

Abstract
Viruses commonly manipulate cell cycle progression to create cellular conditions that are most beneficial to their replication. To accomplish this feat, viruses often target critical cell cycle regulators in order to have maximal effect with minimal input. One such master regulator is the large, multisubunit E3 ubiquitin ligase anaphase-promoting complex (APC) that targets effector proteins for ubiquitination and proteasome degradation. The APC is essential for cells to progress through anaphase, exit from mitosis, and prevent a premature entry into S phase. These far-reaching effects of the APC on the cell cycle are through its ability to target a number of substrates, including securin, cyclin A, cyclin B, thymidine kinase, geminin, and many others. Recent studies have identified several proteins from a number of viruses that can modulate APC activity by different mechanisms, highlighting the potential of the APC in driving viral replication or pathogenesis. Most notably, human cytomegalovirus (HCMV) protein pUL21a was recently identified to disable the APC via a novel mechanism by targeting APC subunits for degradation, both during virus infection and in isolation. Importantly, HCMV lacking both viral APC regulators is significantly attenuated, demonstrating the impact of the APC on a virus infection. Work in this field will likely lead to novel insights into viral replication and pathogenesis and APC function and identify novel antiviral and anticancer targets. Here we review viral mechanisms to regulate the APC, speculate on their roles during infection, and identify questions to be addressed in future studies.
INTRODUCTION
Viruses often modulate the cell cycle to their advantage in order to provide the proper resources and conditions for their replication (1). As viral genomes are limited in their coding capacity, viruses target cellular processes where they can exert a maximal effect with relatively few activities. Studies of viral interactions with the host cell cycle have not only shed light on the mechanisms of virus replication and pathogenesis, they have also led to the identification and characterization of essential cell cycle regulators, such as p53 and Rb (2–5). Another important protein complex involved in cell cycle control is the anaphase-promoting complex (APC), an E3 ubiquitin ligase required for ubiquitination and subsequent proteasome degradation of multiple cell cycle regulator and effector proteins. Without the APC, cells cannot separate sister chromatids during anaphase, exit mitosis, or properly enter S phase (6). Due to its essential role in cell cycle progression, it is not surprising that several viruses have been recently reported to target the APC. Perhaps the most notable example is human cytomegalovirus (HCMV). A recent study has identified the HCMV protein pUL21a as a novel viral regulator that induces degradation of APC subunits during virus infection (7). Abrogation of the viral ability to disable APC activity results in a marked attenuation in HCMV infection, thus demonstrating for the first time the role of a viral APC regulator in the context of virus infection (7). Interestingly, some viruses that encode APC regulators are known to cause cancer or are associated with cancer, suggesting that virus-altered APC activity may be involved in the development of virus-induced tumors. Studies of these proteins will likely impart insights into viral replication and pathogenesis and lead to novel targets for antiviral therapeutics. Perhaps equally important, as was the case with p53 and Rb, these viral proteins may prove to be excellent tools to study APC biology. As the APC is now emerging as an attractive anticancer drug target (8), these studies may ultimately allow us to exploit these viral regulators and identify novel mechanisms targeting the APC for cancer therapies. Here we will review what is known about viral APC regulators, speculate as to why viruses might disarm the APC, and discuss questions which need to be addressed in future research. A particular emphasis is given to the most recent discovery of the novel APC regulator in HCMV.
ANAPHASE-PROMOTING COMPLEX
Structure.
The APC is a multisubunit E3 ubiquitin ligase that targets more than 30 proteins for ubiquitin-dependent proteasome degradation (9). E3 ubiquitin ligases are a large class of enzymes which catalyze the final step in the ubiquitin transfer reaction, namely, the transfer of ubiquitin from an E2 ubiquitin ligase to a target protein. The majority of APC substrates are involved in cell cycle regulation, making the APC an essential regulator of the cell cycle.
The human APC is an approximately 1.5-MDa complex that contains at least 12 subunits and two coactivators (Fig. 1). It can be broken down into three subcomplexes: the enzymatic unit, the specificity arm, and the bridge. The major components of the enzymatic unit are APC2, APC10 (also called Doc1), and APC11. The APC is a cullin-RING E3 ubiquitin ligase in which the cullin and RING domains reside within APC2 and APC11, respectively (10, 11). APC10/Doc1 plays an important role in identifying specific substrates (12, 13). On the opposite side of the enzymatic unit is the specificity arm that is made up of APC3 (also called CDC27), APC6, APC7, APC8, APC12 (CDC26), APC13, and APC16. These proteins contain multiple copies of the tetratricopeptide repeat (TPR) that likely facilitate complex assembly as well as substrate binding (6). Consequently, the specificity arm is also known as the TPR subunits. These two subcomplexes are held together by the bridge that is composed of APC1, APC4, and APC5. Why the APC is so large and contains so many subunits remains elusive, but it is sensible to speculate that its large size allows the APC the flexibility to target a wide range of substrates for ubiquitin conjugation.
Fig 1.
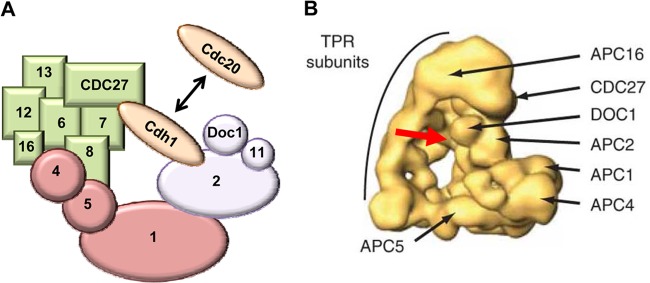
Structure of the APC. (A) Schematic structure of the APC. The APC is composed of three distinct subcomplexes that are color-coded for easy identification. The enzymatic unit (purple) contains the cullin and RING domains, which reside in APC2 and APC11, respectively. The specificity arm (also called the TPR subunits [green]) facilitates complex assembly as well as substrate binding. The bridge (pink) holds the other subcomplexes together. The coactivators (brown) bind to both the enzymatic unit and the specificity arm, where they interact with substrates. For clarity, the diagram is not drawn to scale and not all subunits are shown. (B) The human APC structure derived from single-particle electronic microscopic analysis and three-dimensional (3D) reconstruction (73). Shown are the triangular architecture of the APC and the locations of representative subunits of the specificity arm (i.e., APC3/CDC27, APC16, and other TPR subunits), enzymatic unit (i.e., APC2 and APC10/Doc1), and bridge (i.e., APC1, APC4, and APC5). The red arrow depicts the binding site of the APC coactivator (e.g., Cdh1, which is not shown in the image).
Regulation.
APC activity is regulated by two coactivator proteins, cell division cycle protein 20 (Cdc20) and Cdc20 homologue 1 (Cdh1). These two activator proteins act at different times to regulate APC activity, allowing it to exert distinct functions at different phases of the cell cycle (Fig. 2). Cdc20 functions from metaphase through anaphase to promote separation of sister chromatids, while Cdh1 is active from the end of mitosis until the G1-to-S transition to prevent premature entry into S phase. The precise mechanism of how the activator proteins function is unknown, but it is thought that they enable the APC to bind to its specific substrates (14).
