

Abstract
Enterovirus 71 (EV71) causes hand, foot, and mouth disease and severe neurological disorders in children. Human scavenger receptor class B member 2 (hSCARB2) and P-selectin glycoprotein ligand-1 (PSGL-1) are identified as receptors for EV71. The underling mechanism of PSGL-1-mediated EV71 entry remains unclear. The endocytosis required for EV71 entry were investigated in Jurkat T and mouse L929 cells constitutively expressing human PSGL-1 (PSGL-1-L929) or human rhabdomyosarcoma (RD) cells displaying high SCARB2 but no PSGL-1 by treatment of specific inhibitors or siRNA. We found that disruption of clathrin-dependent endocytosis prevented EV71 infection in RD cells, while there was no influence in Jurkat T and PSGL-1-L929 cells. Disturbing caveolar endocytosis by specific inhibitor or caveolin-1 siRNA in Jurkat T and PSGL-1-L929 cells significantly blocked EV71 infection, whereas it had no effect on EV71 infection in RD cells. Confocal immunofluorescence demonstrated caveola, and EV71 was directly colocalized. pH-dependent endosomal acidification and intact membrane cholesterol were important for EV71 infection, as judged by the pretreatment of inhibitors that abrogated the infection. A receptor-dominated endocytosis of EV71 infection was observed: PSGL-1 initiates caveola-dependent endocytosis and hSCARB2 activates clathrin-dependent endocytosis.
INTRODUCTION
Enterovirus 71 (EV71), a positive-strand RNA virus, belongs to the Picornaviridae family (1). EV71 is a causative factor for hand, foot, and mouth disease (HFMD), which has symptoms of persistent fever, herpangina, and lymphopenia (2–4). The main complication of EV71 infection is neurological disorder, caused by inflammation in the central nervous system (CNS) and leading to encephalitis, acute flaccid paralysis, pulmonary edema, hemorrhage, and possible fatality, especially in young children (2–4). Since its first identification in California in 1969, several countries have reported an increase in the numbers of EV71 cases and sporadic outbreaks (5, 6). The current clinical course for control of EV71 infection relies on symptomatic treatment (7). An effective medication or vaccination against EV71 infection has yet to be developed.
Previous studies identified hSCARB2 (8) and PSGL-1 (CD162) (9) as cellular receptors for EV71. The scavenger receptor class B receptor is a type III glycoprotein also known as lysosome integral membrane protein 2 (LIMP2). It is ubiquitously expressed in several cell types, including the liver, spleen, testes, retinal pigment epithelium cells, macrophages, osteoblasts, and brain (10–12), and predominantly in the limiting membranes of cell lysosomes and endosomes. Although mouse SCARB2 shares 85.8% homology with human SCARB2, it does not serve as a receptor for EV71 infection (13). Previous studies have demonstrated that hSCARB2 expression can enable normally unsusceptible cell lines to support EV71 propagation and develop cytopathic effects (8). P-selectin glycoprotein ligand-1 is a sialomucin membrane protein restrictively expressed in leukocytes, dendritic cells, tissue macrophages (those in the liver, lung, bowel, and Langerhans cells in the skin), and progenitor myeloid cells (14). It plays a role in the binding of leukocytes to endothelial cells and platelets and in the early stages of inflammation (14, 15). The expression of human PSGL-1 in normally unsusceptible cell lines can also facilitate EV71 infection, leading to the development of cytopathic effects (9, 16).
Previous research has identified several types of endocytosis involved in virus entry following the binding to a receptor, including clathrin and caveola dependent, as well as clathrin- and caveola-independent endocytosis. In clathrin-dependent endocytosis, the virus-bound receptors are targeted to clathrin-coated pits (CCPs), which mature into clathrin-coated vesicles (CCVs), resulting in the internalization of the viruses and their receptors. Adenovirus type 2/5, vesicular stomatitis virus (VSV), and dengue virus all use clathrin-mediated endocytosis for viral entry into the host cells (17–19). Caveola-dependent endocytosis involves the formation of glycolipid rafts in caveolin-1 (CAV-1)-enriched plasma membranes, resulting in the internalization of the membrane-bound viruses. Unlike clathrin-dependent endocytosis, studies have reported the involvement of caveolae in the internalization of selected bacterial toxins (cholera toxin B [CT-B]) (20) and several nonenveloped viruses such as hepatitis B virus, simian virus 40 (SV40), and polyomavirus (21–23).
Our previous study on the mechanism of SCARB2-mediated EV71 infection showed that clathrin-dependent endocytosis is required for EV71 infection in a mouse NIH 3T3 line constitutively expressing human SCARB2 cells (24). Similar report confirmed the activation of clathrin-dependent endocytosis after EV71 infection of RD cells expressing SCARB2 but not PSGL-1 (9, 25). In contrast, the mechanism of human PSGL-1-mediated EV71 infection remains unclear. In the present study, we demonstrate the entry mechanism of EV71 in human PSGL-1-expressing cells, comparing it to the mechanism of EV71 entry in SCARB2-expressing cells. Mouse L929 cells expressing human PSGL-1 (PSGL-1-L929 cells) are able to support infection by the EV71/E59 strain, a B4 subgenotype isolated in Taiwan in 2002. Using pharmacological inhibitors to block the endocytic pathway or small interfering RNA (siRNA) to specifically downregulate cellular clathrin or caveolin-1, we showed that the activation of a specific route for endocytosis in EV71 infection is receptor dependent. PSGL-1 mediates caveola-dependent endocytosis in human Jurkat T and PSGL-1-L929 cells, which occurs synchronously with clathrin-dependent EV71 entry in RD cells. The mechanisms of EV71 entry and the activation of multiple pathways are therefore determined by host cell receptor expression.
MATERIALS AND METHODS
Cell culture.
Human rhabdomyosarcoma (RD) cells originally purchased from American Type Culture Collection (ATCC no. CCL-136) were provided by the Taiwan Centers of Disease Control (Taiwan CDC). Jurkat T cells, a line of human T lymphoma (ATCC no. CRL-8163) were kindly obtained from Charles Sia, Institute of Infectious Disease and Vaccinology, National Health Research Institute, Zhunan, Taiwan. pEF-human PSGL-1−, an expression vector of pEF6-Flag-3S (Belgian Coordinated Collections of Microorganisms) carrying a cDNA of human PSGL-1, transfected mouse L929 cells (PSGL-1-L929 cells), which are human PSGL-1 expressing EV71-susceptible stable cell clones, and the pEF-bsd−, a empty vector of pEF6-Flag-3S, transfected mouse L929 cells (bsd cells), an EV71-insusceptible control cells, were generously provided by Yorihiro Nishimura, Department of Virology II, National Institute of Infectious Diseases, Tokyo, Japan (9).
The RD and Jurkat T cells were cultured in a Dulbecco modified Eagle medium (DMEM) and RPMI medium (Gibco), respectively, containing 10% fetal bovine serum (Biological Industries) and 1% penicillin-streptomycin (Gibco). The PSGL-1-L929 and bsd cells were maintained in the previously described DMEM to which 5 μg of blasticidin S-HCl (Sigma-Aldrich)/ml was added.
Virus preparation.
