

Abstract
Alphaviruses are small enveloped viruses whose surface is covered by spikes composed of trimers of E2/E1 glycoprotein heterodimers. During virus entry, the E2/E1 dimer dissociates within the acidic endosomal environment, freeing the E1 protein to mediate fusion of the viral and endosome membranes. E2 is synthesized as a precursor, p62, which is cleaved by furin in the late secretory pathway to produce mature E2 and a small peripheral glycoprotein, E3. The immature p62/E1 dimer is acid resistant, but since p62 is cleaved before exit from the acidic secretory pathway, low pH-dependent binding of E3 to the spike complex is believed to prevent premature fusion. Based on analysis of the structure of the Chikungunya virus E3/E2/E1 complex, we hypothesized that interactions of E3 residues Y47 and Y48 with E2 are important in this binding. We then directly tested the in vivo role of E3 in pH protection by alanine substitutions of E3 Y47 and Y48 (Y47/48A) in Semliki Forest virus. The mutant was nonviable and was blocked in E1 transport to the plasma membrane and virus production. Although the Y47/48A mutant initially formed the p62/E1 heterodimer, the dimer dissociated during transport through the secretory pathway. Neutralization of the pH in the secretory pathway successfully rescued dimer association, E1 transport, and infectious particle production. Further mutagenesis identified the critical contact as the cation-π interaction of E3 Y47 with E2. Thus, E3 mediates pH protection of E1 during virus biogenesis via interactions strongly dependent on Y47 at the E3-E2 interface.
INTRODUCTION
Enveloped viruses use membrane fusion to deliver their genomes into host cells (reviewed in references 1, 2, and 3). These viral fusion reactions can be triggered by various combinations of receptor/coreceptor binding and/or by the low-pH environment of the endocytic pathway. Fusion is mediated by specialized viral fusion proteins, which are highly regulated for deployment at the correct time and place during entry. Suppression of the fusion reaction during virus biogenesis is as crucial as its correct triggering during virus entry. Viruses with low-pH-triggered fusion reactions have evolved several mechanisms to protect their membrane fusion proteins from the low pH of the exocytic pathway. Influenza virus (4) and hepatitis C virus (5) express small membrane proteins, termed viroporins, that act as ion channels and can neutralize the exocytic pathway as a protection mechanism. The alphavirus and flavivirus fusion proteins are synthesized with “companion” proteins whose dimeric interactions protect the fusion protein from low pH (reviewed in reference 6). Maturation of the companion protein by furin cleavage primes the virus to respond to low pH during entry (7, 8).
Alphaviruses such as Semliki Forest virus (SFV) are plus-strand RNA viruses enveloped in a lipid bilayer containing a lattice of trimeric spikes of E1 and E2 glycoprotein heterodimers (9). The companion protein E2 binds receptors on the cell surface, leading to clathrin-mediated endocytosis of the virus particle (10, 11). The E2/E1 dimer dissociates in the low-pH environment of the endosome, allowing the hydrophobic fusion loop on E1 to insert into the endosomal membrane. E1 then refolds to a trimeric hairpin-like structure and drives membrane fusion (11, 12).
The alphavirus E2 protein is synthesized in the endoplasmic reticulum (ER) as the precursor p62 (9, 13). p62 acts as a chaperone during glycoprotein synthesis and transport and is necessary for proper folding of E1. p62 is processed by furin in the late secretory pathway to produce the mature E2 protein and E3, a peripheral glycoprotein with a predicted molecular mass (without glycosylation) of about 7 kDa (9, 14, 15). The immature p62/E1 dimer is more acid stable than the mature form (16), and the immature SFV fusion threshold is ∼pH 5.0 while that of mature SFV is ∼pH 6.2 (8, 14, 17). Since the late secretory pathway has a pH ranging from ∼5.5 to 6.0 (18), the mature E2/E1 heterodimer is exposed to conditions sufficient to trigger premature fusion and E1 inactivation.
Following p62 processing, SFV E3 can remain bound to the virus particle at low pH and can be released at neutral pH (19). The bound E3 inhibits virus fusion and E1 trimerization. The crystal structures of the alphavirus p62/E1 and E2/E1 heterodimers indicate that E3 interacts only with E2 (13, 20). Since E3 does not interact with the fusion protein directly, it may act by stabilizing the E2-E1 dimer and thus maintain pH protection until the virus buds from the plasma membrane (19). While this is a compelling model, it has not been tested in vivo in infected cells, and the key interactions within the E3-E2 interface are undefined.
Here, we used site-directed mutagenesis of SFV E3 to test the role of two tyrosine residues, Y47 and Y48, in heterodimer stability and envelope protein biogenesis. The E3 Y47A Y48A (Y47/48A) mutant heterodimer was destabilized during spike transport, and the mutant was blocked in E1 cell surface expression and virus production. Neutralization of low pH in the secretory pathway rescued heterodimer transport to the plasma membrane and budding of infectious particles. Our data indicate that E3 plays a critical role in pH protection during alphavirus biogenesis and that disruption of key E3 Y47 interactions leads to the loss of pH protection and to the inactivation of E1 during transport.
(The data in this paper are from theses to be submitted by W.F. and by O.U. in partial fulfillment of the requirements for a Ph.D. in the Graduate Division of Medical Sciences, Albert Einstein College of Medicine, Yeshiva University.)
MATERIALS AND METHODS
Cells and viruses.
BHK-21 cells were cultured at 37°C in complete BHK medium (Dulbecco's modified Eagle medium containing 5% fetal bovine serum, 10% tryptose phosphate broth, 100 U penicillin/ml, and 100 μg of streptomycin/ml). The CHO-K1 cell line and its derivative, the furin-deficient FD11 CHO cell line (21), were kindly provided by Stephen H. Leppla at the National Institutes of Health. The cells were cultured at 37°C in alpha-minimal essential medium containing 10% fetal bovine serum, 100 U penicillin/ml, and 100 μg of streptomycin/ml.
Construction of the SFV mutant infectious clones.
Note that we use the Chikungunya virus (CHIKV) numbering throughout the manuscript; the SFV numbering is E3 Y48/Y49. Site-directed mutagenesis was performed using the plasmid DG-1 encoding the viral envelope proteins, as previously described (22). Briefly, circular mutagenesis was used to introduce the E3 mutation(s) Y47/48A, Y47A, Y47F, or Y48A into plasmid DG-1 using primeSTAR HS DNA polymerase (TaKaRa Bio Inc., Madison WI). The mutated DG-1 NsiI/SpeI fragments were subcloned into the pSP6-SFV4 infectious clone (23). The infectious clones obtained were sequenced to confirm the presence of the desired mutation and absence of other mutations (Genewiz, Inc., North Brunswick, NJ). Two independent clones of each mutant were generated from two independent PCRs and used to confirm initial results. Infectious RNAs were generated by in vitro transcription and electroporated into BHK-21 cells to produce virus infection (23). Plaque assays were performed on BHK cells.
