

Abstract
Reactivation of Epstein-Barr virus (EBV) from latency is dependent on expression of the viral transactivator BZLF1 protein, whose promoter (Zp) normally exhibits only low basal activity but is activated in response to chemical or biological inducers. Using a reporter assay system, we screened for factors that can activate Zp and isolated genes, including those encoding MEF2B, KLF4, and some cellular b-Zip family transcription factors. After confirming their importance and functional binding sites in reporter assays, we prepared recombinant EBV-BAC, in which the binding sites were mutated. Interestingly, the MEF2 mutant virus produced very low levels of BRLF1, another transactivator of EBV, in addition to BZLF1 in HEK293 cells. The virus failed to induce a subset of early genes, such as that encoding BALF5, upon lytic induction, and accordingly, could not replicate to produce progeny viruses in HEK293 cells, but this restriction could be completely lifted by exogenous supply of BRLF1, together with BZLF1. In B cells, induction of BZLF1 by chemical inducers was inhibited by point mutations in the ZII or the three SP1/KLF binding sites of EBV-BAC Zp, while leaky BZLF1 expression was less affected. Mutation of MEF2 sites severely impaired both spontaneous and induced expression of not only BZLF1, but also BRLF1 in comparison to wild-type or revertant virus cases. We also observed that MEF2 mutant EBV featured relatively high repressive histone methylation, such as H3K27me3, but CpG DNA methylation levels were comparable around Zp and the BRLF1 promoter (Rp). These findings shed light on BZLF1 expression and EBV reactivation from latency.
INTRODUCTION
Epstein-Barr virus (EBV) is a human gammaherpesvirus that predominantly establishes latent infection in B lymphocytes. Only a small percentage of infected cells switch from the latent stage into the lytic cycle and produce progeny viruses (1). Although the mechanism of EBV reactivation in vivo is not fully understood, it is known to be elicited in vitro by treatment of latently infected B cells with some chemical or biological reagents, such as 12-O-tetradecanoylphorbol-13-acetate (TPA), calcium ionophores, sodium butyrate, immunoglobulin (Ig), or transforming growth factor beta. Stimulation of the EBV lytic cascade with such reagents leads to the expression of two viral transcriptional regulatory genes, encoding BZLF1 (also known as Zta, EB1, ZEBRA, or Z) and BRLF1 (Rta or R) (2, 3).
The BZLF1 protein is a transcriptional activator that shares structural similarities with the basic leucine zipper (b-Zip) family of transcriptional factors, and BZLF1 expression alone can trigger the entire reactivation cascade (1–3). It forms homodimers directly binding to BZLF1-responsive elements in the lytic gene promoters and thereby induces lytic gene expression (4, 5). Expression of the BZLF1 gene is tightly controlled at the transcriptional level. The BZLF1 promoter (Zp) normally exhibits low basal activity and is activated in response to TPA or the other reagents described above by transcriptional factors, including myocyte enhancer factor 2D (MEF2D) (6) and SP1/3 (7). Cellular b-Zip-type transcription factors, such as the cyclic AMP response element-binding protein (CREB), activating transcription factor (ATF), activator protein 1 (AP-1) (8–10), or a spliced form of the X-box binding protein 1 [XBP-1(s)] (11–13), also play crucial roles. We previously showed the importance of CREB and its calcineurin-dependent activation by transducer of regulated CREB 2 (TORC2) (9). Once produced, BZLF1 itself can bind to and activate its own promoter (14, 15). Most of the positive factors have been demonstrated or are presumed to upregulate the BZLF1 promoter by recruiting transcriptional coactivators, such as histone acetylases. In fact, BZLF1 promoter activation is associated with epigenetic modifications, including high histone acetylation and H3K4me3 levels (16). On the other hand, the activity of Zp is restricted by repressive factors, including Jun dimerization protein 2 (JDP2) (8), zinc finger E-box binding factor (ZEB) (17), yin yang 1 (YY1) (18), and sumoylation of BZLF1 (15, 19), since these factors facilitate the access of repressive transcriptional cofactors, such as histone deacetylase (HDAC), to the promoter and/or block the binding or functions of the transcriptional activators noted above.
In addition to BZLF1, BRLF1 has also been implicated in transcriptional activation of viral lytic genes by binding directly to BRLF1-responsive elements (20) and/or by indirectly binding to DNA through other transcription factors (21), or even by simply enhancing certain signal transduction cascades, such as that associated with MAPK signaling (22). The BRLF1 gene is situated adjacent to the BZLF1 gene in the EBV genome. The BZLF1 gene can be transcribed from its own promoter, Zp, producing short monocistronic mRNA, or from the upstream BRLF1 gene promoter (Rp), creating long, bicistronic mRNA. However, translation of the second cistron, the BZLF1 gene of the bicistronic mRNA, is not very efficient (23), and thus, the BZLF1 promoter rather than the BRLF1 promoter is believed to be the main regulator of BZLF1 expression in EBV reactivation from latency.
In the present study, we carried out a screen for genes that enhance transcription from Zp. MEF2, SP1/KLF family, and b-Zip-type transcription factors were found to activate the promoter efficiently, as well as several other factors. cis-regulatory elements in Zp were also defined and mutated in the context of the virus genome. Thus, we could demonstrate that MEF2 binding to Zp enhances the expression of not only BZLF1, but also another regulatory gene, encoding BRLF1. These results provide a clear overview of how the BZLF1 gene is induced in EBV reactivation from latency.
MATERIALS AND METHODS
Cell culture and reagents.
HeLa, HEK293T, and HEK293 EBV-BAC cells were maintained in Dulbecco's modified Eagle medium (Sigma) and Raji, B95-8, and Akata(−) cells and lymphoblastoid cell lines (LCLs) in RPMI 1640 medium, both supplemented with 10% fetal bovine serum. Anti-tubulin (number 2148) and anti-MEF2C (number 5030) antibodies were from Cell Signaling, anti-histone H3 (ab1791) antibody was from Abcam, and anti-H3K4me3 (no. 17-614) antibody was from Millipore. Anti-acetylated H3K9 (H3K9Ac) (39137), anti-H3K9me2 (39375), and anti-H3K9me3 (39161) antibodies were purchased from Active Motif. Anti-MEF2A (TA303858) antibody was obtained from OriGene Technologies, and anti-MEF2B (sc-98594) and anti-MEF2D (sc-271153) antibodies were from Santa Cruz Biotechnology. Rabbit anti-BZLF1, anti-BRLF1, anti-BMRF1, anti-BALF2, and anti-BALF5 antibodies were generated as reported previously (24). Horseradish peroxidase (HRP)-linked goat antibodies to mouse/rabbit IgG were from Amersham Biosciences.
Plasmid construction.
The expression vector for BZLF1 was constructed previously (9). For library screening, a SuperScript Bone Marrow (mixed-population) Premade cDNA Library was purchased from Invitrogen. The library is supplied as Escherichia coli glycerol stock, and each bacterium contains one plasmid of unknown cDNA from bone marrow cells. The bacteria were adequately diluted and colonized on LB plates with ampicillin. Independent colonies were randomly picked up and cultured overnight in LB solution with the antibiotic. Full-grown culture medium was collected from 10 tubes and mixed to prepare one pool, which thus contained 10 different species of cDNA, followed by plasmid purification using a GeneElute Plasmid Miniprep Kit (Sigma). pCMV-RL and pGL4.70(hRluc) were from Promega. pCMV-SP1 was supplied by G. Suske (25) and pcDNA3-Flag-CREB by K. Shimotohno (26). To make pcDNAFlagATF-1, the coding region for human ATF-1 was amplified by reverse transcription (RT)-PCR and cloned into the EcoRI and NotI sites of the pcDNAFlag vector (24). The primers used for ATF-1 amplification were 5′-AGAGAATTCATGGAAGATTCCCACAAGAG-3′ and 5′-ATGAGCGGCCGCTCAAACACTTTTATTGGAATAAAG-3′. pcDNAFlagXBP1s was made previously (8) by cloning the PCR fragment amplified from pCMV-Myc-XBP1s, obtained from K. Mori. As for derivatives of pZp-luc, point mutations were introduced as shown in Fig. 1 by the inverse-PCR method using appropriate primers (9).
Fig 1.

Alignment of wild-type and mutated sequences of the EBV minimal BZLF1 promoter. cis-regulatory element sequences are shown in capitals. Mutated nucleotides are boxed and shown in capitals.
Transfection, luciferase assay, and immunoblotting.
Plasmid DNA was transfected into HEK293T cells using Lipofectamine 2000 reagent (Invitrogen) or Microporator (Digital Bio). The total amounts of plasmid DNAs were standardized by the addition of an empty vector. Proteins were extracted from cells with the lysis buffer supplied in a Dual-Luciferase Reporter Assay System (Promega) kit, and luciferase activity was measured using the kit. Counts for firefly luciferase were normalized to those for Renilla luciferase. For immunoblotting (IB), protein samples were subjected to SDS-PAGE, followed by IB with the indicated antibodies as described previously (24).
Genetic manipulation of EBV-BAC DNA, cloning of HEK293 cells, and preparation of LCLs with EBV-BAC.
EBV-BAC DNA was provided by W. Hammerschmidt (27). Homologous recombination was carried out in E. coli as described previously (24).
To prepare mutants of EBV-BAC, at the BZLF1 promoter, a transfer DNA fragment for the first recombination was generated by PCR using the rpsL-neo plasmid (Gene Bridges) as the template, with Neo/StFor (5′-TGAAATCTTGGATACATTTCTAAATGATGAATGTCTGCTGCATGCCATGCATATTTCAACTGGCCTGGTGATGATGGCGGGATC-3′) and NeoStRev (5′-GAGTTACCTGTCTAACATCTCCCCTTTAAAGCCAAGGCACCAGCCTCCTCTGTGATGTCATCAGAAGAACTCGTCAAGAAGG-3′) primers. After recombination, kanamycin-resistant colonies were selected and checked by colony PCR using the primers 5′-AATGTCTGCTGCATGCCATG-3′ and 5′-GTTTGGGTCCATCATCTTCAG-3′ to make intermediate DNA. The Neo/St cassette in the intermediate DNA was then replaced using the next transfer vector DNA, containing each mutation in the BZLF1 promoter. These transfer vector DNAs were prepared by PCR using derivatives of pZp-luc, point mutated as in Fig. 1, and the following primers: for SP1 site mutation, 5′-ATTTGAATCTGGACTCCCCCCTGACCCCCGAACTTAATGAAATCTTGGATACATTTCTAAATGATGAATGTCTGCATGCCATGCATATTTCAACTAA-3′ and 5′-AATGTTTAGTGAGTTACCTGTCTAACATCT-3′; for MEF2 site mutation, 5′-ATTTGAATCTGGACTCCCCCCTGACCCCCGAACTTAATGAAATCTTGGATACATTTCTAAATGATGAATGTCTGCATGCCATGCATATTTCAACTGG-3′ and 5′-AATGTTTAGTGAGTTACCTGTCTAACATCT-3′. Electroporation of E. coli was performed using a Gene Pulser III (Bio-Rad), and purification of EBV-BAC DNA was achieved with NucleoBond Bac100 (Macherey-Nagel). Recombination was confirmed with PCR products of the promoter region, by electrophoresis of the BamHI- or EcoRI-digested viral genome, and by sequencing analysis.