Fig 2.
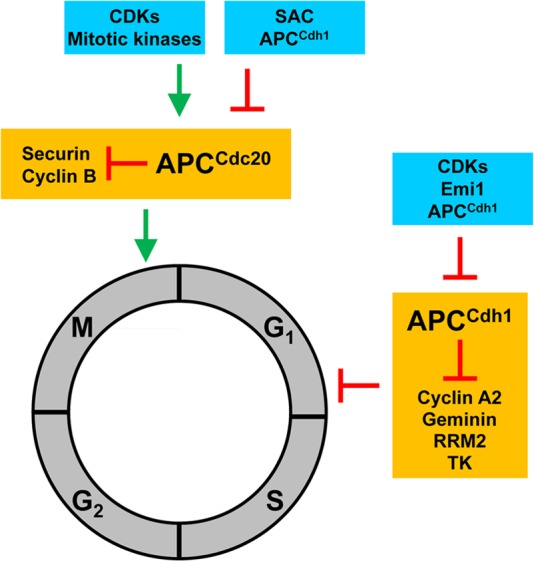
General function and key targets of the APC. The eukaryotic cell cycle is divided into four phases, mitosis (M), Gap1 (G1), DNA synthesis (S), and Gap2 (G2). The APC interacts with the coactivator Cdc20 or Cdh1 to form APCCdc20 or APCCdh1, respectively, the two of which act on different phases of the cell cycle. APCCdc20 starts to form toward the end of G2 phase as CDK activity increases, but its activity is inhibited by the spindle assembly checkpoint (SAC) until anaphase. Once active, APCCdc20 degrades securin, cyclin B1, and others to promote the completion of M phase. APCCdc20 activity then diminishes in late M phase with the drop in CDK activity and degradation of Cdc20 by APCCdh1. In contrast, APCCdh1 is formed in late M phase and maintains high activity throughout G1. It targets a number of proteins, including cyclin A2, geminin, ribonucleotide reductase (RRM2), thymidine kinase (TK), and others, for degradation to prevent premature entry into S phase. APCCdh1 must be inhibited before S phase can begin. It is inhibited by Emi1, CDK activity, and a negative feedback loop in which it targets Cdh1 for degradation.
Cdc20 and Cdh1 are both tightly regulated to ensure that they function precisely at different times during the cell cycle (15–20). Mitotic kinases phosphorylate Cdc20 during metaphase to promote its association with the APC. However, the Cdc20-containing APC complex (APCCdc20) is kept inactive by the spindle assembly checkpoint (SAC) until the spindle poles align (21). On the other hand, Cdh1 is inactivated by phosphorylation and remains inactive until cyclin-dependent kinase (CDK) activity diminishes in late mitosis. The Cdh1 complex (APCCdh1) is highly active in G1 phase and is then inhibited by early mitotic inhibitor 1 (Emi1) during the G1-to-S transition (22). Finally, the APC also undergoes feedback regulation, as APCCdh1 itself can target Cdc20 and Cdh1 for proteasome degradation (23–27). As a result, during the late stages of mitosis, the APCCdh1-mediated degradation of Cdc20, together with the diminishment of CDK activity, is thought to facilitate the transition from APCCdc20 to APCCdh1. Conversely, the APCCdh1-mediated degradation of Cdh1 and its E2 enzyme UbcH10, together with Emi1 induction and increased CDK activity, diminishes and inactivates APCCdh1 upon entering S phase.
Targets.
The APC targets more than 30 proteins for polyubiquitination and proteasome degradation. The two best-known motifs that the APC recognizes in its target proteins are the D box (RXXLXXXXN/D/E) (28–30) and the KEN domain (KEN) (31, 32). Some of the well-known substrates include cyclin A, cyclin B, and securin. The degradation of the cyclins during mitosis reduces CDK activity. This promotes the disassembly of the mitotic spindle, chromosome decondensation, reformation of the nuclear envelope, and formation of a cytokinetic groove (33–35). Securin binds and inhibits separase, an enzyme that cleaves cohesin to separate sister chromatids. Thus, degradation of securin by APCCdc20 allows for activation of separase, segregation of sister chromatids, and initiation of anaphase (reviewed in reference 36). In addition, the APC has many other targets involved in a variety of functions central to the cell cycle. These include but are not limited to mitotic kinases (aurora kinase A and B, polo-like kinase [PLK-1]), DNA replication (Cdc6 and geminin), nucleotide biosynthesis (thymidine kinase [TK], ribonucleotide reductase [RRM2], and deoxythymidylate kinase), glycolysis (6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase isoform 3 [PFKFB3]), and glutaminolysis (glutaminase 1) (37) (Fig. 3).
Fig 3.
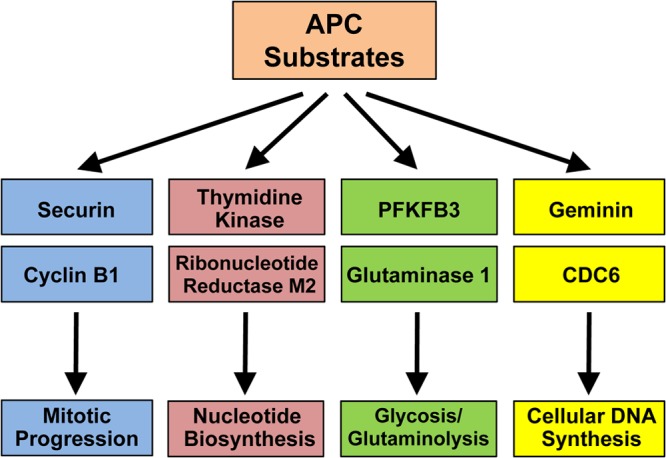
APC substrates that may impact virus replication. There are more than 30 known protein substrates of the APC, and selected candidates that have potential to impact viral infection due to their specific role in host cell cycle progression are listed.
MODULATION OF THE APC BY VIRUSES
As many viruses manipulate the cell cycle during infection and some of them cause tumors, it is not surprising that the APC would be targeted by viruses. An accumulating body of literature in recent years has identified multiple viral proteins that can modulate the function of the APC. They do so via a diverse array of mechanisms, highlighting the fact that viruses have gained the ability to modulate this important host factor through convergent evolution. Most recently, the HCMV pUL21a protein was found to disrupt the APC by proteasome-dependent degradation of APC subunits. Other documented viral APC regulators include adenovirus (AdV) E4orf4 and E1A, human T-lymphotropic virus type 1 (HTLV-1) Tax, human papillomavirus (HPV) E2, hepatitis B virus (HBV) X, chicken anemia virus (CAV) apoptin, orf virus poxvirus APC/cyclosome regulator (PACR), and HCMV pUL97 (summarized in Table 1 and Fig. 4). Intriguingly, a number of these viruses are either oncogenic (HTLV-1, HPV, HBV) or potentially associated with cancer (HCMV). Here we discuss the mechanisms that these viral proteins use to control the APC and identify questions that need to be addressed in future studies in order to understand the role of APC regulation in the biology and pathogenesis of these viruses.