Pulse-chase analysis.
Pulse-chase experiments were used to evaluate envelope protein biosynthesis and virus assembly as previously described (14, 24). In brief, BHK-21 cells were electroporated with wild-type (WT) or mutant RNA and incubated for 6 h at 37°C. The cells were then pulse-labeled with [35S]methionine-cysteine (PerkinElmer) for 30 min and chased in medium without label. The cell lysates and chase medium were collected at the times indicated on the figures and analyzed via immunoprecipitation with a rabbit polyclonal antibody specific to the SFV envelope proteins. Samples were analyzed by SDS-PAGE and phosphorimaging. Endoglycosidase H (Endo H; New England BioLabs) digestion was used to study the trafficking of the E1 protein from the ER to the Golgi compartment. Lysate samples collected at the 0- and 1-h chase times were digested with Endo H for 90 min at 37°C in 50 mM citrate buffer at pH 5.0 (25).
Pulse-chase assays in the presence of inhibitors of secretion and acidification were used to evaluate envelope protein transport and virus assembly. BHK-21 cells were electroporated with either WT or mutant RNA and incubated for 2 h at 37°C. Medium with or without 10 μM monensin (Sigma-Aldrich) or 100 nM bafilomycin A-1 (bafilomycin; Kamiya Biomedical) was added, and the cells were incubated for an additional 4 h at 37°C. Monensin blocks envelope protein transport from the medial to the trans-Golgi compartment (26) while bafilomycin is a specific inhibitor of the vacuolar ATPase (27). The cells were then labeled with [35S]methionine-cysteine for 10 min and chased in medium without label for 1, 2, and 4 h, maintaining the presence of inhibitor throughout. Lysate samples were immunoprecipitated with the E1 monoclonal antibody (MAb) E1-1 (25) to evaluate the E1-E2 dimer (16, 28) or with a rabbit polyclonal antibody to the SFV envelope proteins. Samples were analyzed by SDS-PAGE and phosphorimaging. To evaluate E1 surface expression in bafilomycin-treated cells, the inhibitor was added at 2 h postelectroporation, and the cells were cultured 12 h at 37°C. Indirect immunofluorescence was performed using MAb E1-1. The production of infectious virus was evaluated using parallel control or bafilomycin-treated cultures that were not radiolabeled.
RNA transfection into CHO and FD11 cells.
Viral RNA transfection was performed in Opti-MEM serum-free medium (Life Technologies) using Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocol. At 6 h posttransfection, the cells were washed four times with serum-free medium and incubated at 37°C for the time points indicated in Fig. 4. Viral titers were calculated by plaque assay on BHK cells. To test viral protein expression on the cell surface, cells were washed and incubated in growth medium, and indirect immunofluorescence was performed at 24 h posttransfection using E1 MAb E1-1.
Fig 4.
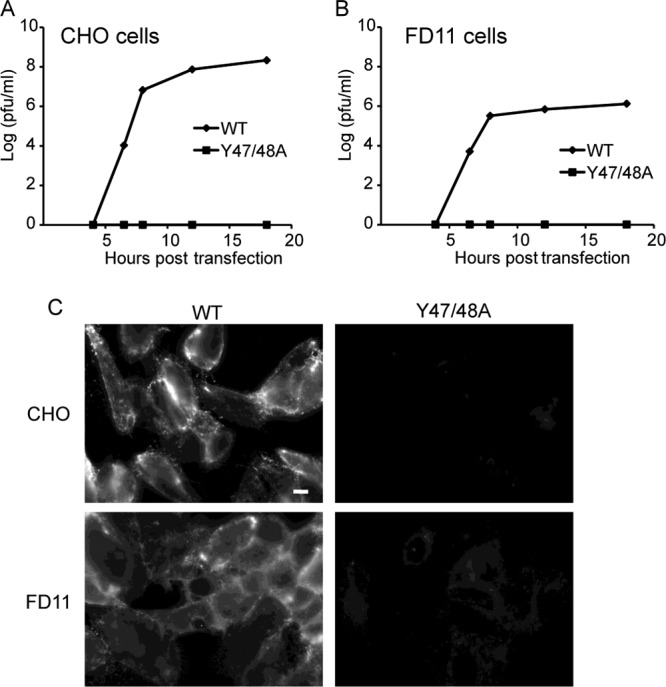
Furin dependence of Y47/48A growth and E1 surface expression. WT or mutant SFV RNA was transfected into WT (A) or furin-deficient FD11 (B) CHO cells. Cells were cultured for the indicated times, and progeny virus production was quantitated by plaque assay on BHK cells. At 24 h posttransfection, cells were fixed with paraformaldehyde, and E1 surface expression was visualized by indirect immunofluorescence using a MAb to E1 (C). Fluorescence microscopy images were acquired with the same exposure times. Scale bar, 10 μm. Data are representative of the results of two to three independent experiments.
Fusion-infection assay.
Appropriate dilutions of virus stocks were prebound to BHK-21 cells on ice to prevent endocytosis. Fusion with the plasma membrane was triggered by treatment of cells with media of various pHs for 3 min at 37°C. Cells were cultured at 28°C overnight in medium containing 20 mM NH4Cl to prevent secondary infection. Virus-infected cells were quantitated by immunofluorescence as described previously (29).
RESULTS
Analysis of interactions at the CHIKV E3-E2 interface.
We set out to test the role of the E3-E2 interaction in vivo and to identify important contacts that mediate this interaction. The structure of the CHIKV E3-E2-E1 complex (13) highlighted several E3 residues that interact with E2 (Fig. 1). These include E3 E39 and D40, which form salt bridges with E2 R251, and E3 Y48, which can hydrogen bond to E2 E166 and has van der Waals contacts with E2 H256. Y47 is also predicted to hydrogen bond with E2 K254. We analyzed the E3-E2 interface using the contacts of structural units (CSU) and PyMOL programs (30, 31). Based on our results and the previous analysis, we selected E3 Y47 and Y48 for mutational studies (Fig. 1B). While Y47 was not specifically highlighted in the report of the CHIKV E3-E2-E1 structure, we noted that Y47 is completely conserved among the alphaviruses (Fig. 1C) and that its predicted E2 partner, K254, while highly conserved, is an arginine in a fish alphavirus (13), suggesting a potential cation-π interaction.
Fig 1.
