

Abstract
All eight human herpesviruses have a conserved herpesvirus protein kinase (CHPK) that is important for the lytic phase of the viral life cycle. In this study, we show that heat shock protein 90 (Hsp90) interacts directly with each of the eight CHPKs, and we demonstrate that an Hsp90 inhibitor drug, 17-dimethylaminoethylamino-17-demethoxygeldanamycin (17-DMAG), decreases expression of all eight CHPKs in transfected HeLa cells. 17-DMAG also decreases expression the of the endogenous Epstein-Barr virus protein kinase (EBV PK, encoded by the BGLF4 gene) in lytically infected EBV-positive cells and inhibits phosphorylation of several different known EBV PK target proteins. Furthermore, 17-DMAG treatment abrogates expression of the human cytomegalovirus (HCMV) kinase UL97 in HCMV-infected human fibroblasts. Importantly, 17-DMAG treatment decreased the EBV titer approximately 100-fold in lytically infected AGS-Akata cells without causing significant cellular toxicity during the same time frame. Increased EBV PK expression in 17-DMAG-treated AGS-Akata cells did not restore EBV titers, suggesting that 17-DMAG simultaneously targets multiple viral and/or cellular proteins required for efficient viral replication. These results suggest that Hsp90 inhibitors, including 17-DMAG, may be a promising group of drugs that could have profound antiviral effects on herpesviruses.
INTRODUCTION
Human herpesviruses are enveloped viruses containing relatively large, double-stranded DNA genomes. Although all herpesviruses experience both latent and lytic stages of infection, they are grouped into three separate families (alpha-, beta-, and gammaherpesviruses) according to differences in sequence homology and cellular tropisms. The alphaherpesviruses, which comprise herpes simplex virus 1 (HSV-1), HSV-2, and varicella-zoster virus (VZV), cause recurrent skin lesions and meningitis (1, 2). Human cytomegalovirus (HCMV), human herpesviruses 6A and 6B (HHV6), and human herpesvirus 7 (HHV7) are betaherpesviruses, which cause severe disease in patients with compromised immune function (3, 4). The gammaherpesviruses are Epstein-Barr virus (EBV) and Kaposi's sarcoma-associated herpesvirus (KSHV), which are causally associated with mononucleosis (EBV) as well as a variety of human cancers (5, 6).
Each of the eight human herpesviruses encodes a protein kinase (PK) with discernible homology in amino acid sequences and positional similarity in their respective viral genomes. These related protein kinases, termed the conserved herpesvirus protein kinases (CHPKs), are important for viral replication and infection (7–13). They play important roles in multiple processes, including gene expression (8, 11, 14), viral DNA replication (11, 15–17), capsid nuclear egress (7, 11, 18, 19), and the DNA damage response (20, 21). For example, EBV PK (the product of the BGLF4 gene) phosphorylates a number of different viral and cellular proteins, including the viral DNA polymerase processivity factor BMRF1 (7, 22–24); the latent viral proteins EBNA1 (25), EBNA2 (26), and EBNA LP (27); the EBV immediate early (IE) protein BZLF1 (28); the cell cycle regulatory proteins p27 (29) and pRB (30); nuclear lamin A/C (7, 31); and interferon regulatory factor 3 (IRF3) (32). In addition, EBV PK may upregulate the expression of two viral proteins important for nuclear egress, BFRF1 and BFLF2 (11, 33). Both EBV PK and the homologous HCMV kinase, UL97, greatly enhance but are not absolutely required for the release of infectious viral particles in vitro and appear to be intimately involved in the pathogenesis associated with viral infections in vivo (34, 35). Although maribavir, an inhibitor of HCMV UL97, failed a phase III clinical trial in bone marrow transplant patients (36) (possibly due to insufficient dosing), CHPKs nevertheless remain very promising targets for development of novel antiviral therapeutics.
Two guanine nucleoside analogues, ganciclovir (GCV) and acyclovir (ACV), have been used frequently to inhibit replication of various human herpesviruses by targeting viral DNA polymerases (37–40). UL97 mediates the first step of GCV and ACV phosphorylation (41–43). Since the triphosphorylated forms of GCV and ACV are much better substrates for herpesvirus DNA polymerases than cellular DNA polymerases, GCV and ACV inhibit viral DNA replication more effectively than cellular DNA replication (44, 45). It was recently found that EBV PK is required for inhibition of lytic EBV replication mediated by GCV and ACV (46).
Heat shock proteins (Hsps), a group of molecular chaperones, facilitate proper protein folding, stability, interactions, and intracellular trafficking (47, 48). Unlike other Hsps, only a relatively small subset of cellular proteins (numbering in the hundreds) are thought to be clients of Hsp90 (49, 50). Interestingly, cellular kinases make up the bulk of Hsp90 clients; indeed, Hsp90 was recently shown to interact with over half of the known human kinases (49). Hsp90 inhibitors such as 17-DMAG (17-dimethylaminoethylamino-17-demethoxygeldanamycin) (also known as alvespimycin) bind to the ATP-binding motif of Hsp90 and inhibit its protein chaperoning activity, resulting in misfolding and subsequent degradation of client proteins (51, 52). Hsp90 inhibitors are often more toxic to tumor cells than to normal cells (50), since a specific Hsp90 conformation required for inhibitor binding exists more frequently in tumor cells (53), and a variety of Hsp90 client proteins contribute to tumor cell growth, such as EGFR (epidermal growth factor receptor), AKT (also known as protein kinase B), mutant p53, and IKK (IκB kinase) (54–57).
We have previously demonstrated that Hsp90 inhibitors decrease EBNA1 expression in a variety of different EBV-infected cell types and kill EBV-transformed B cells at nontoxic doses in vitro and in a SCID mouse xenograft model (58). Here we show that Hsp90 associates with the CHPKs encoded by each of the 8 human herpesviruses and that 17-DMAG treatment decreases expression of the virally encoded kinases in cells transfected with expression vectors for each kinase. In addition, we find that expression of the EBV-encoded and HCMV-encoded kinases are markedly decreased in EBV-infected and HCMV-infected cells, respectively, when treated with 17-DMAG, while viral IE protein expression is not affected. Finally, we also show that 17-DMAG reduces intracellular lytic viral replication, as well as the release of infectious viral particles, in EBV-infected cells.
MATERIALS AND METHODS
Cell lines and reagents.
AGS-Akata/BX1 cells were a gift from Lindsey Hutt-Fletcher and are gastric carcinoma cells superinfected with the Akata strain of EBV, which contains the green fluorescent protein (GFP) open reading frame and a G418 resistance gene (59). The HONE-Akata cell line is a nasopharyngeal carcinoma line superinfected with the Akata strain of EBV containing the GFP open reading frame and a G418 resistance gene (a gift from Lawrence Young). The HeLa cell line is a malignant human epithelial cervical cancer cell line (obtained from the ATCC). Primary human foreskin fibroblasts (HFFs) were a gift from Thomas Shenk, Princeton University. The Mutu I cell line is a type I EBV-positive Burkitt lymphoma line (a gift from Alan Rickinson at the University of Birmingham and Jeff Sample at Pennsylvania State University).
All cell lines were cultured at 37°C with 5% CO2 and 100% humidity in growth medium (Invitrogen) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin-streptomycin (Sigma-Aldrich), except where indicated. AGS-Akata cells were cultured in Ham's F-12 medium, HeLa cells and HFFs were cultured in Dulbecco's modified Eagle's medium (DMEM), and HONE-Akata and Mutu I cells were cultured in RPMI 1640. HONE-Akata and AGS-Akata cells were maintained under drug selection (G418 sulfate at 0.4 mg/ml), as were HFFs (100 μg/ml streptomycin, 0.292 mg/ml glutamine).
17-DMAG (InvivoGen) and lactacystin (Calbiochem) were dissolved in dimethyl sulfoxide (DMSO). 3-MA (3-methyladenine) (Sigma) was dissolved in medium just before use. Acyclovir (Sigma) was dissolved in 1 M HCl. Transforming growth factor β (TGF-β) was purchased from Biolegend (catalog number 580704), diluted in 4 mM HCl (with 1 mg/ml bovine serum albumin [BSA]), and given at a dose of 5 ng/ml of medium.
Plasmids.
Plasmid DNA was purified through columns (Qiagen) as described by the manufacturer. The pSG5-Z construct contains the full-length BZLF1 gene sequence under the control of the simian virus 40 (SV40) promoter in the pSG5 vector (Stratagene) and was a gift from S. Diane Hayward, John Hopkins University. Hemagglutinin (HA)-tagged HSV-2 PK in pSG5 was a gift from Lynda Morrison, Saint Louis University. Since we previously found that the activity of the HCMV IE promoter is decreased by Hsp90 inhibitors (58), HA-tagged HCMV UL97, KSHV PK, HHV6 PK, HHV7 PK, VZV PK, HSV-1 PK, and EBV PK vectors used in this paper were constructed by removing each HA-tagged viral kinase gene from the pCGN vector and inserting it into the BamHI site in the pSG5 vector by using an In-Fusion HD cloning kit (Clontech). Viral kinase sequences were PCR amplified by using KOD Hot Start DNA polymerase (EMD). The templates for HCMV PK (UL97), KSHV PK, HHV6 PK, HHV7 PK, VZV PK, HSV-1 PK, and EBV PK were their respective HA-tagged PK genes in pCGN vectors (30). The forward primer for these 7 viral PKs is 5′-GGGCGAATTCGGATCGCGCGTATGGCTTCTAGCTA. The reverse primers are as follows: 5′-AATAAGATCTGGATCTTACTCGGGGAACAGTTG for HCMV, 5′-AATAAGATCTGGATCTCAGAAAACAAGTCCGCG for KSHV, 5′-AATAAGATCTGGATCTCACATATGAAAGAGAGA for HHV6, 5′-AATAAGATCTGGATCTTACACCCGAAATACAGA for HHV7, 5′-AATAAGATCTGGATCTTATGTCGATCCTATCCA for VZV, 5′-AATAAGATCTGGATCTCACGACAGCGCGTGCCG for HSV-1, and 5′-AATAAGATCTGGATCTCATCCACGTCGGCCATC for EBV. Rat cytomegalovirus (RCMV) PK was constructed by PCR amplification from genomic DNA, kindly provided by Daniel Streblow of Oregon Health Sciences University. The forward primer (encoding an HA tag) is 5′-GGGCGAATTCGGATCATGTACCCATACGATGTTCCGGATTACGCTATGGAGAACACGCCGCCC, and the reverse primer is 5′-AATAAGATCTGGATCTCAGGGGAACAGGGAGAA. Murine gammaherpesvirus 68 (MHV68) PK was constructed by PCR amplification from pORF36 (60). The forward primer (encoding an HA tag) is 5′-GGGCGAATTCGGATCATGTACCCATACGATGTTCCGGATTACGCTATGGATTACCGACAGTTA, and the reverse primer is 5′-AATAAGATCTGGATCTCAAAAAAATCCAGAATA. SG5-HA Z was constructed by amplifying genomic Z from the SG5-Z plasmid using primers that add a BamHI or EcoRI site to the ends of the sequence and also a 5′ HA tag and then inserting the PCR product between the BamHI and EcoRI sites in the SG5 vector. The forward primer (with EcoRI) is 5′-GACGAATTCATGTACCCATACGATGTTCCGGATTACGCTATGATGGACCCAAACTCGAC, and the reverse primer (with BamHI) is 5′-CGTGGATCCTTAGAAATTTAAGAGATCCTCGTG. SG5-HA Daxx was made by cutting pcDNA3-HA Daxx (a gift from M. H. Shih at Academia Sinica, Taiwan [61]) out of the pcDNA3 vector with KpnI and XbaI and ligating it into a modified SG5 vector (a linker with a KpnI/XbaI site had been previously inserted into the SG5 multiple-cloning site). Halo-EBV PK was generated by cloning HA-EBV PK into the expression plasmid pFN21A (Promega), which encodes the Halo protein fused to the N terminus of HA-EBV PK. A kinase-inactive form of the Halo-EBV PK plasmid (in which lysine residue 102 was changed to an isoleucine residue) was also constructed by using site-directed mutagenesis. The HA-EBV PK open reading frame was PCR amplified from pCGN-HA-EBV PK (30) by using the primers 5′-TGTCGCGATCGCCATGGCTTCTAGCTATCCTTATGACGTGCCTGA and 5′-AACTGTTTAAACTCCACGTCGGCCATCTGGACC. The HA-EBV PK PCR fragment and Halo vector (pFN21A) were digested by using PmeI and SgfI enzymes and ligated together to produce Halo-EBV PK. pSG5-BMRF1 (early antigen diffuse [EAD]) was generated in the Kenney laboratory as previously described (62). pGEX-2T (Pharmacia) was used to clone a fragment of the BMRF1 gene for expression of the glutathione S-transferase (GST)-BMRF1 (C terminus) fusion protein in Escherichia coli. A blunt-ended NotI/BglII fragment from pSG5-BMRF1 was ligated into SmaI-digested pGEX-2T to construct pGST-BMRF1 (C terminus), encoding amino acids 303 to 404 of BMRF1, downstream of the GST protein. An N-terminal myc-tagged EBV thymidine kinase (TK) expression vector was a gift from Edward Gershburg (Southern Illinois University School of Medicine) and was constructed by PCR amplifying the TK gene from the EBV genome by using primers 5′-GGATCCGCCGCCATGGAGCAGAAACTCATCTCTGAAGAGGATCTGATGGCTGGATTTCCAGGAAAGGAG-3′ and 5′-CTCGAGCTAGTCCCGATTTCCCCTCTCAAAATCAGAG-3′ and inserting it into the BamHI/XhoI sites in the pTK208 lentiviral vector. A FLAG-tagged Hsp90 expression vector was a gift from Len Neckers at the National Cancer Institute and was constructed as previously described (63). All plasmids constructed by PCR have been sequenced and confirmed to be correct.