The EV71/E59 substrain, a B4 substrain isolated by Taiwan CDC in 2002, was propagated in Vero cells using the microcarrier cell culture bioprocess. Briefly, the Vero cells were infected with EV71 virus at a multiplicity of infection (MOI) of 10−3. EV71 stocks were collected from microcarrier culture supernatants 6 to 8 days postinfection. Cell debris was removed by centrifugation at 3,000 rpm for 15 min, and then the supernatants were filtered through a 0.45-μm-pore-size membrane (Nalgene). Virus stocks were concentrated using an Amicon 100K tube (Millipore) and then stored at −80°C. The titer of virus stocks was tested by using a standard plaque-forming assay.
Pharmacological inhibition during EV71 infection.
Cells (2 × 105/well) were seeded in 12-well plates 1 day prior to exposure to serial concentrations of inhibitors (all from Sigma-Aldrich): chlorpromazine (CPZ), methyl β-cyclodextrin (MβCD), chloroquine, ammonium chloride (NH4Cl), genistein, filipin, and bafilomycin A1 in a serum-free DMEM and incubated for 2 h at 37°C. After drug treatment, the cells were infected with EV71 (MOI = 1) for 30 min at 4°C in the presence of the drugs. The unbound virus and drugs were washed with 1× phosphate-buffered saline (PBS) (pH 7.4) three times. The cells were cultured at 37°C for 3 h for RNA extraction.
Internalization of CT-B and transferrin.
Cells grown on coverslips in 12-well plates after inhibitors treatment as described in the previous paragraph were then cooled to 15°C and incubated with 5 μg of Alexa Fluor 594-conjugated CT-B/ml or 25 μg of Alexa Fluor 594-conjugated transferrin/ml for 30 min, followed by a 4-h additional incubation at 37°C. After washing the unbound indicators, the cells were fixed with cold methanol and then mounted. Images were captured by using a Leica TCS SP5 II confocal microscope.
Transfection of siRNA.
Gene-specific siRNA duplexes (Invitrogen) were diluted to 25 pmol (for RD cells) or 50 pmol (for PSGL-1-L929 cells) in a 200 μl of Opti-MEM I medium (Invitrogen) and then gently mixed with 1.82 μl or 3.7 μl of Lipofectamine RNAi-MAX (Lipo) reagents. The siRNA-Lipo mixtures were added to 800 μl of the appropriate numbers of cells cultured in 12-well plates in a complete culture medium to provide 30 to 50% confluence. Transfection of siRNA into Jurkat T cells was performed using the Neon transfection system (Invitrogen) according to the manufacturer's manual. Cells (2 × 106/reaction) were suspended with 90 μl of resuspension buffer mixed with 100 pmol of siRNA. Take up the mixture of cells and siRNA into the Neon pipette tip, followed by plugging into the pipette holder containing cuvette filled with 3 ml of electroporation buffer. The electroporation tip was pulsed at 1,400 voltage, 10-ms pulse width with three pulse numbers. The mixtures were then cultured at 37°C for 24 h to 48 h prior to whole cellular protein or total RNA isolation.
Real-time reverse transcription-PCR (RT-PCR).
Total RNA was extracted using TRIzol (Invitrogen) according to the manufacturer's manual. To perform the reverse transcription (RT) reaction, 1 μg of isolated RNA was mixed with 10 nM random hexamer (Genomics) and 1 U of SuperScript III reverse transcriptase (Invitrogen), and then incubated at 65°C for 5 min, 55°C for 55 min, and 70°C for 15 min. The synthesized cDNA was subjected to quantitative PCR analysis (The LightCycler 480 real-time PCR system) using respective primer pairs (Genomics BioSci&Tech, Taiwan) (Table 1). Human/mouse β-actin gene expression was also detected as the internal control. The number of cycles required for the amplification of the target gene was obtained and the relative expression of the target gene was calculated as follows: the individual threshold cycle (CT) obtained from the experimental group or control group was normalized by its respective CT (β-actin), and then 2normaliszed CT (experimental cell) was divided by 2normalized CT (control cell).
Table 1.
Sequences of primer pairs specific to target gene
| Target gene | Oligonucleotide |
|
|---|---|---|
| Orientation | Sequence (5′–3′) | |
| EV71 viral protein 1 (VP1) | Forward | GGTACCCCACGTTTGGAGA |
| Reverse | TAGGACACGCTCCATACTCAAG | |
| Mouse clathrin | Forward | GCGCCAAAGTGAACAGGTTGAGAA |
| Reverse | GCTTGCTGCTCTTGGGATTGAAGT | |
| Mouse caveolin-1 | Forward | TGTACCGTGCATCAAGAGCTTCCT |
| Reverse | GTGCTGATGCGGATGTTGCTGAAT | |
| Human clathrin | Forward | AACAAGATCAACAACCGGGCATCC |
| Reverse | GGACACATCTTTGCACTGCTTGCT | |
| Human caveolin-1 | Forward | AGTGCATCAGCCGTGTCTATTCCA |
| Reverse | TCTGCAAGTTGATGCGGACATTGC | |
| Human/mouse actin | Forward | ACCAACTGGGACGACATGGAGAAA |
| Reverse | TAGCACAGCCTGGATAGCAACGTA | |
Antibodies and Western blotting.
MAb979, a mouse monoclonal antibody that specifically recognized EV71 VP0/VP2 capsid proteins (Millipore-Chemicon International), and anti-CAV-1 monoclonal antibody (Cell Signaling Technology), anti-clathrin light-chain monoclonal antibody (Cell Signaling Technology), and anti-β-actin antibody (Sigma-Aldrich) were used. Whole-protein lysates were prepared by lysing cells in a radioimmunoprecipitation assay buffer adding a protease inhibitor cocktail (Roche) and 1 mM phenylmethylsulfonyl fluoride (PMSF) (Sigma-Aldrich). The lysates were centrifuged for 30 min at 14,000 rpm at 4°C to sediment cell debris. The protein concentrations of whole-cell protein lysates were quantified using the Bradford protein assay. Fifty microgram of whole-cell protein was resolved by electrophoresis in an SDS-polyacrylamide gel (Amersham Biosciences) and transferred to a nitrocellulose membrane (Hybond-ECL; Amersham Biosciences). After blocking, the membrane was incubated with the primary antibody: anti-EV71 antibody (diluted 1:1,000), anti-clathrin antibody (1:250), anti-caveolin-1 antibody (1:1,000), or anti-β-actin antibody (1:10,000) at 4°C overnight. After incubation, the diluted secondary antibody, either horseradish peroxidase (HRP)-conjugated anti-mouse (1:10,000; Jackson ImmunoResearch) or anti-rabbit antibody (1:10,000; Jackson ImmunoResearch), was added, followed by incubation for 2 h at 4°C. After four washes with 1× PBS plus 0.1% Tween 20, chemiluminescence on the membrane was developed with substrate (Millipore), and detected using X-ray film (Kodak). The densitometry of specific blots was calculated using Image-Pro Plus 6.0 software.
Immunofluorescence.
Immunofluorescence assays were conducted on cells, and growth on coverslips after EV71 incubation occurred as described previously. After a wash with PBS, the cells were fixed with cold methanol and then incubated with a mixture of primary antibodies, i.e., anti-EV71 antibody (1:1,000) accompanied by anti-CAV-1 (1:500). The fluorescence signals were then examined by hybridization of secondary antibodies—fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG antibody (1:100; BioLegend) accompanied by Alexa Fluor 594-conjugated anti-rabbit IgG antibody (1:200; Invitrogen)—and the nuclei were stained using DAPI (4′,6′-diamidino-2-phenylindole). Signals were detected with using a Leica TCS SP5 II confocal microscope. Measurement of colocalization of viral protein and caveolin-1 in dual-color images was performed using a Delta Vision OMX Super-Resolution imaging system (Genetech Biotech).