EBV-BAC DNA was transfected into HEK293 cells, in order to make HEK293 EBV-BAC, using Lipofectamine 2000 reagent (Invitrogen), followed by culture on 10-cm dishes with 100 to 150 μg/ml of hygromycin B for 10 to 15 days for cloning of green fluorescent protein (GFP)-positive cell colonies. Briefly, for each recombinant virus, we picked up more than 10 hygromycin-resistant, GFP-positive cell colonies to obtain at least 3 typical clones exhibiting minimal spontaneous expression of viral lytic proteins and significant induction of these upon BZLF1 transfection.
For LCLs, wild-type (wt) or mutant EBVs were collected from HEK293 EBV-BAC cell supernatants. Virus titers in the media were determined by infecting Akata(−) cells: viruses in 1 ml of sample medium were adsorbed onto 1 × 106 Akata(−) cells for 2 h with rotation. After centrifugation, the media were exchanged with fresh media, followed by incubation for 2 days. Enhanced-GFP (EGFP)-positive cells were counted by flow cytometry (FACSCalibur; Becton, Dickinson). Titers were normalized according to the percentages by adding control media. Peripheral blood monocytes (PBMCs) were infected with 10-fold dilutions of adjusted culture supernatant medium obtained from wild-type or mutant HEK293 EBV-BAC cells and seeded onto 96-well plates at 1 × 104 cells per well. For PBMCs, blood samples were obtained from healthy adult donors, who gave written informed consent according to protocols approved by the institutional review board of Aichi Cancer Center, Nagoya, Japan. The cells were cultured in the presence of cyclosporine. Half of the medium was exchanged once a week with fresh medium containing cyclosporine. After 5 weeks, 50% transforming doses were calculated.
ChIP assays.
Chromatin immunoprecipitation (ChIP) assays (Upstate Biotechnology, Inc.) were performed essentially as described previously with formaldehyde cross-linked chromatin from 1 × 106 cells for each reaction. The cells were lysed, and chromatin was sonicated to obtain DNA fragments with an average length of 300 bp. Following centrifugation, the chromatin was diluted 10-fold with ChIP dilution buffer and precleared with protein A agarose beads containing salmon sperm DNA (Upstate). Samples were then mixed with antibodies and incubated for 5 h with rotation. Immune complexes were collected by addition of protein A agarose beads, and the DNA was purified using a QIAquick PCR Purification Kit (Qiagen) after uncoupling of the cross-links and proteinase K digestion. The recovered DNA was quantified by real-time PCR using reagents as previously noted (16). Most of the primers were as in the previous report (16), except those for the region about 600 bp upstream of the transcription start site of Rp (Rp-600) (5′-ACAGCATCGGAGTCATCGAG-3′ and 5′-TCCTGAACATGTTGGTCCAC-3′), region around transcription start site of Rp (Rp0) (5′-TCTCTGCTGCCCACTCATAC-3′ and 5′-TTATGAGCCATTGGCATGGG-3′), and terminal-repeat (TR) (5′-GGCAATGGAGCGTGACGAAG-3′ and 5′-GTCAGGGTTGCCTGTGTCAC-3′) sequence.
Real-time RT-PCR.
Total cell RNA was purified using TriPure Isolation Reagent (Roche) and subjected to real-time RT-PCR using a One Step SYBR PrimeScript RT-PCR Kit II (TaKaRa) with the Real Time PCR System 7300 according to the manufacturer's instructions. PCR was performed as described previously (8). The primers used for the RT-PCR were as follows: for BZLF1 mRNA, 5′-AACAGCCAGAATCGCTGGAG-3′ and 5′-GGCACATCTGCTTCAACAGG-3′; for BRLF1 mRNA, 5′-ACAAACAGACGCAGCCATG-3′ and 5′-TCTCAGAGTAATCTCCACAC-3′; for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA, 5′-TGCACCACCAACTGCTAGC-3′ and 5′-GGCATGGACTGTGGTCATGAG-3′.
Bisulfite treatment and methylation analysis.
For bisulfite modification, total DNA was extracted from LCLs using a DNeasy Blood and Tissue Kit (Qiagen). The purified DNA was then subjected to bisulfite modification using an EpiSight Bisulfite Conversion Kit (Wako), followed by PCR with EpiSight Bisul Taq DNA polymerase (Wako). Primer sequences for the PCR were as follows: for the BZLF1 promoter, 5′-GTATGAGTTATAGGTATTGTTAATG-3′ and 5′-AAAAAACACCTAATATAAATCAAA-3′; for the BRLF1 promoter, 5′-GTTAGATGTTTAGGAATTAAAATAA-3′ and 5′-TCCTATATAATATTCTACTTTAAAAAAAC-3. The DNA fragment was cloned into T-Vector pMD20 (TaKaRa) and sequenced using the primer 5′-CGCCAAGCTATTTAGGTGAC-3′.
RESULTS
Screening for factors that can activate transcription from EBV Zp.
In spite of a number of reports published on EBV BZLF1 transcription in vitro or in cultured cells, there still might be unknown cellular factors that induce BZLF1 expression and EBV reactivation. In order to exhaustively search for cellular factors that might enhance BZLF1 transcription, we screened a human bone marrow cDNA expression library for the ability to enhance Zp activity, using a reporter assay system, since we had successfully screened for transcriptional activators of LMP1 previously (28). First, we prepared a reporter construct, pZp-luc, containing Zp to drive the firefly luciferase gene (9). As a control, pCMV-RL, in which the cytomegalovirus (CMV) immediate-early (IE) promoter was placed upstream of the Renilla luciferase gene, was used to normalize for transfection efficiency. To maximize the number of cDNAs that could be assayed while ensuring that no positive clone would be missed, we generated cDNA pools with 10 cDNAs per pool. Each DNA pool was transfected into HEK293T cells with pZp-luc and pCMV-RL. A pool was considered positive when the pZp-luc reporter was activated 2-fold or more compared with the control pCMV-RL. Then, the positive pool was recloned and assayed again to single out the positive clone, followed by sequencing (see reference 28 for more information). So far, we have screened 2,000 pools, or 20,000 genes, and identified at least 9 genes, as shown in Fig. 2A.
Fig 2.
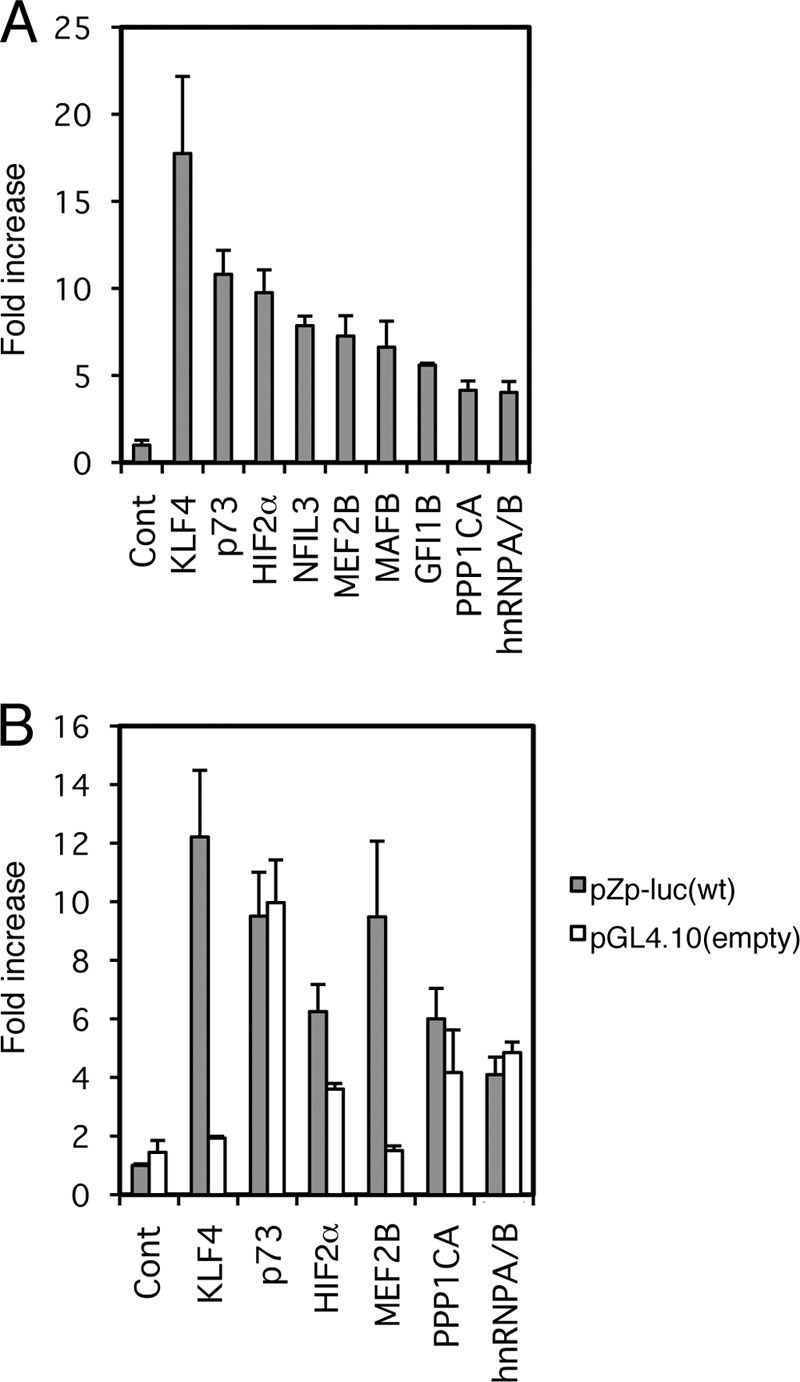
Library screening identified factors involved in BZLF1 promoter activation. (A) HEK293T cells were transfected with 10 ng of the reporter plasmid pZp-luc, 1 ng of pCMV-RL, and 100 ng of expression plasmids for the indicated genes. (B) As in panel A, HEK293T cells were transfected with 10 ng of the reporter plasmid pZp-luc (wt) or its promoterless parental vector, pGL4.10 (empty); 1 ng of pCMV-RL; and 100 ng of expression plasmids for the indicated genes. Luciferase assays were carried out as described in Materials and Methods. Luciferase activity is shown as fold activation over that with the control vector. Each bar represents the mean and standard deviation (SD) of three independent transfections.