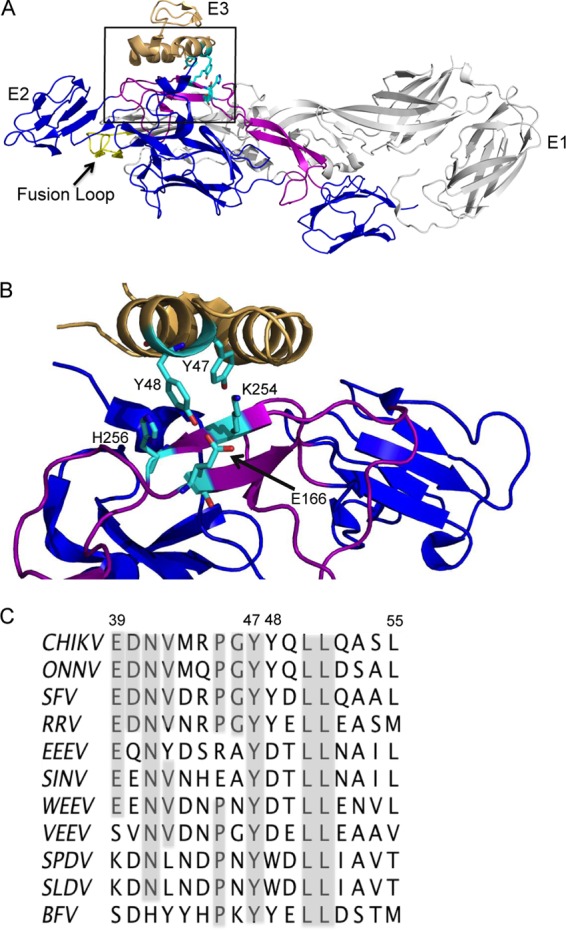
E3 structure, interactions, and sequence conservation. (A) The structure of the CHIKV E3-E2-E1 protein complex. E3 is shown in tan, E2 is in blue with its β-ribbon connector region in purple, and E1 is in light gray with the fusion loop in yellow (Protein Data Bank accession number 3N42) (13). E3 Y47 and Y48 and E2 E166, K254, and H256 are shown in cyan in stick view. The box indicates the magnified region in B. (B) Magnified view of the E3-E2 interface region, rotated to highlight the interactions between E3 Y47 and E2 K254 and between E3 Y48 and E2 E166 and H256. Colors are as in panel A. Panels A and B were prepared using the program PyMOL (31). (C) Amino acid sequence alignment of E3 residues 39 to 55 in alphaviruses, indicating the positions of Y47/Y48 (CHIKV numbering). Alignment was performed using Clustal W software. Virus abbreviations: CHIKV, Chikungunya virus; ONNV, O'nyong-nyong virus; SFV, Semliki Forest virus; RRV, Ross River virus; EEEV, Eastern equine encephalitis virus; SINV, Sindbis virus; WEEV, Western equine encephalitis virus; VEEV, Venezuelan equine encephalitis virus; SPDV, salmon pancreas disease virus; SLDV, sleeping disease virus of trout; BFV, Barmah Forest virus.
Generation and characterization of an E3 Y47/48A mutant.
We used site-directed mutagenesis of the SFV infectious clone to characterize the roles of E3 Y47 and Y48 (CHIKV numbering). Both tyrosines were changed to alanine to abrogate the interactions of the tyrosine side chains. The resultant E3 (Y47A Y48A) double mutant is here referred to as Y47/48A. Viral RNAs were transcribed in vitro and electroporated into BHK cells to characterize the phenotype. Immunofluorescent staining of Y47/48A-infected cells showed that E2, but not E1, was expressed on the surface, while WT-infected cells showed robust surface expression of both E2 and E1 (Fig. 2). Staining of permeabilized Y47/48A-infected cells showed that the steady-state pool of E1 was primarily localized in a perinuclear distribution consistent with the Golgi complex (Fig. 2, bottom panel). Although cell surface expression of some alphavirus envelope protein mutants can be rescued by incubation of infected cells at 28°C (32, 33), these conditions did not rescue E1 transport of the Y47/48A mutant (data not shown). Immunofluorescent staining confirmed that the double mutant did not cause secondary infection of cocultured nonelectroporated BHK cells (data not shown), in keeping with the E1 transport defect.
Fig 2.
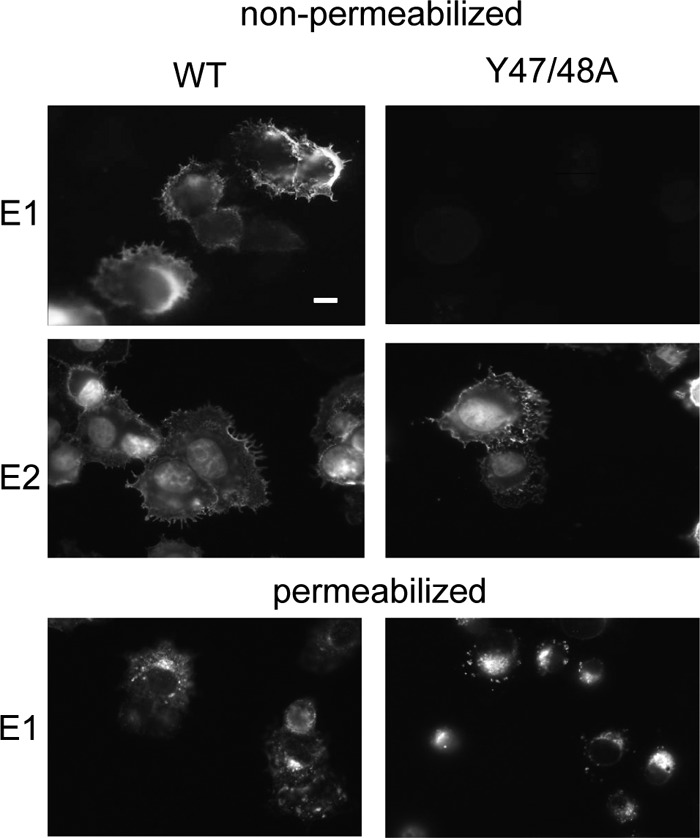
Surface expression of E1 and E2 in WT- and Y47/48A-infected cells. WT or mutant RNA was electroporated into BHK cells, and the cells were cultured at 37°C for 12 h. The cells were then fixed with either paraformaldehyde (nonpermeabilized) or methanol (permeabilized), and indirect immunofluorescence was performed with MAb to the E1 or E2 protein. Fluorescence microscopy images were acquired with the same exposure times. Images are representative examples of multiple (more than three) independent experiments. Scale bar, 10 μm.