Flow cytometry.
A total of 2 × 106 suspended RD cells detached by treating with 1× PBS containing 2% EDTA and Jurkat T cells were preincubated with human Fc receptor binding inhibitor (eBioscience) in 200 μl of 1× PBS with 2% bovine serum at 4°C for 20 min and then biotinylated anti-human SCARB2 antibody (1:50; R&D Systems) or anti-PSGL-1 antibody (clone KPL-1, 1:500; Millipore) were added and continue incubation at 4°C overnight. Cells incubated with the respective isotype IgG (eBioscience) as a control were included. After the incubation, cells were spin down at 5,000 rpm for 3 min and then washed with 1 ml of cold 1× PBS for three times. Cells were stained with FITC-conjugated diluted avidin (1:100 [Biolegend] for biotinylated anti-human SCARB2 antibody) or Alexa Fluor 488-conjugated anti-mouse antibody (1:1,000 [Invitrogen] for anti-PSGL-1 antibody) and incubated at 4°C for 1 h. After three washes in 1× PBS, the stained cells were resuspended in 1× PBS, and signals were detected using a FACScan flow cytometer and analyzed using FCS Express V3 software (De Novo Software).
Assay of cytotoxicity.
The cytotoxicity of treatments in RD and PSGL-1-L929 cells was assessed using the lactate dehydrogenase (LDH) assay (Promega CytoTox 96 kit). Culture supernatants from cells subjected to siRNA transfection or inhibitor treatment for 48 or 2 h, respectively, were harvested for the assay performed according to the manufacturer's instructions. The cytotoxicity of electroporation for siRNA transfection into Jurkat T cells was assessed by dye-exclusion method.
Statistical analysis.
Unpaired two-tailed Student t tests or one-way analysis of variance with the Tukey post test were conducted to compare results obtained from the different experimental groups. A P value of <0.05 was considered statistically significant. The symbols *, **, and §, indicate P values < 0.05, <0.01, and <0.001, respectively.
RESULTS
Expression of human PSGL-1 in mouse L929 cells confers susceptibility to EV71/E59.
We first confirmed that human PSGL-1 expression facilitates EV71 infection in unsusceptible mouse L929 cells and identified the mechanism for EV71 entry into PSGL-1-L929 cells.
We individually inoculated PSGL-1-L929 cells and empty vector-transducing bsd cells with EV71 at various MOIs and compared them to the same infection in EV7-susceptible RD cells. We examined the kinetics of viral entry by evaluating the expression of the EV71 transcripts and capsid proteins in infected cells using quantitative real-time RT-PCR and Western blot analyses. In contrast to the observation of undetectable VP0/VP1 capsid proteins in EV71-infected bsd cells, we observed the expression of capsid proteins or viral RNA in both EV71-infected PSGL-1-L929 and RD cells (Fig. 1A and B). Increases in the levels of the capsid proteins or viral RNA expression were relative to increases in the MOI of EV71 (from 0.1 to 10), in which capsid proteins were detectable at minimum MOI of 1 of EV71 in PSGL-1-L929 cells (the upper panel of Fig. 1A). In the kinetics of infection, capsid proteins were detected in RD cells as early as 3 h postinfection, in contrast to undetected in PSGL-1-L929 cells until 6 h postinfection (the bottom panel of Fig. 1A). Viral RNA was increased at 3 h postinfection in both cell types, whereas almost 3.5-fold (RD cells) and 1.5-fold (PSGL-1-L929 cells) increases were observed and significantly increased at 6 h postinfection (upper panel of Fig. 1B). In addition, viral RNA from infected RD and PSGL-1-L929 but not bsd cells showed a dose-dependent increase (MOI = 1 and up) at 3 h postinfection (bottom panel of Fig. 1B). Then, to examine EV71 viral growth kinetics, we infected bsd, PSGL-1-L929, and RD cells with EV71 at an MOI of 1, and virus production was detected by plaque-forming assay. EV71 grew efficiently in PSGL-1-L929 cells, in which the virus titer was peaked on day 3 after infection compared to the viral loads peaked on day 2 in RD cells. EV71 did not grow in bsd cells (Fig. 1C). The results show that RD cells elicit more susceptibility to EV71 infection than PSGL-1-L929 cells. RD cell susceptibility to EV71 infection was conferred by hSCARB2 but not by PSGL-1 expression on the surface (see Fig. S1A in the supplemental material), whereas pretreatment of hSCARB2 siRNA was able to abrogate EV71 infection (see Fig. S1C in the supplemental material). The downregulation of endogenous hSCARB2 mRNA by specific siRNA was confirmed (see Fig. S1B in the supplemental material). These results indicate that human PSGL-1 expression confers susceptibility to EV71 infection in mouse L929 cells and hSCARB2 supports RD cells to be infected by EV71. Because of the limitation of immunoblot assay in detecting the viral proteins as early as 3 h postinfection, real-time RT-PCR with primers against the VP1 region (Table 1) was used to quantify viral RNA in the following studies.
Fig 1.

Human PSGL-1-expressing mouse L929 cells are susceptible to EV71 infection. (A) Lysates were prepared at 24 h postinfection of bsd, PSGL-1-L929, and RD cells with EV71 (serial MOI), incubated at 4°C for 30 min (upper panel), or infection with EV71 (MOI = 1) and then cultured at 37°C for additional incubation from 0, 0.5, 1, 3, 6, and 24 h (bottom panel). Lysates were subjected to immunoblotting with MAb979 antibody to detect VP capsid proteins as described in the section of Materials and Methods. (B) The total RNA from the infected cells (MOI = 1) at each time point (upper panel) or from the infected cells (serial MOI) at 3 h postinfection (bottom panel) were isolated to detect the viral RNA using real-time RT-PCR with the primers against VP1 region (Table 1). The mean relative viral RNA compared to the expression of the β-actin internal control was calculated as described in Materials and Methods. The data were obtained from three independent experiments. (C) bsd, PSGL-1-L929, and RD cells were infected with EV71 at an MOI of 1. Virus titers in infected cells were determined by standard plaque-forming assay at 6, 24, 48, and 72 h after infection. The data obtained from two independent experiments were shown as the mean PFU ± the standard deviations (SD).
Activation of clathrin-independent EV71 entry in PSGL-1-expressing cells and clathrin-dependent EV71 entry in RD cells.
EV71 infection triggered clathrin-dependent endocytosis in hSCARB2-expressing cells (24, 25). However, it remains unclear whether the same endocytic pathway is used for infection in PSGL-1-expressing cells. We further investigated the mechanism of EV71 infection in different cellular receptor-expressing cells and the tissue tropism of EV71 infection. To address this, we treated cellular proteins participating in the endocytic pathways of RD and PSGL-1-L929 cells with specific inhibitors. Chlorpromazine (CPZ), a cationic amphiphilic reagent that inhibits the assembly of the clathrin adaptor protein AP2, is an inhibitor of clathrin-dependent endocytosis (26). In the present study, we used the internalization of the Alexa Fluor 594-conjugated transferrin (red) as an indicator of clathrin-mediated endocytosis in EV71-infected cells with or without CPZ treatment (27). Alexa Fluor 594-conjugated transferrin was incubated with drug-treated cells at 16°C for 30 min to allow binding and internalization. The unbound fluorescent transferrin was washed away before cell fixing. The fluorescent transferrin was distributed in the cytosol and surface membranes of vehicle-treated RD and PSGL-1-L929 cells. Pretreatment of CPZ significantly inhibited the internalization of the fluorescent transferrin that resulted in the reduction of the fluorescent signals in both cytosol and surface membranes of cells (Fig. 2A). This indicated that CPZ pretreatment effectively disrupted clathrin-dependent endocytosis in the RD and PSGL-1-L929 cells. We then treated RD cells with various concentrations of CPZ prior to EV71 infection (MOI = 1), observing that viral RNA was reduced significantly (ca. 70% inhibition) by 30 μM CPZ treatment at 3 h postinfection. In contrast, PSGL-1-L929 cells pretreated with the same dosage of CPZ showed mild changes (30% inhibition) in viral RNA expression (Fig. 2B). This indicates that the clathrin-mediated pathway contributes to EV71 infection in PSGL-1-expressing cells only partially.