Some of these 9 candidate genes turned out to be false positives. The protein phosphatase 1 catalytic subunit alpha (PPP1CA) and heterogeneous nuclear ribonucleoprotein A/B (hnRNPA/B) did not actually enhance firefly luciferase activity from pZp-luc, instead simply suppressing that from the control pCMV-RL (not shown). As we normalized firefly luciferase activity from pZp-luc using the Renilla luciferase activity from pCMV-RL, the two candidates acted as if they increased firefly luciferase expression from the Zp in the initial screening. Unlike PPP1CA and hnRNPA/B, the p73 gene, a gene related to the p53 tumor suppressor gene, and nuclear factor interleukin 3 regulated (NFIL3) clearly enhanced firefly luciferase production from pZp-luc and did not decrease Renilla luciferase expression from pCMV-RL. However, because the factors enhanced firefly luciferase expression even from pGL4.10, which is the promoterless vector of pZp-luc, as efficiently as from pZp-luc, it is highly likely that Zp activation by p73 or NFIL3 was mediated through one or more sequence motifs in the vector outside Zp (Fig. 2B and 3A, compare wt and empty).
Fig 3.
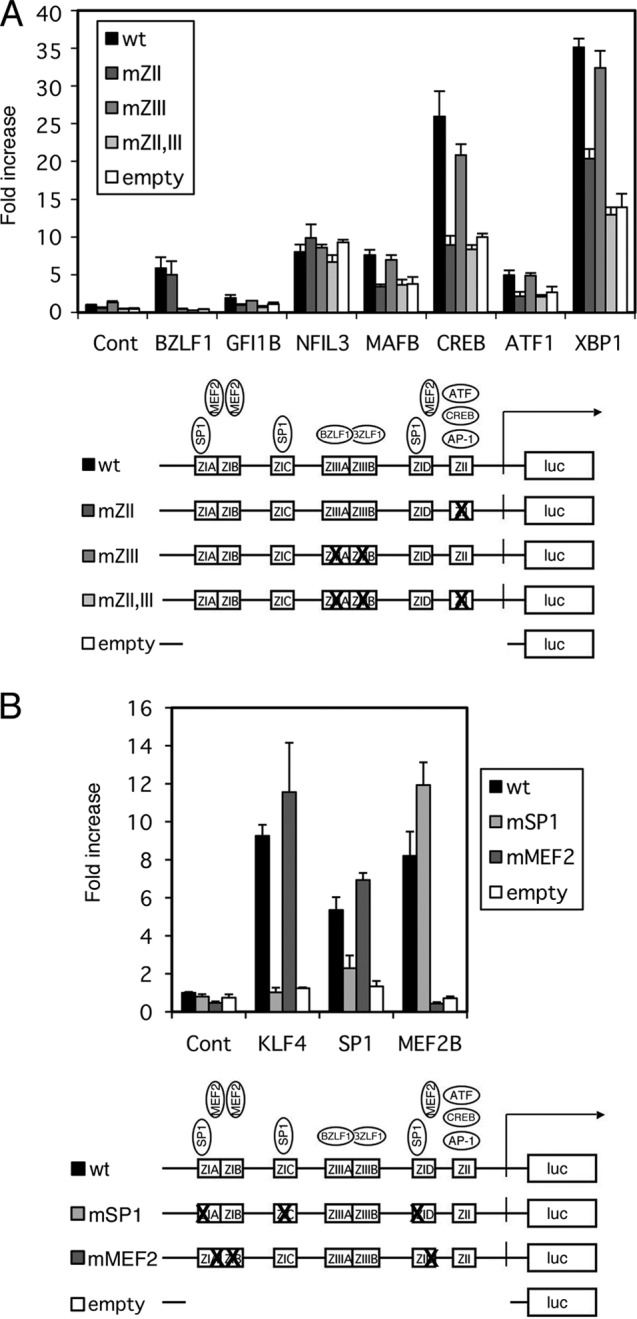
Effects of b-Zip, SP1/KLF, and MEF2 family transcription factors on the BZLF1 promoter. (A) b-Zip family transcription factor activation of the BZLF1 promoter. HEK293T cells were transfected with 10 ng of the reporter plasmid pZp-luc or its derivatives, 1 ng of pCMV-RL, and 100 ng of expression plasmids for the indicated genes. Luciferase assays were carried out as described in Materials and Methods. Luciferase activity is shown as fold activation over that with the control vector (Cont). Each bar represents the mean and SD of three independent transfections. Derivatives of pZp-luc (wt), pZpmZII-luc (mZII), or pZpmZIII-luc (mZIII) contained point mutations in the binding motifs for cellular b-Zip transcription factors or for the viral b-Zip transcription factor BZLF1. The pZpmZII+ZIII-luc vector (mZII,III) contained both mutations. (B) KLF4, SP1, and MEF2B activation of the BZLF1 promoter. HEK293T cells were transfected with 10 ng of the reporter plasmid pZp-luc (wt), 100 ng of pGL4.70(hRluc), and 100 ng of expression plasmid for KLF4, SP1, or MEF2B. pZp-luc reporters with point mutations in SP1-binding sites (mSP1) and MEF2 (mMEF2) binding sites were also used. Luciferase assays were carried out as described in Materials and Methods. Luciferase activity is shown as fold activation over that with control vector and pZp-luc (wt). Each bar represents the mean and SD of three independent transfections. Diagrams below the graphs show constructs of the reporter plasmid pZp-luc and its derivatives.
Enhancement of EBV Zp by cellular b-Zip transcriptional factors.
We realized that some of the clones in Fig. 2A, such as growth factor independent 1B (GFI1B), NFIL3, and MAFB, encode b-Zip-type transcription factors. Previous papers also demonstrated that some b-Zip factors, including CREB/ATF/AP1 (8–10) and XBP-1s (11–13), are involved in BZLF1 promoter activation through the ZII cis-element in the promoter during EBV reactivation from latency (9, 10). Therefore, we examined if these b-Zip transcription factors could stimulate the BZLF1 promoter in our reporter assays using the pZp-luc reporter and its derivatives (Fig. 3A). Here, we used pZp-luc (Fig. 3A, wt), which contains wild-type Zp in pGL4.10; pZpmZII-luc, in which the cellular b-Zip factor binding motif ZII is mutated; pZpmZIII-luc (mZIII), bearing a point mutation in the binding site of the viral b-Zip factor BZLF1; pZpmZII+ZIII-luc (mZII,III), in which both ZII and ZIII are mutated; and the promoterless empty vector pGL4.10 (empty). The sequences of the site-directed mutations of the promoter are aligned in Fig. 1. All mutations were introduced in accordance with previous reports (9, 29, 30). As expected from our previous findings (9), BZLF1 induced its own promoter activity by 5.8-fold (wt) and 5.0-fold (mZII), but the enhancement was diminished by mutations in the ZIII motif (mZIII and mZII,ZIII) or when the promoterless empty vector was used (Fig. 3A, BZLF1). Although this system proved applicable to the viral b-Zip factor BZLF1, that was not the case when it was applied to cellular b-Zip factors, because their expression caused upregulation of luciferase activity even when the promoterless empty vector pGL4.10 was used (Fig. 3A). For example, one of the typical b-Zip factors, CREB, enhanced luciferase gene expression from the promoterless empty vector 10-fold (Fig. 3A, CREB, white bar). We speculate that this unexpected artificial enhancement was mediated through a sequence motif(s) somewhere in the empty vector. At the same time, the wild-type BZLF1 promoter was activated by CREB 26-fold (Fig. 3A, CREB, black bar), and with the point mutation in the ZII cis-element, where cellular b-Zip factors bind, it was induced 8.9-fold (Fig. 3A, CREB, darkest gray bar), almost comparable to the enhancement for the promoterless empty vector. Introduction of mutations at ZIII had little effect on the activation by CREB (Fig. 3A, CREB, intermediate gray bar). Similar patterns were observed for most of the b-Zip factors, GFI1B, MAFB, ATF1, and XBP1s (Fig. 3A), suggesting that although the background level was high in the reporter assays, cellular b-Zip factors are involved in Zp activation, and the activation is mediated through the ZII cis-element. As mentioned above, we assume NFIL3 was an artificial hit, because the response with the promoterless empty vector (Fig. 3A, NFIL3, white bar) reached the same level as with wild-type pZp-luc (Fig. 3A, NFIL3, black bar).
In addition to the reporter assays, we already had confirmed the importance of cellular b-Zip transcription factors for BZLF1 expression in the context of the viral genome in HEK293 cells. Introduction of point mutations into the ZII cis-element of Zp in EBV-BAC caused a loss of BZLF1 production and, accordingly, viral early gene production (8). The levels of early gene expression with the ZII mutant virus were restored to the levels in the wild type or its revertant strain by exogenous supply of BZLF1 (8).
Enhancement of EBV Zp by MEF2 and SP1/KLF family transcriptional factors.
We found expression of Kruppel-like factor 4 (KLF4) and MEF2B enhanced Zp by 17.8- and 7.3-fold, respectively, in our initial screening (Fig. 2).
KLF4 is a member of the SP1/KLF family of zinc finger transcription factors (31) which recognize and specifically bind to GC-rich sites or CACC boxes, and the BZLF1 promoter actually contains binding sites for SP1 in the ZIA, ZIC, and ZID motifs (7). Although we did not clone SP1 as a candidate activator in our screening experiment, we confirmed that SP1 could enhance wild-type Zp by 5.4-fold and KLF4 by 9.3-fold (Fig. 3B, black bars).