Table 1.
Viral regulators of the APC
| Virus | Cancer | Protein | Proposed mechanism(s) | Shown in infection | Role in infection | Reference(s) |
|---|---|---|---|---|---|---|
| HTLV-1 | Yes | Tax | Unknown—binds to and activates the APC; may affect APC3/CDC27 phosphorylation | No | No | 33, 38 |
| HBV | Yes | X | Binds to BubR1—promotes the association of Cdc20 with core APC subunits to activate the complex | No | No | 21 |
| Adenovirus | Noa | E4orf4 | Unknown—requires the interaction with PP2A | No | No | 24, 42 |
| Noa | E1A | Binds to CBP/p300 and displaces the APC from CBP/p300 | No | No | 65 | |
| HPV | Yes | E2 | Binds directly to Cdh1/Cdc20; redistributes Cdh1 to insoluble cytoplasmic structures | No | No | 2 |
| Orf virus | No | PACR | APC11 decoy | No | No | 40, 41 |
| CAV | No | Apoptin | Likely binds to APC1, redistributes the APC to PML bodies, and causes the dissociation of the APC | No | No | 16, 60 |
| HCMV | Potential association | pUL97 | Phosphorylates Cdh1 | Yes | No | 62 |
| Potential association | pUL21a | Binds to the APC; targets APC4 and APC5 for proteasome degradation, leading to dissociation of the APC | Yes | Yes | 8 |
Adenovirus is not known to cause cancer in humans, but it can induce tumors in rodents and transform rodent cells in culture.
Fig 4.
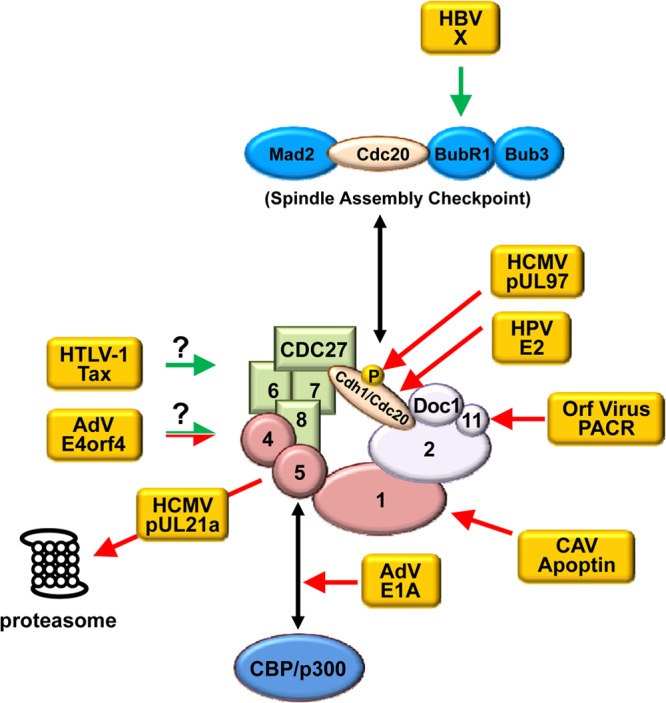
Viral regulators of the APC. Depicted are known viral APC regulators and mechanisms by which they regulate the APC (see also Table 1). HCMV pUL97 phosphorylates Cdh1, rendering it unable to bind the APC. HPV E2 binds directly to Cdh1 and Cdc20 to relocalize them to insoluble cytoplasmic structures. Orf virus PACR acts as a nonfunctional mimic to displace APC11. CAV apoptin interacts with the APC, redistributes it to PML bodies, and causes it to dissociate. Adenovirus E1A likely blocks the interaction of the APC with CBP/p300 by competing with APC5/APC7 for binding to CBP/p300. HCMV pUL21a interacts with the APC and targets the bridge subunits APC4 and APC5 for proteasome-dependent degradation, leading to dissociation of the complex. Adenovirus E4orf4 can either inhibit or activate the APC and does so in a PP2A-dependent manner; however, a detailed mechanism is unknown. HTLV-1 Tax binds to and activates the APC during S phase, but the mechanism by which it activates the APC remains unknown. HBV X protein activates the APC through its interaction with BubR1, which releases the coactivator Cdc20 from the SAC, allowing premature activation of the APC. Arrows in green and red depict the activation and inhibition of the APC by a viral protein, respectively. Question marks indicate that a mechanism is unknown.
Adenovirus E4orf4.
Adenoviruses are a family of double-stranded DNA viruses that cause a variety of clinical illnesses, including upper respiratory infections, conjunctivitis, tonsillitis, and gastroenteritis in mammalian species. Intriguingly, even though it is not known to be oncogenic in humans, in tissue culture, human adenovirus (HAdV) can induce cellular transformation. HAdV encodes the protein E4orf4, which, when overexpressed, can institute a cell cycle block at G2/M phase and induce apoptosis of cancer cells. These effects are dependent on its binding to protein phosphatase 2A (PP2A) (reviewed in reference 38). The E4orf4-PP2A complex also interacts with the APC to alter its activity (39, 40). However, the binding site and mechanism that it uses to regulate the APC remain unknown (39). E4orf4 overexpression alters APC activity in both yeast and human cells, and these effects can be both stimulatory and inhibitory, depending on the stage of the cell cycle in which the protein is expressed (39, 40). However, it has not yet been shown that this regulation of the APC occurs during adenovirus infection. E4orf4 is nonessential for virus replication in cultured human cancer or primary cells, so how this protein impacts adenovirus biology remains elusive (41). Determining whether E4orf4 inhibits the APC during infection, elucidating its consequence on virus replication, and identifying its mechanism of action are key questions to address in future studies.
Adenovirus E1A.
The E1A protein can transform primary rodent cells in tissue culture. It does so by multiple mechanisms, including its ability to bind and inhibit pRb. In fact, the discovery of the interaction between E1A and pRb was the first example of an oncogene and a tumor suppressor gene that physically interact (5). Moreover, E1A binds to the cyclic AMP response element binding protein (CBP) and p300, and these interactions are also required for the transforming capability of E1A (42, 43). CBP/p300 are large lysine acetyltransferases that act as transcriptional coactivators via chromatin modification. Consistently, E1A has been reported to be able to modulate the methylation/acetylation status of E2F promoters and induce transcription of E2F-responsible genes required for S-phase entry (44). It was later discovered that APC5 and APC7 were able to complex with CBP/p300 because they contained E1A-like protein interaction domains (45). The binding of APC5/APC7 to CBP/p300 appeared to be important for their functions, as expression of APC5/APC7 or APC holoenzyme-stimulated CBP/p300 activity and knockdown of CBP inhibited APC ubiquitin ligase activity (45). Importantly, APC5/APC7 overexpression limited the transforming ability of E1A whereas APC5/APC7 knockdown was able to enhance the transforming ability of an otherwise transformation-deficient E1A mutant that was unable to bind to CBP/p300 (45). These studies have led to the model in which E1A disrupts the interaction of the APC and CBP/p300 to disable their functions. This ultimately leads to inhibition of gene transcription involved in G1 arrest and activation of gene transcription required for S-phase entry and cellular DNA synthesis (46). However, no studies have been documented to directly examine how E1A affects APC activity, so the effects of overexpression or knockdown of APC components may be indirect. Biochemical data will be critical to determine if E1A specifically disrupts the activity of a CBP/p300-APC complex.
