

Abstract
The human polyomavirus BK virus (BKV) establishes a latent and asymptomatic infection in the majority of the population. In immunocompromised individuals, the virus frequently (re)activates and may cause severe disease such as interstitial nephritis and hemorrhagic cystitis. Currently, the therapeutic options are limited to reconstitution of the antiviral immune response. T cells are particularly important for controlling this virus, and T cell therapies may provide a highly specific and effective mode of treatment. However, little is known about the phenotype and function of BKV-specific T cells in healthy individuals. Using tetrameric BKV peptide-HLA-A02 complexes, we determined the presence, phenotype, and functional characteristics of circulating BKV VP1-specific CD8+ T cells in 5 healthy individuals. We show that these cells are present in low frequencies in the circulation and that they have a resting CD45RA− CD27+ memory and predominantly CCR7− CD127+ KLRG1+ CD49dhi CXCR3hi T-betint Eomesoderminlo phenotype. Furthermore, their direct cytotoxic capacity seems to be limited, since they do not readily express granzyme B and express only little granzyme K. We compared these cells to circulating CD8+ T cells specific for cytomegalovirus (CMV), Epstein-Barr virus (EBV), and influenza virus (Flu) in the same donors and show that BKV-specific T cells have a phenotype that is distinct from that of CMV- and EBV-specific T cells. Lastly, we show that BKV-specific T cells are polyfunctional since they are able to rapidly express interleukin-2 (IL-2), gamma interferon (IFN-γ), tumor necrosis factor α, and also, to a much lower extent, MIP-1β and CD107a.
INTRODUCTION
In healthy individuals, the polyomavirus BK virus (BKV) establishes a latent, or “smoldering,” but asymptomatic infection. However, in immunocompromised individuals, the virus frequently escapes the normal immunological surveillance to become systemically active, after which it may cause severe pathology. BKV elicits interstitial nephritis of the allograft in about 5% of kidney transplant recipients, making it an important cause of graft failure and graft loss. In up to 30% of hematopoietic stem cell transplant (HSCT) recipients, the virus induces hemorrhagic cystitis, thereby significantly contributing to morbidity and length of hospitalization (1).
Currently, the main mode of therapy for patients suffering from BKV infection comprises reconstitution of the immunological antiviral response. In solid organ transplant recipients, this is achieved through tapering of the immunosuppressive medication. Unfortunately, this comes at the cost of increased allograft rejection, and in HSCT recipients this is an unattractive approach due to a considerable increase in the risk of graft-versus-host disease. So far, antiviral agents, such as cidofovir and leflunomide, have shown little effect on BKV replication in vivo (1). It is therefore crucial to develop new modes of therapy. In this regard, the normal T cell response was shown to be very important for keeping BKV at bay (2). Treatments that involve the infusion of autologous ex vivo-expanded virus-specific T cells, as well as newer vaccination strategies with autologous antigen-presenting cells pulsed ex vivo with viral antigen, are therefore promising candidates that could provide highly specific and effective modes of therapy (3, 4). It is well established that different T cell specificities give rise to different T cell phenotypes, which in turn is indeed also related to T cell function (5, 6). In this regard, little is known about the normal phenotype and function of BKV-specific T cells that are controlling BKV infection in healthy individuals, information that is necessary for the successful design of effective T cell therapies and vaccination strategies.
In the current study, we used fluorescent tetrameric HLA-A02 complexes presenting four different immunodominant BKV epitopes in order to visualize and characterize circulating BKV-specific CD8+ T cells. Phenotype and functional characteristics of these cells were analyzed in 5 healthy HLA-A02-positive adults. Furthermore, these BKV tetramers contain epitopes with a high degree of homology to the corresponding polyomavirus JC virus (JCV) epitopes, varying from a two-amino-acid difference to no difference at all. Indeed, cross-reactivity between the respective BKV and JCV tetramers was demonstrated (7–10). Moreover, antigen-presenting cells pulsed with BKV lysate can activate JCV-specific T cells and vice versa (8). Therefore, it is highly likely that the BKV-specific CD8+ T cells described in the current study are in fact also JCV-specific CD8+ T cells. Since it is well established that CD8+ T cell specificity correlates with phenotype, we compared the phenotypic characteristics of these BKV-specific CD8+ T cells to those of cytomegalovirus (CMV)-, Epstein-Barr virus (EBV)-, and influenza virus (Flu)-specific CD8+ T cells circulating in the same individuals to see how these phenotypes relate to each other (5). We found low frequencies of circulating BKV virion protein 1 (VP1)-specific CD8+ T cells that predominantly displayed an effector-memory cell phenotype. Furthermore, we observed that these cells were phenotypically distinct from circulating CMV- and EBV-specific CD8+ T cells, whereas they resembled Flu-specific T cells in several aspects. Lastly, BKV-specific T cells were polyfunctional with regard to their rapid expression of cytokines upon stimulation but did not express granzyme B.
MATERIALS AND METHODS
Study subjects.
We obtained peripheral blood mononuclear cells (PBMCs) from 25 buffy coats deriving from different HLA-A02-positive healthy blood donors, aged between 18 and 64 years, from Sanquin Blood Supply, Amsterdam, Netherlands. Corresponding plasma samples were not available for serologic or virologic examinations.
Isolation of PBMCs.
PBMCs were isolated using standard density gradient centrifugation, after which they were cryopreserved until the day of analysis.
Tetrameric complexes.
All tetrameric complexes were obtained from Sanquin (Amsterdam, Netherlands). For BKV, four different immunodominant epitopes shared by the majority of BKV strains were selected including two BKV capsid protein VP1 epitopes, BKV VP1-derived AITEVECFL (VP1 p44) and BKV VP1 LLMWEAVTV (VP1 p108) (11), and two large T antigen protein (LTAg) epitopes, BKV LTAg LLLIWFRPV (LTAg p579) and BKV LTAg FLHCIVFNV (LTAg p410) (12, 13). These were incorporated in phycoerythrin (PE)-labeled HLA-A02 tetrameric complexes. The respective epitopes show a high degree of overlap with the corresponding epitopes of JCV and simian virus 40 (SV40): JCV VP1p36-44 SITEVECFL and SV40 VP1p46-54 SFTEVECFL, JCV VP1p100-108 ILMWEAVTL and SV40 VP1p110-118 ILMWEAVTV, JCV LTagp409-417 FLKCIVLNI and SV40 LTAgp408-416 FLKCMVYNI, and JCV LTAgp577-585 LLLIWFRPV and SV40 LTAgp577-585 LMLIWYRPV. Indeed, a high degree of cross-reactivity with the JCV VP1p36 and VP1p100 tetramers and the SV40 VP1p110 peptide has been demonstrated. For the detection of the other circulating antiviral CD8+ T cells, allophycocyanin (APC)-labeled HLA-A2 tetramers loaded with hCMV-pp65 protein-derived NLVPMVATV (CMV pp65 p495), influenza matrix protein 1-derived GILGFVFTL (Flu MP1 p58), and EBV BMLF-1 derived GLCTLVAML (EBV BMLF-1 p259) were used.
PBMC peptide cultures.