The MEF2 family of transcription factors was first identified as a regulator of muscle development and differentiation and later was shown to have diverse functions in various types of cells and tissues (32). MEF2B is a potent transcriptional activator that binds to the same DNA sequence element as other MEF2 family members (33, 34). Liu and others have demonstrated involvement of MEF2D in the activation of the BZLF1 promoter through binding to ZIA, -B, and -D cis-elements (6). Although we isolated MEF2B as a positive candidate activator in our screening, we could not clone MEF2D, which has been reported to bind to the BZLF1 promoter (6, 35). Therefore, we tested if MEF2D expression activates Zp. However, for unknown reasons, MEF2D did not enhance transcription from Zp in our reporter system (data not shown). This result does not contradict the previous report (6), because the authors observed that induction by TPA/ionophore correlated with MEF2D binding to Zp but did not test if expression of MEF2D could increase BZLF1 transcription.
Introduction of mutations into the SP1 binding sites at ZIA, ZIC, and ZID of the pZp-luc vector (Fig. 1) almost completely eliminated SP1- or KLF4-dependent transcriptional activation from the promoter (Fig. 3B, mSP1, light-gray bars), which confirmed that the binding sites for SP1/KLF family transcription factors in the BZLF1 promoter are ZIA, ZIC, and ZID elements, as reported previously (7). In Fig. 3B, we show bars normalized with pGL4.70(hRluc) instead of pCMV-RL because functional SP1 binding sites are also present in the CMV IE promoter (36, 37), and thus, the enhancement of Zp by SP1 did not stand out when normalized with the CMV promoter. Interestingly, KLF4 did not increase transcription from the CMV IE promoter while it did induce Zp activity, which made it possible to clone the factor in our initial screening (Fig. 2).
Point mutations at the MEF2 binding sites in the ZIA, ZIB, and ZID cis-elements (Fig. 1) of pZp-luc clearly diminished the activation by MEF2B (Fig. 3B, mMEF2, dark-gray bar), as expected from the previous report (6).
Because we have identified SP1/KLF family transcriptional factors and MEF2 transcriptional factors as important regulators of Zp, and the binding sites for both factors are located very close together in ZIA and ZID cis-elements of the promoter, we examined whether they could act synergistically. While SP1 and MEF2B alone enhanced BZLF1 promoter activity by 3.3-fold and 5.0-fold, respectively, coexpression of both increased the promoter activity only 3.6-fold (data not shown). In addition, the result in Fig. 3B also suggested that the binding sites for MEF2B and KLF4 or SP1 are functionally separable and do not affect each other. This is the first evidence that SP1 and MEF2 function independently, but not synergistically, even though the binding sites for the two are adjacent to each other in the BZLF1 promoter. However, we admit that SP1 might act synergistically with MEF2D or other members besides MEF2B, especially when inducing reagents, such as TPA, ionophore, butyrate, or anti-Ig, are added.
Point mutations in MEF2 binding sites of Zp cause loss of both BZLF1 and BRLF1 expression in the context of viral genome in HEK293 cells.
Because the above-documented results implicated SP1/KLF and MEF2 family transcription factors in Zp activation, we generated recombinant EBVs with point mutations in the SP1/KLF or MEF2 binding motifs, as shown in Fig. 1, for further verification (Fig. 4A and B). To this end, we first replaced the Zp region with a marker cassette (NeoSt+) and then exchanged it with mutated sequences to prepare EBV-BAC mSP1 or mMEF2. The mutated mSP1 or mMEF2 sequence of the EBV was again swapped with a NeoSt+ cassette, followed by replacement with the wild-type sequence, to generate a revertant strain, EBV-BAC mSP1/R or mMEF2/R. Sequencing analysis confirmed that the EBV-BAC mSP1 or mMEF2 DNA had mutations and that the EBV-BAC revertants had the same sequence as the wild-type virus, as intended. The integrity of the BAC DNA was checked by BamHI and EcoRI digestion, followed by electrophoresis to confirm that the recombinant viruses did not carry obvious deletions or insertions (Fig. 4, BamHI and EcoRI). Shifting of bands by the insertion of the cassette could not be detected by BamHI or EcoRI digestion. We therefore performed PCR using forward (F) and reverse (R) primers located outside the minimal BZLF1 promoter, as indicated in Fig. 4. Since the NeoSt cassette was about 1.6 kb, insertion of the cassette into the minimal BZLF1 promoter region could clearly be detected in the intermediate BAC constructs (Fig. 4, PCR).
Fig 4.

Construction of recombinant EBV featuring point mutations in SP1 binding sites (A) and MEF2 binding sites (B) of the BZLF1 gene promoter. Shown is a schematic arrangement of the recombination of the EBV genome using tandemly arranged neomycin resistance and streptomycin sensitivity genes (NeoSt+). The BZLF1 promoter region was first replaced with the NeoSt+ cassette, which was then replaced with point-mutated sequences (asterisks) to construct EBV-BAC mSP1 or mMEF2. The mutated sequence was replaced again with the NeoSt+ marker cassette and swapped with the wild-type promoter sequence to prepare the revertant clone, EBV-BAC mSP1/R or mMEF2/R. The recombinant EBV genomes were digested with BamHI or EcoRI and separated in an agarose gel. PCR products produced by the indicated primers were electrophoresed to show successful recombinations.
Recombinant EBV-BAC DNA was introduced into HEK293 cells, followed by hygromycin selection, to establish cell lines in which multiple copies were maintained as an episome. More than 10 cell colonies from each recombinant virus were obtained, and viral protein expression levels were examined. Figure 5 shows immunoblotting data for typical cells. In the absence of an exogenous supply of BZLF1, levels of the viral genes, including BZLF1, in EBV-BAC mMEF2 cells were noticeably lower than those in wild-type or revertant cells, while EBV-BAC mSP1 cells exhibited similar levels of BZLF1 or other gene products. Interestingly, even when the BZLF1 expression vector was transfected, levels of early genes, such as the BALF2, BALF5, or BMRF1 gene, were lower than those in wild-type or revertant cells (Fig. 5, mMEF2, BZLF1+). Thus, introduction of point mutations into the SP1 binding elements in the Zp of the viral genome did not influence BZLF1 expression, but mutations in the MEF2 binding motifs caused repression of BZLF1 gene expression in HEK293 cells. Moreover, this appeared to have a greater effect than simply loss of BZLF1 expression, since the exogenous BZLF1 supply did not fully restore early gene expression. In fact, the mMEF2 mutant virus showed markedly lower expression of BRLF1 even with BZLF1 (Fig. 5, mMEF2, BZLF1+).
Fig 5.
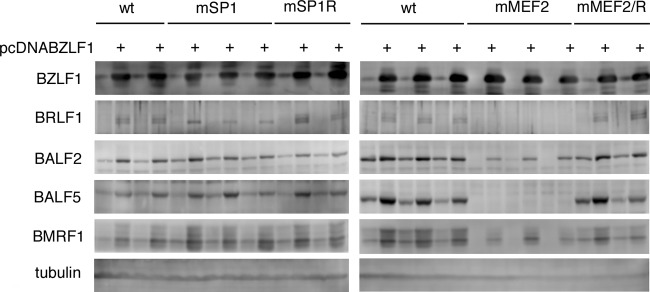
Expression of viral proteins from recombinant viruses in HEK293 cells. The recombinant EBV-BAC DNAs shown in Fig. 5 were introduced into HEK293 cells, followed by hygromycin selection. The resultant cell clones were tested for viral protein expression. Two or three clones of each recombinant virus were transfected with the BZLF1 expression vector (pcDNABZLF1) or its empty control vector. After 24 h, cell proteins were harvested and immunoblotting was performed using anti-BZLF1, -BRLF1, -BALF2, -BALF5, -BMRF1, and -tubulin antibodies.
Suppression of early genes in the MEF2 mutant virus could be restored by coexpression of BRLF1 in HEK293 cells.
In the previous section, we reported that the mMEF2 mutant EBV failed to induce a subset of early genes even when BZLF1 was supplied exogenously, a phenotype reminiscent of BRLF1-deficient viruses (38–40). Heilmann and others clearly showed, employing the ChIP-sequencing (ChIP-seq) technique, that the BALF5 promoter is induced by BRLF1 (41). We therefore checked the levels of BRLF1 in cells with wild-type and mMEF2 viruses (Fig. 5 and 6A). In Fig. 6A, a typical clone of the wt and two clones of mMEF2 were transfected with the BZLF1 and/or BRLF1 expression vector(s). BRLF1 could not be detected in any of the tested cells in the absence of the BZLF1 expression vector (Fig. 5 and 6A, lanes 2, 6, and 10). When BZLF1 was transfected, while BRLF1 was faintly but certainly produced in the wild type, the mMEF2 cells expressed little or no BRLF1 protein (Fig. 5 and 6A, BRLF1 dense, lanes 3, 7, and 11). Expression of early genes from the mMEF2 virus was lower than that from the wild type with BZLF1 overexpression, but coexpression of BZLF1 and BRLF1 markedly restored the expression of early genes from the mMEF2 virus, particularly that of the BALF5 gene (Fig. 6A, lanes 8 and 12). To further verify the effect of exogenous expression of BRLF1, production of progeny virus in the cells was measured, as shown in Fig. 6B. Three days after transfection of BZLF1, alone or with BRLF1, the supernatant was collected from the HEK293 EBV-BAC cells and was then infected with EBV-negative Akata cells. If the supernatant contains EBV, naive Akata cells can be infected and start expressing GFP. BZLF1 overexpression in wild-type virus cells induced high levels of progeny virus production (2.6%), while it did not in mMEF2 cells. However, cotransfection of BZLF1 and BRLF1 rescued the infectivity to comparable levels (2.2 and 2.4%). In addition, viral DNA replication levels were also measured. Transfection of BZLF1 alone to mMEF2 cells did not increase viral DNA (0.86-fold of mock transfection), and coexpression of BRLF1 with BZLF1 restored the levels (15.1-fold) 2 days after transfection. These results suggest that binding of MEF2 proteins to the BZLF1 promoter induces transcription of both the BZLF1 gene and its adjacent IE gene, the BRLF1 gene.
Fig 6.