Pulse-chase studies were used to further define the biosynthesis and assembly of the Y47/48A mutant (Fig. 3A). In the WT-infected cells, radiolabeled p62 was processed to mature E2 during a 1-h chase period, but processing in mutant-infected cells produced a form of E2 that migrated more slowly than that of the WT. Complete removal of the N-linked carbohydrate chains with endoglycosidase F produced equivalent migration of WT and mutant E2 (data not shown). Thus, the difference in electrophoretic migration was due to aberrant E2 glycosylation, in keeping with previous results for mutants with blocks in E1 transport (34). While WT envelope proteins were released in virus particles during the chase time (data not shown), significant levels of WT E1 and E2 also remained in the cells after a 4-h chase (Fig. 3A). In contrast, while the initial levels of radiolabeled envelope proteins in Y47/48A-infected cells were similar to those of the WT, the levels decreased significantly during chase even though no virus budding occurred, suggesting envelope protein degradation. Endo H digestion was then used to characterize the E1 protein in mutant-infected cells (Fig. 3B). After a 1-h chase, a significant proportion of the WT E1 protein became resistant to Endo H digestion, while most of the E1 protein detected in Y47/48A-infected cells was sensitive to Endo H digestion. Thus, while a stable pool of WT E1 was transported past the medial Golgi compartment, most of the detected mutant E1 protein had not transited past the medial Golgi compartment. As previously observed, E2 remains sensitive to Endo H due to an unmodified high-mannose carbohydrate chain (35).
Fig 3.
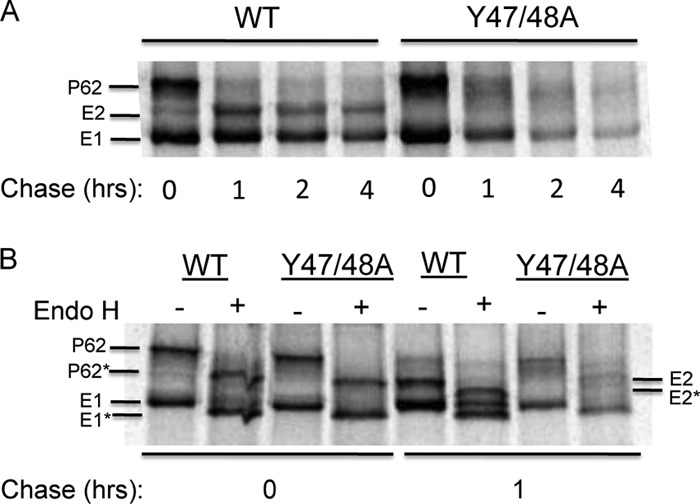
Envelope protein biosynthesis in WT- and Y47/48A-infected cells. BHK cells were electroporated with either WT or mutant RNA and incubated at 37°C for 6 h, pulse-labeled for 30 min with [35S]methionine-cysteine, and then chased for the indicated times. The cell lysates were immunoprecipitated with a polyclonal antibody to the SFV envelope glycoproteins. Samples were analyzed directly by SDS-PAGE (A) or following digestion with Endo H (B). p62*, E1*, and E2* illustrate the new positions of the proteins after Endo H digestion. Representative examples of two independent experiments are shown.
The role of p62 processing in the Y47/48A phenotype.
We hypothesized that the Y47/48A mutations destabilize the E3-E2 interaction, leading to the loss of pH protection for the E1 fusion protein and thus to its degradation. Given the observed enrichment of E1 in the Golgi complex in mutant-infected cells, it was important to examine whether the mutant phenotype correlated with p62 processing or with the exposure of the envelope proteins to mildly acidic pH, both of which occur in the late stages of the secretory pathway. In order to define the relative importance of these two factors, we compared envelope protein biogenesis in control and furin-deficient (FD11) CHO cells. We transfected both cell types with WT or mutant viral RNA and tested for production of infectious progeny virus by plaque assays on indicator BHK cells (14). WT virus was efficiently produced from both CHO and FD11 cells (Fig. 4A and B). The E3 Y47/48A mutant did not produce infectious virus from control CHO cells (Fig. 4A) and was not rescued in FD11 cells (Fig. 4B). Immunofluorescent staining of Y47/48A-infected cells confirmed that E1 was not transported to the surfaces of either control or FD11 CHO cells (Fig. 4C). These results suggested that the mutant phenotype was due to destabilizing effects on the p62-E1 heterodimer (see below), independent of maturation to E3-E2.
The role of transport and acidification in the Y47/48A phenotype.
We then tested the stability of the p62-E1 heterodimer at various times after biosynthesis using a coimmunoprecipitation (co-IP) assay. WT- and Y47/48A-infected BHK cells were pulse-labeled and chased for 1 to 4 h, lysed, and immunoprecipitated with a MAb to the E1 protein. In the WT samples, both p62 and E2 efficiently coimmunoprecipitated with E1 after a 1-h chase, and the mature E2 protein showed co-IP after chase times of up to 4 h (Fig. 5B), in keeping with the known stability of the heterodimer (16). Parallel immunoprecipitation of the samples with a rabbit antibody (Ab) to the envelope proteins (Fig. 5A) showed that the ratio of E2 and E1 in the cell lysates was similar to the ratio observed by co-IP. In contrast, the mutant samples showed co-IP of p62 with E1 after a 1-h chase but very little co-IP after a 2- to 4-h chase (Fig. 5B) even though E2 was readily detectable in the cell lysates at those time points (Fig. 5A). This result suggests that the mutant heterodimer is initially stable but is dissociated or destabilized during transport through the secretory pathway.
Fig 5.
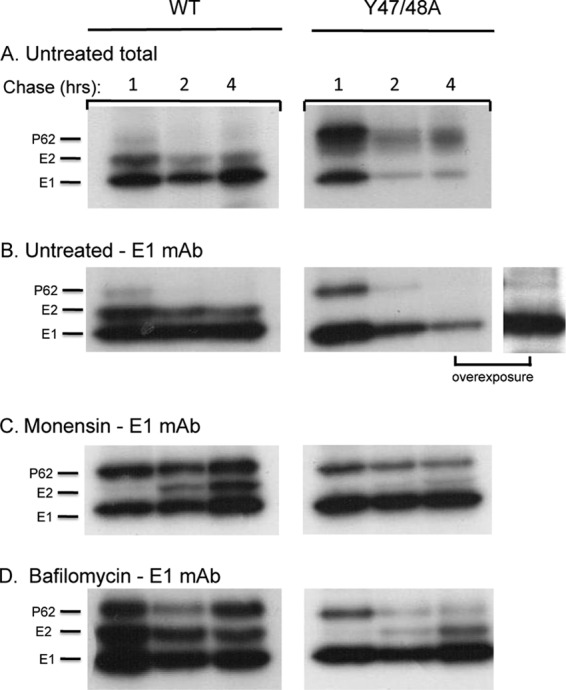
Effects of monensin and bafilomycin treatment on Y47/48A dimer interaction. BHK cells were electroporated with WT or mutant viral RNA and incubated for 2 h at 37°C. Culture medium alone (A and B) or medium containing monensin (C) or bafilomycin (D) was then added to the cells, and the incubation was continued for an additional 4 h. The cells were then pulse-labeled with [35S]methionine-cysteine and chased for the indicated times, all in the presence of monensin or bafilomycin as indicated. The cell lysates were immunoprecipitated with either a rabbit polyclonal antibody to the E1 and E2 proteins to evaluate the total envelope proteins in the cell lysates (A) or with a MAb to E1 to test for dimer stability by co-IP (B to D). The samples were analyzed by SDS-PAGE and fluorography, with the exposure time in the experiment shown in panel A decreased to visualize the ratio of E2 and E1. Panel B shows the results of overexposure of a Y47/48A 4-h sample (right). Data are representative examples of three independent experiments.
