

Abstract
Infectious bursal disease virus (IBDV), a double-stranded RNA virus belonging to the Birnaviridae family, causes immunosuppression in chickens. In this study, we defined the localization of IBDV replication complexes based on colocalization analysis of VP3, the major protein component of IBDV ribonucleoproteins (RNPs). Our results indicate that VP3 localizes to vesicular structures bearing features of early and late endocytic compartments located in the juxtanuclear region. Interfering with the endocytic pathway with a dominant negative version of Rab5 after the internalization step leads to a reduction in virus titer. Triple-immunostaining studies between VP3, the viral RNA-dependent RNA polymerase VP1, and viral double-stranded RNA (dsRNA) showed a well-defined colocalization, indicating that the three critical components of the RNPs colocalize in the same structure, likely representing replication complexes. Interestingly, recombinant expressed VP3 also localizes to endosomes. Employing Golgi markers, we found that VP3-containing vesicles were closely associated with this organelle. Depolymerization of microtubules with nocodazole caused a profound change in VP3 localization, showing a punctate distribution scattered throughout the cytoplasm. However, these VP3-positive structures remained associated with Golgi ministacks. Similarly, brefeldin A (BFA) treatment led to a punctate distribution of VP3, scattered throughout the cytoplasm of infected cells. In addition, analysis of intra- and extracellular viral infective particles after BFA treatment of avian cells suggested a role for the Golgi complex in viral assembly. These results constitute the first study elucidating the localization of IBDV replication complexes (i.e., in endocytic compartments) and establishing a role for the Golgi apparatus in the assembly step of a birnavirus.
INTRODUCTION
Infectious bursal disease virus (IBDV) belongs to the Birnaviridae family, which groups economically important pathogens. IBDV is a nonenveloped, bisegmented, double-stranded-RNA (dsRNA) virus with an icosahedral capsid that belongs to the genus Avibirnavirus (1). Segment A contains two partially overlapping open reading frames (ORFs). The first ORF encodes the nonessential nonstructural viral protein 5 (VP5) (2). The second ORF encodes a polyprotein that is cotranslationally self-cleaved by the viral protease VP4, yielding the precursor pVP2, VP4, and VP3. The resulting intermediate, pVP2, is further processed at the C-terminal region during maturation into VP2 polypeptide and several peptides that remain associated with the capsid (3). VP2 is the single structural component of the viral capsid. VP2 and VP3 are the major structural proteins, constituting 60% and 35% of the virion, respectively (4). Segment B is monocistronic and encodes the viral RNA-dependent RNA polymerase VP1 (5).
The Birnaviridae family lacks the T=2 core structurally conserved among all dsRNA viruses, but their genome is instead integrated into a ribonucleoprotein (RNP) complex (6) functionally competent for RNA synthesis, similarly to the replication and transcription complexes of some positive-sense single-stranded-RNA (ssRNA) viruses (7, 8). IBDV RNPs consist of VP1-dsRNA, “free” VP1, and VP3 (6). VP3 is a multitasking protein that acts as a scaffolding protein for pVP2 during particle morphogenesis (9, 10), participates in the recruitment of VP1 to the capsid, and serves as a transcriptional activator (11). All these well-documented observations predict that VP3 is a protagonist member of the viral replication process, and therefore, its cellular localization becomes a marker of the localization of IBDV replication complexes (RCs).
RNA viruses replicate their genome in intracellular membranes of infected cells (12, 13), integrated into a complex structure known as the “viral factory.” The viral replication machinery is usually inserted into single- or double-membrane vesicles that can be associated with a variety of organelles, such as the rough endoplasmic reticulum (ER), mitochondria, the endolysosomal system, and chloroplasts or vacuolar membranes in plants (13–16). Nowadays, much effort is being made to gain insight into the cellular aspects of the birnaviral replication process. We and others have recently demonstrated that the virus utilizes the endocytic pathway to gain entry into the cell (17, 18). Galloux et al. suggested that uncoating of the virus occurs within the endosome in response to a low-pH environment (19); however, the details of how and where birnaviruses replicate their genomes and translate their proteins remain completely unexplored.