Cell transfection and infection.
AGS-Akata, HONE-Akata, and HeLa cells were transfected by using FuGene 6 (Roche) or Lipofectamine 2000 (Invitrogen) according to the manufacturer's protocols. At 4 h posttransfection, cells were treated with 0.17 μM 17-DMAG or the DMSO control. In some experiments, cells were treated in the absence or presence of lactacystin (20 μM) and 3-MA (10 mM). Lactacystin and 3-MA were added into the medium 16 h and 24 h, respectively, before cell harvesting.
For HCMV infection studies, primary human fibroblasts (HFFs) were seeded onto a plate, and the next day, cells were subjected to serum starvation (0.1% FBS) for 48 h and then infected with HCMV (strain AD169) at a multiplicity of infection (MOI) of 1 or 3 PFU/cell. Cells were harvested after 6, 24, 48, or 72 h. In some experiments, cells were treated in the absence or presence of 17-DMAG (0.17 μM), which was added 1 h prior to infection, and/or lactacystin (7.2 or 10 μM) or 3-MA (10 mM), which were added at 6 h postinfection.
Immunoblot analyses.
Total cellular protein was harvested, separated on 10% or 7.5% SDS-PAGE gels, and subjected to immunoblot analyses. The primary antibodies used were as follows: EBV PK (BGLF4) (catalog number AP8057b, at a 1:1,000 dilution; Abgent), EBV BZLF1 (catalog number sc-53904, 1:200; Santa Cruz), EBV BMRF1 (catalog number vp-E608, 1:250; Vector Laboratories), Cdc2 (catalog number sc-54, 1:200; Santa Cruz), Cdc37 (catalog number sc-13129, 1:200; Santa Cruz), HA (catalog number sc-805, 1:200; Santa Cruz) (catalog number MMS-101P, 1:1,000; Covance), FLAG M2 antibody resin (catalog number A2220; Sigma), Hsp90 (catalog number sc-7947, 1:1,000; Santa Cruz), UL44 (catalog number CA006-100, 1:1,000; Virusys), β-actin (catalog number A5441, 1:5,000; Sigma), lamin A/C (catalog number sc-7292, 1:500; Santa Cruz), phospho-lamin A/C Ser22 (catalog number 2026, 1:1,000; Cell Signaling), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (catalog number AM4300, 1:1,000; Ambion). A monoclonal antibody used to detect EBV PK expression in lytically infected cells was derived as previously described (64) and was a gift from M. R. Chen at the College of Medicine, National Taiwan University. A monoclonal primary antibody, 1B12, was a gift from Tom Shenk at Princeton University and was raised against the C terminus of IE1, as described previously (65), and used at a 1:100 dilution. A primary antibody raised against HCMV-UL97 in rabbits was a gift from Donald Coen at Harvard University (66) and was used at a 1:1,000 dilution. Rabbit anti-EBV TK (used at a 1:200 dilution) was prepared and analyzed as described previously (67). Briefly, full-length recombinant EBV TK was expressed in bacteria and purified as described previously (68), and the GST moiety was removed as described previously for HHV8 TK (69). Preimmune rabbit serum was collected, and rabbits were immunized with purified recombinant EBV TK that was then provided to a commercial vendor (Harlan Bioproducts) for antibody production. Serum was harvested at different time intervals, and antibodies from both preimmune and immune sera were purified by protein A chromatography (Pierce) and analyzed for titer and specificity (67). The secondary antibodies used were horseradish peroxidase-conjugated secondary anti-mouse IgG (catalog number 31430, 1:10,000; Pierce Biotechnology), anti-rabbit IgG (catalog number 31460, 1:5,000; Pierce Biotechnology), and anti-rat IgG (catalog number 31230, 1:5,000; Pierce Biotechnology). Bound antibodies were detected by using the ECL system (Pierce Biotechnology). Image quantifications were performed by using ImageQuant software (GE Healthcare).
siRNA experiments.
HONE-Akata cells were transfected with 80 pmol small interfering RNA (siRNA) against the human Cdc37 message (catalog number sc-29255; Santa Cruz) or a negative-control siRNA (catalog number sc-37007; Santa Cruz) by using X-tremeGENE (Roche). Two days after delivery of the siRNA, the cells were transfected with 20 pmol Cdc37 siRNA (or control siRNA) and 400 ng of the pSG5-EBV PK plasmid by using Lipofectamine 2000 (Invitrogen). After 4 h, cells were then treated with 0.17 μM 17-DMAG or the DMSO control. Two days later, total protein was harvested, separated on 10% SDS-PAGE gels, and subjected to immunoblot analyses.
EBV replication assays.
AGS-Akata cells were transfected in six-well plates with a BZLF1 expression plasmid (pSG5-Z, at 0.1 μg/well) to induce lytic replication in the presence or absence of 0.17 μM 17-DMAG. Cell medium was replaced at 2 days posttransfection with medium free of 17-DMAG or DMSO. Twenty-four hours later, virus supernatants were harvested and filtered through a 0.8-μm filter (3 days after transfection). Viral titers were determined by the green Raji cell assay (70). Raji cells (4 × 105 cells in 1 ml of medium/well) were infected in 24-well plates with a serial dilution of virus supernatant. Phorbol-12-myristate-13-acetate (TPA) (20 ng/ml) and sodium butyrate (3 mM) were added 24 h later. The next day, GFP-positive Raji cells were scored by using a fluorescence microscope. The number of green Raji cells per milliliter (green Raji units [GRU]/ml) was calculated to determine the concentration of infectious virus particles. All experiments were performed in duplicate.
Preparation of EBV PK.
Active (or kinase-dead) purified EBV PK was prepared by using HaloTag technology (Promega), according to the manufacturer's instructions. Briefly, expression plasmids for HaloTagged EBV PK were transfected into 293T cells, and protein lysates were prepared 48 h later. HaloTagged PK was covalently captured by HaloLink resin (catalog number G6200) during incubation overnight. After three washes, the kinase was liberated from the resin and the attached HaloTag by the addition of tobacco etch virus (TEV) protease (a gift from Richard Burgess, University of Wisconsin—Madison [UW-Madison]). After 2 h, centrifugation was done to remove the beads and HaloTag, generating a supernatant containing purified and active PK. The full lysate and the purified PK were separated on SDS-PAGE gels and stained with Coomassie to estimate purity.
In vitro kinase assays.
To express GST and GST-tagged BMRF1 (C terminus), GST plasmids were transformed into Escherichia coli DH5α cells. The bacteria were grown in 10 ml of LB medium containing 50 mg/ml ampicillin at 37°C until the optical density at 600 nm reached 0.6 to 0.8. Protein expression was induced with 1 nM isopropylthiogalactopyranoside (IPTG; Sigma-Aldrich) at 37°C for 2 h. Bacteria were pelleted and lysed in cold phosphate-buffered saline (PBS) containing protease inhibitors (Roche) by sonication. After sonication, Triton X-100 was added to a final concentration of 1%, and bacterial debris was removed by centrifugation. Crude extracts were incubated with 50 μl of a 50% slurry of glutathione-agarose beads (Sigma) for 1 h at 4°C. Beads were washed three times with cold PBS and one time with incomplete kinase assay buffer (50 mM HEPES [pH 7.4], 5 mM MgCl2, 300 mM KCl, 0.1% NP-40). The beads were split into three aliquots. One aliquot of the beads was resuspended in SDS sample buffer, and protein expression was analyzed on 10% SDS-PAGE gels and detected by Coomassie blue staining. The other aliquots of the beads were incubated with kinase buffer containing 50 mM HEPES (pH 7.4), 5 mM MgCl2, 300 mM KCl, 0.1% NP-40, 5 μCi of [γ-32P]ATP (Amersham), and either purified GST or the GST-BGLF4 fusion protein at 30°C for 30 min. After the reaction, the beads were washed with buffer (50 mM HEPES [pH 7.4], 5 mM MgCl2, 300 mM KCl, 0.5% NP-40) three times. After the proteins were resolved by 10% SDS-PAGE, the proteins were then transferred onto a nitrocellulose membrane and subjected to autoradiography.
Mass spectrometry.
Halo-EBV PK was overexpressed in 293T cells, and protein complexes were pulled down by using HaloLink resin, as described above. The resulting samples were digested with trypsin overnight and analyzed on the ThermoElectron Finnigan LTQ mass spectrometer with XCalibur software at the UW-Madison Human Proteomics Program Core Facility. Data were analyzed with Bioworks software using SEQUEST searches with human parameters. The resulting hits were filtered and compared with a control sample expressing only the HaloTag protein.
Coimmunoprecipitation assays.
For coimmunoprecipitation experiments using the various HA-tagged viral kinases or the HaloTagged EBV PK, cells were transfected with expression plasmids in 10-cm dishes. After 24 h, cells were harvested by scraping into 1 ml of PBS. After removal of PBS by centrifugation, cells were resuspended in 300 μl of mammalian lysis buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 1% Triton X-100, 0.1% Na deoxycholate, protease inhibitors [catalog number G652A; Promega]), followed by incubation on ice for 5 min. The lysates were sonicated for 1 min using 10-s on and 10-s off intervals at 30% amplitude. After centrifugation at 14,000 × g for 5 min at 4°C, the supernatants were transferred into new tubes and stored on ice until ready for binding to resin or antibody. After three washes using washing buffer (PBS, 0.0005% IGEPAL CA-630), either HaloLink resin or HA antibody was added to the protein lysates, followed by incubation overnight with shaking. After centrifugation at 800 × g for 5 min at 4°C, supernatants were removed, and beads were washed 4 times using PBS. Proteins were eluted from the beads by resuspending the beads in 20 μl of 2× sample buffer (125 mM Tris-HCl [pH 6.8], 4% SDS, 20% [vol/vol] glycerol, 0.004% bromophenol blue) and boiling for 3 min. Experiments using FLAG-tagged Hsp90 and cotransfected EBV and CMV kinases were performed in a similar manner, except that the immunoprecipitations were performed by using the M2 FLAG resin (Sigma), as suggested by the manufacturer.
EBV DNA terminus assay.
To evaluate fused and linear structures of the EBV genome, DNA was isolated from AGS-Akata cells after 48 h of treatment with 17-DMAG (0.17 μM), the DMSO control, or acyclovir (50 μg/ml). DNA was cut with BamHI, run on a 0.8% agarose gel, blotted onto a Hybond nylon membrane, and hybridized with a 32P-labeled riboprobe targeting the EBV termini, as previously described (71).
RESULTS
Hsp90 and Cdc37 associate with EBV PK.