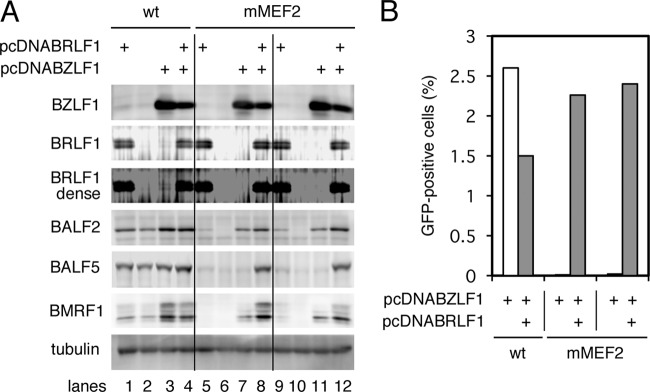
Suppression of early gene expression with MEF2 binding site mutation of the BZLF1 promoter could be reversed with exogenous BRLF1. (A) HEK293 cells latently infected with the wt or the mMEF2 mutant of recombinant EBV-BAC DNA were transfected with the BZLF1 expression vector (pcDNABZLF1), the BRLF1 expression vector (pcDNABRLF1), and/or its empty vector. After 24 h, cell proteins were harvested and immunoblotting was performed using anti-BZLF1, -BRLF1, -BALF2, -BALF5, -BMRF1, and -tubulin antibodies. (B) HEK293 cells latently infected with the wt or the mMEF2 mutant of recombinant EBV-BAC DNA were transfected with BZLF1 expression vector (pcDNABZLF1) (white bars) or BZLF1 expression vector plus BRLF1 expression vector (pcDNABRLF1) (gray bars). The total amount of transfected DNA was adjusted with the empty vector, and 72 h after transfection, the culture supernatants were collected and cocultured with Akata(−) cells for 48 h, and then GFP-positive cells were counted by fluorescence-activated cell sorter (FACS).
Point mutations in MEF2 binding sites of Zp cause loss of both BZLF1 and BRLF1 expression in the context of the viral genome in B cells.
As documented above, mutagenesis experiments were carried out in HEK293 cells, since they serve as an efficient tool in producing progeny virus. However, since HEK293 is a very artificial cell line, prepared from the kidney, which is not a natural host for EBV, here, we established LCLs latently infected with recombinant EBVs. To this end, we infected human PBMCs from a healthy donor with recombinant viruses prepared from HEK293 EBV-BAC cells and cultured them in the presence of cyclosporine. Recombinant viruses from HEK293 EBV-BAC cells were prepared by inducing lytic infection by transfecting BZLF1, but in regard to mMEF2 mutant EBV, the lytic replication cycle was induced by transfecting BRLF1 and BZLF1, as described above (Fig. 6B). All the mutant and repaired strains of EBV we tested here could transform B cells.
In Fig. 7, viral BZLF1 (Fig. 7A and C) and BRLF1 (Fig. 7B and D) levels were measured by real-time RT-PCR. As for Fig. 7A and B, here, we examined IE gene expression of the wild-type, SP1 mutant, MEF2 mutant, and revertant strains, since these LCLs were prepared simultaneously and were cultured side by side. Apart from these LCLs, the mZII mutant and the revertant (Fig. 7C and D) are shown separately from Fig. 7A and B, as they were made on another occasion.
Fig 7.

Expression of viral IE genes in B cells. (A to E) LCLs latently infected with recombinant EBV-BAC prepared from HEK293 EBV-BAC cells were treated (Induction) or not (Control) with TPA (200 ng/ml), A23187 (0.1 μM), and sodium butyrate (10 mM). After 24 h, cell RNAs were harvested and subjected to real-time RT-PCR using specific primers for BZLF1, BRLF1, and GAPDH. Relative BZLF1mRNA (A and C) and BRLF1 mRNA (B and D) levels are shown after normalization to GAPDH mRNA levels. Each bar represents the mean and SD of three independent treatments. (E) LCLs were treated likewise, and cell proteins were subjected to Western blotting using anti-BZLF1 and -tubulin antibodies.
Without induction, LCLs latently infected with mMEF2 mutant EBV exhibited severely suppressed expression of BZLF1 and BRLF1 (only 29 and 14% of the wild type, respectively) and could not increase the levels even after induction with chemical reagents (Fig. 7A and B, green bars), when wild-type or revertant viruses caused about 4- to 8-fold induction. Therefore, the MEF2 binding sites in the BZLF1 promoter play a crucial role, not only in induced expression, but also in leaky spontaneous expression of both of the IE genes in B cells. On the other hand, the mSP1 virus was not associated with any reduction in BZLF1 and BRLF1 levels, and reinforcement by chemical inducers failed to increase IE gene transcription (Fig. 7A and B, red bars).
We previously prepared point-mutated EBV at the ZII site of the BZLF1 promoter, which is bound by cellular b-Zip transcription factors, and confirmed that ZII motif mutation decreased BZLF1 expression in HEK293 cells (8). In order to further verify the significance of the motif, we made LCLs featuring the recombinant viruses. Levels of BZLF1 and BRLF1 were slightly lower (78 and 39%, respectively) without lytic induction and stayed low (45 and 39%, respectively), even with induction of lytic replication (Fig. 7C and D, orange bars).
To extend this, BZLF1 protein levels were examined (Fig. 7E). This experiment yielded results similar to those for mRNA quantification (Fig. 7A and C). Among the clones, we found BZLF1 protein in the mMEF2 strain was most severely restricted.
Although here we show only one representative item of data, we obtained two LCL lines for each recombinant, and these lines all behaved in similar manners. These results indicate that all the cis-motifs (MEF2, SP1, and b-Zip binding motifs) are needed for enforced expression of BZLF1 and BRLF1 by TPA, ionophore, and butyrate. Among the motifs, MEF2 binding sites most profoundly influence the expression of the IE genes, since leaky expression was also tightly blocked in the MEF2 mutant.
CpG DNA methylation levels in the Zp and Rp of MEF2 mutant EBV did not correlate with low expression levels of BZLF1 and BRLF1.
Our results so far demonstrated the importance of cellular MEF2, SP1/KLF, and b-Zip-type family transcription factor binding for positive regulation of BZLF1 and BRLF1 gene expression, with a strong emphasis on the MEF2 family. The question then arises as to how the host transcription factors regulate BZLF1/BRLF1 transcription, which eventually leads to EBV reactivation. Because CpG dinucleotide DNA methylation could be one possible cause of BZLF1 promoter repression, as it is frequently associated with constitutive heterochromatin, we analyzed levels of epigenetic DNA modification at the BZLF1 promoter in LCLs latently infected with recombinant EBVs. There are only three CpG dinucleotides in the minimal BZLF1 promoter loci (Fig. 8A), two of which appear downstream of the TATA box (TTTAAA for the BZLF1 promoter) and one in the coding region. Although CpG methylation levels in the BZLF1 promoter are much lower than in other lytic promoters (42), the modification is still present (43, 44). Our bisulfite modification analysis revealed that there are no preferences in CpG methylation levels of MEF2 mutant or SP1 mutant EBVs at the Zp loci (Fig. 8B). The mZII mutant virus was an exception in that the CpG methylation level was somewhat higher (20/30), than that of the wild type (15/30) or the repaired virus (13/30), suggesting the possibility that the mutation at the ZII element might cause a slight loss of BZLF1 through increased CpG methylation. Taken together, our bisulfite data indicate that CpG methylation of the BZLF1 promoter does not account for the repression of BZLF1 transcription in the MEF2 sites of mutant viruses.
Fig 8.

CpG DNA methylation of the BZLF1 promoter in mutant LCLs. (A) Schematic illustration of the minimal BZLF1 promoter. The distribution of three CpG dinucleotides analyzed in panel B is shown with circled numbers. cis-element motifs and the BZLF1 coding region are also depicted. (B) CpG DNA methylation of the BZLF1 promoter in B cells. DNA from LCLs latently infected with wild-type or mutant recombinant EBV-BAC was subjected to bisulfite modification, followed by sequencing. Filled circles, methylated; open circles, unmethylated.
Interestingly, the CpG dinucleotide between the TATA box and the start codon (CpG 2 in Fig. 8A) had a discernible tendency toward more intense methylation, in agreement with the previous literature (44).
We then analyzed the levels of epigenetic DNA CpG methylation at the BRLF1 promoter in LCLs latently infected with recombinant EBVs (Fig. 9). All six of the CpG dinucleotides in the BRLF1 promoter locus (Fig. 9A) were more severely methylated (Fig. 9B) than the BZLF1 promoter (Fig. 8B). Because more than 95% of the CpGs were methylated even in the wild type and the levels did not increase in point-mutated strains, it is suggested that CpG methylations were not involved in inhibition of BRLF1 expression in the mutant viruses.
Fig 9.

CpG DNA methylation of the BRLF1 promoter in mutant LCLs. (A) Schematic illustration of the BRLF1 promoter. The distribution of six CpG dinucleotides analyzed in panel B is shown with circled numbers. cis-element motifs and BRLF1 coding region are also depicted. (B) CpG DNA methylation of the BRLF1 promoter in B cells. DNA from LCLs latently infected with wild-type or mutant recombinant EBV-BAC was subjected to bisulfite modification, followed by sequencing. Filled circles, methylated; open circles, unmethylated.
Suppressive histone methylation levels in the Zp and Rp of MEF2 mutant EBV correlated with the low expression levels of BZLF1 and BRLF1.
Possible epigenetic modifications that might silence the promoter, besides CpG DNA methylation, include histone changes. The existence of both active and suppressive epigenetic histone modifications of the BZLF1 promoter during latency, such as H3K9Ac or H3K27me3, was reported previously (16). In order to obtain insights into histone situations, levels of typical epigenetic histone modifications in the LCLs were assessed by ChIP assays (Fig. 10 and 11).
Fig 10.

Histone modification of BZLF1/BRLF1 promoters in mSP1 and mMEF2 mutant LCLs. LCLs latently infected with recombinant EBV-BAC were cross-linked and subjected to ChIP experiments as described in Materials and Methods using normal IgG and anti-histone H3, -H3K9Ac, -H3K4me3, -H3K9me2, -H3K27me3, -H3K9me3, and -H4K20me3 antibodies, followed by DNA extraction and real-time PCR to detect DNA fragments using the indicated primers. The Zp/Rp numbers indicate the sequence positions relative to the transcription start site. As controls, the TR region of EBV, the β-globin promoter (Globinp), and the GAPDH promoter (GAPDHp) were also quantified.
Fig 11.