Fig 2.

Inhibitors specific to clathrin-dependent endocytosis abrogate EV71 infection in RD but not PSGL-1-L929 cells. The PSGL-1-L929 and RD cells cultured on coverslips were treated with 30 μM CPZ and 75 μM MβCD in a serum-free DMEM at 37°C for 2 h. Then, 5 μg of Alexa Fluor 594-conjugated transferrin (red) was then added to the culture medium in the presence of inhibitors and incubated at 16°C for 30 min. (A) After washing with PBS and fixing with cold methanol, the cell nuclei were stained with DAPI (blue), and the fluorescence signal was detected using a Leica TCS SP5 II confocal microscope. (B) The EV71-infected cells (MOI = 1) were subjected to 2-h drug treatment at 4°C for 30 min, and the drug's cytotoxicity was evaluated using an LDH assay. Cells were incubated for 3 h postinfection, prior to the isolation of total RNA and the performance of real-time RT-PCR to detect viral RNA. The mean relative viral RNA was calculated as described previously.
To further confirm the occurrence of clathrin-mediated endocytosis in EV71 infection in RD cells but not in PSGL-1-L929 cells, we used clathrin light-chain B (CLTB)-specific siRNA to knockdown endogenous clathrin expression in both cell types. We observed that clathrin mRNA in RD and PSGL-1-L929 cells was significantly inhibited at 24 h (data not shown) or 48 h after siRNA transfection compared to the control siRNA-transfected cells (Fig. 3A). The significant inhibition of clathrin protein was also observed in RD cells at 24 or 48 h after siRNA transfection but was only observed in PSGL-1-L9292 cells at 48 h posttransfection (Fig. 3B). After EV71 infection, the expression of viral RNA at 3 h postinfection (Fig. 3C) and capsid proteins at 24 h postinfection (Fig. 3D) in CLTB siRNA-treated RD cells were reduced significantly compared to control siRNA-treated RD cells. In contrast, PSGL-1-L929 cells revealed no differences in viral RNA (Fig. 3C) or capsid proteins (Fig. 3D) expression while transfected with CLTB siRNA. These results indicate that the mechanism of EV71 infection in PSGL-1-L929 cells is clathrin independent; in contrast, clathrin-dependent endocytosis occurred in RD cells.
Fig 3.

Clathrin light-chain B-specific siRNA inhibits EV71 infection in RD but not PSGL-1-L929 cells. Cells were transfected with 50 pmol of human (for RD cells) or mouse (for PSGL-1-L929 cells) CLTB-specific siRNA and then incubated at 37°C for 48 h. (A) Clathrin mRNA expression was quantified by real-time RT-PCR using specific primers (Table 1). (B) Protein expression was quantified by Western blotting with anti-clathrin antibody. Changes in clathrin expression at 24 and 48 h after transfection were compared to the expression in control siRNA-transfected cells (as 100%). The cytotoxicity of siRNA was evaluated using LDH assay after 48 h transfection. After the cells were infected with EV71 (MOI = 1) and incubated at 4°C for 30 min. After washing three times with PBS, cells were recovered in a culture medium at 37°C for an additional 3 h for total RNA extraction or 24 h for lysate preparation. Total RNA (C) and lysates (D) were extracted and subjected to real-time RT-PCR and Western blotting with MAb979 antibody. The cellular β-actin internal control was detected by blotting the same membrane with monoclonal anti-β-actin antibody. The mean relative expression of RNA and protein levels of clathrin and EV71 were calculated as described previously. The data were quantified using Image-Pro Plus 6.0 software and are representative of three independent experiments.
Requirement for intact membrane cholesterol in EV71 entry.
Receptor-mediated endocytosis for the entry of viruses, such as herpes simplex virus (27) and poliovirus (28), requires the presence of cholesterol in the lipid raft of the cell membrane. To confirm the requirement for intact membrane cholesterol in both clathrin- and caveola-mediated endocytosis, we removed cholesterol from the plasma membrane of cells by treating them with methyl-β-cyclodextrin (MβCD), a cyclic oligosaccharide with seven hydrophobic molecule-binding pockets. In RD and PSGL-1-L929 cells pretreated with 5 mM MβCD, the internalization of Alexa Fluor 594-conjugated transferrin was markedly suppressed compared to vehicle-treated cells (Fig. 2A). At 3 h after EV71 infection, the expression of viral RNA in both MβCD-treated RD and PSGL-1-L929 cells was inhibited significantly but remained unchanged in vehicle-treated cells (Fig. 4). These data suggest that PSGL-1- and SCARB2-mediated EV71 infection both require the presence of membrane cholesterol on the cell surface.
Fig 4.
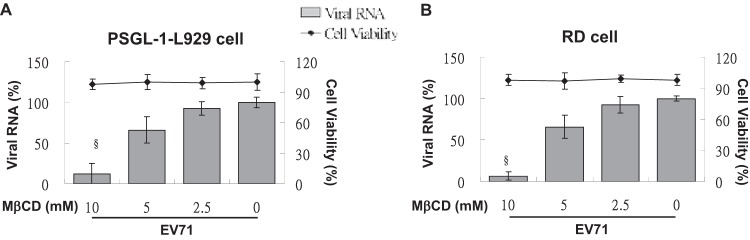
Treatment with MβCD disrupts EV71 infection. PSGL-1-L929 and RD cells were treated with various concentrations of MβCD in a serum-free medium at 37°C for 2 h, and then the cytotoxicity of drug treatment was evaluated using an LDH assay. After EV71 (MOI = 1) infection, the cells were incubated at 37°C for 3 h and lysed prior to RNA isolation. The level of viral RNA in each group was detected using real-time RT-PCR, and the mean relative expression compared to untreated cells was calculated as described previously. The data were obtained from three independent experiments.
Caveola-dependent endocytosis is required for EV71 infection in PSGL-1-L929 cells but not in RD cells.