In this work, we have studied the subcellular localization of the replication complexes of IBDV, analyzing the distribution of the IBDV components VP3, VP1, and dsRNA. We determined their localization by using well-established subcellular markers and confocal laser scanning microscopy (CLSM). Surprisingly, our data strongly suggest that IBDV replication occurs on endosomal membrane compartments, and functionally interfering with the endocytic pathway affected intra- and extracellular virus yields. In addition, we employed the Golgi-disrupting agents nocodazole (ND) and brefeldin A (BFA) to assess the functional role of this organelle. Our results with avian cells indicate that the Golgi apparatus plays an important role in IBDV assembly. To the best of our knowledge, this is the first study describing the cellular compartments involved in the establishment of the replication complexes of IBDV and in the assembly step of an important member of the Birnaviridae family.
MATERIALS AND METHODS
Cells, viruses, and antibodies.
The IBDV Soroa strain, a virulent serotype 1 virus, was propagated in QM7 quail muscle cells (ATCC CRL-1962) as previously described (20). Human epithelial HeLa cells and avian fibroblast QM7 cells were grown at 37°C in an atmosphere of 95% air and 5% CO2 in Dulbecco's modified Eagle's medium (DMEM; Life Technologies, Argentina) containing 10% fetal calf serum (FCS; Internegocios, Argentina). Western blot and confocal laser scanning microscopy (CLSM) analyses were carried out by using rabbit anti-VP3 specific sera as previously described (21), whole rabbit antiserum immunized with recombinant VP1, or whole rat antiserum immunized with recombinant VP3 obtained by the laboratory of José F. Rodríguez. Mouse monoclonal dsRNA antibody clone J2 was purchased from English & Scientific Consulting Bt. (Hungary). Monoclonal antibody J2 has been shown to specifically recognize dsRNAs >40 bp in length, and antibody binding is independent of sequence and nucleotide composition (22). Moreover, Weber et al. clearly demonstrated that J2 efficiently detected the presence and localization of dsRNA in cells infected with a positive-strand RNA or a dsRNA genome as well as DNA viruses by employing immunofluorescence (IF) analysis (23). Purified mouse anti-early endosome antigen 1 (EEA1) and anti-GM130 were obtained from BD Biosciences (Becton, Dickinson, Argentina). Monoclonal rat anti-lysosome-associated membrane protein 2 (LAMP-2) and mouse anti-β-tubulin were obtained from the Developmental Studies Hybridoma Bank (DSHB, USA). Monoclonal mouse anti-lysobisphosphatidic acid (LBPA) was a gift from J. Gruenberg (Department of Biochemistry, University of Geneva, Switzerland), and it was used as described previously (24). Mouse monoclonal antibody CTR433 was a gift from M. Bornens (Institut Curie, Paris, France) (25). Secondary goat anti-rabbit antibodies conjugated with Alexa 488 or Cy3, donkey anti-mouse antibodies conjugated with Cy3, and goat anti-rat antibodies conjugated with Cy3 and Hoechst 33342 were purchased from Molecular Probes (Life Technologies, Argentina). Peroxidase-conjugated secondary antibodies were purchased from Sigma-Aldrich (Buenos Aires, Argentina).
Plasmids and reagents.