To identify proteins that interact with EBV PK, we fused either wild-type EBV PK or a kinase-dead PK mutant (K102I) in frame with the HaloTag and transfected it into 293T cells. EBV PK protein was isolated from the cells 2 days later, and the HaloTag was then removed by using HaloTag technology (Promega). The purity of the final isolated EBV PK product was estimated to be >95% based on Coomassie staining (Fig. 1A). To determine if purified wild-type EBV PK is active, an in vitro kinase assay was performed by using a GST-BMRF1 (C terminus) fusion protein that contains several known EBV PK phosphorylation sites (7, 22–24). As shown in Fig. 1B, purified EBV PK is active based on its ability to autophosphorylate itself and to phosphorylate the GST-BMRF1 protein but not the GST protein alone. As expected, the kinase-dead PK mutant did not phosphorylate either GST-BMRF1 or itself.
Fig 1.
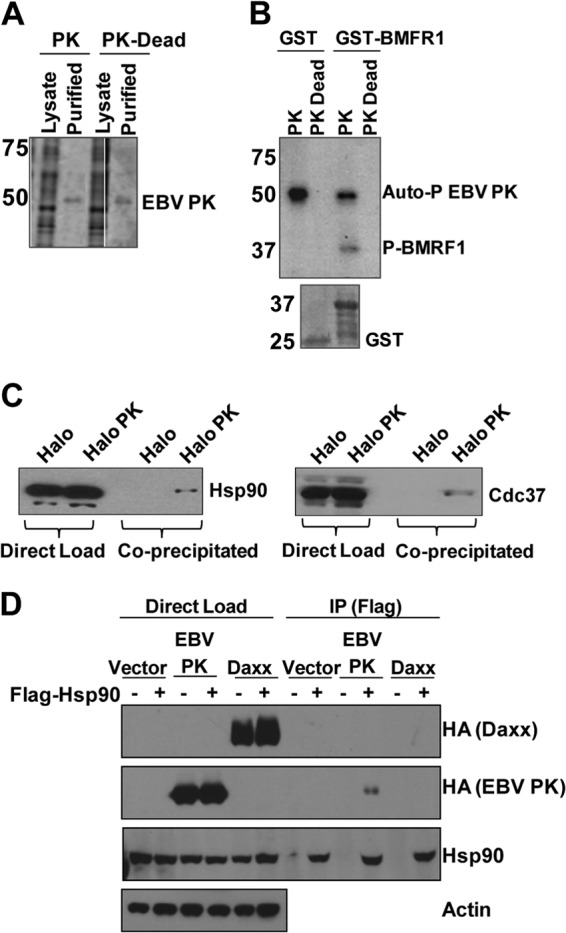
EBV PK associates with Hsp90 and Cdc37. (A) 293T cells were transfected with a vector expressing wild-type EBV PK fused in frame to a HaloTag or a kinase-dead mutant (PK-Dead), and the EBV PK proteins were then purified from the cell lysates (and cleaved from the tag). A Coomassie-stained gel showing the total cell lysate and purified EBV PK proteins is shown. (B) In vitro kinase assays using [32P]ATP were performed by using GST or GST-BMRF1 (C terminus) fusion protein and purified EBV PK proteins. (Top) Autoradiography showed phosphorylation of BMRF1 and EBV PK autophosphorylation, as indicated. (Bottom) A Coomassie stain of the GST proteins used for the kinase assay is also shown. Molecular weights (in thousands) are indicated on the left of panels A and B. (C) 293T cells were transfected with the Halo control vector or Halo-EBV PK, purified as described above for panel A, and immunoblotted with antibodies against Hsp90 (left) or Cdc37 (right). (D) HeLa cells were transfected with various combinations of the vector control, HA-tagged EBV PK, HA-tagged Daxx, and Flag-tagged Hsp90 expression vectors, as indicated, and then immunoprecipitated (IP) by using a FLAG antibody. Immunoblot analyses were performed to examine the expression of transfected proteins (direct load) or immunoprecipitated proteins by using HA, Hsp90, and actin antibodies.
The purified wild-type EBV PK protein was then subjected to mass spectrometry analysis, since this approach has been used to identify proteins that interact with more than 100 yeast protein kinases (72). The results of this analysis showed that Hsp90 and Cdc37 were specifically pulled down by the Halo-EBV PK fusion protein, but not the HaloTag alone, in two separate mass spectrometry analyses (data not shown), suggesting that Hsp90 and/or Cdc37 may form a complex with EBV PK in cells.
To further confirm the mass spectrometry results, the HaloTag control vector (Halo) and the Halo-EBV PK vector were transfected into 293T cells purified by using Halo technology, and the purified proteins (or unpurified transfected-cell extracts) were then subjected to immunoblot analysis using anti-Hsp90 or anti-Cdc37 antibodies. As shown in Fig. 1C, the Hsp90 and Cdc37 proteins were copurified with the Halo-EBV PK protein but not the HaloTag alone, suggesting that both Hsp90 and Cdc37 associate with EBV PK. Furthermore, FLAG antibody coimmunoprecipitated HA-tagged EBV PK protein when it was cotransfected with a FLAG-tagged Hsp90 expression vector (Fig. 1D).
Cdc37 knockdown does not affect EBV PK stability.
Hsp90 and Cdc37 are known to act as cochaperones for many cellular kinases (49, 73). To determine if Cdc37 affects the expression level of EBV PK, we treated HONE-Akata cells with control siRNA or Cdc37-targeted siRNA and then examined the level of transfected EBV PK protein by immunoblot analysis 2 days later (Fig. 2). Although treatment of HONE-Akata cells with Cdc37 siRNA decreased the expression levels of Cdc37 and Cdc2 (a known substrate of Cdc37 [74]), it did not affect the level of EBV PK. These results suggest that Cdc37 may not be required for EBV PK stability, even though EBV PK and Cdc37 can be coprecipitated from 293T cells (Fig. 1C). However, it remains possible that the small amount of Cdc37 expressed in the presence of the siRNA is sufficient to stabilize EBV PK.
Fig 2.
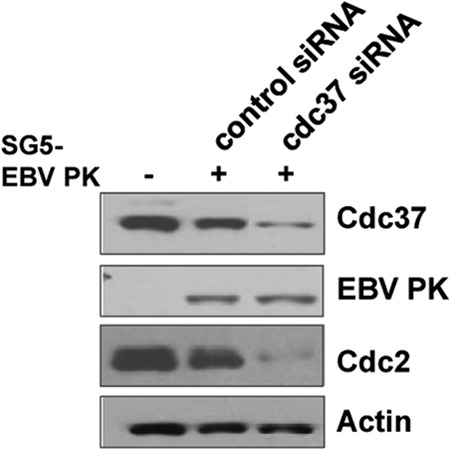
Knockdown of Cdc37 does not affect EBV PK expression. HONE-Akata cells were transfected with pSG5-EBV PK in the presence of Cdc37 siRNA or equivalent amounts of a control siRNA. Whole-cell extracts were prepared, and immunoblot analysis was performed to analyze the expression of Cdc37, EBV PK, Cdc2 (a known cellular substrate of Cdc37), and actin.
The Hsp90 inhibitor 17-DMAG decreases expression of EBV PK in EBV-positive cells.
To determine if Hsp90 is required for EBV PK stability, we examined the effect of the Hsp90 inhibitor drug 17-DMAG on EBV PK expression in two different EBV-positive (EBV+) cell lines (HONE-Akata and AGS-Akata). HONE-Akata cells were transfected with an expression vector for the EBV immediate early (IE) protein BZLF1 (to induce the lytic form of viral infection) and then treated with either 17-DMAG or the DMSO control beginning at 4 h posttransfection. Immunoblot analyses were performed 2 days later to quantitate the expression levels of EBV PK, the BMRF1 early lytic protein (which encodes the viral DNA polymerase processivity factor), BZLF1, Cdc2 (a known substrate of Hsp90 [75]), and actin (a loading control). As shown in Fig. 3A, 17-DMAG treatment reduced the expression level of EBV PK derived from the endogenous viral genome in response to BZLF1 transfection, although the level of transfected BZLF1 protein was not affected. Furthermore, 17-DMAG treatment almost completely abolished the hyperphosphorylated form of the BMRF1 protein (PP-BMRF1), which is known to be generated in an EBV PK-dependent manner (7). As expected, 17-DMAG also decreased the expression of Cdc2. Similar results were obtained with BZLF1-transfected AGS-Akata cells (Fig. 3B).
Fig 3.

17-DMAG decreases the expression level of EBV PK in EBV-positive cells. (A) HONE-Akata cells were transfected with an empty vector or a BZLF1 expression vector (SG5-Z), followed by a 48-h treatment with 17-DMAG (0.17 μM) or the DMSO control beginning at 4 h posttransfection. After 2 days, whole-cell extracts were prepared, and immunoblot analysis was performed to analyze the expression of EBV PK, BMRF1 (both derived from the endogenous viral genome), transfected BZLF1, Cdc2, and actin, as indicated. (B) SG5-Z-transfected AGS-Akata cells were treated with or without 17-DMAG (0.17 μM), as indicated, and immunoblot analyses were performed to detect the level of EBV PK, transfected BZLF1, or actin. (C) Different amounts of the protein extracts from the experiment shown in panel B were loaded onto a gel, as indicated, and examined by immunoblotting using antibodies directed against EBV PK and actin. (D) Latent EBV-positive HONE-Akata cells were transfected with SG5, SG5-EBV PK (HA tagged) (top), or SG5-Z (bottom), as indicated, followed by a 48-h treatment with 17-DMAG (0.17 μM) or the DMSO control beginning at 4 h posttransfection. Whole-cell extracts were prepared after 2 days, and immunoblot analysis was performed to analyze the expression of EBV PK, Cdc2, actin, and BZLF1, as indicated. (E) HeLa cells were transfected with SG5, SG5-EBV PK (HA-tagged), or SG5-Z (HA-tagged) expression vectors, and immunoblot analysis was performed by using an anti-HA antibody to detect the expression of the transfected EBV PK and BZLF1 proteins, Cdc2, or actin. (F) HeLa cells were transfected with the SG5-EBV PK (HA-tagged) vector in the presence or absence of 17-DMAG, and different amounts of the protein extracts were loaded onto a gel, as indicated, and examined by immunoblotting using antibodies directed against EBV PK (HA antibody) and actin.
To obtain more quantitative information about the effect of 17-DMAG treatment on EBV PK expression, we also performed immunoblotting in which the level of EBV PK expression derived from various different amounts of protein (2 to 20 μg) harvested from DMSO-treated AGS-Akata cells was compared to the level of EBV PK expression in 20 μg of protein harvested from 17-DMAG-treated cells. As shown in Fig. 3C, the EBV PK expression level in 17-DMAG-treated cells appeared to be approximately 10-fold lower than that in DMSO-treated cells. These results suggest that 17-DMAG inhibits expression of the EBV PK protein during lytic viral infection.
To determine whether 17-DMAG also alters EBV PK expression when the protein is driven by a constitutively active promoter, HONE-Akata cells or EBV-negative HeLa cells were transfected with an EBV PK expression vector (SG5-EBV PK) driven by the SV40 promoter and then treated with or without 17-DMAG beginning at 4 h posttransfection. 17-DMAG treatment decreased the expression of the transfected EBV PK protein (Fig. 3D, top), while the expression of transfected BZLF1 protein cloned into the same vector (Fig. 3D, bottom) was not affected. The expression of the cellular protein Cdc2 was decreased in cells treated with 17-DMAG, whereas actin expression was not affected. Similarly, 17-DMAG treatment also decreased the expression of HA-tagged EBV PK, but not that of the HA-tagged BZLF1 protein, in HeLa cells (Fig. 3E). Quantification of the 17-DMAG effect on transfected HA-tagged PK expression in HeLa cells suggested that the drug decreases EBV PK expression levels by approximately 10-fold (Fig. 3F). Together, these results suggest that Hsp90 enhances EBV PK protein expression in both EBV-positive and EBV-negative cells.
17-DMAG treatment decreases phosphorylation of EBV PK substrates.