Several studies have shown that clathrin-independent caveolar endocytosis can be disrupted by genistein, a tyrosine kinase inhibitor (29, 30), or filipin, a cholesterol-binding agent (30). To investigate the role of caveolar endocytosis in EV71 infection, we treated RD and PSGL-1-L929 cells with various concentrations of genistein or filipin prior to EV71 infection. Because cholera toxin B (CT-B) uptake is caveola dependent, we used the internalization of Alexa Fluor 594-conjugated CT-B as an indicator of endogenous caveolar endocytosis (20, 30). The procedure to assay internalization for Alexa Fluor 594-conjugated CT-B was similar to the experiment for Alexa Fluor 594-conjugated transferrin internalization (Fig. 2A). After pretreatment of the cells with an effective dose of genistein (40 μg/ml) or filipin (2.5 μg/ml), internalization of Alexa Fluor 594-conjugated CT-B into the cytosol was reduced significantly in RD and PSGL-1-L929 cells compared to vehicle-pretreated cells (Fig. 5A). We determined the influence of this inhibition on early EV71 infection in PSGL-1-L929 and RD cells by measuring the viral RNA (Fig. 5B and D) and capsid proteins using immunohistochemistry (brown color, Fig. 5C and E), compared to the cells treated with vehicle. We observed that pretreatment of PSGL-1-L929 cells with an effective dose of 20 μg of genistein (Fig. 5B)/ml or 0.75 μg of filipin (Fig. 5D)/ml showed significant inhibition in contrast to the no inhibition observed in RD cells even when pretreated with a maximum dose of genistein (40 μg/ml) or filipin (3 μg/ml). The result from the immunohistochemistry staining of viral capsid proteins consistently showed that genistein or filipin at a maximum dose of significantly inhibited EV71 infection in PSGL-1-L929 cells but not in RD cells (Fig. 5C and E).
Fig 5.

Inhibitors specific to caveolar endocytosis abrogate EV71 infection in PSGL-1-L929 but not RD cells. The PSGL-1-L929 and RD cells were cultured on coverslips and treated with 40 μg of genistein/ml and 2.5 μg of filipin/ml in a serum-free DMEM at 37°C for 2 h. Alexa Fluor 594-conjugated CT-B (2.5 μg; red) was then added to the culture medium in the presence of inhibitors and incubated at 16°C for 30 min, followed by 37°C incubation for 4 h. (A) After washing with PBS and fixing with cold methanol, cell nuclei were stained with DAPI (blue), and fluorescence was detected using a confocal microscope Leica TCS SP5 II. (B and D) Genistein (B)- and filipin (D)-pretreated cells were further infected with EV71 (MOI = 1). The cytotoxicity of the inhibitors was evaluated using an LDH assay. At 3 h after EV71 infection, the total RNA was isolated from the drug-treated cells and subjected to real-time RT-PCR to detect viral RNA. The mean expression of viral RNA relative to untreated cells was calculated as described previously. The data were obtained from three independent experiments. (C and E) Immunohistochemistry of PSGL-1-L929 and RD cells individually pretreated with or without 40 μg of genistein/ml (C) and 3 μg of filipin/ml (E) after EV71 infection was performed with MAb979 antibody and DAB for colorization. The pictures were taken at ×200 magnification from three independent experiments, and the results for one of the experiments are shown.
To further confirm the role of caveolae in EV71 infection, RD and PSGL-1-L929 cells pretreated with caveolin-1 (CAV-1)-specific siRNA prior to EV71 infection were performed. The results showed that the levels of CAV-1 mRNA (Fig. 6A) and protein (Fig. 6B) expression in RD or PSGL-1-L929 cells were downregulated significantly at 48 h after siRNA transfection, compared to the control siRNA-transfected cells. Both cell types displayed no evidence of transfection-induced cytotoxicity. After EV71 infection, the expression of EV71 RNA (Fig. 6C) and its capsid proteins (Fig. 6D) were markedly reduced in CAV-1 siRNA-treated PSGL-1-L929 cells compared to the control siRNA transfectants. In contrast, CAV-1 siRNA and control siRNA-transfected RD cells displayed no differences in viral RNA and capsid proteins expression (Fig. 6C and D). These results suggest that the PSGL-1-mediated pathway for EV71 infection is caveola dependent, unlike the SCARB2-mediated clathrin-dependent pathway.
Fig 6.

Caveolin-1-specific siRNA abrogates EV71 infection in PSGL-1-L929 but not RD cells. Cells were transfected with mouse (for PSGL-1-L929 cells)- and human CAV-1 (for RD cells)-specific siRNA or control siRNA. The cells were then cultured for 24 and 48 h prior to the detection of CAV-1 mRNA using real-time RT-PCR with CAV-1-specific primers (Table 1) (A) and protein with Western blotting with anti-CAV-1 antibody (B). After 48 h of incubation, the cytotoxicity of siRNA transfection was evaluated using LDH assay. After EV71 (MOI = 1) infection, the levels of viral RNA (C) and capsid proteins (D) in siRNA-treated cells were evaluated by real-time RT-PCR and Western blotting after 3 or 24 h of cell culture, respectively, as described previously. The mean relative expression of viral RNA and capsid proteins compared to untreated cells was calculated. The data were obtained from three independent experiments.
Confocal microscopy of immunofluorescence staining showed that both VP1 (green) and CAV-1 (red) distributed throughout the whole cells at 3 h postinfection. Merging of VP1 and CAV-1 revealed many colocalizations of CAV-1 and EV71 (yellow) observed in the cytosol and submembrane sites of EV71-infected PSGL-1-L929 cells but few in EV71-infected RD cells (Fig. 7A). The colocalizations were quantified, and the score of Pearson coefficient of correlation (31), which correlated to the colocalization of CAV-1 and EV71, was calculated. More colocalizations in PSGL-1-L929 cells versus RD cells (0.5377 versus 0.2814 coefficient factor) were observed (Fig. 7B). These data, combined with results from inhibitory assays (Fig. 5 and 6), support that caveola-dependent endocytosis is essential for PSGL-1-mediated EV71 early infection.
Fig 7.

EV71 and caveosomes are colocalized in PSGL-1-L929 but not in RD cells. (A) Immunofluorescence analysis of EV71 particles (green) was performed using VP-specific antibody (MAb979), followed by FITC-coupled secondary antibody. Caveosome immunofluorescence (red) was stained with caveolin-1-specific antibody, followed by Alexa Fluor 594-coupled secondary antibody. Analyses were conducted on PSGL-1-L929 and RD cells infected with EV71 (MOI = 5) and incubated for 3 h at 37°C. The pictures were taken at ×200 magnification from three independent experiments, and the results for one of the experiments are shown. (B) Quantification of colocalized EV71 and caveosomes in the cells was performed, and the results are plotted by using a Delta Vision OMX Super-Resolution imaging system (Genetech Biotech). The colocalization of the Pearson coefficient factor was calculated.
EV71 infection mediates a caveola-dependent but not clathrin-dependent pathway in PSGL-1-expressing human T cells.
Human T cells such as Jurkat T cells only expressing PSGL-1 on the surface was confirmed by previous report (9) and by our flow cytometric analysis using specific antibody against PSGL-1 compared to SCARB2 antibody (Fig. 8A). In the kinetic infection, Jurkat T cells was susceptible for EV71 infection (MOI = 0.05) was judged by the detection of viral RNA expression as early as at 6 h and significant increase at 24 h postinfection (Fig. 8B). Pretreatment of Jurkat T cells with either CLTB or CAV-1 siRNA all downregulated the respective mRNA (Fig. 8C) and protein (Fig. 8D) expression after 48 h transfection. At 24 h after EV71 infection, viral RNA (Fig. 8E) and their capsid protein (Fig. 8F) expression were measured, and all were reduced significantly in CAV-1 siRNA-transfected cells but remained unchanged in the CLTB siRNA- or control siRNA-transfected cells. These results indicate that the pathway of EV71 infection in Jurkat T cells is caveola dependent and clathrin independent.
Fig 8.