Plasmids encoding enhanced green fluorescent protein-tagged wild-type Rab5 (EGFP-Rab5wt) and EGFP-Rab5S34N were kindly provided by Philip D. Stahl (Washington University, St. Louis, MO, USA). A plasmid encoding EGFP-Rab7 was a gift from Bo van Deurs (University of Copenhagen, Copenhagen, Denmark), and a plasmid encoding EGFP–LAMP-1 was kindly provided by Renato Mortara (Escuola Paulista de Medicina, Sao Paulo, Brazil). The plasmids encoding the N-terminal domain of the glycolipid glycosyltransferases SialT2 (GM3_2,8-sialyltransferase or GD3 synthase) and GalNAcT (GM3/GD3_1,4-N-acetylgalactosaminyltransferase) fused to yellow fluorescent protein (YFP) were kindly provided by H. J. Maccioni (CIQUIBIC, UNC-CONICET, Universidad Nacional de Córdoba, Córdoba, Argentina). pcDNAVP3wt is a derivative of the pcDNA3 eukaryotic expression vector (Invitrogen) that contains the VP3 coding sequence. The acidic compartment marker LysoTracker Red DND-99 and the self-quenched Bodipy dye conjugated to bovine serum albumin (Bodipy-DQ-BSA) were obtained from Molecular Probes (Life Technologies, Argentina). Brefeldin A (BFA) and nocodazole (ND) were purchased from Sigma-Aldrich (Buenos Aires, Argentina) and were prepared according to the manufacturer's instructions.
Transfections and infections.
HeLa or QM7 cells grown on coverslips were transfected with Lipofectamine Plus (Invitrogen, Life Technologies, Argentina) according to the manufacturer's recommendations. Viral infections were carried out with IBDV Soroa strain virions at a multiplicity of infection (MOI) of 1 PFU/cell. After 60 min of virus adsorption, the cells were washed, fresh medium was added, and the infection was allowed to proceed at 37°C for 36 h. When indicated, BFA or ND was added to the infected and mock-infected cells at 24 or 12 h postinfection (p.i.), respectively.
Labeling with LysoTracker.
Acidic compartments were labeled with LysoTracker by incubating the cells with 1 μM LysoTracker at 37°C for 60 min, and the cells were washed and left an additional 60 min with fresh medium without the probe. Cells were fixed and analyzed by CLSM.
Test of lysosomal function by degradation of chromogenic BSA.
The ability of cells to endocytose and degrade the self-quenched red Bodipy dye conjugated to BSA (DQ-BSA) was used to measure lysosomal degradative function. Red DQ-BSA requires enzymatic cleavage in an acidic intracellular compartment to generate a highly fluorescent product, which can be monitored by confocal microscopy. Cells were incubated for 12 h at 37°C with DQ-BSA (10 μg/ml in complete culture medium) to ensure that the reagent reached the lysosomal compartment, washed, and left an additional 60 min with fresh medium without the probe. Cells were fixed and analyzed by CLSM.
SDS-PAGE and Western blotting.
Protein samples of a total cell lysate from mock-infected or infected HeLa and QM7 cells were run on a 12% polyacrylamide gel and transferred onto Hybond-ECL (GE Healthcare Argentina S.A.) nitrocellulose membranes. The membranes were blocked for 1 h with blocking solution (5% nonfat milk, 0.1% Tween 20, and phosphate-buffered saline [PBS]), washed twice with PBS, and incubated with anti-VP3 or anti-β-tubulin primary antibody and the corresponding peroxidase-conjugated secondary antibodies. Membranes were washed extensively, the corresponding bands were detected by using an enhanced chemiluminescence detection kit from Healthcare (catalog number RPN2109; Amersham), and the data were collected with an LAS-4000 imaging system (Fujifilm, Japan). The protein band intensities from three independent experiments were quantitated with Adobe Photoshop CS5 software and normalized against tubulin. Results are expressed as relative units representing a ratio between VP3 and tubulin band intensities. A paired Student t test was performed by using Ky-Plot software.
Indirect immunofluorescence and quantification of colocalization.
