher A, Vile RG. 2008. Use of biological therapy to enhance both virotherapy and adoptive T-cell therapy for cancer. Mol. Ther. 16:1910–1918 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Qiao J, Wang H, Kottke T, Diaz RM, Willmon C, Hudacek A, Thompson J, Parato K, Bell J, Naik J, Chester J, Selby P, Harrington K, Melcher A, Vile RG. 2008. Loading of oncolytic vesicular stomatitis virus onto antigen-specific T cells enhances the efficacy of adoptive T-cell therapy of tumors. Gene Ther. 15:604–616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Labib M, Zamay AS, Muharemagic D, Chechik A, Bell JC, Berezovski MV. 2012. Electrochemical sensing of aptamer-facilitated virus immunoshielding. Anal. Chem. 84:1677–1686 [DOI] [PubMed] [Google Scholar]
- 62. Tesfay MZ, Kirk AC, Hadac EM, Griesmann GE, Federspiel MJ, Barber GN, Henry SM, Peng KW, Russell SJ. 2013. PEGylation of vesicular stomatitis virus extends virus persistence in blood circulation of passively immunized mice. J. Virol. 87:3752–3759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Peng KW, Myers R, Greenslade A, Mader E, Greiner S, Federspiel MJ, Dispenzieri A, Russell SJ. 2013. Using clinically approved cyclophosphamide regimens to control the humoral immune response to oncolytic viruses. Gene Ther. 20:255–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Willmon C, Diaz RM, Wongthida P, Galivo F, Kottke T, Thompson J, Albelda S, Harrington K, Melcher A, Vile R. 2011. Vesicular stomatitis virus-induced immune suppressor cells generate antagonism between intratumoral oncolytic virus and cyclophosphamide. Mol. Ther. 19:140–149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Leveille S, Samuel S, Goulet ML, Hiscott J. 2011. Enhancing VSV oncolytic activity with an improved cytosine deaminase suicide gene strategy. Cancer Gene Ther. 18:435–443 [DOI] [PubMed] [Google Scholar]
- 66. Porosnicu M, Mian A, Barber GN. 2003. The oncolytic effect of recombinant vesicular stomatitis virus is enhanced by expression of the fusion cytosine deaminase/uracil phosphoribosyltransferase suicide gene. Cancer Res. 63:8366–8376 [PubMed] [Google Scholar]
- 67. Ebert O, Shinozaki K, Kournioti C, Park MS, Garcia-Sastre A, Woo SL. 2004. Syncytia induction enhances the oncolytic potential of vesicular stomatitis virus in virotherapy for cancer. Cancer Res. 64:3265–3270 [DOI] [PubMed] [Google Scholar]
- 68. Shin EJ, Chang JI, Choi B, Wanna G, Ebert O, Genden EM, Woo SL. 2007. Fusogenic vesicular stomatitis virus for the treatment of head and neck squamous carcinomas. Otolaryngol. Head Neck Surg. 136:811–817 [DOI] [PubMed] [Google Scholar]
- 69. Chang G, Xu S, Watanabe M, Jayakar HR, Whitt MA, Gingrich JR. 2010. Enhanced oncolytic activity of vesicular stomatitis virus encoding SV5-F protein against prostate cancer. J. Urol. 183:1611–1618 [DOI] [PubMed] [Google Scholar]
- 70. Altomonte J, Braren R, Schulz S, Marozin S, Rummeny EJ, Schmid RM, Ebert O. 2008. Synergistic antitumor effects of transarterial viroembolization for multifocal hepatocellular carcinoma in rats. Hepatology 48:1864–1873 [DOI] [PubMed] [Google Scholar]
- 71. Breitbach CJ, De Silva NS, Falls TJ, Aladl U, Evgin L, Paterson J, Sun YY, Roy DG, Rintoul JL, Daneshmand M, Parato K, Stanford MM, Lichty BD, Fenster A, Kirn D, Atkins H, Bell JC. 2011. Targeting tumor vasculature with an oncolytic virus. Mol. Ther. 19:886–894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Melcher A, Parato K, Rooney CM, Bell JC. 2011. Thunder and lightning: immunotherapy and oncolytic viruses collide. Mol. Ther. 19:1008–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Boudreau JE, Bridle BW, Stephenson KB, Jenkins KM, Brunelliere J, Bramson JL, Lichty BD, Wan Y. 2009. Recombinant vesicular stomatitis virus transduction of dendritic cells enhances their ability to prime innate and adaptive antitumor immunity. Mol. Ther. 17:1465–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bridle BW, Hanson S, Lichty BD. 2010. Combining oncolytic virotherapy and tumour vaccination. Cytokine Growth Factor Rev. 21:143–148 [DOI] [PubMed] [Google Scholar]