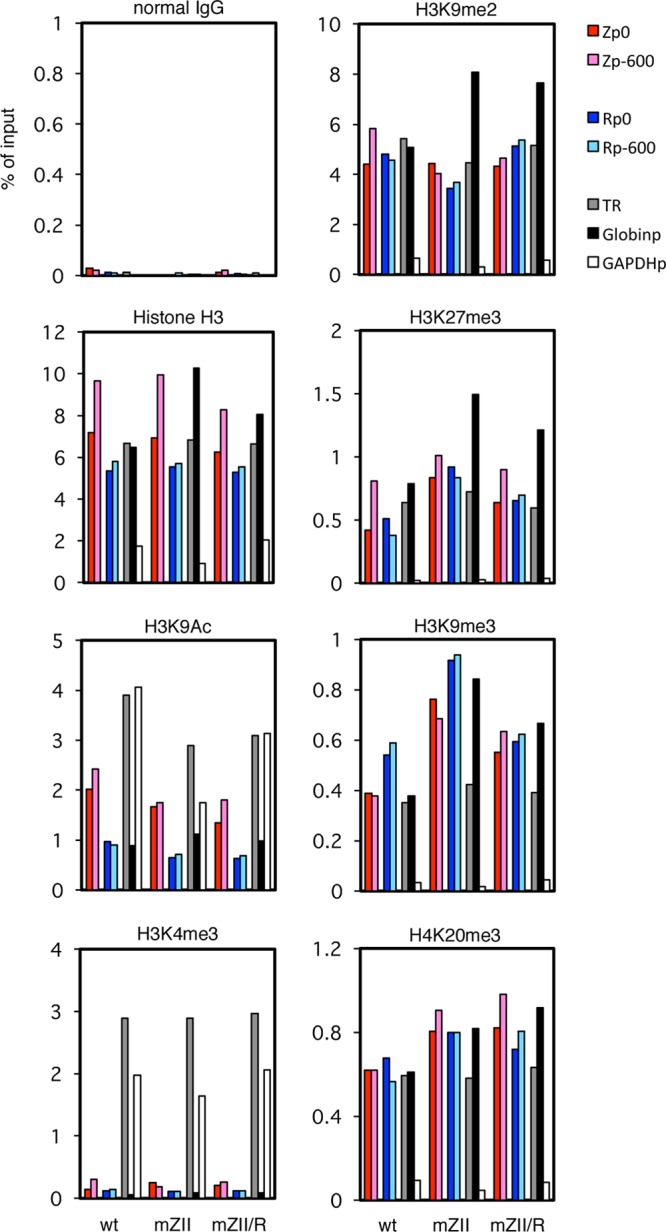
Histone modification of BZLF1/BRLF1 promoters in mZII mutant LCLs. LCLs latently infected with recombinant EBV-BAC were cross-linked and subjected to ChIP experiments as described in Materials and Methods using normal IgG and anti-histone H3, -H3K9Ac, -H3K4me3, -H3K9me2, -H3K27me3, -H3K9me3, and -H4K20me3 antibodies, followed by DNA extraction and real-time PCR to detect DNA fragments using the indicated primers. The Zp/Rp numbers indicate the sequence positions relative to the transcription start site. As controls, the TR region of EBV, the β-globin promoter (Globinp), and the GAPDH promoter (GAPDHp) were also quantified.
Levels of background precipitation with normal IgG could be ignored (Fig. 10 and 11, normal IgG). Histone H3 levels are fairly constant throughout the EBV genome of latently infected LCLs (16) (Fig. 10 and 11, Histone H3). In addition to Zp and Rp loci (colored bars), we measured the levels at the TR of EBV (gray bars) and some cellular loci as controls (β-globin promoter [Fig. 10 and 11, Globinp] and GAPDH promoter [Fig. 10 and 11, GAPDHp]). We used these loci here because it is known that the β-globin promoter locus is silenced in nonerythroid cells by high levels of facultative chromatin histone modifications, such as H3K9me2 and H3K27me3, and the GAPDH gene, a typical housekeeping gene, is ubiquitously and abundantly transcribed with high levels of active markers, such as histone acetylation and H3K4me3. Interestingly, anti-histone H3 antibody precipitated the GAPDHp loci very weakly (Fig. 10 and 11, Histone H3, white bars) for unknown reasons. It is speculated that because the GAPDH promoter is highly active, the chromatin structure of the loci may be open and loose.
One of the active chromatin markers, H3K9 acetylation, was present at slightly lower levels at Zp, Rp (Fig. 10 and 11, colored bars), and Globinp (Fig. 10 and 11, black bars) than at the TR (Fig. 10 and 11, gray bars) or active GAPDHp (Fig. 10 and 11, white bars). However, the patterns were quite similar among the LCL lines, and mZII or mMEF2 appeared to exhibit no obvious features (Fig. 10 and 11, H3K9Ac).
On the other hand, levels of another active histone marker, H3K4me3, were very low at Zp, Rp (Fig. 10 and 11, H3K4me3, colored bars) and Globinp (Fig. 10 and 11, H3K4me3, black bars), while the levels were markedly high at the TR (Fig. 10 and 11, H3K4me3, gray bars) and GAPDHp (Fig. 10 and 11, H3K4me3, white bars). Here again, however, we found no obvious differences in patterns of the modification between the LCLs (Fig. 10 and 11, H3K4me3).
The suppressive facultative chromatin marker H3K9me2 was checked next (Fig. 10 and 11, H3K9me2). While GAPDHp (white bars) exhibited appreciably weak modification, Zp, Rp (colored bars), and TR (gray bars) were marked by methylation in all LCLs almost as efficiently as Globinp (black bars), which is silenced in LCLs.
Another suppressive facultative chromatin marker, H3K27me3, showed similar patterns in that the level of methylation of the GAPDHp promoter was low (Fig. 10 and 11, H3K27me3,white bars), but we found a striking difference in that BZLF1 and BRLF1 promoters in the mMEF2 strain were more intensely associated with H3K27me3 (Fig. 10, H3K27me3, colored bars). Because MEF2 mutation resulted in suppression of BZLF1 transcription, increased H3K27me3 methylation may play a role in this.
Two constitutive heterochromatin markers, H3K9me3 and H4K20me3, were found in the BZLF1 and BRLF1 promoters at higher levels in the mMEF2 mutant than in wild-type or the repaired viruses (Fig. 10, H3K9me3 and H4K20me3, colored bars).
The Zp and Rp of the mZII mutant, which has a mutation in the binding sites of cellular b-Zip transcription factors, showed slightly higher levels of H3K9me3 than the wild-type or revertant viruses but comparable levels of other modifications (Fig. 11, H3K9me3, colored bars).
The results suggested that suppressive markers, especially H3K27me3, play an important role in blocking activation of the BZLF1 and BRLF1 promoters in the MEF2 mutant LCLs. However, we still cannot rule out involvement of other epigenetic modifications in the process.
DISCUSSION
In this study, an exhaustive library screening isolated a number of cellular factors, including KLF4, MEF2B, and some b-Zip transcription factors, as activators of the BZLF1 promoter. We believe this screening system was appropriate, since some of the clones we identified proved to be family members of transcription factors that have been reported previously to enhance expression of BZLF1, or at least to bind to Zp. So far, we have checked more than 20,000 clones from the library. One might argue that more clones must be tested, but we assume this number is sufficient, because junk sequences accounted for only a few percent of the total in our library: we substantially reduced the number of junk genes in the library before use and confirmed more than 95% of our library clones to contain full-length cDNAs. Also, we examined the effects of the family or related proteins in the hit clones, and thus, it was not necessary to isolate the exact ones that actually function in vivo. Interestingly, we cloned KLF4 and MEF2B, family members of SP1 and MEF2D transcription factors, respectively, both of which have been reported to bind to Zp in B cells (6, 7). However, while KLF4 and MEF2B were specifically isolated as inducers of the BZLF1 promoter over the CMV IE promoter in our screen system, SP1 and MEF2D were not, because SP1 also efficiently induced the CMV IE promoter and MEF2D did not enhance pZp-luc, for unknown reasons. Although our screening system is artificial and has limitations, we are confident in the success of our screen and feel that our system has its own advantages, since we applied a method different from electrophoretic mobility shift assay (EMSA), upon which most previous reports relied.
By introducing point mutations into the binding sites of cellular b-Zip factors (the ZII cis-element), we previously demonstrated that the ZII mutant EBV exhibited impaired expression of BZLF1 and thereby restricted the production of downstream early genes in HEK293 cells (8). This restriction could easily be removed by exogenous overexpression of BZLF1. In B cells, enhancement by chemical inducers was also discouraged in LCLs by the introduction of a mutation at the ZII element in the context of the virus genome (Fig. 7C).
Besides the ZII cis-element, we prepared point mutant EBV at SP1 or MEF2 binding sites of Zp (Fig. 4). Although point mutation in the SP1 site in EBV-BAC did not appreciably influence the BZLF1 expression level in HEK293 cells (Fig. 5), enhancement of the promoter activity was inhibited by the SP1 mutation in B cells (Fig. 7A).
It is of particular interest that point mutations in MEF2 family transcription factor sites of the BZLF1 promoter profoundly affect not only spontaneous, but also induced, expression of the BZLF1 gene in both HEK293 cells and LCLs (Fig. 5 and 7A). Our results here are not in agreement with the previous report that MEF2 recruits class II HDAC to the latent BZLF1 promoter during latency and functions to suppress the spontaneous expression of the BZLF1 gene, thereby maintaining virus latency (45). Because the earlier authors relied solely on artificial methods, such as reporter assays, we believe our conclusion that MEF2 acts to increase BZLF1 promoter activity even in latency is more reliable.
In addition, in B cells, all three mutants we examined here (ZII, SP1, and MEF2) somehow influenced transcription, not only on the BZLF1 gene, but also on the BRLF1 gene, which is another EBV transcriptional-regulatory gene (Fig. 7). Because activation of a promoter mediated through binding motifs downstream of the transcription start site of the promoter is not rare (28), it is likely that these factors in Zp somehow help the region of the two regulatory genes to remain active. We speculate that the BZLF1 and BRLF1 genes are located adjacent to one another for a reason and not just incidentally; if they are close, the virus can save an area of relatively open chromatin. There may be one window of relatively open chromatin for the two neighboring IE genes, which is tightly silenced during latency but not completely inactivated by constitutive heterochromatinization. Tight, but not too tight, silencing like this must be dependent on the consummate balancing of active and suppressive factors and thus may easily be disrupted for various reasons, including point mutations or changes in cell signaling. Another possibility we propose, especially when we take into consideration the heavy CpG methylation at the BRLF1 promoter (Fig. 9) and the fact that BZLF1 binds to and enhances CpG methylated sequences (46), is that BRLF1 levels simply reflect the abundance of BZLF1, since BZLF1 and BRLF1 levels were found to be roughly parallel (Fig. 7), although there are some minor exceptions.