All immunofluorescence procedures were carried out at room temperature. Cells were fixed with a 3% paraformaldehyde solution in PBS for 15 min at room temperature and quenched by incubation with 50 mM NH4Cl in PBS for 20 min. Subsequently, transfected cells were permeabilized with 0.05% saponin in PBS containing 0.2% BSA and incubated with primary antibodies and the corresponding Alexa 488-, Cy3-, or Cascade Blue-conjugated secondary antibodies. Cells were mounted with Mowiol (plus Hoechst stain) and analyzed by CLSM. Images were captured by using an Olympus FluoView TM FV1000 confocal microscope (Olympus, Argentina) with FV10-ASW (version 01.07.00.16) software and processed by using Adobe CS5 (Adobe Systems). A modification to Pearson's correlation coefficient developed by Manders et al. (26) was used to quantitate the degree of colocalization for the proteins, known commonly as the “overlap coefficient” (26, 27). In this work, we determined the fraction of pixels corresponding to VP3, VP1, or dsRNA that overlapped those corresponding to the subcellular marker expressed as a percentage. We employed ImageJ software (National Institutes of Health). The data in the figures represent the means and standard deviations obtained after analyzing 50 cells per condition from three independent experiments. Representative images are shown for each case.
2D and 3D reconstruction with confocal laser scanning microscopy.
Images were computerized to automatically make 10 serial optical sections of each cell at intervals of around 39 μm. The two-dimensional (2D) and 3D reconstructions were performed with ImageJ software (National Institutes of Health). For 3D reconstruction, images were opened with ImageJ, and a 3D surface model was created based on the appropriate intensity threshold.
Virus titration.
We used the 50% tissue culture infective dose (TCID50) titration method based on the endpoint dilution of the virus at which a cytopathic effect (CPE) is detected in 50% of the cell culture infected by a given amount of virus suspension, as described previously (28). CPE induced by IBDV could be observed under a microscope after 5 days. To titrate extracellular infective viral particles, we used the infected-cell supernatants. Instead, when intracellular infective viral particles were titrated, infected cells were collected with 1 ml of DMEM, subjected to three freeze-thaw cycles, and then centrifuged to eliminate cellular detritus. The supernatants corresponding to a viral suspension were employed for titration. The titers obtained from three independent experiments were statistically analyzed by a paired Student t test using Ky-Plot software.
RNA isolation, cDNA synthesis, and qRT-PCR assays.
Total RNA was extracted from 1 × 107 cells by employing TRIzol reagent (Invitrogen, Life Technologies, USA) according to the manufacturer's recommendations. Total RNA (1 μg) was reverse transcribed in a final volume of 20 μl containing 1× PCR buffer II (Applied Biosystems, Life Technologies, USA), 5 mM MgCl2, 1 mM deoxynucleoside triphosphate (dNTP), 20 U RNase inhibitor, 50 U murine leukemia virus (MuLV) reverse transcriptase (Applied Biosystems, Life Technologies, USA), 5 μM random primers (Gibco), and diethyl pyrocarbonate (DEPC)-treated distilled H2O. Transcript levels were determined by real-time quantitative PCR (qRT-PCR), using a StepOne real-time PCR system (Applied Biosystems, Life Technologies, USA) and SYBR green dye (Applied Biosystems, Life Technologies, USA). Reactions were performed with a final volume of 20 μl containing 10 μl of 2× Power SYBR green PCR Master Mix (including AmpliTaq Gold DNA polymerase, dNTPs, and SYBR green dye), 250 nM forward and reverse specific primers, and a 1:10 dilution of cDNA. After enzyme activation at 95°C for 10 min, amplifications were carried out by using a two-step PCR procedure with 40 cycles of 15 s at 95°C for denaturation and 1 min at 60°C for annealing/extension. Gene-specific primers for viral and control RNA were designed by using Oligo Explorer 1.2 software (Gene Link) based on sequence information deposited at the National Center for Biotechnology Information. Primer sequences used in the qRT-PCR analyses were described previously (29). Nontemplate controls were included for each primer pair, and each PCR was completed in triplicate. Relative quantification was performed by means of the ΔΔCT method using StepOne software v2.2.2 (Applied Biosystems, Life Technologies, USA).
RESULTS
Time course analysis of IBDV-infected cells.