EV71 infection is caveola dependent in Jurkat T lymphocytes. (A) The Jurkat T cells were incubated with biotinylated anti-SCARB2 antiserum or anti-PSGL-1 antiserum (black curve) prior stained with FITC-conjugated avidin or anti-mouse antibody, respectively. The Jurkat T cells incubated with the respective isotype IgG (gray curve) as controls were included. Stained cells were suspended in 1× PBS before run on a FACScan flow cytometer and analyzed using FCS Express V3 (De Novo Software). (B) Cells were infected with EV71 (MOI = 0.05) and then cultured at 37°C for additional incubation from 0, 0.5, 1, 3, 6, and 24 h. Total RNA were extracted at each time point postinfection and used to detect the viral RNA by real-time RT-PCR as described previously. Cells were transfected with 100 pmol of human CLTB-, CAV-1-specific siRNA or control siRNA and then cultured at 37°C for 48 h. After incubation, total RNAs were isolated to assay clathrin or caveolin-1 mRNA by real-time RT-PCR using the respective primers (Table 1) (C) and protein expression by immunoblotting with anti-clathrin or anti-caveolin-1 antibody (D). Changes in clathrin or caveolin-1 expression were compared to the expression in control siRNA-transfected cells (as 100%). After EV71 (MOI = 0.05) infection, total RNA and lysates were prepared at 24 h postinfection to detect viral RNA (E) and capsid proteins (F) as described previously. The mean relative expression of RNA and protein levels of clathrin, caveolin-1, and EV71 were calculated as described previously. The cellular β-actin detected as internal control for the findings shown in panels E and F was included. The data were quantified using Image-Pro Plus 6.0 software. Three independent experiments were performed, and one of them were shown.
EV71 entry into PSGL-1-expressing cells requires lysosomal acidification.
After the endocytosis of viral particles, acidification in the endosome is the key step for rearrangement of the viral capsid structure, resulting in the release of viral RNA from the particles for replication (32, 33). To elucidate the role of endosomal acidification in PSGL-1-mediated EV71 infection, we used lysosomotropic agents, the compounds ammonium chloride (NH4Cl) and chloroquine, which have weak bases and can inhibit endosomal acidification by increasing intracellular pH (34–36), and bafilomycin A1, which acts by inhibiting vacuolar H+ ATPase (V-ATPase) that prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes (37) in our study. Pretreatment of PSGL-1-L929 and RD cells with various doses of NH4Cl (Fig. 9A), chloroquine (Fig. 9B), and bafilomycin A1 (Fig. 9C) prior to EV71 infection was performed. No cytotoxicity of NH4Cl, chloroquine, and bafilomycin A1 treatments in both infected cells was noted. The results of real-time RT-PCR analysis indicated that pretreatment of RD and PSGL-1 cells with NH4Cl, chloroquine, and bafilomycin A1 significantly inhibited EV71 infection in a dose-dependent manner. These data confirmed that endosome-lysosomal acidification is required for clathrin-dependent EV71 infection in RD cells and also for caveola-dependent infection in PSGL-1-L929 cells.
Fig 9.

EV71 infection is pH dependent in PSGL-1-L929 and RD cells. Various concentrations of chloroquine (A), NH4Cl (B), and bafilomycin A1 (C) were individually added to PSGL-1-L929 and RD cells prior to infection with EV71 (MOI = 1). Cells were incubated for 3 h, and total RNA was isolated prior to the evaluation of the viral transcripts using real-time RT-PCR. The cytotoxicity of inhibitors was evaluated using LDH assay. The mean relative viral RNA compared to untreated cells was calculated. The data were obtained from three independent experiments.
DISCUSSION
Beside enterovirus 71, several viruses also use multiple cellular receptors for host entry, such as PVRL4 (nectin 4) and SLAM (CDw150) in the measles virus (38, 39), angiotensin-converting enzyme 2 and CD209L (L-SIGN) in the severe acute respiratory syndrome coronavirus (40, 41), and ubiquitous glucose transporter GLUT-1 and neurophilin-1 in human T-cell lymphotropic virus type 1 (42, 43). Previous studies identified the requirement for clathrin-dependent endocytosis in EV71 infection of human SCARB2-expressing mouse NIH 3T3 cells and human RD cells (24, 25). In the present study, we demonstrated that caveola-mediated endocytosis is required for EV71 entry in human PSGL-1-expressing cells. These results suggest that differential cellular receptors determine the pathway of endocytosis in EV71 infection. Investigators have observed a similar phenomenon in herpes simplex virus (HSV) infection, which uses nectin-1 receptors and the TNF/NGF receptor family for host entry (44, 45). The differential expression of cellular receptors in different cells might also determine the tissue tropism of EV71 infection in the host. Similarly, HSV infection uses distinct pathways for entry and eliciting tissue tropism (46), suggesting that receptor-determining factors in a target cell with multiple cellular receptors have a major influence on viral infection.
In contrast to the ubiquitous expression of SCARB2 in somatic cells, PSGL-1 is restrictedly expressed in myeloid, lymphoid, and dendritic cells and serves as a common ligand for P-, L-, or E-selectin in platelets, leukocytes, or endothelium cells. It plays a role in leukocyte and inflammatory cell infiltration in inflammatory tissues (14, 15). P-selectin glycoprotein ligand-1 has been identified as one of the receptors to which EV71 binds when it infects myeloid, lymphoid, and dendritic cells. Nishimura et al. identified that a monoclonal antibody (KLP1) can neutralize the binding between EV71 and PSGL-1 by blocking the recognition sites for P- and L-selectin (9). The binding site for the viral particle on PSGL-1 is identical to the selectin binding site that is responsible for cellular signal transduction (9), suggesting that the binding of EV71 might initiate signals for the recruitment of EV71-infected leukocytes or neutrophils to infiltrate the relevant tissues and release the replicating viruses. Clinical studies have detected large amounts of neutrophil-like inflammatory cells (CD68+, CD15−) and three types of lymphocytes (B cells, CD4+, and CD8+ T cells) in the CNS of severe EV71-infected patients (47, 48). These studies also observed EV71 particles in the CNS of these patients. However, it remains unclear whether PSGL-1 expressed on inflammatory cells mediates EV71 infection and also triggers infiltration into the CNS to result in severe neurological diseases.
Previous studies have shown that the binding of the PSGL-1 ligand (PSGL-1L) to PSGL-1 activates phosphoinositide 3-kinase, Src, or mitogen-activated protein kinase signaling, resulting in actin redistribution and interleukin secretion (49–51). Recent studies described the recruitment and activation of leukocytes and the generation of CD4+ CD25+ Foxop3+ regulatory T cells and tolerogenic dendritic cells in tolerogenic immune responses after PSGL-1–PSGL-1L engagement (52, 53). The role of PSGL-1 in EV71 infection, such as the induction of tolerogenic immune responses following the binding of EV71 to PSGL-1 that enable the virus to escape host defenses, has yet to be fully elucidated. In the present, we demonstrate that the mechanism of EV71 entry is receptor dependent, in which human PSGL-1 initiates caveola-dependent endocytosis in parallel with the activation of clathrin-dependent endocytosis by human SCARB2. These findings might facilitate the development of anti-EV71 medications.
Y.-H.C. conceived and designed the experiments. H.-Y.L., Y.-T.Y., K.-N.H., and S.-L.Y. performed the experiments. Y.-H.C. and H.-Y.L. analyzed the data. C.-C.L. contributed reagents, materials, and analysis tools. Y.-H.C. and H.-Y.L. wrote the paper. C.S. edited the manuscript.