To further confirm that loss of EBV PK expression in 17-DMAG treated cells results in decreased phosphorylation of EBK-PK-phosphorylated substrates in lytically infected cells, we examined the effect of 17-DMAG on phosphorylation of the EBV BMRF1, cellular lamin A, and EBV thymidine kinase proteins. As shown in Fig. 4A, similar to the effect of 17-DMAG on HONE-Akata cells (Fig. 3A), the amount of hyperphosphorylated BMRF1 protein was greatly decreased by 17-DMAG treatment of AGS-Akata cells. Since we previously showed that EBV PK also phosphorylates lamin A serine residue 22 (7), we examined the amount of the phosphorylated lamin A S22 residue in vector control-transfected, versus BZLF1-transfected, AGS-Akata cells in the presence and absence of 17-DMAG (Fig. 4B). BZLF1-transfected cells had an increased amount of phosphorylated lamin A S22, and this effect was decreased by 17-DMAG treatment. Finally, since we serendipitously noted that cotransfection of an EBV thymidine kinase (EBV TK) expression vector with the EBV PK expression vector in HeLa cells resulted in a higher-migrating form of EBV TK, which was not observed in the absence of EBV PK (even when larger amounts of EBV TK were transfected [Fig. 4C]), we determined if the higher-migrating form of EBV TK expressed in lytically infected Mutu I Burkitt lymphoma cells was also decreased by 17-DMAG treatment. As shown in Fig. 4D, treatment of Mutu I cells with TGF-β induced both EBV TK and EBV PK expression (consistent with its known ability to activate lytic protein expression in type I Burkitt cells [76]). EBV PK expression was greatly decreased by 17-DMAG treatment, as was the expression of the higher-migrating form of the EBV TK protein. These results suggest that phosphorylation of EBV PK substrates is highly inhibited by 17-DMAG treatment of lytically infected cells.
Fig 4.

17-DMAG decreases phosphorylation of EBV PK substrates. (A) AGS-Akata cells were treated with 17-DMAG or the DMSO control for 48 h. Whole-cell extracts were prepared 2 days later, and immunoblot analysis was performed to analyze the expression of BMRF1, BZLF1, Cdc2, and actin, as indicated. (B) AGS-Akata cells transfected with the vector control or BZLF1 (in the presence or absence of 17-DMAG) were examined by immunoblotting using antibodies to detect phosphorylated serine 22 of lamin A, total lamin A, EBV PK, transfected BZLF1, and tubulin. (C) HeLa cells were transfected with the vector control, a myc-tagged expression vector for EBV thymidine kinase (EBV TK) (150 ng), or the EBV TK expression vector (50 ng) and the EBV PK expression vector (50 ng) in the presence or absence of 17-DMAG. Immunoblot analyses were performed to examine EBV TK, EBV PK, and actin expression. (D) Mutu I Burkitt lymphoma cells were treated with and without TGF-β to induce lytic infection (in the presence or absence of 17-DMAG), and immunoblot analyses were performed to detect the expression of EBV PK, EBV TK, and actin.
Hsp90 associates with each of the eight CHPKs.
The conserved herpesvirus protein kinases (CHPKs) share discernible homology in amino acid sequences and positional similarity in viral genomes. Since Hsp90 forms a complex with EBV PK, we next determined whether Hsp90 also associates with the CHPKs of HCMV, KSHV, VZV, HSV-1, HSV-2, HHV6, and HHV7. SG5 vectors expressing each of the eight known viral CHPKs (HA tagged), or the appropriate HA vector control, were transfected into HeLa cells and immunoprecipitated with anti-HA antibody. Immunoblots were then performed by using an anti-HA antibody to detect each of the different transfected viral kinases (Fig. 5A, top two panels), and an anti-Hsp90 antibody (bottom two panels). As shown in Fig. 5A, although the expression levels of the various different CHPKs were somewhat variable (as previously described [30]), Hsp90 protein was coimmunoprecipitated with each of the HA-tagged CHPKs but was not coimmunoprecipitated in cells transfected with the HA-tag-only control vector. In addition, HA-tagged UL97 kinase was shown to be coprecipitated with FLAG-tagged Hsp90 (Fig. 5B). These results suggest that the ability to associate with Hsp90 is conserved across each of the CHPKs.
Fig 5.
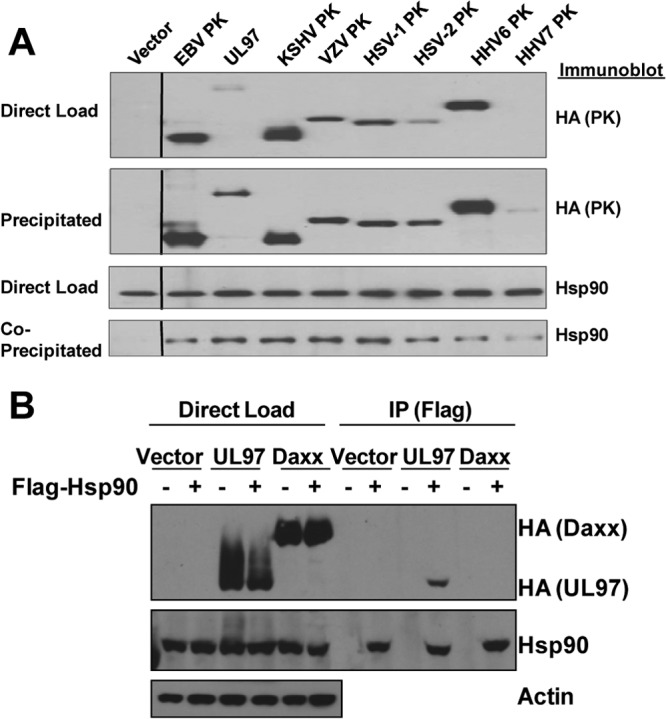
Hsp90 is coprecipitated with multiple different CHPKs. (A) HeLa cells were transfected with the empty vector control (expressing the HA tag only) or vectors expressing different HA-tagged CHPKs, as indicated. After 28 h, cells were harvested, and the cell lysate was immunoprecipitated with an anti-HA antibody or loaded directly onto the gel (direct-load control). The precipitates and direct-load control were immunoblotted with antibodies against Hsp90 or HA, as indicated. (B) HeLa cells were transfected with various combinations of the vector control, HA-tagged HMCV UL97 or HA-tagged Daxx expression vectors, or a FLAG-tagged Hsp90 expression vector, as indicated, and then immunoprecipitated by using a FLAG antibody. Immunoblot analyses were performed to examine the expression of transfected proteins under each condition (direct load) or immunoprecipitated proteins by using antibodies against HA, Hsp90, and actin.
17-DMAG decreases expression of multiple different herpesvirus protein kinases.
Since Hsp90 associates with each of the CHPKs, we next determined if the Hsp90 inhibitor drug 17-DMAG reduces the expression of these other CHPKs, similar to its effect on EBV PK. HeLa cells were transfected with the control vector or vectors expressing HA-tagged HCMV PK (UL97), HHV6 PK, HHV7 PK, KSHV PK, rat CMV (RCMV) PK, mouse MHV68 (MHV68) PK, and EBV PK. Similar to its effect on the EBV PK, 17-DMAG treatment also decreased the expression of each of the other CHPKs examined (Fig. 6A). As expected, the expression of Cdc2 was decreased in 17-DMAG-treated cells, whereas actin expression was not affected. Quantification of the effect of 17-DMAG on HA-UL97-transfected HeLa cells showed that 17-DMAG decreased the expression of UL97 by approximately 10-fold (Fig. 6B). These results indicate that 17-DMAG inhibits the expression of many different CHPKs.
Fig 6.
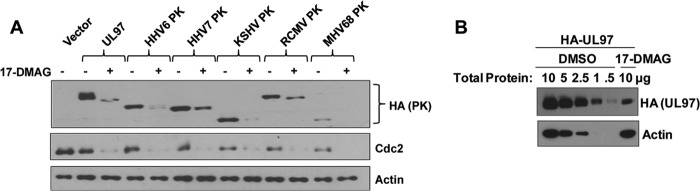
17-DMAG decreases expression of multiple CHPKs. (A) HeLa cells were transfected with the empty vector control or vectors expressing UL97, HHV6 PK, HHV7 PK, KSHV PK, RCMV PK, MHV68 PK, or EBV PK (all tagged with HA). A 48-h treatment with 17-DMAG (0.17 μM) or the DMSO control began at 4 h posttransfection. Whole-cell extracts were prepared, and immunoblot analyses were performed to analyze the expression of PKs using antibodies against HA, Cdc2, or actin, as indicated. (B) HeLa cells were transfected with the SG5-UL97 (HA-tagged) vector in the presence or absence of 17-DMAG, and different amounts of the protein extracts were loaded onto a gel, as indicated, and examined by immunoblotting using antibodies directed against UL97 (HA antibody) and actin.
17-DMAG reduces HCMV PK (UL97) expression during infection of primary human fibroblasts.
To determine if Hsp90 is required for efficient expression of the HCMV kinase during normal infection, primary human fibroblasts were infected with HCMV (strain AD169) and treated with no drug or 17-DMAG (0.17 μM) for 6, 24, 48, or 72 h. Whole-cell extracts were prepared, and immunoblot analysis was performed to analyze the expression of HCMV PK (UL97), the viral immediate early 1 (IE1) protein, the early lytic viral protein UL44, and cellular GAPDH (loading control), as indicated. As shown in Fig. 7A, 17-DMAG treatment dramatically decreased the expression of the HCMV PK protein, while the expression of the viral IE1 protein and the UL44 early lytic protein was much less affected, and the cellular GAPDH protein was not affected. Similar results were obtained with cells infected for up to 72 h with HCMV in the presence of 17-DMAG (Fig. 7B). These results suggest that Hsp90 inhibitors might be used to prevent expression of the viral kinase in HCMV-infected, as well as EBV-infected, cells.
Fig 7.
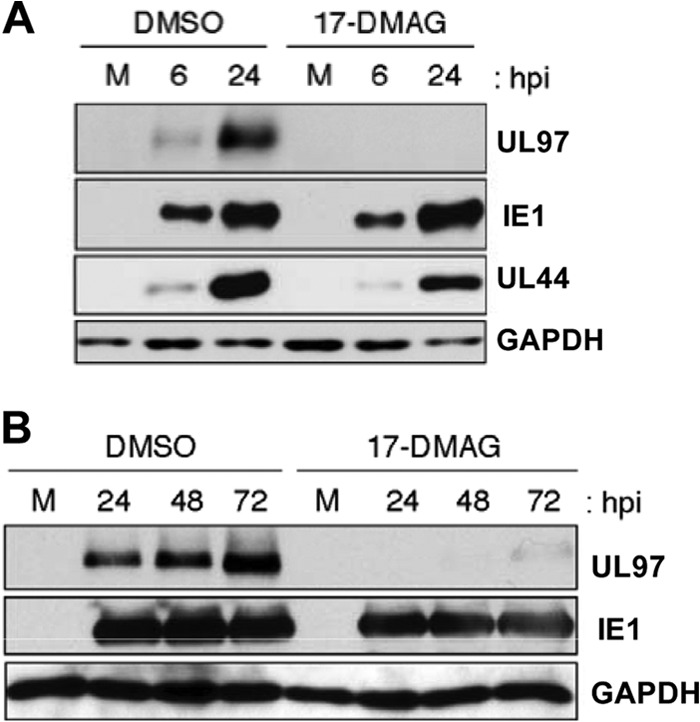
17-DMAG inhibits expression of HCMV PK in HCMV-infected fibroblasts. (A) Primary human fibroblasts were infected with HCMV (strain AD169) and treated with 17-DMAG (0.17 μM) or the DMSO control for 6 or 24 h. Whole-cell extracts were prepared, and immunoblot analysis was performed to analyze the expression of UL97, IE1, UL44, and GAPDH, as indicated. M, mock infection. (B) Primary human fibroblasts were infected with HCMV in the presence or absence of 17-DMAG, and extracts were harvested at 24, 48, and 72 h postinfection (hpi). Immunoblot analysis was performed to analyze the expression of UL97, IE1, and GAPDH, as indicated.
17-DMAG treatment reduces the number of infectious viral particles released from EBV-infected cells.