After thawing, PBMCs were labeled with carboxyfluorescein succinimidyl ester (CFSE). Thereafter, 2 × 10E6 PBMCs were incubated with one of four BKV peptides (1.25 × 10E−3 mg/ml) and recombinant human interleukin-2 (IL-2) (50 U/ml; Biotest, Solihull, United Kingdom) in culture medium that consisted of Iscove's modified Dulbecco's medium (IMDM), 10% human pool plasma (PAA, Piscataway, NJ, USA), penicillin/streptomycin, and β-mercaptoethanol (Sigma, Zwijndrecht, Netherlands) for the duration of 10 days. Extra IL-2 was added on day 3 and day 6 of culture. Controls comprised PBMCs cultured in medium with IL-2 alone, PBMCs cultured in medium containing the respective peptide alone, or PBMCs in culture medium alone without the addition of the respective peptide or IL-2. After culture, PBMCs were first incubated with the respective PE-labeled tetramer for 30 min in the dark at 4°C, after which they were incubated with the following antibodies: CD3-Alexa Fluor 700, CD8 PercP-eFluor 710, CD27 APC-Alexa Fluor 780 (eBioscience Inc., San Diego, CA, USA), and CD45RA PE-Cy7 (BD Biosciences, San Jose, CA, USA) for 30 min in the dark at 4°C. The Live/Dead fixable red cell stain kit (Invitrogen Ltd., Paisley, United Kingdom) was used in every staining to exclude dead cells from the analysis. Thereafter, measurements were done on the BD Biosciences' FACSCanto II flow cytometer. BKV-specific CD8+ CTLs were defined as being live CD3+ CD8+ tetramer+ events that had become CFSE dim due to cell division. Analyses were done using FlowJo software v9.6 (FlowJo, Ashland, OR, USA).
Tetramer staining and antibodies used for determining T cell phenotype.
Before surface staining, up to 10 × 10E6 PBMCs were incubated with the respective tetramer for 30 min in the dark at 4°C. Live/Dead fixable red cell staining was utilized during all flow cytometry measurements and analyses in order to gate out dead cells. Monoclonal antibodies used for the determination of T cell phenotype and function included the following: CD3, V500; CCR7, PE-Cy7; CD45RA, PE-Cy7; CD45RA, fluorescein isothiocyanate (FITC); CD8, brilliant violet 421 (all from BD Biosciences); CD49d, PE-Cy7; CXCR3, Alexa Fluor 647; CD38, brilliant violet 421; CD8, Alexa Fluor 700; CD127, APC-Alexa Fluor 780; CD27, PerCP-eFluor 710; CD45RA, eFluor605; CD8, eFluor 605; CD27, APC-Alexa Fluor 780; CX3CR1, APC; HLA-DR, Alexa Fluor 700 (all from eBioscience); CXCR3, PE (R&D systems, Abingdon, United Kingdom); and KLRG1, Alexa Fluor 488 (14).
After extracellular staining, antibodies were fixed and the cells were permeabilized using fluorescence-activated cell sorter (FACS) Lysing solution and FACS Permeabilizing solution 2 (BD Biosciences) according to the manufacturer's instructions. Cells were subsequently incubated with a combination of the following intracellular antibodies for 30 min in the dark at 4°C: T-bet, brilliant violet 421 (BioLegend, San Diego, CA, USA; eomesodermin, PerCP-eFluor 710; eomesodermin, Alexa Fluor 647; Ki-67, PE-Cy7; granzyme B, Alexa Fluor 700 (BD Biosciences); and granzyme K, FITC (Immunotools, Friesoythe, Germany). All flow cytometry measurements for these experiments were done using BD Biosciences' LSRFortessa flow cytometer. Analysis of the results was performed with FlowJo software v9.6 (FlowJo, Ashland, OR, USA), always gating out duplets by using forward scatter width/height and sideward scatter width/height event characteristics.
Stimulation assays and cytokine production.
Cytokine release after peptide or phorbol 12-myristate 13-acetate (PMA)/ionomycin stimulation was performed as described by Lamoreaux et al. (15). PBMCs were thawed and rested overnight in suspension flasks (Greiner-Bio-One, Alphen a/d Rijn, Netherlands) in RPMI supplemented with 10% fetal calf serum (FCS), penicillin, and streptomycin (culture medium). Two million cells per well were stimulated with PMA (10 ng/ml) and ionomycin (1 μg/ml) or with the viral peptides in culture medium in the presence of anti-CD107a FITC (eBioscience), αCD29 (TS2/16, 1 μg/ml), brefeldin A (10 μg/ml; Invitrogen), and GolgiStop (BD Biosciences) in a final volume of 200 μl for 4 h or 5 h (peptides) at 37°C and 5% CO2. Stimulations were performed in untreated, round-bottom, 96-well plates (Corning, Amsterdam, Netherlands). Subsequently, cells were incubated with the appropriate tetramers, followed by incubation with CD3 V500, CD8 brilliant violet 421, CD4 PerCP efluor 710 (Ebioscience), and Live/Dead fixable red cell stain for 30 min at 4°C. The cells were then washed twice, fixed, and permeabilized (Cytofix/Cytoperm reagent; BD Biosciences) and were subsequently incubated with the following intracellular monoclonal antibodies: gamma interferon (IFN-γ), APC-Alexa Fluor 750 (Invitrogen); tumor necrosis factor alpha (TNF-α,) Alexa Fluor 700; IL-2, APC; and anti-Mip-1β, PE-Cy7 (BD Biosciences) for 30 min at 4°C. Cells were washed twice and measured on an LSRFortessa flow cytometer and analyzed with FlowJo Version 9.6 software, always gating out dead cells and duplets.
Gating strategy.
Lymphocytes were gated based on the forward scatter and sideward scatter event characteristics. Duplets were then gated out as described above. Live/Dead fixable red cell staining was used to gate out dead or dying cells. CD3+ events were then gated, after which the expression of CD8 was plotted against tetramer-positive events (Fig. 1 and 2). Total CD3+ CD8+ (CD8+ T cells) events were then gated separately from total CD3+ CD8+ tetramer+ (virus-specific CD8+ T cells) events. Gates were drawn on total CD8+ T cells or a major CD8+ T cell population (naive, effector, or memory), after which they were copied to the virus-specific CD8+ T cells for the following markers: CD45RA was plotted against CD27 in order to define CD45RA+ CD27+ naive, CD45RA− CD27+ memory, and CD45RA+/− CD27− effector cell populations (16); the expression of CCR7 was determined by plotting the former against CD45RA, and gates were placed based on the double-positive naive CD8+ T cell population (not shown); CD127 was plotted against KLRG1, and gates were placed based on the CD127+ KLRG1− naive CD8+ T cell population (see Fig. 4); CD49d was plotted against CD45RA, and gates were placed based on the CD45RA+ CD49d− naive CD8+ T cell population (see Fig. 5); CD38 was plotted against HLA-DR, and gates were placed based on the double-negative CD8+ T cell populations (not shown). Ki-67 was plotted against CD38, and gates were placed based on the double-negative CD8+ T cell population (not shown); granzyme B was plotted against granzyme K, and gates were placed based on the double-negative naive CD8+ T cell population (see Fig. 7); T-bet was plotted against Eomes, and gates were placed based on the double-negative naive CD8+ T cell population (see Fig. 6). Gates for the following markers were placed based on their expression on total alive lymphocytes: CXCR3 was plotted against CD3 within total alive lymphocytes, and gates were placed based on the CXCR3hi CD3− natural killer cell population (see Fig. 5); PD-1 was plotted against CD3, and gates were placed based on the PD-1lo CD3− population (not shown).
Fig 1.