The furin-deficient cell results shown in Fig. 4 suggested that p62 cleavage alone was not responsible for destabilization of the mutant dimer. Studies of mutant-infected WT and FD11 cells showed that mutant co-IP was not rescued by the inhibition of p62 processing (data not shown). We therefore tested the effects of blocking transport through the secretory pathway using monensin, an ionophore that inhibits transport from the medial to the trans-Golgi network (26, 36). Monensin was added to cells at 2 h postelectroporation and maintained during pulse-labeling and chase. In WT-infected cells, p62 cleavage was significantly inhibited by monensin treatment, but efficient co-IP with E1 was observed at all chase times (Fig. 5C). Processing of p62 was also inhibited in Y47/48A-infected cells, while co-IP of p62 with E1 was increased (compare 2-h chase times in Fig. 5C versus B), suggesting stabilization of the mutant heterodimer.
Monensin treatment inhibits envelope protein transport, modifications such as glycosylation events, and exposure of the dimer to the mildly acidic pH of the late secretory pathway. To more directly test for the effects of low-pH exposure on heterodimer stability, we treated cells with bafilomycin, an inhibitor of the vacuolar ATPase (27, 37). Bafilomycin was added at 2 h postelectroporation, and pulse-labeling, chase, and co-IP analyses were performed as described above. Bafilomycin caused a moderate decrease in the kinetics of p62 processing in both WT- and mutant-infected cells (Fig. 5D). This decrease is most likely due to slower transport through the secretory pathway as no requirement for low pH in p62 processing has been observed (38). As expected, the WT heterodimer was efficiently retrieved by co-IP. Bafilomycin treatment significantly increased co-IP of the Y47/48A heterodimer at both the 2- and 4-h chase times (compare Fig. 5D versus B). Strikingly, mature E2 from the mutant-infected cells comigrated with WT E2, indicating that it was no longer aberrantly glycosylated (Fig. 5D, compare 2- to 4-h WT versus mutant chase samples). Together, these results suggested that neutralization of the secretory pathway significantly stabilized the mutant heterodimer.
We then tested if bafilomycin treatment could rescue mutant E1 transport and virus particle assembly. WT- and Y47/48A-infected cells were incubated in the presence of bafilomycin and immunostained to detect cell surface E1 (Fig. 6A). Significant rescue of E1 transport was observed in the mutant-infected cells. Medium samples from untreated and bafilomycin-treated cells were collected, and titers were determined by plaque assay on BHK cells (Fig. 6B). Neutralization of the secretory pathway significantly decreases protein secretion (39, 40), and, indeed, viral production from WT-infected cells was decreased ∼3 logs by bafilomycin treatment. While no mutant virus was produced from untreated cultures, virus production was rescued in the presence of bafilomycin, with mutant- and WT-infected cells producing similar titers under these conditions. These WT and mutant virus stocks showed similar pH dependence in tests of virus fusion with the plasma membrane of BHK cells (Fig. 6C). Thus, once mutant E1 transport was rescued and virus particles were produced, the E3 Y47/48A mutations had no detectable effects on virus pH dependence. Together, our results suggest that the mutations act by disrupting the E3-E2 interaction, destabilizing the mutant heterodimer, and causing a loss in the pH protection of E1 during transit through the secretory pathway.
Fig 6.
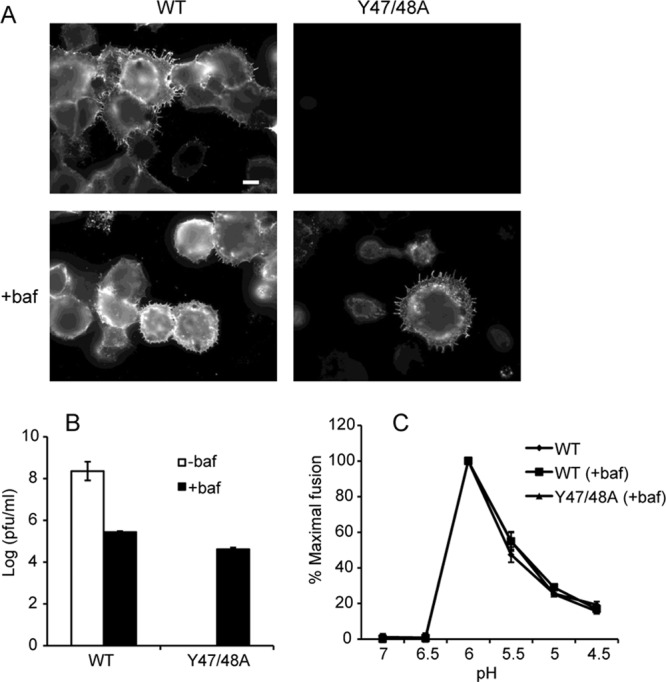
Assembly properties of Y47/48A after neutralization of exocytic pH. BHK cells were electroporated with WT or mutant viral RNA and incubated for 2 h at 37°C. Medium with (+) or without (−) bafilomycin (baf) was added, and the incubation was continued for 8 to 12 h. (A) After 12 h, the surface expression of E1 was visualized as described in the legend of Fig. 4. Fluorescence microscopy images were acquired with the same exposure times. Scale bar, 10 μm. (B) After 8 h, medium samples were collected, and titers were determined by plaque assay on BHK cells. (C) Virus stocks from the experiment described in panel B were also used to determine the pH dependence of virus fusion with the plasma membrane of BHK cells, as described in Materials and Methods. The images in panel A are representative of three independent experiments. Data in panels B and C represent the average and standard deviation of three independent experiments.
Defining the residues responsible for the Y47/48A phenotype.
Further mutagenesis was performed to determine whether E3 Y47 and/or Y48 was responsible for the loss in pH protection. Each tyrosine was changed to an alanine separately to create Y47A and Y48A virus mutants. Y47 was also changed to a phenylalanine (Y47F) to test the role of an aromatic residue at this position. We compared the growth kinetics of these viruses in BHK cells, as shown in Fig. 7. The Y47F and Y48A mutants had similar growth kinetics as those of WT SFV, while the Y47A mutation was lethal (Fig. 7). Thus, the tyrosine at position 47 is the critical residue at the E3-E2 interface for pH protection of E1. Although this tyrosine is completely conserved in alphavirus E3 proteins, a phenylalanine substitution was viable.