With the aim of determining the adequate time point after infection for viral replication analysis, HeLa or QM7 cells were infected for different time periods with the IBDV Soroa strain at a multiplicity of infection (MOI) of 1 PFU/cell. We used HeLa cells to perform some experiments because they are flat and provide high-quality images for fluorescence microscopy. However, when possible, we also carried out the experiments with QM7 cells, because they are closer to the natural IBDV host cellular system. We analyzed the progression of the infection by determining the levels of VP3 accumulation by Western blotting, the presence and subcellular distribution of VP3 by immunofluorescence, and extracellular viral titers at different time points after infection. As shown in Fig. 1, we were able to detect VP3 only after 8 h postinfection (p.i.) in both cell types. Subsequently, the specific signal became stronger, and it was present in a higher percentage of the cells. At 36 h p.i., almost all of the cells were clearly infected without a widespread cytopathic effect. A similar scenario was observed by Western blotting. Virus titration revealed the presence of infective virions in the extracellular milieu from 8 h p.i., which increased over time. Therefore, these results pointed to 36 h p.i. as a suitable time point to study viral replication, avoiding the interference of viral internalization step.
Fig 1.

Time course of IBDV infection. HeLa (top) or QM7 (bottom) cells grown on coverslips in an M6 multiwell plate were infected with IBDV virions at an MOI of 1. After 4, 8, 12, 24, and 36 h p.i., coverslips were recovered and processed for IF analysis. Primary anti-VP3 and Alexa 488-conjugated secondary antibodies were employed to detect the VP3 distribution (green signal). The remaining cells in each well were harvested and analyzed by Western blotting to determine the VP3 accumulation levels (bottom right corners in HeLa and QM7 panels). The tissue culture supernatants were also collected, and IBDV titers were determined at each time point (results are in red below the Western blot image). Images are representative of three independent experiments.
The IBDV VP3 protein colocalizes with endocytic markers at the perinuclear region in infected cells.
In order to assess the subcellular localization of VP3 in host cells, infection of human HeLa or avian QM7 cells was carried out with the virus at a MOI of 1 PFU/cell, as indicated above. At 36 h p.i., cells were processed for immunofluorescence (IF) analysis using anti-VP3 antibodies and analyzed by confocal laser scanning microscopy (CLSM). As shown in Fig. 2a, e, i, m, q, and u, VP3 presented a punctate distribution with a marked localization in the juxtanuclear region. Since endocytosis has been demonstrated to be an entry pathway for IBDV (17, 18), we assessed whether the virus uses these organelles to carry out its own replication process. Therefore, antibodies or overexpressed proteins were used in double-labeling IF studies. To label early endosomes, antibodies against early endosome antigen 1 (EEA1) as well as overexpression of the early endosomal protein Rab5 (30) as a green fluorescent protein construct (i.e., pEGFP-Rab5) were used. Untransfected HeLa cells or cells overexpressing EGFP-Rab5 were infected, and viral VP3 expression was detected. As shown in Fig. 2, we found a marked colocalization of the VP3 polypeptide with both EEA1 [(71.6 ± 17.8)%] (Fig. 2a to d) and Rab5 [(53.0 ± 15.0)%] (Fig. 2e to h). To label the late endocytic compartment, we analyzed the colocalization of VP3 with Rab7, a protein specifically related to degradative compartments such as late endosomes and lysosomes (31). In addition, we employed antibodies against lysobisphosphatidic acid (LBPA), an acidic lipid found in late endosomes, particularly in the membrane of intraluminal vesicles of multivesicular bodies (32). Cells overexpressing EGFP-Rab7 or untransfected HeLa cells were infected, and viral VP3 expression was detected. As shown in Fig. 2i to l, the level of colocalization of the VP3 polypeptide with EGFP-Rab7 was relatively high [(47.8 ± 15.1)%], whereas very little colocalization was observed with LBPA [(8.2 ± 2.6)%] (Fig. 2m to p). In addition, since lysosome-associated membrane proteins (LAMPs) are well-known markers of late endocytic structures as well as lysosomes (33, 34), antibodies to LAMP-2 or an EGFP-fused LAMP-1 construct were used. We found colocalization of VP3 with both LAMP markers [(33.6% ± 15.9)% for endogenous LAMP-2 (Fig. 2q and t) and (81.2 ± 11.4)% for overexpressed EGFP–LAMP-1 (Fig. 2u to x)]. In all cases, a normal distribution of the endocytic markers was observed in infected cells.