Although not expressed during viral latency, EBV PK is required for efficient production of infectious viral particles in at least some cell types (7, 11). Since 17-DMAG decreases the expression of EBV PK, we examined its effect on the amount of infectious EBV particles released from AGS-Akata cells. Cells were transfected with a control vector or the SG5-Z vector (to increase virion production over baseline levels) and treated with 17-DMAG or the DMSO control. Transfected cell extracts were examined by immunoblotting to detect the expression of BZLF1, BMRF1, and actin, and the infectious viral titer in each supernatant was determined by using the green Raji cell assay. 17-DMAG inhibited virus production by approximately 100-fold in both BZLF1-transfected and control vector-transfected AGS-Akata cells (Fig. 8A). In contrast, the number of viable AGS-Akata cells in the same experiment (determined by trypan blue staining) was reduced by only 50% (Fig. 8B). This reduction in the total number of AGS-Akata cells was expected given the known ability of 17-DMAG to reduce the expression of cellular proteins such as Cdc2 that are required for cellular replication. Consistent with this, when AGS-Akata cells were plated at a high density (resulting in decreased cellular replication), the same course of 17-DMAG treatment did not affect cell numbers (data not shown). Immunoblot analysis of transfected AGS-Akata cell extracts confirmed that 17-DMAG treatment decreased the phosphorylation of BMRF1 but not the levels of transfected BZLF1 and actin (Fig. 8C). These results show that 17-DMAG treatment greatly impairs production of infectious viral particles during lytic EBV infection.
Fig 8.

17-DMAG reduces the EBV titer in lytically infected cells. AGS-Akata cells were transfected with SG5 or SG5-Z expression vectors, followed by a 48-h treatment with 17-DMAG (0.17 μM) or the DMSO control beginning at 4 h posttransfection. The cell medium was replaced after 48 h with medium free of drug. At 72 h posttransfection, cells and supernatant were harvested. (A) The virus titer was quantitated by infecting Raji cells with various amounts of the supernatant at 72 h posttransfection and counting the number of GFP-positive cells (green Raji units [GRU]) by using a fluorescence microscope. The results from two independent experiments are shown. (B) The number of viable AGS-Akata cells under each condition shown in panel A was determined by counting the cells (with trypan blue) at the end of the experiment. (C) Whole-cell extracts were prepared from the AGS-Akata cells used to derive the virus titer shown in panel A, and immunoblot analysis was performed to analyze the expression of BMRF1, BZLF1, and actin.
17-DMAG treatment inhibits intracellular lytic viral replication in EBV-infected cells.
Since we previously reported that a PK-deleted EBV mutant is defective for the release of infectious viral particles in 293T cells but is not defective for intracellular viral DNA replication (7, 11), we examined whether 17-DMAG treatment decreases the amount of lytically replicated EBV DNA in AGS-Akata cells. AGS-Akata cells were treated with 17-DMAG and/or acyclovir (which prevents lytic viral DNA replication) for 48 h, and DNA was then isolated and examined by Southern blotting using the EBV terminus assay, as previously described (71). This assay can distinguish between EBV genomes that contain fused terminal repeats (a combination of latent and lytically replicated DNA) and linear viral genomes in which the terminal repeats have been cleaved (derived only from lytically replicated viral DNA). As shown in Fig. 9, both 17-DMAG and acyclovir treatment decreased the amount of linear (replicated) EBV genomes (Fig. 9, left), although neither drug affected the levels of BMRF1 and BZLF1 in AGS-Akata cell extracts (Fig. 9, right). These results indicate that 17-DMAG treatment inhibits EBV lytic DNA replication as well as EBV virion production in AGS-Akata cells.
Fig 9.
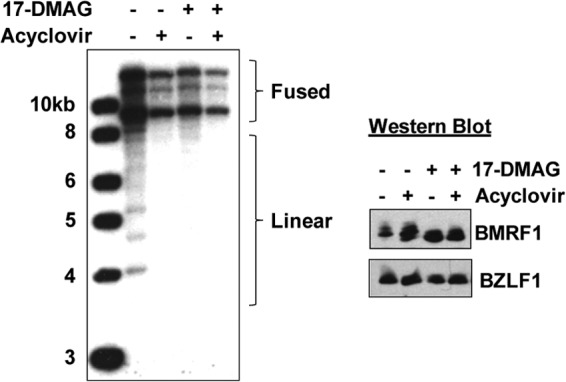
17-DMAG decreases intracellular lytic EBV DNA replication. (Left) AGS-Akata cells were treated with 17-DMAG (0.17 μM), the DMSO control, and/or acyclovir (50 μg/ml) for 2 days, and the DNA was harvested. The DNA was cut with BamHI and hybridized to a probe spanning the EBV termini for Southern blotting. The forms of the EBV genome containing fused viral termini (a mixture of latent and lytically replicated genomes) as well as the cleaved linear forms of the genome (produced only by lytically replicated viral DNA) are indicated. (Right) Immunoblotting was performed to ascertain the protein levels of BMRF1 and BZLF1 in the AGS-Akata samples used in the left panel.
Increasing the EBV PK expression level does not rescue virus production in 17-DMAG-treated cells.
To determine if the 17-DMAG effect on EBV virus production is due primarily to the loss of EBV PK expression, versus the combined effect of the drug on potentially multiple different viral and/or cellular proteins, AGS-Akata cells were transfected with SG5-Z in the presence or absence of a cotransfected EBV PK expression vector and treated with 17-DMAG or the DMSO control. Viral titers were examined 3 days later by using the green Raji cell assay, and the AGS-Akata cell extracts were examined by immunoblotting for BMRF1, BZLF1, and actin expression. While overexpression of EBV PK increased the level of BMRF1 hyperphosphorylation in 17-DMAG-treated cells (Fig. 10B), suggesting that the EBV PK expression level was increased, it did not rescue EBV virion production (Fig. 10A). This result suggests that in addition to EBV PK, 17-DMAG reduces the expression of other viral and/or cellular proteins required for efficient EBV lytic replication.
Fig 10.
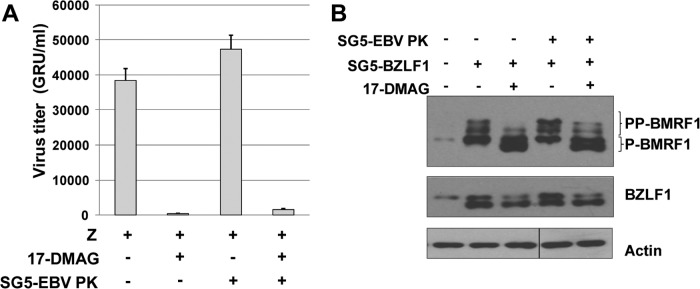
Overexpression of EBV PK does not rescue viral replication in 17-DMAG-treated cells. AGS-Akata cells were transfected with SG5-Z, SG5-EBV PK, or both, followed by a 48-h treatment with 17-DMAG (0.17 μM) or the DMSO control beginning at 4 h posttransfection. The cell medium was replaced after 48 h with medium free of drug. At 72 h posttransfection, cells and supernatant were harvested. (A) The virus titer was quantitated by infecting Raji cells with various amounts of the supernatant at 72 h posttransfection and counting the number of GFP-positive cells (GRU) by using a fluorescence microscope. The results from two independent experiments are shown. (B) Whole-cell extracts were prepared from the AGS-Akata cells used to derive the virus in the experiments in panel A, and immunoblot analysis was performed to analyze the expression of BMRF1, BZLF1, and actin.
The effect of 17-DMAG on EBV PK and HCMV UL97 expression is not completely reversed by proteasomal or autophagy inhibitors.
Many Hsp90 client proteins are degraded via the ubiquitin-proteasome pathway in the absence of Hsp90 (77–79), suggesting that proteasomal inhibitors might attenuate the effect of the Hsp90 inhibitor on EBV PK and/or HCMV UL97 expression. In addition to the ubiquitin-proteasome pathway, the autophagy pathway is another important protein degradation pathway and has been shown to mediate the degradation of a subset of Hsp90 client proteins (80–83). To examine this in EBV-infected cells, AGS-Akata cells were treated with 17-DMAG or the DMSO control in the presence or absence of the proteasomal inhibitor lactacystin or an autophagy inhibitor, 3-methyladenine (3-MA). As shown in Fig. 11A, 3-MA somewhat increased the EBV PK expression level in the presence of 17-DMAG, while lactacystin had no effect. The effect of 17-DMAG on UL97 kinase expression in HCMV-infected primary human foreskin fibroblasts (HFFs) was slightly reversed by both 3-MA and lactacystin (Fig. 11B). These results suggest that in addition to its effect on protein stability, 17-DMAG may decrease the expression of both EBV PK and HCMV UL97 by inhibiting their synthesis.
Fig 11.
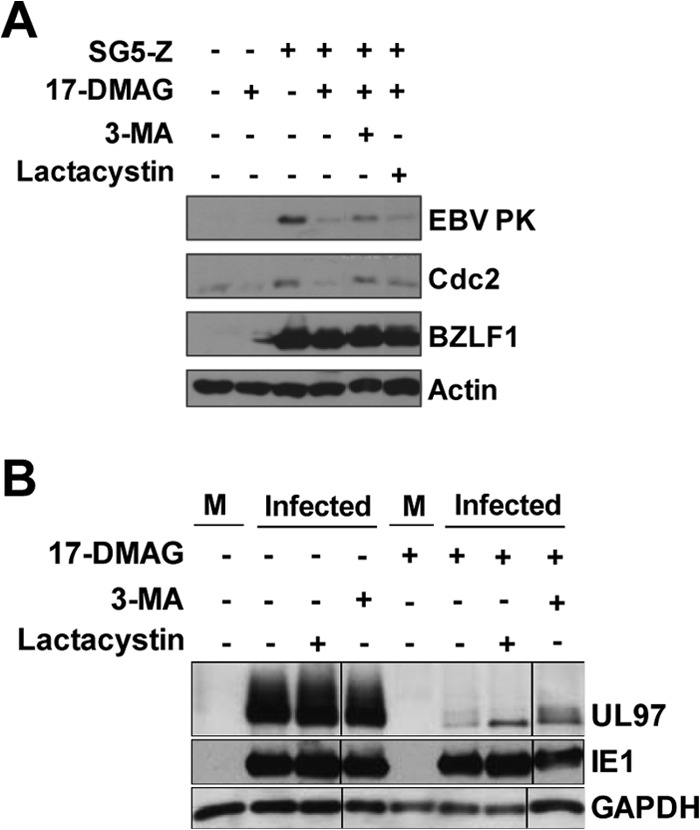
EBV PK and HCMV UL97 expression is not completely rescued by proteasomal or autophagy inhibitors in the presence of 17-DMAG. (A) AGS-Akata cells were transfected with SG5 or SG5-Z, as indicated, followed by a 24-h treatment with 17-DMAG (0.17 μM) or the DMSO control beginning at 12 h posttransfection in the presence or absence of the proteasomal inhibitor lactacystin or the autophagy inhibitor 3-MA. Immunoblot analysis was performed to analyze the expression of EBV PK, Cdc2, transfected BZLF1, and actin. (B) Primary human foreskin fibroblasts were subjected to serum starvation for 48 h and then infected with HCMV (strain AD169) at an MOI of 1. Before infection, cells were pretreated with 0.17 μM 17-DMAG for 1 h and incubated with the same amount of this drug until harvesting. Under some conditions, lactacystin or 3-MA was added to the medium at 6 h postinfection. Cells were harvested at 24 h postinfection, and immunoblot analysis performed to analyze the expression of UL97, IE1, and GAPDH. M, mock infection.
DISCUSSION
Although a number of drugs, such as acyclovir and ganciclovir, have been used as antiherpetic agents (84), the prolonged use of these antivirals in patients can result in the generation of resistant viral mutants as well as severe side effects. Therefore, the identification of novel drug candidates with newly defined targets and mechanisms of action may result in the development of improved treatments for various forms of herpesvirus infections. Since our laboratories, and others, have shown that EBV PK as well as HCMV UL97 are required for efficient production of infectious viral particles in many (but not all) cell types (7, 11, 12, 18, 85, 86), the development of drugs that can block the activity of these viral kinases may be therapeutically useful. In the present study, we show that heat shock protein 90 (Hsp90) is coprecipitated by all eight conserved herpesvirus protein kinases (CHPKs) and that the Hsp90 inhibitor 17-DMAG reduces the expression, and activity, of the EBV and HCMV kinases in virally infected cells. Furthermore, we find that 17-DMAG efficiently inhibits intracellular EBV lytic DNA replication, as well as the release of infectious viral particles, at a nontoxic dose in AGS-Akata cells.