Fig 7.
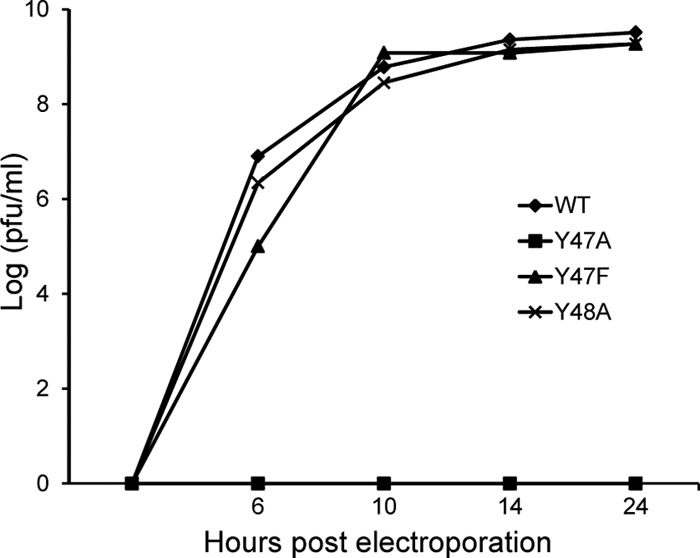
Infectious particle production by WT and mutants. BHK cells were electroporated with WT or mutant RNA as indicated and incubated at 37°C. At the indicated times, medium was collected, and progeny virus titers were determined by plaque assays. Data are representative of two independent experiments.
DISCUSSION
Roles of alphavirus E3.
E3 is a small glycoprotein that provides the signal sequence for p62 translocation into the ER (9). However, E3 plays additional roles since its replacement by an artificial signal sequence inhibits spike protein heterodimer formation and the transport of both E2 and E1 to the plasma membrane (41). This finding and other data have led to a general model in which p62, perhaps via the E3 region, initially acts as a chaperone to promote the correct folding and transport of E1 to the plasma membrane (see, e.g., references 9, 42, and 43). Following spike biosynthesis and folding, p62 is believed to protect E1 from low pH during transport. In keeping with this idea, the interactions of p62 with E1 are resistant to the pH range of the secretory pathway and become sensitive after cleavage to the mature E2 form (8, 14, 16).
The three-dimensional structures of the p62/E2-E1 dimer of the alphaviruses CHIKV and Sindbis virus were recently solved (13, 20). These heterodimer structures reveal that E3 interacts only with E2 and has no direct contacts with E1. In addition, the only change between the structures of the p62-E1 dimer and E2-E1 dimer is within the loop containing the furin cleavage site (13). Thus, the change in pH sensitivity between the immature and mature spike proteins appears to be due not to structural rearrangements but to the release of the E3 protein itself. This is in keeping with the finding that E3 remains bound to virus at low pH and is released at neutral pH (19) and provides a possible mechanism to protect E1 during its transit through the secretory pathway. Since dynamic p62-E1 contacts during protein folding in the ER could differ from the static p62-E1 structure, a direct chaperone role for E3 remains unclear.
Here, we identified a key interaction at the E3-E2 interface that conferred pH protection of E1 during secretory pathway transport. Alanine substitutions of SFV E3 Y47/48 were predicted to disrupt their interactions with E2 and proved lethal to the virus. While the mutant heterodimer was initially stable, it dissociated during chase, and E1 was not delivered to the plasma membrane. Inhibition of p62 cleavage did not rescue E1 transport or virus production, strongly suggesting that the mutant E3 moiety did not properly interact with E2 even prior to p62 processing. However, neutralization of the pH of the secretory pathway rescued mutant dimer association, E1 transport to the plasma membrane, and virus particle production. We propose that the critical E3 Y47A mutation caused the loss of E1 pH protection as the mutant heterodimer was transported through the acidic environment of the late secretory pathway (trans-Golgi cisternae and beyond), leading to E1's inactivation and the subsequent degradation of E1 and E2.
Y47 interactions.
Based on structural studies, the tyrosines at E3 positions 47 and 48 were predicted to form hydrogen bonds and van der Waals contacts with E2 K254 and E2 (E166 and H256), respectively (CHIKV numbering) (13). Our mutagenesis data demonstrated that the key interaction was via E3 Y47 but that it could be replaced by phenylalanine, another aromatic residue. E3 Y47 is less than 3.5 Å from E2 K254, which is either lysine or arginine in all of the reported alphavirus sequences (13). Thus, our data support an important cation-π interaction between the aromatic residue at position Y47 and E2 lysine 254.
The strength of a cation-π interaction is of the same order of magnitude as a hydrogen bond or salt bridge (44). The Y47-K254 interaction would presumably promote E3-E2 stability at low pH but would need to be destabilized at neutral pH. One explanation could involve effects of the local environment on the pKa of lysine. In solution, the lysine side chain has a pKa of ∼10.8, but pKa values as low as 5.3 and 6.7 have been observed in protein structures (45, 46). If the pKa of E2 K254 is decreased due to the local environment, then this could allow deprotonation of the side chain at neutral pH, destabilizing the cation-π interaction and promoting E3 dissociation.
Although in our experimental system E3 Y48 did not play an important role, studies of alphavirus E3/E2 chimeras have implicated this residue in envelope protein biogenesis and virus assembly (47). E3 Y48 is conserved within the SFV clade, where it interacts with E2 E166 and H256. In contrast, within the Sindbis virus clade, E3 contains an aspartate residue in the equivalent position and a corresponding tyrosine in place of E2 E166 (13, 47). Swaps of these residues across clades produced strong effects on virus envelope protein transport and particle infectivity (47). It will be interesting to evaluate if such substitutions cause effects on E1 pH protection in the E3/E2 chimeras.
The role of pr protein in flavivirus pH protection.
Flaviviruses, which have membrane fusion proteins that are structurally similar to those of the alphaviruses, go through an analogous maturation process (48–50). The flavivirus prM protein interacts with the E fusion protein during virus biogenesis (51). Late in the secretory pathway, furin cleaves prM into the peripheral pr protein and transmembrane M protein (7). The pr protein remains bound to the virus at low pH, and its release at neutral pH allows the virus to fuse and infect a new cell (48–50, 52). However, pr and E3 have dissimilar structures and different mechanisms of action. While the alphavirus E3 protein acts by stabilizing the E2-E1 dimer, the flavivirus pr protein binds directly to the fusion loop at the tip of the E protein. This interaction is stabilized by electrostatic interactions between pr and E (48, 49). Mutation of a key histidine residue at the tip of E reduces binding to pr and makes the virus susceptible to low-pH inactivation during virus exit (52).