- 75. Diaz RM, Galivo F, Kottke T, Wongthida P, Qiao J, Thompson J, Valdes M, Barber G, Vile RG. 2007. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 67:2840–2848 [DOI] [PubMed] [Google Scholar]
- 76. Galivo F, Diaz RM, Thanarajasingam U, Jevremovic D, Wongthida P, Thompson J, Kottke T, Barber GN, Melcher A, Vile RG. 2010. Interference of CD40L-mediated tumor immunotherapy by oncolytic VSV. Hum. Gene Ther. 21:439–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kottke T, Galivo F, Wongthida P, Diaz RM, Thompson J, Jevremovic D, Barber GN, Hall G, Chester J, Selby P, Harrington K, Melcher A, Vile RG. 2008. Treg depletion-enhanced IL-2 treatment facilitates therapy of established tumors using systemically delivered oncolytic virus. Mol. Ther. 16:1217–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Qiao J, Kottke T, Willmon C, Galivo F, Wongthida P, Diaz RM, Thompson J, Ryno P, Barber GN, Chester J, Selby P, Harrington K, Melcher A, Vile RG. 2008. Purging metastases in lymphoid organs using a combination of antigen-nonspecific adoptive T cell therapy, oncolytic virotherapy and immunotherapy. Nat. Med. 14:37–44 [DOI] [PubMed] [Google Scholar]
- 79. Willmon CL, Saloura V, Fridlender ZG, Wongthida P, Diaz RM, Thompson J, Kottke T, Federspiel M, Barber G, Albelda SM, Vile RG. 2009. Expression of IFN-beta enhances both efficacy and safety of oncolytic vesicular stomatitis virus for therapy of mesothelioma. Cancer Res. 69:7713–7720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Wongthida P, Diaz RM, Galivo F, Kottke T, Thompson J, Pulido J, Pavelko K, Pease L, Melcher A, Vile R. 2010. Type III IFN interleukin-28 mediates the antitumor efficacy of oncolytic virus VSV in immune-competent mouse models of cancer. Cancer Res. 70:4539–4549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wongthida P, Diaz RM, Galivo F, Kottke T, Thompson J, Melcher A, Vile R. 2011. VSV oncolytic virotherapy in the B16 model depends upon intact MyD88 signaling. Mol. Ther. 19:150–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wongthida P, Diaz RM, Pulido C, Rommelfanger D, Galivo F, Kaluza K, Kottke T, Thompson J, Melcher A, Vile R. 2011. Activating systemic T-cell immunity against self tumor antigens to support oncolytic virotherapy with vesicular stomatitis virus. Hum. Gene Ther. 22:1343–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Heiber JF, Barber GN. 2011. Vesicular stomatitis virus expressing tumor suppressor p53 is a highly attenuated, potent oncolytic agent. J. Virol. 85:10440–10450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Cobleigh MA, Bradfield C, Liu Y, Mehta A, Robek MD. 2012. The immune response to a vesicular stomatitis virus vaccine vector is independent of particulate antigen secretion and protein turnover rate. J. Virol. 86:4253–4261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Goel A, Carlson SK, Classic KL, Greiner S, Naik S, Power AT, Bell JC, Russell SJ. 2007. Radioiodide imaging and radiovirotherapy of multiple myeloma using VSV(Delta51)-NIS, an attenuated vesicular stomatitis virus encoding the sodium iodide symporter gene. Blood 110:2342–2350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Tumilasci VF, Oliere S, Nguyen TL, Shamy A, Bell J, Hiscott J. 2008. Targeting the apoptotic pathway with BCL-2 inhibitors sensitizes primary chronic lymphocytic leukemia cells to vesicular stomatitis virus-induced oncolysis. J. Virol. 82:8487–8499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Samuel S, Tumilasci VF, Oliere S, Nguyen TL, Shamy A, Bell J, Hiscott J. 2010. VSV oncolysis in combination with the BCL-2 inhibitor obatoclax overcomes apoptosis resistance in chronic lymphocytic leukemia. Mol. Ther. 18:2094–2103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schache P, Gurlevik E, Struver N, Woller N, Malek N, Zender L, Manns M, Wirth T, Kuhnel F, Kubicka S. 2009. VSV virotherapy improves chemotherapy by triggering apoptosis due to proteasomal degradation of Mcl-1. Gene Ther. 16:849–861 [DOI] [PubMed] [Google Scholar]
- 89. Nowak AK, Robinson BW, Lake RA. 2002. Gemcitabine exerts a selective effect on the humoral immune response: implications for combination chemo-immunotherapy. Cancer Res. 62:2353–2358 [PubMed] [Google Scholar]