Ex vivo detectable circulating BKV-specific CD8+ T cells in 5 healthy adults. Dot plots of BKV VP1 p44 and p108 tetramer-positive populations detected ex vivo. Tetramer fluorescence intensity is depicted on the y axis and anti-CD8 fluorescence intensity on the x axis. Percentages of tetramer+ T cells are shown.
Fig 2.
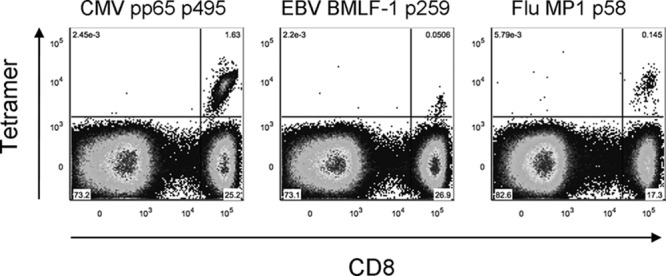
Ex vivo detection of circulating CMV-, EBV-, and Flu-specific CD8+ T cells in the same subjects. Dot plots of circulating CMV pp65, EBV BMLF1 (subject 6), and Flu MP1 (subject 4) tetramer-positive populations as detected ex vivo. Tetramer fluorescence intensity is plotted on the y axis and anti-CD8 fluorescence intensity on the x axis. Percentages of tetramer+ T cells are shown.
Fig 4.

BKV-specific CD8+ T cells are predominantly CD127+ KLRG1+. (A) Dot plots displaying the expression of KLRG1 and CD127 by BKV VP1 p108 (subject 4), BKV VP1 p44 (subject 9), CMV, EBV (subject 6), and Flu (subject 4) tetramer+ cells in black presented as an overlay on total CD8+ T cells in gray. Anti-KLRG1 fluorescence intensity is plotted on the y axis and anti-CD127 fluorescence intensity on the x axis. Percentages of tetramer+ T cells are shown. (B) Percentages of CD8+ T cells, CD8+ CD45RA− CD27+ memory T cells, and BKV VP1 (n = 5), CMV PP65 (n = 3), EBV BMLF1 (n = 3), and Flu MP1 (n = 3) specific CD8+ T cells expressing CD127 (n = 5 subjects; median, IQR). Asterisks mark statistically significant differences between BKV VP1-specific T cells and the respective T cell population, as defined by a P value of <0.05. (C) Percentages of CD8+ T cells, CD8+ CD45RA− CD27+ memory T cells, and BKV VP1 (n = 5), CMV PP65 (n = 3), EBV BMLF1 (n = 3), and Flu MP1 (n = 3) specific CD8+ T cells expressing KLRG1 (median, IQR).
Fig 5.
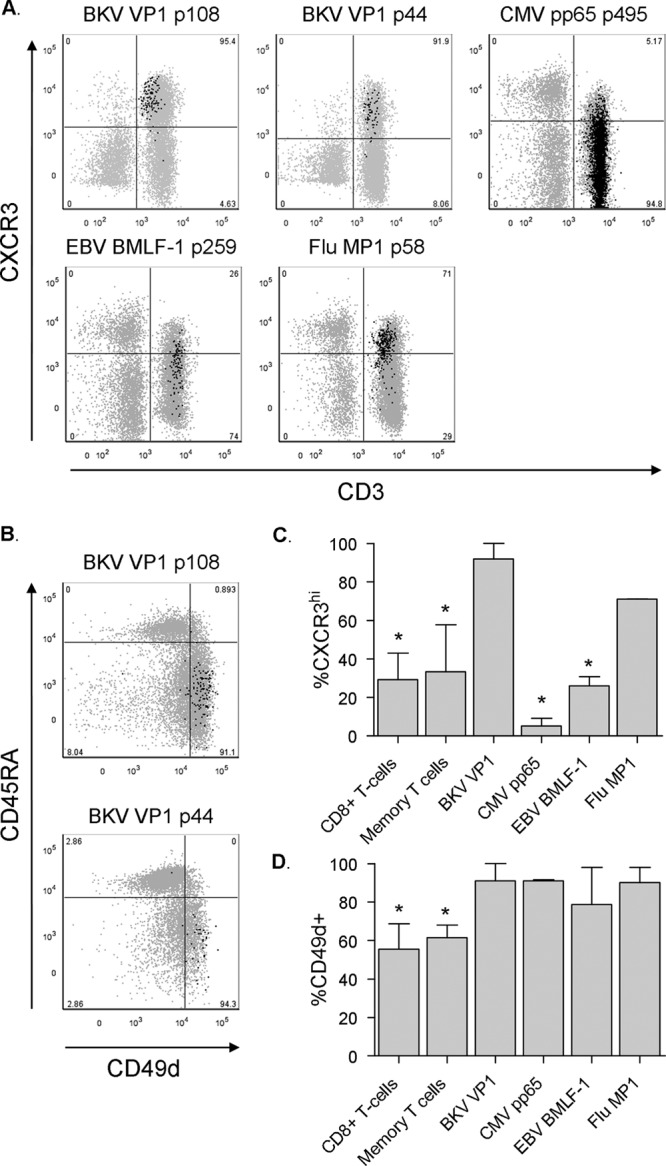
BKV-specific CD8+ T cells highly express CXCR3 and CD49d. (A) Dot plots displaying the expression of CXCR3 by BKV VP1 p108 (subject 4), BKV VP1 p44 (subject 9), CMV, EBV (subject 6), and Flu (subject 4) tetramer-positive events in black presented as an overlay on total live lymphocytes in gray. Anti-CXCR3 fluorescence intensity is plotted on the y axis and anti-CD3 fluorescence intensity on the x axis. Percentages of tetramer+ T cells are shown. (B) Dot plots displaying the expression of CD49d by BKV VP1 p108 (subject 4) and BKV VP1 p44 (subject 9) presented as an overlay on total CD8+ T cells in gray. Anti-CD45RA fluorescence intensity is plotted on the y axis and anti-CD49d fluorescence intensity on the x axis. Percentages of tetramer+ T cells are shown. (C) Percentages of CD8+ T cells, CD8+ CD45RA− CD27+ memory T cells, and BKV VP1 (n = 5), CMV PP65 (n = 3), EBV BMLF1 (n = 3), and Flu MP1 (n = 3) specific CD8+ T cells highly expressing CXCR3 (median, IQR). Asterisks mark statistically significant differences between BKV VP1-specific T cells and the respective T cell population, as defined by a P value of <0.05. (D) Percentages of CD8+ T cells, CD8+ CD45RA− CD27+ memory T cells, and BKV VP1 (n = 5), CMV PP65 (n = 3), EBV BMLF1 (n = 3), and Flu MP1 (n = 3) specific CD8+ T cells highly expressing CD49d (median, IQR). Asterisks mark statistically significant differences between BKV VP1-specific T cells and the respective T cell population, as defined by a P value of <0.05.
Fig 7.