Hsp90 inhibitors are a potentially promising new pool of antiviral drugs. Heat shock proteins (Hsps) are a key group of cellular proteins that serve as molecular chaperones, facilitating proper protein folding and stability. They have also been shown in some cases to aid in protein-protein interactions and intracellular trafficking (48, 87). The cochaperone Cdc37 is also necessary for the stability of some cellular protein kinases, including Raf1 (88) and Cdk4 (89), and Cdc37 binds at the mouth of the ATP-binding pocket of Hsp90 (73, 90). Interestingly, a recent report studying the interaction of Hsp90 with cellular proteins revealed that Hsp90 associates with approximately 60% of cellular kinases, versus only 7% of cellular transcription factors, and that the Cdc37 cochaperone protein primarily mediates this kinase-specific recognition (49). We show here that Hsp90 also interacts with each of the conserved herpesvirus protein kinases and stabilizes their expression in cells. However, in the case of EBV PK at least, knockdown of Cdc37 expression did not reduce the expression of the viral kinase, while it decreased the expression of the cellular kinase Cdc2. Therefore, the virally encoded protein kinases (which have not yet been crystallized) may have a somewhat different interaction with Hsp90/Cdc37 than the cellular kinases. Nevertheless, since Cdc37 was not completely knocked down in these experiments, it remains possible that only a small amount of Cdc37 is needed to stabilize EBV PK.
Hsp90 inhibitors often cause Hsp90 clients to be degraded via the proteasomal (77–79) or (less commonly) the autophagy (91) pathways. We found here that the autophagy inhibitor 3-MA partially increased EBV PK expression levels in the presence of 17-DMAG, while the proteasomal inhibitor had no effect. Both the autophagy and proteasomal inhibitors slightly increased the level of HCMV UL97 in the presence of 17-DMAG. These results suggest that in addition to inhibiting the stability of the EBV PK and HCMV UL97 kinases, Hsp90 inhibitors may also decrease the translation of these kinases. Of note, Hsp90 inhibitors were previously shown to decrease the synthesis of at least two different viral proteins, including the EBV EBNA1 protein (58, 92).
CHPKs are key players in viral infection, as they are involved in multiple critical processes, including retinoblastoma (Rb) inactivation (30), gene expression (8, 11, 14), viral DNA replication (11, 15–17, 86), DNA damage response activation (20, 21), and capsid nuclear egress (7, 11, 18, 19). Thus, by targeting CHPKs, Hsp90 inhibitors can potentially disrupt several essential viral pathways simultaneously. Although we previously reported that an EBV mutant with a deletion of the EBV PK gene (BGLF4) is not defective for intracellular lytic DNA replication in 293T cells but is defective for infectious virion production (7), here we found that treatment of AGS-Akata cells (an EBV-infected gastric cell line) with 17-DMAG prevented both intracellular lytic EBV DNA replication and the release of infectious viral particles. As the phenotypes of kinase-deficient HCMV and EBV mutants have been shown to be cell type dependent (7, 86) and can be partially reversed by the expression of viral oncoproteins, such as SV40 T antigen and papillomavirus E7, that inhibit Rb function (7, 86), it is possible that while EBV PK is not essential for intracellular lytic EBV DNA replication in 293T cells (which express adenovirus E1A protein), it is indeed essential for intracellular lytic DNA replication in AGS-Akata cells.
In addition to the effect on EBV PK, Hsp90 inhibitors may also repress the expression, or nuclear localization, of other EBV proteins required for intracellular lytic viral DNA replication. Of note, the Hsp90 inhibitor geldanamycin has been shown to block HSV-1 viral DNA synthesis and prevent nuclear translocation of the viral DNA polymerase (93). In addition, while the manuscript was being reviewed, another group reported that Hsp90 inhibitors decrease the ability of the EBV-encoded DNA polymerase to enter the nucleus (94). Hsp90 inhibitors may also inhibit viral replication at least partially via their effects on cellular proteins. Of particular interest to our findings here, geldanamycin has been reported to inhibit HCMV replication by disruption of the phosphatidylinositol 3-kinase (PI3-K) signaling pathway, thereby leading to decreased expression levels of the viral IE genes (95). However, in contrast to the previous study (95), we did not find that 17-DMAG treatment affects expression of HCMV IE1 protein, or its ability to activate the UL44 early lytic gene, during infection of primary human fibroblasts. Cell surface Hsp90 has also been reported to be necessary for mitogen-activated protein kinase (MAPK) activation and efficient viral gene expression during de novo KSHV infection (96).
In the case of the oncogenic gammaherpesviruses EBV and KSHV, there is increasing evidence that Hsp90 inhibitors might also be used to prevent the expression of essential viral transforming proteins during latent forms of infection. We previously reported that 17-DMAG decreases the expression of the EBV EBNA1 protein, which is required for the maintenance of the viral genome during latent infection, and showed that a nontoxic dose of the Hsp90 inhibitor 17-allylamino-17-demethoxygeldanamycin (17-AAG) inhibits the growth of EBV-induced lymphoproliferative disease in SCID mice (58). Interestingly, the KSHV homologue of EBNA1, LANA, was recently shown to be a Hsp90 client protein that is degraded in a proteasome-dependent manner by Hsp90 inhibitors (97). In addition, the expression level of another KSHV latency protein that promotes cellular transformation, K1, has also been shown to be reduced in the presence of Hsp90 inhibitors (98), as was the essential EBV transforming protein LMP1 (99). Thus, Hsp90 inhibitors might be useful for treatment of both the latent and lytic forms of gammaherpesvirus infection.
The fact that Hsp90 inhibitors likely abrogate lytic EBV and HCMV replication via multiple mechanisms actually enhances the attractiveness of these agents as antiviral drugs, as it suggests that EBV and HCMV are not likely to escape the effects of these drugs through a simple mutation. This is even more important given the recent failure of the drug maribavir (an inhibitor of the enzymatic activity of HCMV UL97) in a phase III clinical trial (36). There are several reasons why maribavir may have failed, including the use of an insufficient dose of drug in the trial, the development of specific viral gene mutations that abrogated the antiviral effect of the drug, and previous findings in vitro suggesting that the activity of the various CHPKs is more important in some cell types (and growth states) than in others (7, 86). Multiple Hsp90 inhibitors have been tested in or are currently undergoing clinical trials (phases I to III) for cancers such as HER2-positive breast cancer, ALK-mutated non-small-cell lung cancer, and melanoma (100). These studies and other research have revealed that tumor cells utilize Hsp90 differently than noncancerous cells and require more of it, which is why inhibition of Hsp90 targets abnormal cells more than their normal counterparts (53, 101–103). Although our in vitro results here suggest that Hsp90 inhibitors might be used to inhibit EBV and HCMV lytic replication (and potentially the replication of other herpesviruses) in humans by using doses that are not toxic to normal cells, the safety and efficacy of this approach will next need to be assessed in animal models of these infections.
ACKNOWLEDGMENTS
We thank Todd Seaman for his work constructing the GST-BMRF1 (C terminus) plasmid and Coral Wille for her help constructing the SG5-HA-Z vector. We also thank Ying Ge and Matthew Lawrence from the UW-Madison Human Proteomics Program for use of their mass spectrometry facility and help with analysis of data.
This work was supported by NIH grants P01-CA022443 and R01-AI080675 and a scholarship from the Japan Herpesvirus Infections Forum awarded to S.I. R.F.K. is a Burroughs Wellcome Fund investigator in the pathogenesis of infectious disease.
Footnotes
Published ahead of print 10 July 2013
REFERENCES
- 1. Roizman B, Knipe DM. 2006. Herpes simplex viruses, p 2502–2601 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2. Cohen J, Straus S, Arvin A. 2006. Varicella-zoster virus replication, pathogenesis, and management, p 2774–2818 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 3. Mocarski E, Shenk T, Pass R. 2006. Cytomegaloviruses, p 2702–2772 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 4. Yamanashi K, Mori Y, Pellett P. 2006. Human herpesviruses 6 and 7, p 2819–2845 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 5. Ganem D. 2006. Kaposi's sarcoma-associated herpesvirus, p 2847–2888 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 6. Rickinson A, Kieff E. 2006. Epstein-Barr virus, p 2655–2700 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 7. Meng Q, Hagemeier SR, Kuny CV, Kalejta RF, Kenney SC. 2010. Simian virus 40 T/t antigens and lamin A/C small interfering RNA rescue the phenotype of an Epstein-Barr virus protein kinase (BGLF4) mutant. J. Virol. 84:4524–4533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Purves FC, Ogle WO, Roizman B. 1993. Processing of the herpes simplex virus regulatory protein alpha 22 mediated by the UL13 protein kinase determines the accumulation of a subset of alpha and gamma mRNAs and proteins in infected cells. Proc. Natl. Acad. Sci. U. S. A. 90:6701–6705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hu H, Cohen JI. 2005. Varicella-zoster virus open reading frame 47 (ORF47) protein is critical for virus replication in dendritic cells and for spread to other cells. Virology 337:304–311 [DOI] [PubMed] [Google Scholar]
- 10. Moffat JF, Zerboni L, Sommer MH, Heineman TC, Cohen JI, Kaneshima H, Arvin AM. 1998. The ORF47 and ORF66 putative protein kinases of varicella-zoster virus determine tropism for human T cells and skin in the SCID-hu mouse. Proc. Natl. Acad. Sci. U. S. A. 95:11969–11974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gershburg E, Raffa S, Torrisi MR, Pagano JS. 2007. Epstein-Barr virus-encoded protein kinase (BGLF4) is involved in production of infectious virus. J. Virol. 81:5407–5412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Prichard MN, Gao N, Jairath S, Mulamba G, Krosky P, Coen DM, Parker BO, Pari GS. 1999. A recombinant human cytomegalovirus with a large deletion in UL97 has a severe replication deficiency. J. Virol. 73:5663–5670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Stevenson D, Colman KL, Davison AJ. 1994. Characterization of the putative protein kinases specified by varicella-zoster virus genes 47 and 66. J. Gen. Virol. 75(Part 2):317–326 [DOI] [PubMed] [Google Scholar]
- 14. Long MC, Leong V, Schaffer PA, Spencer CA, Rice SA. 1999. ICP22 and the UL13 protein kinase are both required for herpes simplex virus-induced modification of the large subunit of RNA polymerase II. J. Virol. 73:5593–5604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Krosky PM, Baek M-C, Jahng WJ, Barrera I, Harvey RJ, Biron KK, Coen DM, Sethna PB. 2003. The human cytomegalovirus UL44 protein is a substrate for the UL97 protein kinase. J. Virol. 77:7720–7727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Marschall M, Freitag M, Suchy P, Romaker D, Kupfer R, Hanke M, Stamminger T. 2003. The protein kinase pUL97 of human cytomegalovirus interacts with and phosphorylates the DNA polymerase processivity factor pUL44. Virology 311:60–71 [DOI] [PubMed] [Google Scholar]