Fig 2.

IBDV VP3 colocalizes with endocytic markers. Transfected or untransfected HeLa cells were infected and processed for indirect immunofluorescence analysis with anti-VP3 (1:500) and the corresponding Alexa 488-conjugated (green signal in untransfected cells) or Cy3-conjugated (red signal in EGFP-Rab5 [e to h]-, EGFP-Rab7 [i to l]-, and EGFP–LAMP-1 [u to x]-transfected cells) secondary antibodies. In untransfected HeLa cells, double immune staining was performed with the corresponding endosomal marker as indicated. The nuclei are stained with Hoechst stain (blue). The insets show the colocalization of VP3 with the cellular marker in the perinuclear region. Images are representative of three independent experiments. Scale bars represent 10 μm.
We next determined if the VP3-containing vesicles were acidic (using LysoTracker) or degradative by employing DQ-BSA, as described in Materials and Methods. We observed that the majority of the VP3-containing vesicles were not labeled by LysoTracker, indicating their nonacidic nature. Additionally, the VP3 protein-specific signal did not colocalize with DQ-BSA, suggesting that these vesicles are nondegradative compartments (data not shown).
Taken together, our colocalization studies indicate that at 36 h p.i., VP3 is associated with endocytic structures, likely representing the site for the replication complexes in infected cells.
The three components of IBDV RNPs (i.e., VP3, VP1, and dsRNA) colocalize in the same structure.
In order to corroborate that VP3 is a marker of IBDV replication complexes (RCs), we analyzed the subcellular localization of two additional IBDV RNP elements: the polymerase VP1 and the viral dsRNA. The subcellular distribution of VP1 in infected HeLa and QM7 cells was analyzed by using a specific rabbit antiserum. For comparative purposes, VP3 was also analyzed in the same experiment (Fig. 3Aa to d and Ba to d). As shown in Fig. 3Ae, the VP1-specific fluorescence presented a punctate distribution, with marked localization detected in the juxtanuclear region in HeLa cells similar to that exhibited by VP3. In addition, anti-EEA1 antibodies were used in double-labeling IF studies to characterize the intracellular location of VP1. As shown in Fig. 3Ad and h, we found a high degree of colocalization of both VP3 and VP1 with EEA1 in HeLa cells [(52.8 ± 8.3)% and (63.2 ± 6.9)%, respectively]. These results strongly suggest that VP1, similarly to VP3, localizes in endosomal structures, likely constituting the IBDV RCs.
Fig 3.

Colocalization analysis of IBDV RNP components. (A) Infected HeLa cells were processed for double immune staining with anti-VP1 (1:300) or anti-VP3 (1:500) as well as the corresponding Alexa 488 (green signal)-conjugated secondary antibodies and anti-EEA1 antibody and the corresponding Cy3 (red signal)-conjugated secondary antibodies. (B) Infected HeLa cells were processed for double immune staining with anti-VP1 (1:300) or anti-VP3 (1:500) as well as the corresponding Alexa 488-conjugated (green signal) secondary antibodies and anti-dsRNA antibody and the corresponding Cy3-conjugated (red signal) secondary antibodies. The nuclei were stained with Hoechst stain (blue). (C) Triple immune staining of infected HeLa cells with mouse anti-dsRNA, rat anti-VP3, and rabbit anti-VP1 and the Cascade Blue-, Alexa 488-, and Cy3-conjugated secondary antibodies, respectively. The insets show colocalization of the proteins in the perinuclear region. Images are representative of three independent experiments. Scale bars represent 10 μm. DIC, differential interference contrast.