HTLV-1 Tax.
Human T-lymphotropic virus type 1 (HTLV-1) is the etiologic agent of T cell leukemia and lymphoma. Pathological findings have shown that mitotic aberrations accompany HTLV-1 viral replication, which may promote tumorigenesis (47, 48). HTLV-1 transformed T cells are delayed in their progression through the S/G2/M phases of the cell cycle, and this delay correlates with a decrease in several APC substrates (49). Moreover, APCCdc20 is prematurely activated in Tax-expressing cells. Tax interacts with both Cdc20- and Cdh1-containing APC complexes, suggesting that it likely binds to a core subunit of the APC, but the mechanism of its action remains unknown (49). Tax expression in Saccharomyces cerevisiae cells results in the reduction of APC substrates and severe chromosomal abnormalities and can lead to senescence (50, 51). Furthermore, point mutants of Tax that have specifically lost the ability to regulate the APC are also unable to induce senescence in HeLa and yeast cells (52). Collectively, these studies suggest that Tax enhances APC activity, which may be linked to its ability to induce mitotic aberrations. Further experiments with these mutants are needed to determine if Tax-mediated APC regulation is important for HTLV-1-induced tumors.
HPV E2.
Human papillomavirus (HPV) is a small DNA virus that causes cervical cancers. Different strains of HPV can be divided into low-risk and high-risk groups for their ability to promote cervical cancer. Bellanger and colleagues found that HPV E2 proteins from high-risk but not low-risk strains were capable of inducing a mitotic block and subsequent apoptosis (53). Interestingly, cells that overcame this block developed severe genomic instability. Furthermore, they showed that E2 proteins from high-risk strains can bind directly to Cdh1 and Cdc20, induce the accumulation of APC substrates, and redistribute Cdh1 to insoluble cytoplasmic aggregates (53). This suggests that inhibition of the APC may be one mechanism for HPV to promote tumors. It is not known, however, whether E2-induced genomic instability is the direct result of APC inhibition or can be attributed to other functions of this multifunctional protein. As E2 is required for viral DNA synthesis, APC regulation may also be important for the virus to replicate its genome (54). A mutant E2 protein that specifically abrogates its ability to inhibit the APC is crucial to elucidate the role of APC regulation in HPV replication and carcinogenesis.
HBV X.
Hepatitis B virus (HBV) is one of the primary etiological factors for the development of hepatocellular carcinoma (HCC), the possible outcome of virus-induced chromosome instability. Kim and colleagues reported that the HBV X protein bound to BubR1, a component of the spindle assembly checkpoint (SAC) (55). The binding of BubR1 was mapped to the C terminus of X, which was also critical for the X protein to promote aberrant chromosomes (55). The binding of X to BubR1 prevented Cdc20 from binding to BubR1. This released Cdc20 from the spindle assembly checkpoint and allowed it to prematurely activate the APC prior to spindle pole alignment, thus having the potential to induce aberrant chromosome segregation. However, the X protein has also been shown to bind the E3 ubiquitin ligase damaged DNA binding protein 1 (DDB1), and this activity may play a prominent role in its induction of chromosome instability (56). Thus, more-definitive evidence is needed to determine if X-mediated APC regulation plays a role in HBV-mediated carcinogenesis.
CAV apoptin.
Chicken anemia virus (CAV) is a small, single-stranded DNA virus that causes severe anemia and immunodeficiency in young chickens by inducing apoptosis of erythroblastoid cells and cortical thymocytes. Apoptin, one of the three proteins encoded by this virus, induces a strong G2/M arrest and subsequent apoptosis of cancer cells (57). Its ability to specifically kill cancer cells has made it an attractive candidate for novel cancer therapies (1). Teodoro and colleagues identified APC1 as a binding partner of apoptin by coimmunoprecipitation (co-IP)-mass spectrometry analysis (57). Although it has not yet been confirmed, APC1 is likely to be a direct binding partner, as it was the only protein identified from this analysis. Apoptin expression led to dissociation of the APC and stabilization of APC substrates (57). A subsequent study showed that apoptin induced the formation of promyelocytic leukemia (PML) nuclear bodies and relocalized the APC to PML bodies and that these activities were dependent on its ability to shuttle between the nucleus and cytoplasm (58). The APC binding domain was mapped to the C-terminal 40 amino acids of apoptin, which is also sufficient to induce apoptosis (57). This suggests that the induction of apoptosis by apoptin may depend on its ability to regulate the APC. To support this hypothesis, small interfering RNA (siRNA) knockdown of APC1 induced G2/M arrest and apoptosis in a manner similar to that by apoptin (57). Future work should address how apoptin induces the dissociation of the APC and whether this requires its redistribution to PML bodies. Interestingly, while apoptin-null CAV is defective in DNA synthesis, an apoptin point mutant of CAV is capable of synthesizing viral DNA but fails to produce virus particles (59). Thus, it will also be important to determine whether APC depletion by siRNA restores DNA replication or particle formation in these mutants in order to define the potential role of APC regulation in CAV replication.
Orf virus PACR.
Orf virus is a parapoxvirus which causes an exanthemous disease in sheep and goats. A recent study by Mo and colleagues identified a poxvirus protein with significant sequence homology to APC11 (60). Termed PACR (poxviral APC/C regulator), this protein is present only in the parapoxvirus family. It binds APC2 in a manner that mimics APC11, but it lacks the catalytic residues of an E3 ligase, and thus, its enzymatic domain is nonfunctional and does not contain ubiquitin ligase activity (60, 61). Importantly, swapping the enzymatic domains of APC11 and PACR also reverses their ubiquitin ligase activity in vitro. PACR overexpression leads to a G2/M arrest with an increase in APC substrates, supporting the notion that PACR inhibits the APC by mimicking a nonfunctional APC11. PACR-null orf virus has a nearly 100-fold growth defect. While this defect is likely due to the ability of PACR to inhibit the APC, analyzing domain swap recombinant viruses or examining orf virus infection in APC-depleted cells will provide definitive evidence to test this hypothesis.
HCMV pUL97.