The role of alphavirus E3 in pH protection.
Our studies provide new information on the chaperone and pH protection functions of the alphavirus p62 protein. While p62 appears to be required for helping to chaperone the folding of E1, our experiments suggest that E3 Y47/Y48 are not critical for this step in virus biogenesis. In particular, our data demonstrate that when the Y47/48A mutant is rescued by neutralization of the low pH during exit, the resultant virus shows similar growth and pH dependence for fusion as the WT virus, suggesting that both E1 and E2 are correctly folded. Further studies may define direct chaperone functions of p62 distinct from its role in E1 pH protection.
The alphaviruses and flaviviruses have both evolved mechanisms in which a small viral protein provides protection from low pH in the exocytic pathway. While the protective activity of E3 or pr is critical to the virus, this strategy has the potential to inhibit viral infectivity if the protein fails to dissociate. Not all of the fusion proteins need to be activated for virus fusion to occur (8), which probably provides some flexibility in this regulatory process. For both viruses, the strength of the interaction at the E3-E2 (or pr-E) interface needs to be carefully balanced in order for the protective peptide to stay bound after furin cleavage but still allow for release in the neutral pH outside the cell. Our studies identify a key residue in the alphavirus E3-E2 interaction and demonstrate the important role that E3 plays in pH protection. Such regulation of viral fusion proteins may have implications for the future development of antiviral therapies.
ACKNOWLEDGMENTS
We thank all the members from our lab for helpful discussions and experimental suggestions and Claudia Sanchez San Martin, Katie Stiles, and Aihua Zheng for their comments on the manuscript. We thank Youqing Xiang for excellent technical assistance.
This work was supported by a grant to M.K. from the National Institute of Allergy and Infectious Diseases (R01-AI075647) and by Cancer Center Core Support grant NIH/NCI P30-CA13330. O.U. was supported by NIH MSTP training grant T32 GM007288 and Training Grant in Geographic Medicine and Emerging Infections T32 AI070117. W.F. was supported by the Training Program in Cellular and Molecular Biology and Genetics, T32 GM007491.
The contents of this paper are solely our responsibility and do not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
Footnotes
Published ahead of print 17 July 2013
REFERENCES
- 1. Mercer J, Schelhaas M, Helenius A. 2010. Virus entry by endocytosis. Annu. Rev. Biochem. 79:803–833 [DOI] [PubMed] [Google Scholar]
- 2. Harrison SC. 2008. Viral membrane fusion. Nat. Struct. Mol. Biol. 15:690–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. White JM, Delos SE, Brecher M, Schornberg K. 2008. Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme. Crit. Rev. Biochem. Mol. Biol. 43:189–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pinto LH, Lamb RA. 2006. The M2 proton channels of influenza A and B viruses. J. Biol. Chem. 281:8997–9000 [DOI] [PubMed] [Google Scholar]
- 5. Wozniak AL, Griffin S, Rowlands D, Harris M, Yi M, Lemon SM, Weinman SA. 2010. Intracellular proton conductance of the hepatitis C virus p7 protein and its contribution to infectious virus production. PLoS Pathog. 6:e1001087. 10.1371/journal.ppat.1001087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sanchez-San Martin C, Liu CY, Kielian M. 2009. Dealing with low pH: entry and exit of alphaviruses and flaviviruses. Trends Microbiol. 17:514–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Stadler K, Allison SL, Schalich J, Heinz FX. 1997. Proteolytic activation of tick-borne encephalitis virus by furin. J. Virol. 71:8475–8481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Salminen A, Wahlberg JM, Lobigs M, Liljeström P, Garoff H. 1992. Membrane fusion process of Semliki Forest virus II: cleavage-dependent reorganization of the spike protein complex controls virus entry. J. Cell Biol. 116:349–357 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kuhn RJ. 2007. Togaviridae: The Viruses and Their Replication, p 1001–1022 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 10. Helenius A, Kartenbeck J, Simons K, Fries E. 1980. On the entry of Semliki Forest virus into BHK-21 cells. J. Cell Biol. 84:404–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kielian M, Chanel-Vos C, Liao M. 2010. Alphavirus entry and membrane fusion. Viruses 2:796–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gibbons DL, Vaney M-C, Roussel A, Vigouroux A, Reilly B, Lepault J, Kielian M, Rey FA. 2004. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature 427:320–325 [DOI] [PubMed] [Google Scholar]
- 13. Voss JE, Vaney MC, Duquerroy S, Vonrhein C, Girard-Blanc C, Crublet E, Thompson A, Bricogne G, Rey FA. 2010. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468:709–712 [DOI] [PubMed] [Google Scholar]
- 14. Zhang X, Fugere M, Day R, Kielian M. 2003. Furin processing and proteolytic activation of Semliki Forest virus. J. Virol. 77:2981–2989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. de Curtis I, Simons K. 1988. Dissection of Semliki Forest virus glycoprotein delivery from the trans-Golgi network to the cell surface in permeabilized BHK cells. Proc. Natl. Acad. Sci. U. S. A. 85:8052–8056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wahlberg JM, Boere WA, Garoff H. 1989. The heterodimeric association between the membrane proteins of Semliki Forest virus changes its sensitivity to low pH during virus maturation. J. Virol. 63:4991–4997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Smit JM, Klimstra WB, Ryman KD, Bittman R, Johnston RE, Wilschut J. 2001. PE2 cleavage mutants of Sindbis virus: correlation between viral infectivity and pH-dependent membrane fusion activation of the spike heterodimer. J. Virol. 75:11196–11204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Demaurex N, Furuya W, D'Souza S, Bonifacino JS, Grinstein S. 1998. Mechanism of acidification of the trans-Golgi network (TGN). In situ measurements of pH using retrieval of TGN38 and furin from the cell surface. J. Biol. Chem. 273:2044–2051 [DOI] [PubMed] [Google Scholar]