BKV-specific CD8+ T cells do not readily carry granzyme B. (A) Dot plots displaying the expression of granzyme B and granzyme K by BKV VP1 p108 (subject 4), BKV VP1 p44 (subject 9), and CMV, EBV (subject 6), and Flu (subject 4) tetramer-positive events in black presented as an overlay on total CD8+ T cells in gray. Anti-granzyme B fluorescence intensity is plotted on the y axis and anti-granzyme K fluorescence intensity on the x axis. Percentages of tetramer+ T cells are shown. (B) Percentages of CD8+ T cells, CD8+ CD45RA− CD27+ memory T cells, and BKV VP1 (n = 4), CMV PP65 (n = 3), EBV BMLF1 (n = 3), and Flu MP1 (n = 3) specific CD8+ T cells expressing granzyme B. Due to insufficient BKV tetramer-positive events after intracellular staining, BKV tetramer data from subject 6 were excluded from this analysis (median, IQR). Asterisks mark statistically significant differences between BKV VP1-specific T cells and the respective T cell population, as defined by a P value of <0.05. (C) Percentages of CD8+ T cells, CD8+ CD45RA− CD27+ memory T cells, and BKV VP1 (n = 4), CMV PP65 (n = 3), EBV BMLF1 (n = 3), and Flu MP1 (n = 3) specific CD8+ T cells expressing granzyme K. Due to insufficient BKV tetramer-positive events after intracellular staining, BKV tetramer data from subject 6 were excluded from this analysis (median, IQR). CD8+ memory T cells showed a trend toward higher expression of granzyme K than did BKV-specific T cells, P = 0.06.
Fig 6.

Distinct T-bet/Eomes patterns found in different virus-specific T cells. (A) Dot plots displaying the expression of T-bet and Eomes by BKV VP1 p108 (subject 4), BKV VP1 p44 (subject 9), CMV, EBV (subject 6), and Flu (subject 4) tetramer-positive events in black presented as an overlay on total CD8+ T cells in gray. Anti-Eomes fluorescence intensity is plotted on the y axis and anti-T-bet fluorescence intensity on the x axis. Percentages of tetramer+ T cells are shown. (B) Percentages of CD8+ T cells, CD8+ CD45RA− CD27+ memory T cells, and BKV VP1 (n = 4), CMV PP65 (n = 3), EBV BMLF1 (n = 3), and Flu MP1 (n = 3) specific CD8+ T cells expressing T-bet. Due to insufficient BKV tetramer-positive events after intracellular staining, BKV tetramer data from subject 6 were excluded from this analysis (median, IQR). CMV pp65-specific T cells showed a trend toward a higher expression of T-bet than BKV-specific T cells, P = 0.057. (B) Percentages of CD8+ T cells, CD8+ CD45RA− CD27+ memory T cells, and BKV VP1 (n = 4), CMV PP65 (n = 3), EBV BMLF1 (n = 3), and Flu MP1 (n = 3) specific CD8+ T cells expressing Eomes. Due to insufficient BKV tetramer-positive events after intracellular staining, BKV tetramer data from subject 6 were excluded from this analysis (median, IQR). CMV pp65- and EBV BMLF-1-specific T cells showed a trend toward a higher expression of Eomes than did BKV-specific T cells; for both, P = 0.057.
Statistical analysis.
The two-tailed Mann-Whitney test was used for analysis of differences between T cell populations. P values of less than 0.05 were considered statistically significant.
RESULTS
Circulating BKV VP1-specific CD8+ T cells are detectable after in vitro expansion.
In view of the low frequencies in which BKV-specific CD8+ T cells are found in the circulation of healthy individuals, we first cultured CFSE-labeled PBMCs from 25 healthy HLA-A02-positive adults with four different BKV peptides, two derived from the capsid protein VP1, and two from the large T antigen (LTAg) protein, for 10 days in the presence of IL-2, in order to determine in which of these donors BKV-specific CD8+ T cells were detectable (7, 11). This yielded expanded BKV-specific CD8+ T cell populations that were readily detectable by flow cytometry in 15 of 25 subjects (60%). Seven of these were found to be positive for the VP1 p44 tetramer (28%), eight for the VP1 p108 tetramer (32%), five for the LTAg p579 tetramer (20%), and none for the LTAg p410 tetramer. Four subjects were positive for multiple BKV tetramers (16%) (Table 1). In contrast to the peptide/IL-2 condition, IL-2-only and peptide-only controls never yielded expanded BKV-specific CD8 T cell populations in this 10-day time window (data not shown).
Table 1.
BKV-specific CD8+ T cells detectable after culturea
| Subject no. | % of BKV tetramer-positive events with peptide: |
|||
|---|---|---|---|---|
| VP1 p44 | VP1 p108 | LTAg p579 | LTAg p410 | |
| 1 | 4.8 | |||
| 2 | 4.5 | |||
| 3 | 0.7 | |||
| 4 | 21.3 | |||
| 5 | 0.02 | 0.03 | ||
| 6 | 1.95 | |||
| 7 | <0.01 | 0.03 | ||
| 8 | 0.02 | 6.9 | 0.02 | |
| 9 | 4.16 | 0.32 | ||
| 10 | 0.2 | |||
| 11 | 0.05 | |||
| 12 | 0.01 | |||
| 13 | <0.01 | |||
| 14 | 0.01 | |||
| 15 | 0.03 | |||
Expanded after 10 days of culture of PBMCs in the presence of IL-2.
Circulating BKV VP1-specific CD8+ T cells can be detected directly ex vivo.
The low frequencies of these cells in the circulation complicate a direct ex vivo detection with tetramers, and prior expansion of CD8+ T cells by culture will hamper a reliable determination of their phenotype and function. Therefore, we measured a substantial amount of PBMCs (∼10 × 10E6) per antibody panel ex vivo only in those subjects whose PBMC fraction contained a particularly large BKV-specific CD8+ T cell population after 10-day expansion. As shown in Table 1, subjects 1, 2, 4, 6, 8, and 9 all had VP1 tetramer-positive populations that exceeded 1% of total CD8+ T cells. Indeed, BKV VP1-specific CD8+ T cells were detectable directly ex vivo in 5 of these 6 individuals (Fig. 1).
Circulating BKV VP1-specific CD8+ T cells are resting memory cells.
We then measured the expression of a wide variety of T cell markers in order to determine the phenotype of the BKV-specific CD8+ T cells. For comparison, the expression of the same markers was determined on HLA-A02 epitope-restricted CMV-, EBV- and Flu-specific CD8+ T cells circulating in the same five subjects (Table 2 and Fig. 2). As shown in Fig. 3A, we found that BKV VP1-specific T cells were CD45RA−, denoting them as antigen-experienced cells. Moreover, the majority of them expressed CD27 but lacked CCR7, marking them as being predominantly memory, and more specifically as “effector-memory” T cells (TEM) (Fig. 3C) (16, 17). The memory phenotype was supported by a high expression of CD127, the IL-7-receptor α chain that is important for maintaining memory cell homeostasis in the absence of antigen (Fig. 4A and B) (18). The majority of BKV-specific T cells expressed killer cell lectin-like receptor G1 (KLRG1), an inhibitory receptor that was previously found to be highly expressed by TEM cells (Fig. 4A and C) (14). However, as shown in Fig. 4C, KLRG1 expression levels varied notably between individuals. Flu-specific T cells were also predominantly CD45RA− CD27+ CCR7− TEM cells with a high expression of CD127 (Fig. 3 and 4B). The expressions of KLRG1 by the different virus-specific T cells did not differ significantly from each other (Fig. 4C). In contrast, the expression of CD127 by CMV- and EBV-specific T cells was significantly lower than that in BKV- and Flu-specific T cells (Fig. 4B).
Table 2.
Other antiviral CD8+ T cells detectable ex vivoa
| Subject no. | % of circulating CD8+ T cells with positive tetramers |
||
|---|---|---|---|
| CMV pp65 p495 | EBV BMLF-1 p259 | Flu MP1 p58 | |
| 2 | 0.07 | 0.03 | |
| 4 | 0.14 | ||
| 6 | 1.64 | 0.06 | |
| 8 | 3.6 | 0.03 | |
| 9 | 0.02 | 0.03 | |
Detected ex vivo in the same five individuals in whom BKV-specific T cells were detected ex vivo.