HCMV is a large, double-stranded DNA virus that establishes persistent and latent infection in human hosts. It is a common source of severe infectious complications in immunocompromised individuals and the leading viral cause of birth defects. Moreover, accumulating evidence suggests a possible link of HCMV with certain human cancers (62–64). HCMV uses multiple mechanisms to modulate the function of the APC (65–67). One mechanism is to induce Cdh1 phosphorylation. Tran and colleagues reported that the viral protein kinase pUL97 was responsible for Cdh1 phosphorylation during HCMV infection (65). pUL97 is a CDK mimic (68, 69), so it is not surprising that it is able to phosphorylate Cdh1, a natural target of CDKs. In cells infected with a UL97 deletion virus, Cdh1 was not phosphorylated and APC substrate accumulation was delayed at early times compared to cells infected with wild-type virus (65). However, by the times when viral DNA synthesis occurred (i.e., 24 to 36 h postinfection), the APC substrates accumulated efficiently even in the absence of pUL97. This indicates that HCMV must have additional mechanisms to regulate the APC. It is currently unclear whether the inhibition of the APC by pUL97 alone plays a role in the ability of HCMV to replicate or cause disease.
HCMV pUL21a.
In addition to pUL97, it is clear that HCMV encodes at least one other factor that can regulate the APC by inducing its dissociation. Tran and colleagues analyzed APC subunits and found that APC4 and APC5 were degraded independently of pUL97 in a proteasome-dependent manner during HCMV infection (65). APC4 and APC5 degradation is likely to lead to dissociation of the APC complex, as this dissociation is prevented by proteasome inhibitors. In a proteomics screen for interacting partners of the HCMV protein pUL21a, multiple subunits of the APC, including APC3/CDC27, APC7, and APC8, were identified (7). pUL21a is a 15-kDa protein that shares no significant homology with any known protein (70), and pUL21a-null virus had a marked defect in viral DNA synthesis (71). Subsequent functional analysis indicated that pUL21a was both necessary and sufficient to target APC4 and APC5 for proteasome degradation, leading to dissociation of the complex (7). A pUL21a point mutant, PR109-110AA (herein termed pUL21aPR-AA), that was not able to bind the APC failed to target APC4/APC5 for degradation, indicating that the ability of pUL21a to degrade APC4/APC5 is dependent on its binding to the APC. Incidentally, pUL21a itself is highly unstable, undergoing rapid turnover in a proteasome-dependent, ubiquitin-independent manner (70). It is possible that pUL21a recruits an E3 ligase to the APC to promote ubiquitination of APC4 and APC5. Alternatively, it is intriguing to speculate that pUL21a may act as a ubiquitin-like protein and directly target these APC subunits to the proteasome for degradation. In future studies, it will be important to identify the APC subunit that pUL21a directly interacts with, elucidate the mechanism by which pUL21a degrades APC4 and APC5, and determine whether the intrinsic instability of pUL21a plays a role in its ability to regulate the APC. It is interesting to note that both CAV apoptin and HCMV pUL21a regulate the APC by targeting the bridge subcomplex and inducing its dissociation. This is consistent with the critical function of the bridge subcomplex in maintaining APC stability and suggests a conserved strategy evolved by different viruses converging on this subcomplex as an efficient means to regulate APC activity.
What is the role of pUL21a-mediated APC regulation in HCMV infection? Compared to cells infected with wild-type HCMV, cells infected with UL21a-null virus or the pUL21aPR-AA point mutant virus had much-reduced levels of a subset of APC substrates, but not all APC substrates were affected. This may result from two potential mechanisms which are not mutually exclusive. Modulating the APC complex by dissolving the bridge may allow viruses to alter substrate specificity of the APC but not completely abolish its activity, as the enzymatic portion of the APC is known to have activity in vitro (10, 60). HCMV does not appear to directly destroy the enzymatic unit of APC, so it is of interest to determine if the APC retains some activity or is directed to target different substrates during virus infection. Alternatively, as Cdh1 is phosphorylated by pUL97 during infection, UL21a mutant virus may still inhibit the APC to some extent. This supports the model that HCMV uses two distinct mechanisms, one mediated by pUL21a and the other mediated by pUL97, to regulate the APC. This is further exemplified by the growth kinetics of the pUL21aPR-AA point mutant virus. This mutant virus grew at wild-type levels, but a double mutant virus that carried both the pUL21aPR-AA point mutation and a UL97 deletion was significantly more attenuated than a UL97 single-deletion virus (7). Therefore, the consequence of pUL21a-mediated APC regulation to HCMV replication is apparent only when both viral APC regulators are removed from the virus. These data support the hypothesis that the APC is a restriction factor for HCMV, but this restriction is effectively antagonized by pUL97 or pUL21a, a model that will be fully tested in future studies.
WHY TARGET THE APC?
Even though definitive evidence is still lacking in most viral systems, identification of a number of viral APC regulators from diverse virus families, together with the recent study on HCMV (7), provides appealing evidence that APC activity plays an important role in virus replication and pathogenesis. What are the benefits for viruses to modulate the APC? For HTLV-1 and HBV, premature activation of APCCdc20 may induce chromosome instability and contribute to their tumorigenic ability. Other viruses may inhibit APCCdh1 to promote an S-phase-like environment or APCCdc20 to prevent reformation of the nuclear envelope, so nuclear factors become enriched or accessible for virus replication. As the APC targets more than 30 proteins for ubiquitination and degradation, inhibition of this complex would stabilize its substrates, any one of which may be important for virus replication (Fig. 3). Interestingly, within poxvirus and herpesvirus families, the only viruses that have been found to modulate the APC are parapoxviruses and HCMV. These two viruses do not encode their own thymidine kinase (TK) and ribonucleotide reductase (RRM2) enzymes (60, 67). Both enzymes are APC substrates and critical for the production of deoxyribonucleotides. It seems plausible that APC regulation may be one mechanism used by these viruses to accumulate sufficient nucleotides for viral genome amplification, even though this has not been experimentally tested. Finally, the APC may also restrict viral replication by targeting viral proteins for ubiquitination and degradation. This is exemplified by the facts that the bovine papillomavirus replicative helicase E1 is targeted by the APC (72) and that several HCMV proteins contain the consensus D box, an APC recognition signal commonly found in its substrates (65). The precise mechanism may differ for different viruses, but a common theme is that these viral regulators are key components of the invasive strategy for viruses to overcome direct and indirect effects of the APC on their infection.
FUTURE PERSPECTIVE
At least nine proteins from diverse viral families have now been reported to modulate the function of the APC, and more are likely to be discovered, highlighting the potential importance of this cell cycle master regulator in virus infection. The major limitation is that the data from most of these studies are correlative at best. In many cases, it has not been shown that the APC is regulated during virus infection, and in several cases, the mechanism has not been clearly defined. The potential role of virus-mediated APC regulation during infection is largely unknown, an issue further complicated by the fact that most of the identified viral APC regulators have other known functions. Future studies should focus on identifying the mechanisms that these proteins use to regulate the APC, determining whether or not they function during virus infection, and providing definitive evidence of whether and how the APC is involved in virus replication and pathogenesis. In cases in which the APC restricts virus replication, it will be important to determine whether the APC plays its role by acting on viral or cellular substrates. It will be important to identify the APC substrates that conversely act to promote virus replication. For instance, it would be informative to determine whether overexpression of TK or RRM2 can restore virus replication of HCMV, orf virus, HPV, or CAV mutants that lack their APC regulators. Finally, the fact that multiple tumor-inducing viruses (HPV, HBV, and HTLV-1) encode proteins that regulate the APC raises an important question of whether these viruses use APC regulation as a mechanism to promote tumorigenesis, which should be addressed definitively in future investigations.