- 17. Wolf DG, Courcelle CT, Prichard MN, Mocarski ES. 2001. Distinct and separate roles for herpesvirus-conserved UL97 kinase in cytomegalovirus DNA synthesis and encapsidation. Proc. Natl. Acad. Sci. U. S. A. 98:1895–1900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Krosky PM, Baek M-C, Coen DM. 2003. The human cytomegalovirus UL97 protein kinase, an antiviral drug target, is required at the stage of nuclear egress. J. Virol. 77:905–914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hamirally S, Kamil JP, Ndassa-Colday YM, Lin AJ, Jahng WJ, Baek M-C, Noton S, Silva LA, Simpson-Holley M, Knipe DM, Golan DE, Marto JA, Coen DM. 2009. Viral mimicry of Cdc2/cyclin-dependent kinase 1 mediates disruption of nuclear lamina during human cytomegalovirus nuclear egress. PLoS Pathog. 5:e1000275. 10.1371/journal.ppat.1000275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Li R, Zhu J, Xie Z, Liao G, Liu J, Chen M-R, Hu S, Woodard C, Lin J, Taverna SD, Desai P, Ambinder RF, Hayward GS, Qian J, Zhu H, Hayward SD. 2011. Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe 10:390–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Li R, Wang L, Liao G, Guzzo CM, Matunis MJ, Zhu H, Hayward SD. 2012. SUMO binding by the Epstein-Barr virus protein kinase BGLF4 is crucial for BGLF4 function. J. Virol. 86:5412–5421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yang P-W, Chang S-S, Tsai C-H, Chao Y-H, Chen M-R. 2008. Effect of phosphorylation on the transactivation activity of Epstein-Barr virus BMRF1, a major target of the viral BGLF4 kinase. J. Gen. Virol. 89:884–895 [DOI] [PubMed] [Google Scholar]
- 23. Chen MR, Chang SJ, Huang H, Chen JY. 2000. A protein kinase activity associated with Epstein-Barr virus BGLF4 phosphorylates the viral early antigen EA-D in vitro. J. Virol. 74:3093–3104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gershburg E, Pagano JS. 2002. Phosphorylation of the Epstein-Barr virus (EBV) DNA polymerase processivity factor EA-D by the EBV-encoded protein kinase and effects of the L-riboside benzimidazole 1263W94. J. Virol. 76:998–1003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhu J, Liao G, Shan L, Zhang J, Chen M-R, Hayward GS, Hayward SD, Desai P, Zhu H. 2009. Protein array identification of substrates of the Epstein-Barr virus protein kinase BGLF4. J. Virol. 83:5219–5231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Yue W, Gershburg E, Pagano JS. 2005. Hyperphosphorylation of EBNA2 by Epstein-Barr virus protein kinase suppresses transactivation of the LMP1 promoter. J. Virol. 79:5880–5885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kato K, Yokoyama A, Tohya Y, Akashi H, Nishiyama Y, Kawaguchi Y. 2003. Identification of protein kinases responsible for phosphorylation of Epstein-Barr virus nuclear antigen leader protein at serine-35, which regulates its coactivator function. J. Gen. Virol. 84:3381–3392 [DOI] [PubMed] [Google Scholar]
- 28. Asai R, Kato A, Kato K, Kanamori-Koyama M, Sugimoto K, Sairenji T, Nishiyama Y, Kawaguchi Y. 2006. Epstein-Barr virus protein kinase BGLF4 is a virion tegument protein that dissociates from virions in a phosphorylation-dependent process and phosphorylates the viral immediate-early protein BZLF1. J. Virol. 80:5125–5134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Iwahori S, Murata T, Kudoh A, Sato Y, Nakayama S, Isomura H, Kanda T, Tsurumi T. 2009. Phosphorylation of p27Kip1 by Epstein-Barr virus protein kinase induces its degradation through SCFSkp2 ubiquitin ligase actions during viral lytic replication. J. Biol. Chem. 284:18923–18931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuny CV, Chinchilla K, Culbertson MR, Kalejta RF. 2010. Cyclin-dependent kinase-like function is shared by the beta- and gamma-subset of the conserved herpesvirus protein kinases. PLoS Pathog. 6:e1001092. 10.1371/journal.ppat.1001092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee C-P, Huang Y-H, Lin S-F, Chang Y, Chang Y-H, Takada K, Chen M-R. 2008. Epstein-Barr virus BGLF4 kinase induces disassembly of the nuclear lamina to facilitate virion production. J. Virol. 82:11913–11926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wang J-T, Doong S-L, Teng S-C, Lee C-P, Tsai C-H, Chen M-R. 2009. Epstein-Barr virus BGLF4 kinase suppresses the interferon regulatory factor 3 signaling pathway. J. Virol. 83:1856–1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Feederle R, Mehl-Lautscham AM, Bannert H, Delecluse H-J. 2009. The Epstein-Barr virus protein kinase BGLF4 and the exonuclease BGLF5 have opposite effects on the regulation of viral protein production. J. Virol. 83:10877–10891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hume AJ, Finkel JS, Kamil JP, Coen DM, Culbertson MR, Kalejta RF. 2008. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 320:797–799 [DOI] [PubMed] [Google Scholar]
- 35. Jacob T, Van den Broeke C, Favoreel HW. 2011. Viral serine/threonine protein kinases. J. Virol. 85:1158–1173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Marty FM, Ljungman P, Papanicolaou GA, Winston DJ, Chemaly RF, Strasfeld L, Young J-AH, Rodriguez T, Maertens J, Schmitt M, Einsele H, Ferrant A, Lipton JH, Villano SA, Chen H, Boeckh M. 2011. Maribavir prophylaxis for prevention of cytomegalovirus disease in recipients of allogeneic stem-cell transplants: a phase 3, double-blind, placebo-controlled, randomised trial. Lancet Infect. Dis. 11:284–292 [DOI] [PubMed] [Google Scholar]
- 37. Cheng YC, Huang ES, Lin JC, Mar EC, Pagano JS, Dutschman GE, Grill SP. 1983. Unique spectrum of activity of 9-[(1,3-dihydroxy-2-propoxy)methyl]-guanine against herpesviruses in vitro and its mode of action against herpes simplex virus type 1. Proc. Natl. Acad. Sci. U. S. A. 80:2767–2770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Datta AK, Colby BM, Shaw JE, Pagano JS. 1980. Acyclovir inhibition of Epstein-Barr virus replication. Proc. Natl. Acad. Sci. U. S. A. 77:5163–5166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lin JC, DeClercq E, Pagano JS. 1987. Novel acyclic adenosine analogs inhibit Epstein-Barr virus replication. Antimicrob. Agents Chemother. 31:1431–1433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Matthews T, Boehme R. 1988. Antiviral activity and mechanism of action of ganciclovir. Rev. Infect. Dis. 10(Suppl 3):S490–S494 [DOI] [PubMed] [Google Scholar]
- 41. Littler E, Stuart AD, Chee MS. 1992. Human cytomegalovirus UL97 open reading frame encodes a protein that phosphorylates the antiviral nucleoside analogue ganciclovir. Nature 358:160–162 [DOI] [PubMed] [Google Scholar]
- 42. Sullivan V, Talarico CL, Stanat SC, Davis M, Coen DM, Biron KK. 1992. A protein kinase homologue controls phosphorylation of ganciclovir in human cytomegalovirus-infected cells. Nature 359:85. [DOI] [PubMed] [Google Scholar]
- 43. Talarico CL, Burnette TC, Miller WH, Smith SL, Davis MG, Stanat SC, Ng TI, He Z, Coen DM, Roizman B, Biron KK. 1999. Acyclovir is phosphorylated by the human cytomegalovirus UL97 protein. Antimicrob. Agents Chemother. 43:1941–1946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. De Clercq E. 2004. Antivirals and antiviral strategies. Nat. Rev. Microbiol. 2:704–720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gershburg E, Pagano JS. 2005. Epstein-Barr virus infections: prospects for treatment. J. Antimicrob. Chemother. 56:277–281 [DOI] [PubMed] [Google Scholar]
- 46. Meng Q, Hagemeier SR, Fingeroth JD, Gershburg E, Pagano JS, Kenney SC. 2010. The Epstein-Barr virus (EBV)-encoded protein kinase, EBV-PK, but not the thymidine kinase (EBV-TK), is required for ganciclovir and acyclovir inhibition of lytic viral production. J. Virol. 84:4534–4542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nahleh Z, Tfayli A, Najm A, El Sayed A, Nahle Z. 2012. Heat shock proteins in cancer: targeting the “chaperones.” Future Med. Chem. 4:927–935 [DOI] [PubMed] [Google Scholar]
- 48. Whitesell L, Lindquist SL. 2005. HSP90 and the chaperoning of cancer. Nat. Rev. Cancer 5:761–772 [DOI] [PubMed] [Google Scholar]
- 49. Taipale M, Krykbaeva I, Koeva M, Kayatekin C, Westover KD, Karras GI, Lindquist S. 2012. Quantitative analysis of Hsp90-client interactions reveals principles of substrate recognition. Cell 150:987–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hong DS, Banerji U, Tavana B, George GC, Aaron J, Kurzrock R. 2013. Targeting the molecular chaperone heat shock protein 90 (HSP90): lessons learned and future directions. Cancer Treat. Rev. 39:375–387 [DOI] [PubMed] [Google Scholar]
- 51. Grenert JP, Sullivan WP, Fadden P, Haystead TA, Clark J, Mimnaugh E, Krutzsch H, Ochel HJ, Schulte TW, Sausville E, Neckers LM, Toft DO. 1997. The amino-terminal domain of heat shock protein 90 (hsp90) that binds geldanamycin is an ATP/ADP switch domain that regulates hsp90 conformation. J. Biol. Chem. 272:23843–23850 [DOI] [PubMed] [Google Scholar]
- 52. Prodromou C, Roe SM, O'Brien R, Ladbury JE, Piper PW, Pearl LH. 1997. Identification and structural characterization of the ATP/ADP-binding site in the Hsp90 molecular chaperone. Cell 90:65–75 [DOI] [PubMed] [Google Scholar]
- 53. Kamal A, Thao L, Sensintaffar J, Zhang L, Boehm MF, Fritz LC, Burrows FJ. 2003. A high-affinity conformation of Hsp90 confers tumour selectivity on Hsp90 inhibitors. Nature 425:407–410 [DOI] [PubMed] [Google Scholar]
- 54. Kobayashi N, Toyooka S, Soh J, Yamamoto H, Dote H, Kawasaki K, Otani H, Kubo T, Jida M, Ueno T, Ando M, Ogino A, Kiura K, Miyoshi S. 2012. The anti-proliferative effect of heat shock protein 90 inhibitor, 17-DMAG, on non-small-cell lung cancers being resistant to EGFR tyrosine kinase inhibitor. Lung Cancer 75:161–166 [DOI] [PubMed] [Google Scholar]
- 55. Tatokoro M, Koga F, Yoshida S, Kawakami S, Fujii Y, Neckers L, Kihara K. 2012. Potential role of Hsp90 inhibitors in overcoming cisplatin resistance of bladder cancer-initiating cells. Int. J. Cancer 131:987–996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Li D, Marchenko ND, Schulz R, Fischer V, Velasco-Hernandez T, Talos F, Moll UM. 2011. Functional inactivation of endogenous MDM2 and CHIP by HSP90 causes aberrant stabilization of mutant p53 in human cancer cells. Mol. Cancer Res. 9:577–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Hertlein E, Wagner AJ, Jones J, Lin TS, Maddocks KJ, Towns WH, III, Goettl VM, Zhang X, Jarjoura D, Raymond CA, West DA, Croce CM, Byrd JC, Johnson AJ. 2010. 17-DMAG targets the nuclear factor-kappaB family of proteins to induce apoptosis in chronic lymphocytic leukemia: clinical implications of HSP90 inhibition. Blood 116:45–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Sun X, Barlow EA, Ma S, Hagemeier SR, Duellman SJ, Burgess RR, Tellam J, Khanna R, Kenney SC. 2010. Hsp90 inhibitors block outgrowth of EBV-infected malignant cells in vitro and in vivo through an EBNA1-dependent mechanism. Proc. Natl. Acad. Sci. U. S. A. 107:3146–3151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Molesworth SJ, Lake CM, Borza CM, Turk SM, Hutt-Fletcher LM. 2000. Epstein-Barr virus gH is essential for penetration of B cells but also plays a role in attachment of virus to epithelial cells. J. Virol. 74:6324–6332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Mounce BC, Mboko WP, Bigley TM, Terhune SS, Tarakanova VL. 2013. A conserved gammaherpesvirus protein kinase targets histone deacetylases 1 and 2 to facilitate viral replication in primary macrophages. J. Virol. 87:7314–7325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Lin D-Y, Huang Y-S, Jeng J-C, Kuo H-Y, Chang C-C, Chao T-T, Ho C-C, Chen Y-C, Lin T-P, Fang H-I, Hung C-C, Suen C-S, Hwang M-J, Chang K-S, Maul GG, Shih H-M. 2006. Role of SUMO-interacting motif in Daxx SUMO modification, subnuclear localization, and repression of sumoylated transcription factors. Mol. Cell 24:341–354 [DOI] [PubMed] [Google Scholar]