Fig 3.

BKV-specific CD8+ T cells have a memory phenotype. (A) Dot plots displaying the expression of CD45RA and CD27 by BKV VP1 p108 (subject 4), BKV VP1 p44 (subject 9), CMV pp65, EBV BMLF1 (subject 6), and Flu MP1 (subject 4) tetramer+ T cells in black presented as an overlay on total CD8+ T cells in gray. Anti-CD45RA fluorescence intensity is plotted on the y axis and anti-CD27 fluorescence intensity on the x axis. Percentages of tetramer+ T cells are shown. (B) Distribution of CD45RA− CCR7+ central-memory and CD45RA− CCR7− effector-memory T cell subsets among BKV VP1 (n = 5), CMV pp65 (n = 3), EBV BMLF1 (n = 3), and Flu MP1 (n = 3) specific T cells, represented as a percentage of total tetramer+ T cells (median, interquartile range [IQR]). (C) Distribution of CD45RA+/− CD27− effector and CD45RA− CD27+ memory T cell subsets among BKV VP1 (n = 5), CMV pp65 (n = 3), EBV BMLF1 (n = 3) and Flu MP1 (n = 3) specific cells represented as a percentage of total tetramer+ T cells (median, IQR).
With regard to their activation status, BKV-specific T cells did not express the activation markers CD38 or HLA-DR or the marker of active proliferation, Ki-67 (data not shown). PD-1, a marker of T cell exhaustion or activation, was expressed by nearly 20% of BKV-specific T cells but did not differ significantly from total CD8+ T cells, CD8+ memory T cells, or the other virus-specific T cells (data not shown).
In conclusion, circulating BKV VP1-specific CD8+ T cells in healthy adults have a memory phenotype and can be considered to be in a “resting” state since they did not express activation markers and were not actively proliferating.
Circulating BKV VP1-specific CD8+ T cells highly express the homing markers CD49d and CXCR3.
BKV is generally accepted to latently reside in epithelial cells lining the urogenital tract. However, in immunocompetent individuals, the virus has also been detected in the central nervous system, stool samples, and saliva (19–21). Since BKV-specific T cells are present in the circulation, one could theorize that they are trafficking to a site of (re)activation. Thus, we determined the expression of several specific T cell homing markers, in order to elucidate to which sites BKV-specific CD8+ T cells can migrate.
BKV-specific cells lacked expression of CD103, the alpha E integrin chain, a marker for tissue-resident T cells that is also expressed by a small proportion of circulating regulatory CD8+ T cells (22). The cells did highly express CD49d (Fig. 5B), the integrin α4 chain that mediates T cell migration through endothelial and epithelial barriers into various tissues and also through the blood-brain barrier (23, 24). However, since CD49d was also highly expressed on T cells specific for other viruses, this appeared not to be a specific trait of BKV-specific T cells (Fig. 5B and D). BKV-specific T cells expressed high levels of CXCR3, a chemokine receptor which has been shown to mediate T cell homing to reactive lymph nodes, stressed epithelium, and sites of inflammation in a general sense (25, 26). Interestingly, Flu-specific T cells, but not CMV- and EBV-specific T cells, displayed a similarly high expression of CXCR3 (Fig. 5A and C). BKV-specific T cells did not express the chemokine receptors CCR4 or CCR9 (data not shown).
Circulating BKV VP1-specific CD8+ T cells display a particularly low expression of Eomes.
The T-box transcription factors T-bet and eomesodermin (Eomes) are key regulators of CD8+ T cell differentiation and have been shown to cooperatively steer not only important CD8+ T cell functions such as the production of IFN-γ, expression of the IL-12 receptor, and expression of granzyme B but also the overall differentiation process of naive CD8+ T cells toward an effector or memory phenotype (27–31). Interestingly, the different virus-specific CD8+ T cell populations displayed distinct and specific patterns of T-bet and Eomes expression (Fig. 6). BKV-specific T cells displayed an intermediate T-bet expression with a markedly low expression of Eomes (Fig. 6). Similarly, Flu-specific T cells also displayed a T-betint Eomeslo expression, with almost no cells in the double-positive population. In contrast, CMV- and EBV BMLF1-specific T cells were highly expressing mainly both T-bet and Eomes (Fig. 6).
Circulating BKV VP1-specific CD8+ T cells do not express granzyme B.
To extend the phenotypic analysis of BKV-specific T cells, we determined the expression of the cytotoxic effector molecules granzyme B and granzyme K. Interestingly, the immediate cytotoxic capability of BKV VP1-specific CD8+ T cells appears to be limited since we found them not to express granzyme B (Fig. 7A and B). In contrast, CMV- and EBV-specific CD8+ T cells comprised a substantial population of granzyme B+ T cells (Fig. 7A and B). Also, a small population of Flu-specific T cells expressed granzyme B; however, this was lower than for CMV- and EBV-specific T cells (Fig. 7A and B). The virus-specific T cells did not differ from each other with regard to the expression of granzyme K (Fig. 7A and C).
Functionality of circulating BKV VP1-specific CD8+ T cells.
Next, we wanted to determine whether the BKV VP1-specific CD8+ T cells are capable of producing cytokines immediately upon stimulation. Therefore, we stimulated the cells with PMA/ionomycin to subsequently measure the intracellular presence of IL-2, IFN-γ, TNF-α, MIP-1β, and CD107a. Due to stimulation-induced T cell receptor (TCR) internalization, there is an inverse correlation between the degree of stimulation and the ability to detect virus-specific T cells with tetramers. As a consequence, we were able to detect BKV-specific T cells only in the PBMC fractions of subjects 4 and 9 after PMA/ionomycin stimulation. In the other three subjects, the already few tetramer-positive events had become undetectable after stimulation (data not shown). Upon PMA/ionomycin stimulation, BKV VP1-specific CD8+ T cells from both subjects displayed a polyfunctional profile: nearly 11% of BKV VP1 p108-specific CD8+ T cells in subject 4 were able to rapidly express all five proteins (IL-2, IFN-γ, TNF-α, and to a lesser extent, also MIP-1-β and CD107a) upon stimulation. About 23% excreted a combination of four, and 52% excreted a combination of three of these (Fig. 8A and B). In subject 9, about 4% of VP1p44-specific CD8+ T cells excreted all five proteins, 26% excreted a combination of four, and about 56% produced a combination of three of these proteins. In both subjects, the T cells most often produced the combination of IL-2, IFN-γ, and TNF-α (Fig. 8B).
Fig 8.