Further studies of these viral regulators will also promise to shed light on the biology of the APC. As has been shown in many masterpieces of seminal work, viruses are extremely useful tools to probe the biology of critical cellular proteins (e.g., p53, Rb). Elucidating how these viral proteins interact with the APC will undoubtedly enhance our understanding of APC structure, assembly, and regulation. On a more applied side, such studies will likely have important implications for cancer research, exemplified by the fact that drugs are now being developed to target the APC for anticancer therapy (8). These viral proteins are invaluable tools to identify key features of the APC that can be used as additional targets for novel drugs. Apoptin is currently being investigated as a potential cancer therapy due to its ability to selectively kill tumor cells, and other viral APC regulators are likely to have similar qualities. For instance, expression of E4orf4, E2, or PACR also has a dramatic effect on cell cycle progress (39, 40, 53, 60, 61). Furthermore, pUL21a overexpression prevented the proliferative ability of a transformed cell line (7), suggesting that pUL21a regulation of the APC may inhibit cancer cell growth. Conversely, HPV E2 and HBV X proteins may facilitate tumorigenesis in their respective virus systems and thus may be attractive drug targets to prevent certain types of viral-induced cancer. Future work should strive to gain an understanding of intricate interplays among the APC, virus replication, cell cycle, and virus-induced cancer.
ACKNOWLEDGMENTS
We thank the members of the Yu lab for critical readings of the manuscript.
This study was supported by a Public Health Service grant (R01CA120768). D.Y. holds an Investigators in the Pathogenesis of Infectious Disease award from the Burroughs Wellcome Fund.
Footnotes
Published ahead of print 12 June 2013
REFERENCES
- 1. Heilman DW, Green MR, Teodoro JG. 2005. The anaphase promoting complex: a critical target for viral proteins and anti-cancer drugs. Cell Cycle 4:560–563 [PubMed] [Google Scholar]
- 2. DeCaprio JA. 2009. How the Rb tumor suppressor structure and function was revealed by the study of Adenovirus and SV40. Virology 384:274–284 [DOI] [PubMed] [Google Scholar]
- 3. Lane DP, Crawford LV. 1979. T antigen is bound to a host protein in SV40-transformed cells. Nature 278:261–263 [DOI] [PubMed] [Google Scholar]
- 4. Linzer DI, Levine AJ. 1979. Characterization of a 54K dalton cellular SV40 tumor antigen present in SV40-transformed cells and uninfected embryonal carcinoma cells. Cell 17:43–52 [DOI] [PubMed] [Google Scholar]
- 5. Whyte P, Buchkovich KJ, Horowitz JM, Friend SH, Raybuck M, Weinberg RA, Harlow E. 1988. Association between an oncogene and an anti-oncogene: the adenovirus E1A proteins bind to the retinoblastoma gene product. Nature 334:124–129 [DOI] [PubMed] [Google Scholar]
- 6. Thornton BR, Ng TM, Matyskiela ME, Carroll CW, Morgan DO, Toczyski DP. 2006. An architectural map of the anaphase-promoting complex. Genes Dev. 20:449–460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fehr AR, Gualberto NC, Savaryn JP, Terhune SS, Yu D. 2012. Proteasome-dependent disruption of the E3 ubiquitin ligase anaphase-promoting complex by HCMV protein pUL21a. PLoS Pathog. 8:e1002789. 10.1371/journal.ppat.1002789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zeng X, Sigoillot F, Gaur S, Choi S, Pfaff KL, Oh DC, Hathaway N, Dimova N, Cuny GD, King RW. 2010. Pharmacologic inhibition of the anaphase-promoting complex induces a spindle checkpoint-dependent mitotic arrest in the absence of spindle damage. Cancer Cell 18:382–395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Peters JM. 2006. The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat. Rev. Mol. Cell. Biol. 7:644–656 [DOI] [PubMed] [Google Scholar]
- 10. Tang Z, Li B, Bharadwaj R, Zhu H, Ozkan E, Hakala K, Deisenhofer J, Yu H. 2001. APC2 cullin protein and APC11 RING protein comprise the minimal ubiquitin ligase module of the anaphase-promoting complex. Mol. Biol. Cell 12:3839–3851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Vodermaier HC, Gieffers C, Maurer-Stroh S, Eisenhaber F, Peters JM. 2003. TPR subunits of the anaphase-promoting complex mediate binding to the activator protein CDH1. Curr. Biol. 13:1459–1468 [DOI] [PubMed] [Google Scholar]
- 12. Carroll CW, Enquist-Newman M, Morgan DO. 2005. The APC subunit Doc1 promotes recognition of the substrate destruction box. Curr. Biol. 15:11–18 [DOI] [PubMed] [Google Scholar]
- 13. Carroll CW, Morgan DO. 2002. The Doc1 subunit is a processivity factor for the anaphase-promoting complex. Nat. Cell Biol. 4:880–887 [DOI] [PubMed] [Google Scholar]
- 14. Kraft C, Vodermaier HC, Maurer-Stroh S, Eisenhaber F, Peters JM. 2005. The WD40 propeller domain of Cdh1 functions as a destruction box receptor for APC/C substrates. Mol. Cell 18:543–553 [DOI] [PubMed] [Google Scholar]
- 15. Kraft C, Herzog F, Gieffers C, Mechtler K, Hagting A, Pines J, Peters JM. 2003. Mitotic regulation of the human anaphase-promoting complex by phosphorylation. EMBO J. 22:6598–6609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kramer ER, Scheuringer N, Podtelejnikov AV, Mann M, Peters JM. 2000. Mitotic regulation of the APC activator proteins CDC20 and CDH1. Mol. Biol. Cell 11:1555–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rudner AD, Murray AW. 2000. Phosphorylation by Cdc28 activates the Cdc20-dependent activity of the anaphase-promoting complex. J. Cell Biol. 149:1377–1390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Shteinberg M, Protopopov Y, Listovsky T, Brandeis M, Hershko A. 1999. Phosphorylation of the cyclosome is required for its stimulation by Fizzy/cdc20. Biochem. Biophys. Res. Commun. 260:193–198 [DOI] [PubMed] [Google Scholar]
- 19. Visintin R, Craig K, Hwang ES, Prinz S, Tyers M, Amon A. 1998. The phosphatase Cdc14 triggers mitotic exit by reversal of Cdk-dependent phosphorylation. Mol. Cell 2:709–718 [DOI] [PubMed] [Google Scholar]
- 20. Zachariae W, Schwab M, Nasmyth K, Seufert W. 1998. Control of cyclin ubiquitination by CDK-regulated binding of Hct1 to the anaphase promoting complex. Science 282:1721–1724 [DOI] [PubMed] [Google Scholar]