DNA methylome analysis of latent EBV genomes previously demonstrated that the Zp region features exclusively weak CpG methylation when the rest of the lytic gene promoters are heavily CpG methylated, except some promoters of latent genes, such as the LMP1 gene (42, 44). In fact, methylation at Zp is markedly weaker than at Rp (Fig. 8 and 9). We believe CpG methylation does not account for the restricted expression of BZLF1 in MEF2 mutant virus, since CpG methylation levels of the BZLF1 promoter were comparable between the wild-type, mutant, and repaired viruses (Fig. 8). Analysis of posttranslational modifications of histones suggested that the BZLF1 promoter is silenced by suppressive histone modifications (16, 47). Our results here suggested involvement of histones H3K27me3, H3K9me3, and H4K20me3. Further analysis is needed to dissect the molecular mechanisms of the promoter regulation. Moreover, a certain type of chromatin boundary factor, such as CCCTC-binding factor (CTCF), may also play a role in regulation of the BZLF1 promoter (48), as has already been shown for Kaposi's sarcoma-associated herpesvirus (KSHV) (49).
Among MEF2 family transcription factors, Liu and others demonstrated, using EMSA supershift assays, that MEF2D is the major example that binds to ZIA, -B, and -D motifs (6). We, on the other hand, cloned MEF2B, but not MEF2D, as an activator of Zp in our library screen. Immunoblotting data here and those of others indicated that all four members (MEF2A, -B, -C, and -D) of the MEF2 family of transcription factors are present in B cells (Fig. 12) (50). We concur with the proposal, based on the paper by Liu et al., that MEF2D obviously plays an important role in BZLF1 promoter activation, but because all MEF2 family transcription factors are present in B cells (Fig. 12) (50), involvement of other examples besides MEF2D cannot be precluded.
Fig 12.
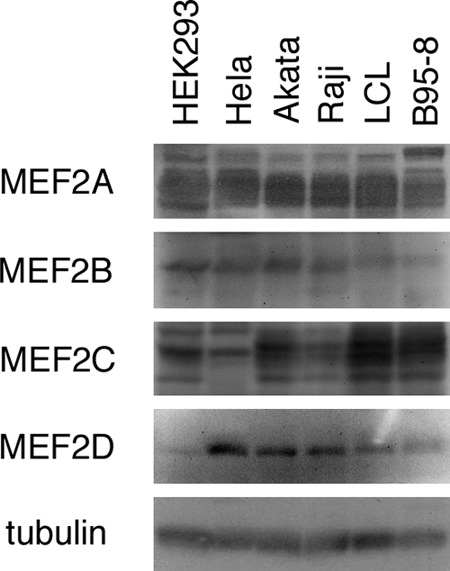
Expression profile of MEF2 family transcription factors. Cell proteins from HEK293, HeLa, Akata, Raji, and B95-8 cells and LCLs were subjected to Western blotting using anti-MEF2A, -MEF2B, -MEF2C, -MEF2D, and -tubulin antibodies.
In addition, our screen identified HIF-2α as a strong inducer of the BZLF1 promoter, causing 9.8-fold elevation of promoter activity (Fig. 2A). HIF-1α and HIF-2α are basic helix-loop-helix (bHLH) transcriptional factors that stimulate, under hypoxic conditions, a large number of genes important for cellular processes, including angiogenesis, cell survival, and metabolism. HIF-2α might be of great interest, because we found a report that the lytic cycle of EBV is induced by hypoxia (51). Actually, we also could confirm that hypoxia (1% oxygen; 36 h) induced BZLF1 levels approximately 10-fold, not only in the B95-8 cells used by earlier authors, but also in Akata cells and LCLs. However, we had to judge that this isolation of HIF-2α as an inducer of Zp must be pseudopositive in our screen, since the HIF-2α clone identified in our screen consisted of only the C-terminal half of the coding region, which lacks the DNA binding motif, while all other clones in Fig. 2A contain full-length open reading frames of the genes.
Similar mutagenesis experiments with IE gene promoters have been reported for cytomegaloviruses. Mutation of all the CREB/ATF or NF-κB sites has little or no effect on human cytomegalovirus replication, while SP1 binding sites in the minimal enhancer elements are essential (36, 52). IE gene expression of the herpes simplex virus appears to be largely dependent on the TAATGARAT sequence, present only in IE promoters, which is bound by VP16, mediated by Oct-1 and HCF (53). Besides herpesviruses, the long terminal repeat (LTR) of human immunodeficiency virus contains cis-acting NF-κB and SP1 sites, both differentially contributing to the transcription of viral gene expression (54). Because an NF-κB binding site has not been reported for EBV Zp to our knowledge, it is likely that, unlike the human immunodeficiency virus case, activation of NF-κB signaling by cytokines or other stimulations may not induce EBV reactivation. Actually, we and others have observed that NF-κB signaling functions to maintain latency and does not enhance reactivation of the virus (55, 56).
In conclusion, we have demonstrated the importance of MEF2 transcription factors for reactivation of EBV from latency. Specific small-molecule inhibition of MEF2, by agents such as cyclosporine (6), may be an alternative possibility to regulate EBV reactivation.
ACKNOWLEDGMENTS
We thank W. Hammerschmidt and H. J. Delecluse for providing the EBV-BAC DNA and HEK293 cells. We also are grateful to G. Suske and K. Mori for pCMV-SP1 and pCMV-Myc-XBP1s, respectively. pcDNA3-Flag-CREB was a generous gift from K. Shimotohno. We also express our appreciation to H. Kimura, T. Watanabe, N. Hotta, C. Noda, and T. Gamano for technical assistance.
This work was supported by grants-in-aid for Scientific Research from the Ministry of Education, Science, Sports, Culture and Technology (no. 20390137, 23390118, and 23114512 to T.T. and no. 20790362, 22790448, and 24590566 to T.M.) and the Ministry of Health, Labor and Welfare (to T.T.) and partly by the Uehara Memorial Research Fund (to T.T. and T.M.); the Takeda Science Foundation (to T.T. and T.M.); and the Yasuda Medical Foundation, the Mochida Memorial Foundation for Medical and Pharmaceutical Research, the Senshin Medical Research Foundation, the Kanae Foundation for Promotion of Medical Science, and the Kitamura Memorial Research Fund (to T.M.).
Footnotes
Published ahead of print 10 July 2013
REFERENCES
- 1. Tsurumi T, Fujita M, Kudoh A. 2005. Latent and lytic Epstein-Barr virus replication strategies. Rev. Med. Virol. 15:3–15 [DOI] [PubMed] [Google Scholar]
- 2. Amon W, Farrell PJ. 2005. Reactivation of Epstein-Barr virus from latency. Rev. Med. Virol. 15:149–156 [DOI] [PubMed] [Google Scholar]
- 3. Speck SH, Chatila T, Flemington E. 1997. Reactivation of Epstein-Barr virus: regulation and function of the BZLF1 gene. Trends Microbiol. 5:399–405 [DOI] [PubMed] [Google Scholar]
- 4. Miller G, El-Guindy A, Countryman J, Ye J, Gradoville L. 2007. Lytic cycle switches of oncogenic human gammaherpesviruses. Adv. Cancer Res. 97:81–109 [DOI] [PubMed] [Google Scholar]
- 5. Sinclair AJ. 2003. bZIP proteins of human gammaherpesviruses. J. Gen. Virol. 84:1941–1949 [DOI] [PubMed] [Google Scholar]
- 6. Liu S, Liu P, Borras A, Chatila T, Speck SH. 1997. Cyclosporin A-sensitive induction of the Epstein-Barr virus lytic switch is mediated via a novel pathway involving a MEF2 family member. EMBO J. 16:143–153 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Liu S, Borras AM, Liu P, Suske G, Speck SH. 1997. Binding of the ubiquitous cellular transcription factors Sp1 and Sp3 to the ZI domains in the Epstein-Barr virus lytic switch BZLF1 gene promoter. Virology 228:11–18 [DOI] [PubMed] [Google Scholar]
- 8. Murata T, Noda C, Saito S, Kawashima D, Sugimoto A, Isomura H, Kanda T, Yokoyama KK, Tsurumi T. 2011. Involvement of Jun dimerization protein 2 (JDP2) in the maintenance of Epstein-Barr virus latency. J. Biol. Chem. 286:22007–22016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Murata T, Sato Y, Nakayama S, Kudoh A, Iwahori S, Isomura H, Tajima M, Hishiki T, Ohshima T, Hijikata M, Shimotohno K, Tsurumi T. 2009. TORC2, a coactivator of cAMP-response element-binding protein, promotes Epstein-Barr virus reactivation from latency through interaction with viral BZLF1 protein. J. Biol. Chem. 284:8033–8041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ruf IK, Rawlins DR. 1995. Identification and characterization of ZIIBC, a complex formed by cellular factors and the ZII site of the Epstein-Barr virus BZLF1 promoter. J. Virol. 69:7648–7657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bhende PM, Dickerson SJ, Sun X, Feng WH, Kenney SC. 2007. X-box-binding protein 1 activates lytic Epstein-Barr virus gene expression in combination with protein kinase D. J. Virol. 81:7363–7370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McDonald C, Karstegl CE, Kellam P, Farrell PJ. 2010. Regulation of the Epstein-Barr virus Zp promoter in B lymphocytes during reactivation from latency. J. Gen. Virol. 91:622–629 [DOI] [PubMed] [Google Scholar]