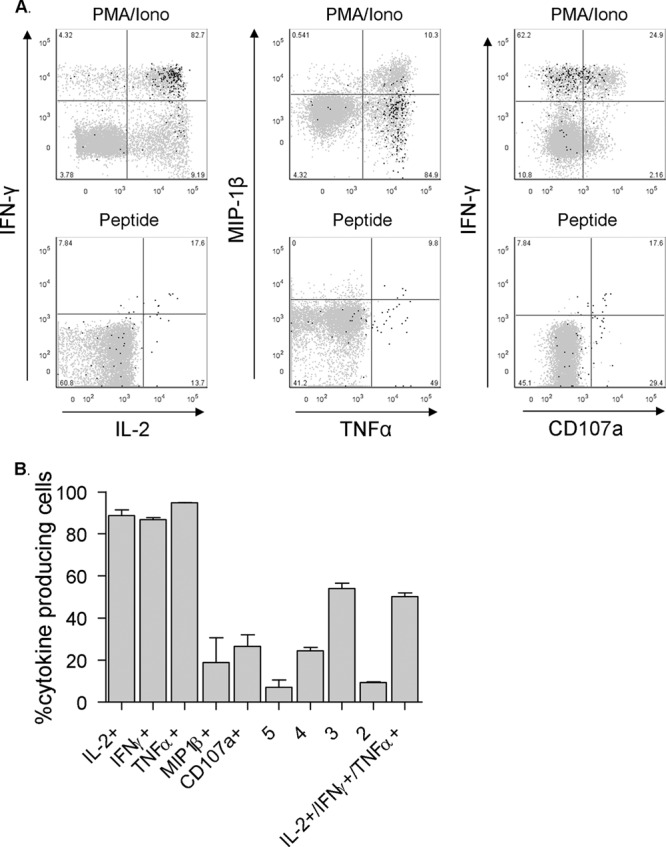
BKV-specific CD8+ T cells are polyfunctional. (A) Dot plots displaying the expression of the intracellular cytokine staining of BKV VP1 p44 tetramer-positive events from subject 9 in black presented as an overlay on total CD8+ T cells in gray, after 4 h of stimulation with PMA/ionomycin or 5 h of stimulation with a cognate peptide. Anti-IFN-γ and MIP-1β fluorescence intensity levels are plotted on the y axis and anti-IL-2, TNF-α, and CD107a fluorescence intensity levels on the x axis. Percentages of tetramer+ T cells are shown. (B) Percentages of BKV VP1-specific CD8+ T cells expressing IL-2, IFN-γ, TNF-α, MIP-1β, and CD107a, as well as quintuple, quadruple, triple, and double expressers, and T cells secreting the combination of IL-2, IFN-γ, and TNF-α after PMA/ionomycin stimulation. (n = 2 subjects; median, IQR).
We then repeated the experiment with cognate peptide stimulation. In order to determine the optimal peptide concentration at which tetramer-positive events were detectable while the cells were also still responding in the sense of cytokine production, we tested peptide concentrations ranging from 1,000 ng/ml to 0.05 ng/ml (data not shown). In subject 4, BKV-specific T cells were detectable and functionally active at 1 ng/ml. The higher concentrations (1,000, 100, and 10 ng/ml) rendered the detection of BKV-specific T cells impossible, again due to TCR internalization and their subsequent inability to bind the tetramer. In subject 9, BKV-specific T cells were detectable and functionally active at 1,000 ng/ml only. In both subjects, the stimulus provided by the lower concentrations proved to be insufficient to trigger cytokine production (data not shown). In contrast to the tetramer-negative CD8+ T cells, a considerable percentage of BKV-specific T cells in both subjects also produced the same proteins in response to their cognate peptide (Fig. 8A). The qualitative and quantitative differences in signaling induced by stimulation with PMA/ionomycin, which circumvents normal outside-in T cell signaling, and the stimulation with this specific concentration of cognate peptide and costimulation, given in this specific time window, offer a likely explanation for the relative differences between PMA/ionomycin- and peptide/costimulation-triggered cytokine release and degranulation.
DISCUSSION
In the current study, we show that BKV-specific CD8+ T cells are present in the circulation of healthy adults in very low frequencies. We show that the BKV VP1-specific CD8+ T cells have a phenotype of resting memory cells, predominantly being CD45RA− CD27+ CD127+ KLRG1+ CCR7− TEM cells that were not activated and did not actively proliferate. Furthermore, these T cells highly express CD49d and CXCR3. Interestingly, they expressed intermediate levels of T-bet but very little or no Eomes. Also, the direct cytotoxic potential of BKV-specific T cells appears to be limited since they are devoid of granzyme B in this resting state. Lastly, stimulation induced rapid excretion of inflammatory cytokines. As such, upon activation these BKV-specific CD8+ T cells are capable of rapidly mobilizing the immunological response by sending out various alarm signals.
The extremely low frequencies of BKV-specific CD8+ T cells in the circulation fit the finding that BKV is normally not detectable in this compartment and is residing at distal sites of viral latency, such as the kidney (32). Indeed, in a minority of healthy persons BKV is episodically shed in the urine (32, 33). Unfortunately, due to their extremely low frequencies in the circulation, we could not perform an ex vivo characterization of BKV LTAg-specific CD8+ T cells. LTAg is a nonstructural protein that is expressed only during the early phase of viral replication and during nonpermissive infection (1). The dominance of VP1-specific T cells over LTAg-specific T cells is in line with LTAg-specific antibody serum titers being lower than VP1-specific antibody titers (34, 35). The discrepancy between VP1 and LTAg expression during the viral replication cycle and, as such, the variations in antigen exposure could explain the differences in population size of VP1-specific and LTAg-specific T cells.
The function of BKV VP1-specific CD8+ T cells in the circulation might be dual: preventing BKV from entering the circulation, which is a common occurrence in severely immunocompromised individuals, and patrolling the circulation for local or distal BKV activity (1). Indeed, BKV-specific cells expressed high levels of CXCR3, known to mediate T cell homing to reactive lymph nodes, stressed epithelium, and sites of inflammation (24–26). Also, a small proportion expressed the central memory marker CCR7, which allows cells to migrate to resting lymph nodes. Furthermore, due to their high expression of CD49d these cells have the capacity to migrate through the blood-brain-barrier, pointing to the central nervous system as a latent niche of BKV infection, as was proposed previously (20). Considering that BKV-specific T cells also recognize epitopes of the JCV, high CD49d expression might be a likely explanation for the association between treatment with the anti-CD49d monoclonal natalizumab and the emergence of progressive multifocal leukoencephalopathy, a severe JCV-mediated neurological disorder (36, 37). However, the similarly high expression of CD49d by all the other circulating virus-specific CD8+ T cells perhaps rather emphasizes the neurotropism of the polyomaviruses, as well as the importance for BKV- or polyomavirus-specific T cells to be present in the central nervous system (23).
It has been demonstrated quite extensively that different pathogens can drive the differentiation of selective CD8+ T cell subsets (5, 6, 38, 39). Indeed, we show here that BKV-specific CD8+ T cells have a phenotype that is distinct from that of CMV- and EBV-specific T cells circulating in these same donors, while sharing some phenotypic aspects with Flu-specific T cells, such as the high expression of CD127 and CXCR3 and the low expression of Eomes. The latter struck us as particularly interesting since both BKV- and Flu-specific CD8+ T cells comprise mainly CD127+ memory cells and Eomes was proposed to be a key inducer of a memory CD8+ T cell phenotype (40). Also, it is remarkable that memory T cells, specific for viruses with two entirely different modes of infection, resemble each other in so many aspects, adding to the model of T cell differentiation as previously proposed by Appay et al. (5). Perhaps such phenotypic similarities could be explained by both T cell populations having abstained from specific extracellular signals for an extended period of time, knowing that BKV is normally not found in the circulation while Flu is thought to be cleared from the body after infection. Previously, we showed that circulating virus-specific T cells differ significantly from their counterparts residing in lymph nodes or the lungs, demonstrating how the tropism of the virus is also an important determinant of the T cell phenotype (41, 42). Local BKV-specific CD8+ T cells could very well display more of an effector phenotype, perhaps readily expressing granzyme B, in order to confer a more specialized defense at sites where the immunogenic pressure is higher. Indeed, if these circulating TEM cells are recruited to a site of BKV reactivation, they may generate or reinforce the local T cell-mediated defense. Therefore, it is crucial to also assess the characteristics of tissue-resident BKV-specific T cells, for example, those residing in the urogenital tract and/or the central nervous system. Lastly, considering the crucial role of CD4+ helper T cells in clearing viral infection, as well as the existence of cytotoxic CD4+ T cells, knowledge about the BKV-specific CD4+ T cell phenotype and function should be a primary research focus in this field (43). Unfortunately, this is currently hindered by specific technical limitations, among which, importantly, is the lack of available major histocompatibility complex (MHC) class II tetramers.