- 21. Hwang LH, Lau LF, Smith DL, Mistrot CA, Hardwick KG, Hwang ES, Amon A, Murray AW. 1998. Budding yeast Cdc20: a target of the spindle checkpoint. Science 279:1041–1044 [DOI] [PubMed] [Google Scholar]
- 22. Hsu JY, Reimann JD, Sorensen CS, Lukas J, Jackson PK. 2002. E2F-dependent accumulation of hEmi1 regulates S phase entry by inhibiting APC(Cdh1). Nat. Cell Biol. 4:358–366 [DOI] [PubMed] [Google Scholar]
- 23. Listovsky T, Oren YS, Yudkovsky Y, Mahbubani HM, Weiss AM, Lebendiker M, Brandeis M. 2004. Mammalian Cdh1/Fzr mediates its own degradation. EMBO J. 23:1619–1626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Prinz S, Hwang ES, Visintin R, Amon A. 1998. The regulation of Cdc20 proteolysis reveals a role for APC components Cdc23 and Cdc27 during S phase and early mitosis. Curr. Biol. 8:750–760 [DOI] [PubMed] [Google Scholar]
- 25. Rape M, Kirschner MW. 2004. Autonomous regulation of the anaphase-promoting complex couples mitosis to S-phase entry. Nature 432:588–595 [DOI] [PubMed] [Google Scholar]
- 26. Shirayama M, Zachariae W, Ciosk R, Nasmyth K. 1998. The Polo-like kinase Cdc5p and the WD-repeat protein Cdc20p/fizzy are regulators and substrates of the anaphase promoting complex in Saccharomyces cerevisiae. EMBO J. 17:1336–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yamanaka A, Hatakeyama S, Kominami K, Kitagawa M, Matsumoto M, Nakayama K. 2000. Cell cycle-dependent expression of mammalian E2-C regulated by the anaphase-promoting complex/cyclosome. Mol. Biol. Cell 11:2821–2831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Araki M, Yu H, Asano M. 2005. A novel motif governs APC-dependent degradation of Drosophila ORC1 in vivo. Genes Dev. 19:2458–2465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Jin L, Williamson A, Banerjee S, Philipp I, Rape M. 2008. Mechanism of ubiquitin-chain formation by the human anaphase-promoting complex. Cell 133:653–665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Littlepage LE, Ruderman JV. 2002. Identification of a new APC/C recognition domain, the A box, which is required for the Cdh1-dependent destruction of the kinase Aurora-A during mitotic exit. Genes Dev. 16:2274–2285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Fang G, Yu H, Kirschner MW. 1998. Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol. Cell 2:163–171 [DOI] [PubMed] [Google Scholar]
- 32. Pfleger CM, Kirschner MW. 2000. The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev. 14:655–665 [PMC free article] [PubMed] [Google Scholar]
- 33. King RW, Peters JM, Tugendreich S, Rolfe M, Hieter P, Kirschner MW. 1995. A 20S complex containing CDC27 and CDC16 catalyzes the mitosis-specific conjugation of ubiquitin to cyclin B. Cell 81:279–288 [DOI] [PubMed] [Google Scholar]
- 34. Murray AW, Solomon MJ, Kirschner MW. 1989. The role of cyclin synthesis and degradation in the control of maturation promoting factor activity. Nature 339:280–286 [DOI] [PubMed] [Google Scholar]
- 35. Sudakin V, Ganoth D, Dahan A, Heller H, Hershko J, Luca FC, Ruderman JV, Hershko A. 1995. The cyclosome, a large complex containing cyclin-selective ubiquitin ligase activity, targets cyclins for destruction at the end of mitosis. Mol. Biol. Cell 6:185–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nasmyth K. 2001. Disseminating the genome: joining, resolving, and separating sister chromatids during mitosis and meiosis. Annu. Rev. Genet. 35:673–745 [DOI] [PubMed] [Google Scholar]
- 37. Manchado E, Eguren M, Malumbres M. 2010. The anaphase-promoting complex/cyclosome (APC/C): cell-cycle-dependent and -independent functions. Biochem. Soc. Trans. 38:65–71 [DOI] [PubMed] [Google Scholar]
- 38. Robert A, Miron MJ, Champagne C, Gingras MC, Branton PE, Lavoie JN. 2002. Distinct cell death pathways triggered by the adenovirus early region 4 ORF 4 protein. J. Cell Biol. 158:519–528 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Kornitzer D, Sharf R, Kleinberger T. 2001. Adenovirus E4orf4 protein induces PP2A-dependent growth arrest in Saccharomyces cerevisiae and interacts with the anaphase-promoting complex/cyclosome. J. Cell Biol. 154:331–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Mui MZ, Roopchand DE, Gentry MS, Hallberg RL, Vogel J, Branton PE. 2010. Adenovirus protein E4orf4 induces premature APCCdc20 activation in Saccharomyces cerevisiae by a protein phosphatase 2A-dependent mechanism. J. Virol. 84:4798–4809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Miron MJ, Blanchette P, Groitl P, Dallaire F, Teodoro JG, Li S, Dobner T, Branton PE. 2009. Localization and importance of the adenovirus E4orf4 protein during lytic infection. J. Virol. 83:1689–1699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Frisch SM, Mymryk JS. 2002. Adenovirus-5 E1A: paradox and paradigm. Nat. Rev. 3:441–452 [DOI] [PubMed] [Google Scholar]
- 43. Gallimore PH, Turnell AS. 2001. Adenovirus E1A: remodelling the host cell, a life or death experience. Oncogene 20:7824–7835 [DOI] [PubMed] [Google Scholar]
- 44. Ghosh MK, Harter ML. 2003. A viral mechanism for remodeling chromatin structure in G0 cells. Mol. Cell 12:255–260 [DOI] [PubMed] [Google Scholar]
- 45. Turnell AS, Stewart GS, Grand RJ, Rookes SM, Martin A, Yamano H, Elledge SJ, Gallimore PH. 2005. The APC/C and CBP/p300 cooperate to regulate transcription and cell-cycle progression. Nature 438:690–695 [DOI] [PubMed] [Google Scholar]
- 46. Turnell AS, Mymryk JS. 2006. Roles for the coactivators CBP and p300 and the APC/C E3 ubiquitin ligase in E1A-dependent cell transformation. Br. J. Cancer 95:555–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fujimoto T, Hata T, Itoyama T, Nakamura H, Tsukasaki K, Yamada Y, Ikeda S, Sadamori N, Tomonaga M. 1999. High rate of chromosomal abnormalities in HTLV-I-infected T-cell colonies derived from prodromal phase of adult T-cell leukemia: a study of IL-2-stimulated colony formation in methylcellulose. Cancer Genet. Cytogenet. 109:1–13 [DOI] [PubMed] [Google Scholar]
- 48. Kinoshita K, Amagasaki T, Ikeda S, Suzuyama J, Toriya K, Nishino K, Tagawa M, Ichimaru M, Kamihira S, Yamada Y. 1985. Preleukemic state of adult T cell leukemia: abnormal T lymphocytosis induced by human adult T cell leukemia-lymphoma virus. Blood 66:120–127 [PubMed] [Google Scholar]