- 13. Sun CC, Thorley-Lawson DA. 2007. Plasma cell-specific transcription factor XBP-1s binds to and transactivates the Epstein-Barr virus BZLF1 promoter. J. Virol. 81:13566–13577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Flemington E, Speck SH. 1990. Autoregulation of Epstein-Barr virus putative lytic switch gene BZLF1. J. Virol. 64:1227–1232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Murata T, Hotta N, Toyama S, Nakayama S, Chiba S, Isomura H, Ohshima T, Kanda T, Tsurumi T. 2010. Transcriptional repression by sumoylation of Epstein-Barr virus BZLF1 protein correlates with association of histone deacetylase. J. Biol. Chem. 285:23925–23935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Murata T, Kondo Y, Sugimoto A, Kawashima D, Saito S, Isomura H, Kanda T, Tsurumi T. 2012. Epigenetic histone modification of Epstein-Barr virus BZLF1 promoter during latency and reactivation in Raji cells. J. Virol. 86:4752–4761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Yu X, Wang Z, Mertz JE. 2007. ZEB1 regulates the latent-lytic switch in infection by Epstein-Barr virus. PLoS Pathog. 3:e194. 10.1371/journal.ppat.0030194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Montalvo EA, Cottam M, Hill S, Wang YJ. 1995. YY1 binds to and regulates cis-acting negative elements in the Epstein-Barr virus BZLF1 promoter. J. Virol. 69:4158–4165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hagemeier SR, Dickerson SJ, Meng Q, Yu X, Mertz JE, Kenney SC. 2010. Sumoylation of the Epstein-Barr virus BZLF1 protein inhibits its transcriptional activity and is regulated by the virus-encoded protein kinase. J. Virol. 84:4383–4394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Gruffat H, Manet E, Rigolet A, Sergeant A. 1990. The enhancer factor R of Epstein-Barr virus (EBV) is a sequence-specific DNA binding protein. Nucleic Acids Res. 18:6835–6843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ragoczy T, Miller G. 2001. Autostimulation of the Epstein-Barr virus BRLF1 promoter is mediated through consensus Sp1 and Sp3 binding sites. J. Virol. 75:5240–5251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Adamson AL, Darr D, Holley-Guthrie E, Johnson RA, Mauser A, Swenson J, Kenney S. 2000. Epstein-Barr virus immediate-early proteins BZLF1 and BRLF1 activate the ATF2 transcription factor by increasing the levels of phosphorylated p38 and c-Jun N-terminal kinases. J. Virol. 74:1224–1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Manet E, Gruffat H, Trescol-Biemont MC, Moreno N, Chambard P, Giot JF, Sergeant A. 1989. Epstein-Barr virus bicistronic mRNAs generated by facultative splicing code for two transcriptional trans-activators. EMBO J. 8:1819–1826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Murata T, Isomura H, Yamashita Y, Toyama S, Sato Y, Nakayama S, Kudoh A, Iwahori S, Kanda T, Tsurumi T. 2009. Efficient production of infectious viruses requires enzymatic activity of Epstein-Barr virus protein kinase. Virology 389:75–81 [DOI] [PubMed] [Google Scholar]
- 25. Hagen G, Muller S, Beato M, Suske G. 1994. Sp1-mediated transcriptional activation is repressed by Sp3. EMBO J. 13:3843–3851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ego T, Tanaka Y, Shimotohno K. 2005. Interaction of HTLV-1 Tax and methyl-CpG-binding domain 2 positively regulates the gene expression from the hypermethylated LTR. Oncogene 24:1914–1923 [DOI] [PubMed] [Google Scholar]
- 27. Delecluse HJ, Hilsendegen T, Pich D, Zeidler R, Hammerschmidt W. 1998. Propagation and recovery of intact, infectious Epstein-Barr virus from prokaryotic to human cells. Proc. Natl. Acad. Sci. U. S. A. 95:8245–8250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Noda C, Murata T, Kanda T, Yoshiyama H, Sugimoto A, Kawashima D, Saito S, Isomura H, Tsurumi T. 2011. Identification and characterization of CCAAT enhancer-binding protein (C/EBP) as a transcriptional activator for Epstein-Barr virus oncogene latent membrane protein 1. J. Biol. Chem. 286:42524–42533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Flemington E, Speck SH. 1990. Identification of phorbol ester response elements in the promoter of Epstein-Barr virus putative lytic switch gene BZLF1. J. Virol. 64:1217–1226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Taylor N, Flemington E, Kolman JL, Baumann RP, Speck SH, Miller G. 1991. ZEBRA and a Fos-GCN4 chimeric protein differ in their DNA-binding specificities for sites in the Epstein-Barr virus BZLF1 promoter. J. Virol. 65:4033–4041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kaczynski J, Cook T, Urrutia R. 2003. Sp1- and Kruppel-like transcription factors. Genome Biol. 4:206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Potthoff MJ, Olson EN. 2007. MEF2: a central regulator of diverse developmental programs. Development 134:4131–4140 [DOI] [PubMed] [Google Scholar]
- 33. Katoh Y, Molkentin JD, Dave V, Olson EN, Periasamy M. 1998. MEF2B is a component of a smooth muscle-specific complex that binds an A/T-rich element important for smooth muscle myosin heavy chain gene expression. J. Biol. Chem. 273:1511–1518 [DOI] [PubMed] [Google Scholar]
- 34. Molkentin JD, Firulli AB, Black BL, Martin JF, Hustad CM, Copeland N, Jenkins N, Lyons G, Olson EN. 1996. MEF2B is a potent transactivator expressed in early myogenic lineages. Mol. Cell. Biol. 16:3814–3824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Bryant H, Farrell PJ. 2002. Signal transduction and transcription factor modification during reactivation of Epstein-Barr virus from latency. J. Virol. 76:10290–10298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Isomura H, Stinski MF, Kudoh A, Daikoku T, Shirata N, Tsurumi T. 2005. Two Sp1/Sp3 binding sites in the major immediate-early proximal enhancer of human cytomegalovirus have a significant role in viral replication. J. Virol. 79:9597–9607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Lang D, Fickenscher H, Stamminger T. 1992. Analysis of proteins binding to the proximal promoter region of the human cytomegalovirus IE-1/2 enhancer/promoter reveals both consensus and aberrant recognition sequences for transcription factors Sp1 and CREB. Nucleic Acids Res. 20:3287–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Feederle R, Kost M, Baumann M, Janz A, Drouet E, Hammerschmidt W, Delecluse HJ. 2000. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 19:3080–3089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lu CC, Jeng YY, Tsai CH, Liu MY, Yeh SW, Hsu TY, Chen MR. 2006. Genome-wide transcription program and expression of the Rta responsive gene of Epstein-Barr virus. Virology 345:358–372 [DOI] [PubMed] [Google Scholar]
- 40. Ragoczy T, Miller G. 1999. Role of the Epstein-Barr virus RTA protein in activation of distinct classes of viral lytic cycle genes. J. Virol. 73:9858–9866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Heilmann AM, Calderwood MA, Portal D, Lu Y, Johannsen E. 2012. Genome-wide analysis of Epstein-Barr virus Rta DNA binding. J. Virol. 86:5151–5164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Fernandez AF, Rosales C, Lopez-Nieva P, Grana O, Ballestar E, Ropero S, Espada J, Melo SA, Lujambio A, Fraga MF, Pino I, Javierre B, Carmona FJ, Acquadro F, Steenbergen RD, Snijders PJ, Meijer CJ, Pineau P, Dejean A, Lloveras B, Capella G, Quer J, Buti M, Esteban JI, Allende H, Rodriguez-Frias F, Castellsague X, Minarovits J, Ponce J, Capello D, Gaidano G, Cigudosa JC, Gomez-Lopez G, Pisano DG, Valencia A, Piris MA, Bosch FX, Cahir-McFarland E, Kieff E, Esteller M. 2009. The dynamic DNA methylomes of double-stranded DNA viruses associated with human cancer. Genome Res. 19:438–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li L, Su X, Choi GC, Cao Y, Ambinder RF, Tao Q. 2012. Methylation profiling of Epstein-Barr virus immediate-early gene promoters, BZLF1 and BRLF1 in tumors of epithelial, NK- and B-cell origins. BMC Cancer 12:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Woellmer A, Arteaga-Salas JM, Hammerschmidt W. 2012. BZLF1 governs CpG-methylated chromatin of Epstein-Barr virus reversing epigenetic repression. PLoS Pathog. 8:e1002902. 10.1371/journal.ppat.1002902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gruffat H, Manet E, Sergeant A. 2002. MEF2-mediated recruitment of class II HDAC at the EBV immediate early gene BZLF1 links latency and chromatin remodeling. EMBO Rep. 3:141–146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bhende PM, Seaman WT, Delecluse HJ, Kenney SC. 2004. The EBV lytic switch protein, Z, preferentially binds to and activates the methylated viral genome. Nat. Genet. 36:1099–1104 [DOI] [PubMed] [Google Scholar]
- 47. Ramasubramanyan S, Osborn K, Flower K, Sinclair AJ. 2012. Dynamic chromatin environment of key lytic cycle regulatory regions of the Epstein-Barr virus genome. J. Virol. 86:1809–1819 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Arvey A, Tempera I, Tsai K, Chen HS, Tikhmyanova N, Klichinsky M, Leslie C, Lieberman PM. 2012. An atlas of the Epstein-Barr virus transcriptome and epigenome reveals host-virus regulatory interactions. Cell Host Microbe 12:233–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Chen HS, Wikramasinghe P, Showe L, Lieberman PM. 2012. Cohesins repress Kaposi's sarcoma-associated herpesvirus immediate early gene transcription during latency. J. Virol. 86:9454–9464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Swanson BJ, Jack HM, Lyons GE. 1998. Characterization of myocyte enhancer factor 2 (MEF2) expression in B and T cells: MEF2C is a B cell-restricted transcription factor in lymphocytes. Mol. Immunol. 35:445–458 [DOI] [PubMed] [Google Scholar]
- 51. Jiang JH, Wang N, Li A, Liao WT, Pan ZG, Mai SJ, Li DJ, Zeng MS, Wen JM, Zeng YX. 2006. Hypoxia can contribute to the induction of the Epstein-Barr virus (EBV) lytic cycle. J. Clin. Virol. 37:98–103 [DOI] [PubMed] [Google Scholar]
- 52. Stinski MF, Isomura H. 2008. Role of the cytomegalovirus major immediate early enhancer in acute infection and reactivation from latency. Med. Microbiol. Immunol. 197:223–231 [DOI] [PubMed] [Google Scholar]
- 53. Wysocka J, Herr W. 2003. The herpes simplex virus VP16-induced complex: the makings of a regulatory switch. Trends Biochem. Sci. 28:294–304 [DOI] [PubMed] [Google Scholar]
- 54. Burnett JC, Miller-Jensen K, Shah PS, Arkin AP, Schaffer DV. 2009. Control of stochastic gene expression by host factors at the HIV promoter. PLoS Pathog. 5:e1000260. 10.1371/journal.ppat.1000260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Brown HJ, Song MJ, Deng H, Wu TT, Cheng G, Sun R. 2003. NF-kappaB inhibits gammaherpesvirus lytic replication. J. Virol. 77:8532–8540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Saito S, Murata T, Kanda T, Isomura H, Narita Y, Sugimoto A, Kawashima D, Tsurumi T. 2013. Epstein-Barr virus deubiquitinase downregulates TRAF6-mediated NF-kappaB signaling during productive replication. J. Virol. 87:4060–4070 [DOI] [PMC free article] [PubMed] [Google Scholar]