- 19. Sjoberg M, Lindqvist B, Garoff H. 2011. Activation of the alphavirus spike protein is suppressed by bound E3. J. Virol. 85:5644–5650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li L, Jose J, Xiang Y, Kuhn RJ, Rossmann MG. 2010. Structural changes of envelope proteins during alphavirus fusion. Nature 468:705–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gordon VM, Klimpel KR, Arora N, Henderson MA, Leppla SH. 1995. Proteolytic activation of bacterial toxins by eukaryotic cells is performed by furin and by additional cellular proteases. Infect. Immun. 63:82–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu CY, Besanceney C, Song Y, Kielian M. 2010. Pseudorevertants of a Semliki Forest virus fusion-blocking mutation reveal a critical interchain interaction in the core trimer. J. Virol. 84:11624–11633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liljeström P, Lusa S, Huylebroeck D, Garoff H. 1991. In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release. J. Virol. 65:4107–4113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chanel-Vos C, Kielian M. 2006. Second-site revertants of a Semliki Forest virus fusion-block mutation reveal the dynamics of a class II membrane fusion protein. J. Virol. 80:6115–6122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kielian M, Jungerwirth S, Sayad KU, DeCandido S. 1990. Biosynthesis, maturation, and acid-activation of the Semliki Forest virus fusion protein. J. Virol. 64:4614–4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Griffiths G, Quinn P, Warren G. 1983. Dissection of the Golgi complex. I. Monensin inhibits the transport of viral membrane proteins from medial to trans Golgi cisternae in baby hamster kidney cells infected with Semliki forest virus. J. Cell Biol. 96:835–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Drose S, Altendorf K. 1997. Bafilomycins and concanamycins as inhibitors of V-ATPases and P-ATPases. J. Exp. Biol. 200:1–8 [DOI] [PubMed] [Google Scholar]
- 28. Zhang X, Kielian M. 2004. Mutations that promote furin-independent growth of Semliki Forest virus affect p62-E1 interactions and membrane fusion. Virology 327:287–296 [DOI] [PubMed] [Google Scholar]
- 29. Liao M, Kielian M. 2005. Domain III from class II fusion proteins functions as a dominant-negative inhibitor of virus-membrane fusion. J. Cell Biol. 171:111–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Sobolev V, Sorokine A, Prilusky J, Abola EE, Edelman M. 1999. Automated analysis of interatomic contacts in proteins. Bioinformatics 15:327–332 [DOI] [PubMed] [Google Scholar]
- 31. DeLano WL. 2002. The PyMOL user's manual. DeLano Scientific, San Carlos, CA [Google Scholar]
- 32. Duffus WA, Levy-Mintz P, Klimjack MR, Kielian M. 1995. Mutations in the putative fusion peptide of Semliki Forest virus affect spike protein oligomerization and virus assembly. J. Virol. 69:2471–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chatterjee PK, Eng CH, Kielian M. 2002. Novel mutations that control the sphingolipid and cholesterol dependence of the Semliki Forest virus fusion protein. J. Virol. 76:12712–12722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Levy-Mintz P, Kielian M. 1991. Mutagenesis of the putative fusion domain of the Semliki Forest virus spike protein. J. Virol. 65:4292–4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hubbard SC. 1988. Regulation of glycosylation. The influence of protein structure on N-Linked oligosaccharide processing. J. Biol. Chem. 263:19303–19317 [PubMed] [Google Scholar]
- 36. Quinn P, Griffiths G, Warren G. 1983. Dissection of the Golgi complex. II. Density separation of specific Golgi functions in virally infected cells treated with monensin. J. Cell Biol. 96:851–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bowman EJ, Siebers A, Altendorf K. 1988. Bafilomycins: A class of inhibitors of membrane ATPases from microorganisms, animal cells, and plant cells. Proc. Natl. Acad. Sci. U. S. A. 85:7972–7976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Jain SK, DeCandido S, Kielian M. 1991. Processing of the p62 envelope precursor protein of Semliki Forest virus. J. Biol. Chem. 266:5756–5761 [PubMed] [Google Scholar]
- 39. Paroutis P, Touret N, Grinstein S. 2004. The pH of the secretory pathway: measurement, determinants, and regulation. Physiology (Bethesda) 19:207–215 [DOI] [PubMed] [Google Scholar]
- 40. Neblock DS, Berg RA. 1982. Lysosomotropic agents ammonium chloride and chloroquine inhibit both the synthesis and secretion of procollagen by freshly isolated embryonic chick tendon cells. Biochem. Biophys. Res. Comm. 105:902–908 [DOI] [PubMed] [Google Scholar]
- 41. Lobigs M, Hongxing Z, Garoff H. 1990. Function of Semliki Forest virus E3 peptide in virus assembly: replacement of E3 with an artificial signal peptide abolishes spike heterodimerization and surface expression of E1. J. Virol. 64:4346–4355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Carleton M, Lee H, Mulvey M, Brown DT. 1997. Role of glycoprotein PE2 in formation and maturation of the Sindbis virus spike. J. Virol. 71:1558–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Snyder AJ, Sokoloski KJ, Mukhopadhyay S. 2012. Mutating conserved cysteines in the alphavirus e2 glycoprotein causes virus-specific assembly defects. J. Virol. 86:3100–3111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dougherty DA. 1996. Cation-pi interactions in chemistry and biology: a new view of benzene, Phe, Tyr, and Trp. Science 271:163–168 [DOI] [PubMed] [Google Scholar]
- 45. Mukouyama EB, Oguchi M, Kodera Y, Maeda T, Suzuki H. 2004. Low pKa lysine residues at the active site of sarcosine oxidase from Corynebacterium sp. U-96. Biochem. Biophys. Res. Comm. 320:846–851 [DOI] [PubMed] [Google Scholar]
- 46. Isom DG, Castaneda CA, Cannon BR, Garcia-Moreno B. 2011. Large shifts in pKa values of lysine residues buried inside a protein. Proc. Natl. Acad. Sci. U. S. A. 108:5260–5265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Snyder AJ, Mukhopadhyay S. 2012. The alphavirus E3 glycoprotein functions in a clade-specific manner. J. Virol. 86:13609–13620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Li L, Lok SM, Yu IM, Zhang Y, Kuhn RJ, Chen J, Rossmann MG. 2008. The flavivirus precursor membrane-envelope protein complex: structure and maturation. Science 319:1830–1834 [DOI] [PubMed] [Google Scholar]
- 49. Yu IM, Zhang W, Holdaway HA, Li L, Kostyuchenko VA, Chipman PR, Kuhn RJ, Rossmann MG, Chen J. 2008. Structure of the immature dengue virus at low pH primes proteolytic maturation. Science 319:1834–1837 [DOI] [PubMed] [Google Scholar]
- 50. Yu IM, Holdaway HA, Chipman PR, Kuhn RJ, Rossmann MG, Chen J. 2009. Association of the pr peptides with dengue virus at acidic pH blocks membrane fusion. J. Virol. 83:12101–12107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Lorenz IC, Allison SL, Heinz FX, Helenius A. 2002. Folding and dimerization of tick-borne encephalitis virus envelope proteins prM and E in the endoplasmic reticulum. J. Virol. 76:5480–5491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Zheng A, Umashankar M, Kielian M. 2010. In vitro and in vivo studies identify important features of dengue virus pr-E protein interactions. PLoS Pathog. 6:e1001157. 10.1371/journal.ppat.1001157 [DOI] [PMC free article] [PubMed] [Google Scholar]