- 49. Liu B, Hong S, Tang Z, Yu H, Giam CZ. 2005. HTLV-I Tax directly binds the Cdc20-associated anaphase-promoting complex and activates it ahead of schedule. Proc. Natl. Acad. Sci. U. S. A. 102:63–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kuo YL, Giam CZ. 2006. Activation of the anaphase promoting complex by HTLV-1 tax leads to senescence. EMBO J. 25:1741–1752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liu B, Liang MH, Kuo YL, Liao W, Boros I, Kleinberger T, Blancato J, Giam CZ. 2003. Human T-lymphotropic virus type 1 oncoprotein tax promotes unscheduled degradation of Pds1p/securin and Clb2p/cyclin B1 and causes chromosomal instability. Mol. Cell. Biol. 23:5269–5281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Merling R, Chen C, Hong S, Zhang L, Liu M, Kuo YL, Giam CZ. 2007. HTLV-1 Tax mutants that do not induce G1 arrest are disabled in activating the anaphase promoting complex. Retrovirology 4:35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Bellanger S, Blachon S, Mechali F, Bonne-Andrea C, Thierry F. 2005. High-risk but not low-risk HPV E2 proteins bind to the APC activators Cdh1 and Cdc20 and cause genomic instability. Cell Cycle 4:1608–1615 [DOI] [PubMed] [Google Scholar]
- 54. Penrose KJ, McBride AA. 2000. Proteasome-mediated degradation of the papillomavirus E2-TA protein is regulated by phosphorylation and can modulate viral genome copy number. J. Virol. 74:6031–6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kim S, Park SY, Yong H, Famulski JK, Chae S, Lee JH, Kang CM, Saya H, Chan GK, Cho H. 2008. HBV X protein targets hBubR1, which induces dysregulation of the mitotic checkpoint. Oncogene 27:3457–3464 [DOI] [PubMed] [Google Scholar]
- 56. Martin-Lluesma S, Schaeffer C, Robert EI, van Breugel PC, Leupin O, Hantz O, Strubin M. 2008. Hepatitis B virus X protein affects S phase progression leading to chromosome segregation defects by binding to damaged DNA binding protein 1. Hepatology 48:1467–1476 [DOI] [PubMed] [Google Scholar]
- 57. Teodoro JG, Heilman DW, Parker AE, Green MR. 2004. The viral protein Apoptin associates with the anaphase-promoting complex to induce G2/M arrest and apoptosis in the absence of p53. Genes Dev. 18:1952–1957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Heilman DW, Teodoro JG, Green MR. 2006. Apoptin nucleocytoplasmic shuttling is required for cell type-specific localization, apoptosis, and recruitment of the anaphase-promoting complex/cyclosome to PML bodies. J. Virol. 80:7535–7545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Prasetyo AA, Kamahora T, Kuroishi A, Murakami K, Hino S. 2009. Replication of chicken anemia virus (CAV) requires apoptin and is complemented by VP3 of human torque teno virus (TTV). Virology 385:85–92 [DOI] [PubMed] [Google Scholar]
- 60. Mo M, Fleming SB, Mercer AA. 2009. Cell cycle deregulation by a poxvirus partial mimic of anaphase-promoting complex subunit 11. Proc. Natl. Acad. Sci. U. S. A. 106:19527–19532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Mo M, Fleming SB, Mercer AA. 2010. Orf virus cell cycle regulator, PACR, competes with subunit 11 of the anaphase promoting complex for incorporation into the complex. J. Gen. Virol. 91:3010–3015 [DOI] [PubMed] [Google Scholar]
- 62. Bhattacharjee B, Renzette N, Kowalik TF. 2012. Genetic analysis of cytomegalovirus in malignant gliomas. J. Virol. 86:6815–6824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Ranganathan P, Clark PA, Kuo JS, Salamat MS, Kalejta RF. 2012. Significant association of multiple human cytomegalovirus genomic loci with glioblastoma multiforme samples. J. Virol. 86:854–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Soroceanu L, Cobbs CS. 2011. Is HCMV a tumor promoter? Virus Res. 157:193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Tran K, Kamil JP, Coen DM, Spector DH. 2010. Inactivation and disassembly of the anaphase-promoting complex during human cytomegalovirus infection is associated with degradation of the APC5 and APC4 subunits and does not require UL97-mediated phosphorylation of Cdh1. J. Virol. 84:10832–10843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Tran K, Mahr JA, Choi J, Teodoro JG, Green MR, Spector DH. 2008. Accumulation of substrates of the anaphase-promoting complex (APC) during human cytomegalovirus infection is associated with the phosphorylation of Cdh1 and the dissociation and relocalization of APC subunits. J. Virol. 82:529–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wiebusch L, Bach M, Uecker R, Hagemeier C. 2005. Human cytomegalovirus inactivates the G0/G1-APC/C ubiquitin ligase by Cdh1 dissociation. Cell Cycle 4:1435–1439 [DOI] [PubMed] [Google Scholar]
- 68. Hume AJ, Finkel JS, Kamil JP, Coen DM, Culbertson MR, Kalejta RF. 2008. Phosphorylation of retinoblastoma protein by viral protein with cyclin-dependent kinase function. Science 320:797–799 [DOI] [PubMed] [Google Scholar]
- 69. Prichard MN, Sztul E, Daily SL, Perry AL, Frederick SL, Gill RB, Hartline CB, Streblow DN, Varnum SM, Smith RD, Kern ER. 2008. Human cytomegalovirus UL97 kinase activity is required for the hyperphosphorylation of retinoblastoma protein and inhibits the formation of nuclear aggresomes. J. Virol. 82:5054–5067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Fehr AR, Yu D. 2010. Human cytomegalovirus gene UL21a encodes a short-lived cytoplasmic protein and facilitates virus replication in fibroblasts. J. Virol. 84:291–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Fehr AR, Yu D. 2011. Human cytomegalovirus early protein pUL21a promotes efficient viral DNA synthesis and the late accumulation of immediate-early transcripts. J. Virol. 85:663–674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Mechali F, Hsu CY, Castro A, Lorca T, Bonne-Andrea C. 2004. Bovine papillomavirus replicative helicase E1 is a target of the ubiquitin ligase APC. J. Virol. 78:2615–2619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Buschhorn BA, Petzold G, Galova M, Dube P, Kraft C, Herzog F, Stark H, Peters JM. 2011. Substrate binding on the APC/C occurs between the coactivator Cdh1 and the processivity factor Doc1. Nat. Struct. Mol. Biol. 18:6–13 [DOI] [PMC free article] [PubMed] [Google Scholar]