Supplementary Material
ACKNOWLEDGMENTS
This study was supported by grants (101-2311-B-400-002- and 101-2325-B-400-024-CC2) from the Taiwan National Science Council (http://web1.nsc.gov.tw/).
None of the authors have any commercial or other association that might pose a conflict of interest.
Footnotes
Published ahead of print 12 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.00573-13.
REFERENCES
- 1. Manki A, Oda M, Seino Y. 1997. Neurologic diseases of enterovirus infections: polioviruses, coxsackieviruses, echoviruses, and enteroviruses type 68 to 72. Nihon Rinsho 55:849–854 (In Japanese.) [PubMed] [Google Scholar]
- 2. Ho M, Chen ER, Hsu KH, Twu SJ, Chen KT, Tsai SF, Wang JR, Shih SR. 1999. An epidemic of enterovirus 71 infection in Taiwan. Taiwan Enterovirus Epidemic Working Group. N. Engl. J. Med. 341:929–935 [DOI] [PubMed] [Google Scholar]
- 3. Liu CC, Tseng HW, Wang SM, Wang JR, Su IJ. 2000. An outbreak of enterovirus 71 infection in Taiwan, 1998: epidemiologic and clinical manifestations. J. Clin. Virol. 17:23–30 [DOI] [PubMed] [Google Scholar]
- 4. Wang SM, Liu CC, Tseng HW, Wang JR, Huang CC, Chen YJ, Yang YJ, Lin SJ, Yeh TF. 1999. Clinical spectrum of enterovirus 71 infection in children in southern Taiwan, with an emphasis on neurological complications. Clin. Infect. Dis. 29:184–190 [DOI] [PubMed] [Google Scholar]
- 5. Blomberg J, Lycke E, Ahlfors K, Johnsson T, Wolontis S, von Zeipel G. 1974. New enterovirus type associated with epidemic of aseptic meningitis and-or hand, foot, and mouth disease. Lancet ii:112 (Letter.) [DOI] [PubMed] [Google Scholar]
- 6. Hagiwara A, Tagaya I, Yoneyama T. 1978. Epidemic of hand, foot and mouth disease associated with enterovirus 71 infection. Intervirology 9:60–63 [DOI] [PubMed] [Google Scholar]
- 7. Qiu J. 2008. Enterovirus 71 infection: a new threat to global public health? Lancet Neurol. 7:868–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yamayoshi S, Yamashita Y, Li J, Hanagata N, Minowa T, Takemura T, Koike S. 2009. Scavenger receptor B2 is a cellular receptor for enterovirus 71. Nat. Med. 15:798–801 [DOI] [PubMed] [Google Scholar]
- 9. Nishimura Y, Shimojima M, Tano Y, Miyamura T, Wakita T, Shimizu H. 2009. Human P-selectin glycoprotein ligand-1 is a functional receptor for enterovirus 71. Nat. Med. 15:794–797 [DOI] [PubMed] [Google Scholar]
- 10. Brodeur MR, Brissette L, Falstrault L, Luangrath V, Moreau R. 2008. Scavenger receptor of class B expressed by osteoblastic cells are implicated in the uptake of cholesteryl ester and estradiol from LDL and HDL3. J. Bone Miner Res. 23:326–337 [DOI] [PubMed] [Google Scholar]
- 11. de Villiers WJ, Smart EJ. 1999. Macrophage scavenger receptors and foam cell formation. J. Leukoc. Biol. 66:740–746 [DOI] [PubMed] [Google Scholar]
- 12. Thilakawardhana S, Everett DM, Murdock PR, Dingwall C, Owen JS. 2005. Quantification of apolipoprotein E receptors in human brain-derived cell lines by real-time polymerase chain reaction. Neurobiol. Aging. 26:813–823 [DOI] [PubMed] [Google Scholar]
- 13. Yamayoshi S, Koike S. Identification of a human SCARB2 region that is important for enterovirus 71 binding and infection. J. Virol. 85:4937–4946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Laszik Z, Jansen PJ, Cummings RD, Tedder TF, McEver RP, Moore KL. 1996. P-selectin glycoprotein ligand-1 is broadly expressed in cells of myeloid, lymphoid, and dendritic lineage and in some nonhematopoietic cells. Blood 88:3010–3021 [PubMed] [Google Scholar]
- 15. Somers WS, Tang J, Shaw GD, Camphausen RT. 2000. Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLe(X) and PSGL-1. Cell 103:467–479 [DOI] [PubMed] [Google Scholar]
- 16. Nishimura Y, Wakita T, Shimizu H. Tyrosine sulfation of the amino terminus of PSGL-1 is critical for enterovirus 71 infection. PLoS Pathog. 6:e1001174. 10.1371/journal.ppat.1001174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cureton DK, Massol RH, Whelan SP, Kirchhausen T. The length of vesicular stomatitis virus particles dictates a need for actin assembly during clathrin-dependent endocytosis. PLoS Pathog. 6:e1001127. 10.1371/journal.ppat.1001127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Gastaldelli M, Imelli N, Boucke K, Amstutz B, Meier O, Greber UF. 2008. Infectious adenovirus type 2 transport through early but not late endosomes. Traffic 9:2265–2278 [DOI] [PubMed] [Google Scholar]
- 19. Peng T, Wang JL, Chen W, Zhang JL, Gao N, Chen ZT, Xu XF, Fan DY, An J. 2009. Entry of dengue virus serotype 2 into ECV304 cells depends on clathrin-dependent endocytosis, but not on caveolae-dependent endocytosis. Can. J. Microbiol. 55:139–145 [DOI] [PubMed] [Google Scholar]
- 20. Le PU, Nabi IR. 2003. Distinct caveolae-mediated endocytic pathways target the Golgi apparatus and the endoplasmic reticulum. J. Cell Sci. 116:1059–1071 [DOI] [PubMed] [Google Scholar]
- 21. Macovei A, Radulescu C, Lazar C, Petrescu S, Durantel D, Dwek RA, Zitzmann N, Nichita NB. Hepatitis B virus requires intact caveolin-1 function for productive infection in HepaRG cells. J. Virol. 84:243–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Pelkmans L, Puntener D, Helenius A. 2002. Local actin polymerization and dynamin recruitment in SV40-induced internalization of caveolae. Science 296:535–539 [DOI] [PubMed] [Google Scholar]
- 23. Richterova Z, Liebl D, Horak M, Palkova Z, Stokrova J, Hozak P, Korb J, Forstova J. 2001. Caveolae are involved in the trafficking of mouse polyomavirus virions and artificial VP1 pseudocapsids toward cell nuclei. J. Virol. 75:10880–10891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lin YW, Lin HY, Tsou YL, Chitra E, Hsiao KN, Shao HY, Liu CC, Sia C, Chong P, Chow YH. Human SCARB2-mediated entry and endocytosis of EV71. PLoS One 7:e30507. 10.1371/journal.pone.0030507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hussain KM, Leong KL, Ng MM, Chu JJ. The essential role of clathrin-mediated endocytosis in the infectious entry of human enterovirus 71. J. Biol. Chem. 286:309–321 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wang LH, Rothberg KG, Anderson RG. 1993. Mis-assembly of clathrin lattices on endosomes reveals a regulatory switch for coated pit formation. J. Cell Biol. 123:1107–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Rothenberger S, Iacopetta BJ, Kuhn LC. 1987. Endocytosis of the transferrin receptor requires the cytoplasmic domain but not its phosphorylation site. Cell 49:423–431 [DOI] [PubMed] [Google Scholar]