In conclusion, these data shed light on the phenotype and function of circulating BKV-specific CD8+ T cells in healthy adults. This information allows for a future comparison with BKV-specific CD8+ T cells circulating in immunocompromised individuals and their tissue-resident counterparts. These data may prove to be helpful for the future design of new immunotherapeutic options for the treatment of BKV-associated disease.
ACKNOWLEDGMENTS
We thank Marjan J. Tempelmans Plat-Sinnige, Gijs van Schijndel, Ester M. M. van Leeuwen, Si La Yong, Simone H. C. Havenith, Daan J. aan de Kerk, and Nelly van der Bom-Baylon for their technical help and useful discussions.
Footnotes
Published ahead of print 17 July 2013
REFERENCES
- 1. van Aalderen MC, Heutinck KM, Huisman C, Ten Berge IJ. 2012. BK virus infection in transplant recipients: clinical manifestations, treatment options and the immune response. Neth. J. Med. 70:172–183 [PubMed] [Google Scholar]
- 2. Schachtner T, Muller K, Stein M, Diezemann C, Sefrin A, Babel N, Reinke P. 2011. BKV-specific immunity kinetics: a predictor of recovery from polyomavirus BK-associated nephropathy. Am. J. Transplant. 11:2443–2452 [DOI] [PubMed] [Google Scholar]
- 3. Blyth E, Clancy L, Simms R, Gaundar S, O'connell P, Micklethwaite K, Gottlieb DJ. 2011. BK virus-specific T cells for use in cellular therapy show specificity to multiple antigens and polyfunctional cytokine responses. Transplantation 92:1077–1084 [DOI] [PubMed] [Google Scholar]
- 4. Micklethwaite KP, Clancy L, Sandher U, Hansen AM, Blyth E, Antonenas V, Sartor MM, Bradstock KF, Gottlieb DJ. 2008. Prophylactic infusion of cytomegalovirus-specific cytotoxic T lymphocytes stimulated with Ad5f35pp65 gene-modified dendritic cells after allogeneic hemopoietic stem cell transplantation. Blood 112:3974–3981 [DOI] [PubMed] [Google Scholar]
- 5. Appay V, Dunbar PR, Callan M, Klenerman P, Gillespie GM, Papagno L, Ogg GS, King A, Lechner F, Spina CA, Little S, Havlir DV, Richman DD, Gruener N, Pape G, Waters A, Easterbrook P, Salio M, Cerundolo V, McMichael AJ, Rowland-Jones SL. 2002. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 8:379–385 [DOI] [PubMed] [Google Scholar]
- 6. van Lier RA, Ten Berge IJ, Gamadia LE. 2003. Human CD8(+) T-cell differentiation in response to viruses. Nat. Rev. Immunol. 3:931–939 [DOI] [PubMed] [Google Scholar]
- 7. Sharma MC, Zhou W, Martinez J, Krymskaya L, Srivastava T, Haq W, Diamond DJ, Lacey SF. 2006. Cross-reactive CTL recognizing two HLA-A*02-restricted epitopes within the BK virus and JC virus VP1 polypeptides are frequent in immunocompetent individuals. Virology 350:128–136 [DOI] [PubMed] [Google Scholar]
- 8. Li J, Melenhorst J, Hensel N, Rezvani K, Sconocchia G, Kilical Y, Hou J, Curfman B, Major E, Barrett AJ. 2006. T-cell responses to peptide fragments of the BK virus T antigen: implications for cross-reactivity of immune response to JC virus. J. Gen. Virol. 87:2951–2960 [DOI] [PubMed] [Google Scholar]
- 9. Tagaram HR, Watson AM, Lemonnier FA, Staveley-O'Carroll K, Tevethia SS, Schell TD. 2008. An SV40 VP1-derived epitope recognized by CD8+ T cells is naturally processed and presented by HLA-A*0201 and cross-reactive with human polyomavirus determinants. Virology 376:183–190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Krymskaya L, Sharma MC, Martinez J, Haq W, Huang EC, Limaye AP, Diamond DJ, Lacey SF. 2005. Cross-reactivity of T lymphocytes recognizing a human cytotoxic T-lymphocyte epitope within BK and JC virus VP1 polypeptides. J. Virol. 79:11170–11178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen Y, Trofe J, Gordon J, Du Pasquier RA, Roy-Chaudhury P, Kuroda MJ, Woodle ES, Khalili K, Koralnik IJ. 2006. Interplay of cellular and humoral immune responses against BK virus in kidney transplant recipients with polyomavirus nephropathy. J. Virol. 80:3495–3505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chen Y, Trofe J, Gordon J, Autissier P, Woodle ES, Koralnik IJ. 2008. BKV and JCV large T antigen-specific CD8+ T cell response in HLA A*0201+ kidney transplant recipients with polyomavirus nephropathy and patients with progressive multifocal leukoencephalopathy. J. Clin. Virol. 42:198–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Provenzano M, Bracci L, Wyler S, Hudolin T, Sais G, Gosert R, Zajac P, Palu' G, Heberer M, Hirsch HH, Spagnoli GC. 2006. Characterization of highly frequent epitope-specific CD45RA+/CCR7+/− T lymphocyte responses against p53-binding domains of the human polyomavirus BK large tumor antigen in HLA-A*0201+ BKV-seropositive donors. J. Transl. Med. 4:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Voehringer D, Koschella M, Pircher H. 2002. Lack of proliferative capacity of human effector and memory T cells expressing killer cell lectinlike receptor G1 (KLRG1). Blood 100:3698–3702 [DOI] [PubMed] [Google Scholar]
- 15. Lamoreaux L, Roederer M, Koup R. 2006. Intracellular cytokine optimization and standard operating procedure. Nat. Protoc. 1:1507–1516 [DOI] [PubMed] [Google Scholar]
- 16. Hamann D, Baars PA, Rep MH, Hooibrink B, Kerkhof-Garde SR, Klein MR, van Lier RA. 1997. Phenotypic and functional separation of memory and effector human CD8+ T cells. J. Exp. Med. 186:1407–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sallusto F, Lenig D, Forster R, Lipp M, Lanzavecchia A. 1999. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401:708–712 [DOI] [PubMed] [Google Scholar]
- 18. Kaech SM, Tan JT, Wherry EJ, Konieczny BT, Surh CD, Ahmed R. 2003. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 4:1191–1198 [DOI] [PubMed] [Google Scholar]
- 19. Vanchiere JA, Abudayyeh S, Copeland CM, Lu LB, Graham DY, Butel JS. 2009. Polyomavirus shedding in the stool of healthy adults. J. Clin. Microbiol. 47:2388–2391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Behzad-Behbahani A, Klapper PE, Vallely PJ, Cleator GM, Bonington A. 2003. BKV-DNA and JCV-DNA in CSF of patients with suspected meningitis or encephalitis. Infection 31:374–378 [DOI] [PubMed] [Google Scholar]