- 62. Zhang Q, Hong Y, Dorsky D, Holley-Guthrie E, Zalani S, Elshiekh NA, Kiehl A, Le T, Kenney S. 1996. Functional and physical interactions between the Epstein-Barr virus (EBV) proteins BZLF1 and BMRF1: effects on EBV transcription and lytic replication. J. Virol. 70:5131–5142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Scroggins BT, Robzyk K, Wang D, Marcu MG, Tsutsumi S, Beebe K, Cotter RJ, Felts S, Toft D, Karnitz L, Rosen N, Neckers L. 2007. An acetylation site in the middle domain of Hsp90 regulates chaperone function. Mol. Cell 25:151–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Wang J-T, Yang P-W, Lee C-P, Han C-H, Tsai C-H, Chen M-R. 2005. Detection of Epstein-Barr virus BGLF4 protein kinase in virus replication compartments and virus particles. J. Gen. Virol. 86:3215–3225 [DOI] [PubMed] [Google Scholar]
- 65. Zhu H, Shen Y, Shenk T. 1995. Human cytomegalovirus IE1 and IE2 proteins block apoptosis. J. Virol. 69:7960–7970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. He Z, He YS, Kim Y, Chu L, Ohmstede C, Biron KK, Coen DM. 1997. The human cytomegalovirus UL97 protein is a protein kinase that autophosphorylates on serines and threonines. J. Virol. 71:405–411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gill MB, Kutok JL, Fingeroth JD. 2007. Epstein-Barr virus thymidine kinase is a centrosomal resident precisely localized to the periphery of centrioles. J. Virol. 81:6523–6535 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Gustafson EA, Chillemi AC, Sage DR, Fingeroth JD. 1998. The Epstein-Barr virus thymidine kinase does not phosphorylate ganciclovir or acyclovir and demonstrates a narrow substrate specificity compared to the herpes simplex virus type 1 thymidine kinase. Antimicrob. Agents Chemother. 42:2923–2931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Gustafson EA, Schinazi RF, Fingeroth JD. 2000. Human herpesvirus 8 open reading frame 21 is a thymidine and thymidylate kinase of narrow substrate specificity that efficiently phosphorylates zidovudine but not ganciclovir. J. Virol. 74:684–692 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Feederle R, Kost M, Baumann M, Janz A, Drouet E, Hammerschmidt W, Delecluse H-J. 2000. The Epstein-Barr virus lytic program is controlled by the cooperative functions of two transactivators. EMBO J. 19:3080–3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Raab-Traub N, Flynn K. 1986. The structure of the termini of the Epstein-Barr virus as a marker of clonal cellular proliferation. Cell 47:883–889 [DOI] [PubMed] [Google Scholar]
- 72. Breitkreutz A, Choi H, Sharom JR, Boucher L, Neduva V, Larsen B, Lin Z-Y, Breitkreutz B-J, Stark C, Liu G, Ahn J, Dewar-Darch D, Reguly T, Tang X, Almeida R, Qin ZS, Pawson T, Gingras A-C, Nesvizhskii AI, Tyers M. 2010. A global protein kinase and phosphatase interaction network in yeast. Science 328:1043–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Pearl LH. 2005. Hsp90 and Cdc37—a chaperone cancer conspiracy. Curr. Opin. Genet. Dev. 15:55–61 [DOI] [PubMed] [Google Scholar]
- 74. García-Morales P, Carrasco-García E, Ruiz-Rico P, Martínez-Mira R, Menéndez-Gutiérrez MP, Ferragut JA, Saceda M, Martínez-Lacaci I. 2007. Inhibition of Hsp90 function by ansamycins causes downregulation of cdc2 and cdc25c and G(2)/M arrest in glioblastoma cell lines. Oncogene 26:7185–7193 [DOI] [PubMed] [Google Scholar]
- 75. Nomura N, Nomura M, Newcomb EW, Zagzag D. 2007. Geldanamycin induces G2 arrest in U87MG glioblastoma cells through downregulation of Cdc2 and cyclin B1. Biochem. Pharmacol. 73:1528–1536 [DOI] [PubMed] [Google Scholar]
- 76. Fahmi H, Cochet C, Hmama Z, Opolon P, Joab I. 2000. Transforming growth factor beta 1 stimulates expression of the Epstein-Barr virus BZLF1 immediate-early gene product ZEBRA by an indirect mechanism which requires the MAPK kinase pathway. J. Virol. 74:5810–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Kang S-A, Cho H-S, Yoon JB, Chung IK, Lee S-T. 2012. Hsp90 rescues PTK6 from proteasomal degradation in breast cancer cells. Biochem. J. 447:313–320 [DOI] [PubMed] [Google Scholar]
- 78. Faresse N, Ruffieux-Daidie D, Salamin M, Gomez-Sanchez CE, Staub O. 2010. Mineralocorticoid receptor degradation is promoted by Hsp90 inhibition and the ubiquitin-protein ligase CHIP. Am. J. Physiol. Renal Physiol. 299:F1462–F1472. 10.1152/ajprenal.00285.2010 [DOI] [PubMed] [Google Scholar]
- 79. Noh H, Kim HJ, Yu MR, Kim W-Y, Kim J, Ryu JH, Kwon SH, Jeon JS, Han DC, Ziyadeh F. 2012. Heat shock protein 90 inhibitor attenuates renal fibrosis through degradation of transforming growth factor-β type II receptor. Lab. Invest. 92:1583–1596 [DOI] [PubMed] [Google Scholar]
- 80. Qing G, Yan P, Qu Z, Liu H, Xiao G. 2007. Hsp90 regulates processing of NF-kappa B2 p100 involving protection of NF-kappa B-inducing kinase (NIK) from autophagy-mediated degradation. Cell Res. 17:520–530 [DOI] [PubMed] [Google Scholar]
- 81. Shen S, Zhang P, Lovchik MA, Li Y, Tang L, Chen Z, Zeng R, Ma D, Yuan J, Yu Q. 2009. Cyclodepsipeptide toxin promotes the degradation of Hsp90 client proteins through chaperone-mediated autophagy. J. Cell Biol. 185:629–639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Liu K-S, Liu H, Qi J-H, Liu Q-Y, Liu Z, Xia M, Xing G-W, Wang S-X, Wang Y-F. 2012. SNX-2112, an Hsp90 inhibitor, induces apoptosis and autophagy via degradation of Hsp90 client proteins in human melanoma A-375 cells. Cancer Lett. 318:180–188 [DOI] [PubMed] [Google Scholar]
- 83. Jinwal UK, Abisambra JF, Zhang J, Dharia S, O'Leary JC, Patel T, Braswell K, Jani T, Gestwicki JE, Dickey CA. 2012. Cdc37/Hsp90 protein complex disruption triggers an autophagic clearance cascade for TDP-43 protein. J. Biol. Chem. 287:24814–24820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Krecmerova M. 2012. Nucleoside and nucleotide analogues for the treatment of herpesvirus infections: current stage and new prospects in the field of acyclic nucleoside phosphonates, p 245–270 In Magel GD. (ed), Herpesviridae—a look into this unique family of viruses. InTech, New York, NY. 10.5772/27995 [DOI] [Google Scholar]
- 85. Murata T, Isomura H, Yamashita Y, Toyama S, Sato Y, Nakayama S, Kudoh A, Iwahori S, Kanda T, Tsurumi T. 2009. Efficient production of infectious viruses requires enzymatic activity of Epstein-Barr virus protein kinase. Virology 389:75–81 [DOI] [PubMed] [Google Scholar]
- 86. Kamil JP, Hume AJ, Jurak I, Münger K, Kalejta RF, Coen DM. 2009. Human papillomavirus 16 E7 inactivator of retinoblastoma family proteins complements human cytomegalovirus lacking UL97 protein kinase. Proc. Natl. Acad. Sci. U. S. A. 106:16823–16828 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Neckers L, Neckers K. 2002. Heat-shock protein 90 inhibitors as novel cancer chemotherapeutic agents. Expert Opin. Emerg. Drugs 7:277–288 [DOI] [PubMed] [Google Scholar]
- 88. Stancato LF, Chow YH, Hutchison KA, Perdew GH, Jove R, Pratt WB. 1993. Raf exists in a native heterocomplex with hsp90 and p50 that can be reconstituted in a cell-free system. J. Biol. Chem. 268:21711–21716 [PubMed] [Google Scholar]
- 89. Dai K, Kobayashi R, Beach D. 1996. Physical interaction of mammalian CDC37 with CDK4. J. Biol. Chem. 271:22030–22034 [DOI] [PubMed] [Google Scholar]
- 90. Roe SM, Ali MMU, Meyer P, Vaughan CK, Panaretou B, Piper PW, Prodromou C, Pearl LH. 2004. The mechanism of Hsp90 regulation by the protein kinase-specific cochaperone p50(cdc37). Cell 116:87–98 [DOI] [PubMed] [Google Scholar]
- 91. Qing G, Yan P, Xiao G. 2006. Hsp90 inhibition results in autophagy-mediated proteasome-independent degradation of IkappaB kinase (IKK). Cell Res. 16:895–901 [DOI] [PubMed] [Google Scholar]
- 92. Castorena KM, Weeks SA, Stapleford KA, Cadwallader AM, Miller DJ. 2007. A functional heat shock protein 90 chaperone is essential for efficient flock house virus RNA polymerase synthesis in Drosophila cells. J. Virol. 81:8412–8420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Burch AD, Weller SK. 2005. Herpes simplex virus type 1 DNA polymerase requires the mammalian chaperone hsp90 for proper localization to the nucleus. J. Virol. 79:10740–10749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Kawashima D, Kanda T, Murata T, Saito S, Sugimoto A, Narita Y, Tsurumi T. 2013. Nuclear transport of Epstein-Barr virus DNA polymerase is dependent on the BMRF1 polymerase processivity factor and molecular chaperone Hsp90. J. Virol. 87:6482–6491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Basha W, Kitagawa R, Uhara M, Imazu H, Uechi K, Tanaka J. 2005. Geldanamycin, a potent and specific inhibitor of Hsp90, inhibits gene expression and replication of human cytomegalovirus. Antivir. Chem. Chemother. 16:135–146 [DOI] [PubMed] [Google Scholar]
- 96. Qin Z, DeFee M, Isaacs JS, Parsons C. 2010. Extracellular Hsp90 serves as a cofactor for MAPK activation and latent viral gene expression during de novo infection by KSHV. Virology 403:92–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Chen W, Sin S-H, Wen KW, Damania B, Dittmer DP. 2012. Hsp90 inhibitors are efficacious against Kaposi sarcoma by enhancing the degradation of the essential viral gene LANA, of the viral co-receptor EphA2 as well as other client proteins. PLoS Pathog. 8:e1003048. 10.1371/journal.ppat.1003048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Wen KW, Damania B. 2010. Hsp90 and Hsp40/Erdj3 are required for the expression and anti-apoptotic function of KSHV K1. Oncogene 29:3532–3544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Murata T, Iwata S, Siddiquey MNA, Kanazawa T, Goshima F, Kawashima D, Kimura H, Tsurumi T. 2013. Heat shock protein 90 inhibitors repress latent membrane protein 1 (LMP1) expression and proliferation of Epstein-Barr virus-positive natural killer cell lymphoma. PLoS One 8:e63566. 10.1371/journal.pone.0063566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Jhaveri K, Modi S. 2012. HSP90 inhibitors for cancer therapy and overcoming drug resistance. Adv. Pharmacol. 65:471–517 [DOI] [PubMed] [Google Scholar]
- 101. Zhang H, Burrows F. 2004. Targeting multiple signal transduction pathways through inhibition of Hsp90. J. Mol. Med. 82:488–499 [DOI] [PubMed] [Google Scholar]
- 102. Dai C, Whitesell L. 2005. HSP90: a rising star on the horizon of anticancer targets. Future Oncol. 1:529–540 [DOI] [PubMed] [Google Scholar]
- 103. Patel HJ, Modi S, Chiosis G, Taldone T. 2011. Advances in the discovery and development of heat-shock protein 90 inhibitors for cancer treatment. Expert Opin. Drug Discov. 6:559–587 [DOI] [PMC free article] [PubMed] [Google Scholar]