- 28. Rahn E, Petermann P, Hsu MJ, Rixon FJ, Knebel-Morsdorf D. Entry pathways of herpes simplex virus type 1 into human keratinocytes are dynamin- and cholesterol-dependent. PLoS One 6:e25464. 10.1371/journal.pone.0025464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Aoki T, Nomura R, Fujimoto T. 1999. Tyrosine phosphorylation of caveolin-1 in the endothelium. Exp. Cell Res. 253:629–636 [DOI] [PubMed] [Google Scholar]
- 30. Parton RG, Joggerst B, Simons K. 1994. Regulated internalization of caveolae. J. Cell Biol. 127:1199–1215 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Manders EMM, FJV, Aten JA. 1993. Measurement of colocalization of objects in dual-colour confocal images. J. Microsc. 169:375–382 [DOI] [PubMed] [Google Scholar]
- 32. Knipe T, Rieder E, Baxt B, Ward G, Mason PW. 1997. Characterization of synthetic foot-and-mouth disease virus provirions separates acid-mediated disassembly from infectivity. J. Virol. 71:2851–2856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li CC, Yang MY, Chen RF, Lin TY, Tsao KC, Ning HC, Liu HC, Lin SF, Yeh WT, Chu YT, Yang KD. 2002. Clinical manifestations and laboratory assessment in an enterovirus 71 outbreak in southern Taiwan. Scand. J. Infect. Dis. 34:104–109 [DOI] [PubMed] [Google Scholar]
- 34. Blanchard E, Belouzard S, Goueslain L, Wakita T, Dubuisson J, Wychowski C, Rouille Y. 2006. Hepatitis C virus entry depends on clathrin-mediated endocytosis. J. Virol. 80:6964–6972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Eash S, Querbes W, Atwood WJ. 2004. Infection of vero cells by BK virus is dependent on caveolae. J. Virol. 78:11583–11590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kolokoltsov AA, Deniger D, Fleming EH, Roberts NJ, Jr, Karpilow JM, Davey RA. 2007. Small interfering RNA profiling reveals key role of clathrin-mediated endocytosis and early endosome formation for infection by respiratory syncytial virus. J. Virol. 81:7786–7800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yamamoto A, Tagawa Y, Yoshimori T, Moriyama Y, Masaki R, Tashiro Y. 1998. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct. 23:33–42 [DOI] [PubMed] [Google Scholar]
- 38. Noyce RS, Bondre DG, Ha MN, Lin LT, Sisson G, Tsao MS, Richardson CD. Tumor cell marker PVRL4 (nectin 4) is an epithelial cell receptor for measles virus. PLoS Pathog. 7:e1002240. 10.1371/journal.ppat.1002240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tatsuo H, Ono N, Tanaka K, Yanagi Y. 2000. SLAM (CDw150) is a cellular receptor for measles virus. Nature 406:893–897 [DOI] [PubMed] [Google Scholar]
- 40. Jeffers SA, Tusell SM, Gillim-Ross L, Hemmila EM, Achenbach JE, Babcock GJ, Thomas WD, Jr, Thackray LB, Young MD, Mason RJ, Ambrosino DM, Wentworth DE, Demartini JC, Holmes KV. 2004. CD209L (L-SIGN) is a receptor for severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. U. S. A. 101:15748–15753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang SM, Lei HY, Huang KJ, Wu JM, Wang JR, Yu CK, Su IJ, Liu CC. 2003. Pathogenesis of enterovirus 71 brainstem encephalitis in pediatric patients: roles of cytokines and cellular immune activation in patients with pulmonary edema. J. Infect. Dis. 188:564–570 [DOI] [PubMed] [Google Scholar]
- 42. Ghez D, Lepelletier Y, Lambert S, Fourneau JM, Blot V, Janvier S, Arnulf B, van Endert PM, Heveker N, Pique C, Hermine O. 2006. Neuropilin-1 is involved in human T-cell lymphotropic virus type 1 entry. J. Virol. 80:6844–6854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Manel N, Kim FJ, Kinet S, Taylor N, Sitbon M, Battini JL. 2003. The ubiquitous glucose transporter GLUT-1 is a receptor for HTLV. Cell 115:449–459 [DOI] [PubMed] [Google Scholar]
- 44. Geraghty RJ, Krummenacher C, Cohen GH, Eisenberg RJ, Spear PG. 1998. Entry of alphaherpesviruses mediated by poliovirus receptor-related protein 1 and poliovirus receptor. Science 280:1618–1620 [DOI] [PubMed] [Google Scholar]
- 45. Montgomery RI, Warner MS, Lum BJ, Spear PG. 1996. Herpes simplex virus-1 entry into cells mediated by a novel member of the TNF/NGF receptor family. Cell 87:427–436 [DOI] [PubMed] [Google Scholar]
- 46. Nicola AV, McEvoy AM, Straus SE. 2003. Roles for endocytosis and low pH in herpes simplex virus entry into HeLa and Chinese hamster ovary cells. J. Virol. 77:5324–5332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Lin YW, Chang KC, Kao CM, Chang SP, Tung YY, Chen SH. 2009. Lymphocyte and antibody responses reduce enterovirus 71 lethality in mice by decreasing tissue viral loads. J. Virol. 83:6477–6483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang Y, Wang H, Gong E, Du J, Zhao X, McNutt MA, Wang S, Zhong Y, Gao Z, Zheng J. 2009. Neuropathology in 2 cases of fatal enterovirus type 71 infection from a recent epidemic in the People's Republic of China: a histopathologic, immunohistochemical, and reverse transcription polymerase chain reaction study. Hum. Pathol. 40:1288–1295 [DOI] [PubMed] [Google Scholar]
- 49. Hidari KI, Weyrich AS, Zimmerman GA, McEver RP. 1997. Engagement of P-selectin glycoprotein ligand-1 enhances tyrosine phosphorylation and activates mitogen-activated protein kinases in human neutrophils. J. Biol. Chem. 272:28750–28756 [DOI] [PubMed] [Google Scholar]
- 50. Luo J, Xu T, Wang X, Ba X, Feng X, Deepak V, Zeng X. PI3K is involved in L-selectin- and PSGL-1-mediated neutrophil rolling on E-selectin via F-actin redistribution and assembly. J. Cell Biochem. 110:910–919 [DOI] [PubMed] [Google Scholar]
- 51. Zarbock A, Abram CL, Hundt M, Altman A, Lowell CA, Ley K. 2008. PSGL-1 engagement by E-selectin signals through Src kinase Fgr and ITAM adapters DAP12 and FcRγ to induce slow leukocyte rolling. J. Exp. Med. 205:2339–2347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Urzainqui A, Martinez del Hoyo G, Lamana A, de la Fuente H, Barreiro O, Olazabal IM, Martin P, Wild MK, Vestweber D, Gonzalez-Amaro R, Sanchez-Madrid F. 2007. Functional role of P-selectin glycoprotein ligand 1/P-selectin interaction in the generation of tolerogenic dendritic cells. J. Immunol. 179:7457–7465 [DOI] [PubMed] [Google Scholar]
- 53. Zarbock A, Muller H, Kuwano Y, Ley K. 2009. PSGL-1-dependent myeloid leukocyte activation. J. Leukoc. Biol. 86:1119–1124 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.