- 21. Robaina TF, Mendes GS, Benati FJ, Pena GA, Silva RC, Montes MA, Janini ME, Camara FP, Santos N. 2013. Shedding of polyomavirus in the saliva of immunocompetent individuals. J. Med. Virol. 85:144–148 [DOI] [PubMed] [Google Scholar]
- 22. Uss E, Rowshani AT, Hooibrink B, Lardy NM, van Lier RA, Ten Berge IJ. 2006. CD103 is a marker for alloantigen-induced regulatory CD8+ T cells. J. Immunol. 177:2775–2783 [DOI] [PubMed] [Google Scholar]
- 23. Ifergan I, Kebir H, Alvarez JI, Marceau G, Bernard M, Bourbonniere L, Poirier J, Duquette P, Talbot PJ, Arbour N, Prat A. 2011. Central nervous system recruitment of effector memory CD8+ T lymphocytes during neuroinflammation is dependent on alpha4 integrin. Brain 134:3560–3577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kurachi M, Kurachi J, Suenaga F, Tsukui T, Abe J, Ueha S, Tomura M, Sugihara K, Takamura S, Kakimi K, Matsushima K. 2011. Chemokine receptor CXCR3 facilitates CD8(22) T cell differentiation into short-lived effector cells leading to memory degeneration. J. Exp. Med. 208:1605–1620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kunkel EJ, Boisvert J, Murphy K, Vierra MA, Genovese MC, Wardlaw AJ, Greenberg HB, Hodge MR, Wu L, Butcher EC, Campbell JJ. 2002. Expression of the chemokine receptors CCR4, CCR5, and CXCR3 by human tissue-infiltrating lymphocytes. Am. J. Pathol. 160:347–355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Guarda G, Hons M, Soriano SF, Huang AY, Polley R, Martin-Fontecha A, Stein JV, Germain RN, Lanzavecchia A, Sallusto F. 2007. L-selectin-negative. Nat. Immunol. 8:743–752 [DOI] [PubMed] [Google Scholar]
- 27. Szabo SJ, Sullivan BM, Stemmann C, Satoskar AR, Sleckman BP, Glimcher LH. 2002. Distinct effects of T-bet in TH1 lineage commitment and IFN-gamma production in CD4 and CD8 T cells. Science 295:338–342 [DOI] [PubMed] [Google Scholar]
- 28. Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, Banica M, DiCioccio CB, Gross DA, Mao CA, Shen H, Cereb N, Yang SY, Lindsten T, Rossant J, Hunter CA, Reiner SL. 2003. Control of effector CD8+ T cell function by the transcription factor Eomesodermin. Science 302:1041–1043 [DOI] [PubMed] [Google Scholar]
- 29. Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, Mullen AC, Gasink CR, Kaech SM, Miller JD, Gapin L, Ryan K, Russ AP, Lindsten T, Orange JS, Goldrath AW, Ahmed R, Reiner SL. 2005. Effector and memory CD8+ T cell fate coupled by T-bet and eomesodermin. Nat. Immunol. 6:1236–1244 [DOI] [PubMed] [Google Scholar]
- 30. Joshi NS, Cui W, Chandele A, Lee HK, Urso DR, Hagman J, Gapin L, Kaech SM. 2007. Inflammation directs memory precursor and short-lived effector CD8(+) T cell fates via the graded expression of T-bet transcription factor. Immunity 27:281–295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Takemoto N, Intlekofer AM, Northrup JT, Wherry EJ, Reiner SL. 2006. Cutting edge: IL-12 inversely regulates T-bet and eomesodermin expression during pathogen-induced CD8+ T cell differentiation. J. Immunol. 177:7515–7519 [DOI] [PubMed] [Google Scholar]
- 32. Egli A, Infanti L, Dumoulin A, Buser A, Samaridis J, Stebler C, Gosert R, Hirsch HH. 2009. Prevalence of polyomavirus BK and JC infection and replication in 400 healthy blood donors. J. Infect. Dis. 199:837–846 [DOI] [PubMed] [Google Scholar]
- 33. Kling CL, Wright AT, Katz SE, McClure GB, Gardner JS, Williams JT, Meinerz NM, Garcea RL, Vanchiere JA. 2012. Dynamics of urinary polyomavirus shedding in healthy adult women. J. Med. Virol. 84:1459–1463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Corallini A, Barbanti-Brodano G, Portolani M, Balboni PG, Grossi MP, Possati L, Honorati C, La PM, Mazzoni A, Caputo A, Veronesi U, Orefice S, Cardinali G. 1976. Antibodies to BK virus structural and tumor antigens in human sera from normal persons and from patients with various diseases, including neoplasia. Infect. Immun. 13:1684–1691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bodaghi S, Comoli P, Bosch R, Azzi A, Gosert R, Leuenberger D, Ginevri F, Hirsch HH. 2009. Antibody responses to recombinant polyomavirus BK large T and VP1 proteins in young kidney transplant patients. J. Clin. Microbiol. 47:2577–2585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Langer-Gould A, Atlas SW, Green AJ, Bollen AW, Pelletier D. 2005. Progressive multifocal leukoencephalopathy in a patient treated with natalizumab. N. Engl. J. Med. 353:375–381 [DOI] [PubMed] [Google Scholar]
- 37. Van Assche G, Van RM, Sciot R, Dubois B, Vermeire S, Noman M, Verbeeck J, Geboes K, Robberecht W, Rutgeerts P. 2005. Progressive multifocal leukoencephalopathy after natalizumab therapy for Crohn's disease. N. Engl. J. Med. 353:362–368 [DOI] [PubMed] [Google Scholar]
- 38. Naito T, Tanaka H, Naoe Y, Taniuchi I. 2011. Transcriptional control of T-cell development. Int. Immunol. 23:661–668 [DOI] [PubMed] [Google Scholar]
- 39. Kaech SM, Cui W. 2012. Transcriptional control of effector and memory CD8(+) T cell differentiation. Nat. Rev. Immunol. 12:749–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Banerjee A, Gordon SM, Intlekofer AM, Paley MA, Mooney EC, Lindsten T, Wherry EJ, Reiner SL. 2010. Cutting edge: the transcription factor eomesodermin enables CD8+ T cells to compete for the memory cell niche. J. Immunol. 185:4988–4992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Remmerswaal EB, Havenith SH, Idu MM, van Leeuwen EM, van Donselaar KA, van der Bom-Baylon N, Bemelman FJ, van Lier RA, Ten Berge IJ. 2012. Human virus-specific effector-type T cells accumulate in blood but not in lymph nodes. Blood 119:1702–1712 [DOI] [PubMed] [Google Scholar]
- 42. Piet B, de Bree GJ, Smids-Dierdorp BS, van der Loos CM, Remmerswaal EB, von der Thusen JH, van Haarst JM, Eerenberg JP, BAten van der Bij W, Timens W, van Lier RA, Jonkers RE. 2011. CD8(+) T cells with an intraepithelial phenotype upregulate cytotoxic function upon influenza infection in human lung. J. Clin. Invest. 121:2254–2263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. van de Berg PJ, van Leeuwen EM, Ten Berge IJ, van Lier R. 2008. Cytotoxic human CD4(+) T cells. Curr. Opin. Immunol. 20:339–343 [DOI] [PubMed] [Google Scholar]