Next, we employed a mouse monoclonal antibody specific for dsRNA to analyze the subcellular distribution of the viral dsRNA in infected cells. We used monoclonal antibody J2, which has been successfully employed to visualize dsRNA in cells infected with a number of positive-strand and dsRNA viruses (35–37). In our system, this antibody allowed us to detect accumulated dsRNA in infected cells, with 36 h p.i. being a time point when most of the cells showed accumulated dsRNA (data not shown). We observed a specific, well-defined, punctate distribution in the juxtanuclear region in both cell types (Fig. 3Bb and f). Furthermore, when double-labeling IF studies were performed, we observed a clear colocalization between the viral proteins and the dsRNA in infected cells (Fig. 3Bc, d, g, and h), with colocalization degrees of (51.7 ± 12.7)% for VP3-dsRNA and (64.8 ± 4.6)% for VP1-dsRNA in HeLa cells. Similar results were obtained with QM7 cells (data not shown). In addition, we employed a new antibody against VP3 raised in rat, which allowed us to perform triple-colocalization studies between VP1, VP3, and the dsRNA. We found a high degree of colocalization, (45.3 ± 13.7)% (Fig. 3C). These results indicate that the viral components VP3, VP1, and dsRNA, involved directly in RNA metabolism, colocalize in endocytic structures located in the juxtanuclear region of infected cells and suggest that replication of IBDV is taking place in these endocytic vesicles.
A functional endocytic pathway is required for virus replication.
Rab5 is a critical regulatory factor of trafficking through the endocytic pathway (38). Key regulatory functions of this protein have been uncovered mainly by the use of mutated proteins in both in vivo and in vitro assays. A mutation that renders a protein defective in GTP binding (i.e., Rab5S34N) leads to small endosomes and inhibition of endocytosis (39). In order to test the functional role of the endocytic pathway in IBDV replication, HeLa cells overexpressing the wild-type form of Rab5, Rab5wt, or the GDP-locked form Rab5S34N as EGFP-fused constructs were used. To avoid affecting virus internalization, HeLa cells were first infected, and at 12 h p.i., cells were transfected with pEGFP, pEGFP-Rab5wt, or pEGFP-Rab5S34N and intra- and extracellular virus titers were determined. As shown in Fig. 4, a significant reduction in virus titer was observed both inside and outside the cells, indicating that a functional endocytic pathway is necessary for IBDV replication.
Fig 4.

Endocytic pathway interference affects viral replication. HeLa cells were infected with IBDV, and at 12 h p.i., the cells were transfected with pEGFP, pEGFP-Rab5wt, or pEGFP-Rab5S34N with a double-heat protocol. At 36 h p.i., both intra- and extracellular IBDV titers were determined, as described in Materials and Methods. Data represent results for the media from two independent experiments.
Recombinant VP3 is able to target endosomes.
To further analyze whether the endosomal localization of VP3 is due to VP3 itself or depends on other viral or cellular structures, we investigated the localization of recombinant VP3 when expressed in the absence of other viral elements. HeLa and QM7 cells were transfected with pcDNA-VP3, and VP3 localization was analyzed by IF at 24 h posttransfection. A punctate pattern dispersed throughout the cytoplasm with a slight accumulation of the VP3 signal at the perinuclear region was observed in both transfected cell types (Fig. 5Aa and b and Bf and j). We then investigated its possible association with endosomes in HeLa cells by employing antibodies against EEA1 (unfortunately, this antibody was unable to label QM7-derived endosomes). As shown in Fig. 5Ab to e, we found strong colocalization between both markers, indicating that VP3 is able to target endosomes without the context of the viral infection. Next, we determined if these vesicles have features of lysosomes by incubating the transfected cells with LysoTracker or DQ-BSA, as described above. As shown in Fig. 5Bf to m, we did not observe colocalization between the two above-mentioned reagents and the VP3 signal. Thus, our results suggest that VP3 is responsible for targeting of the replication machinery to the endocytic vesicles in infected cells.
Fig 5.

Recombinant IBDV VP3 localizes to early endosomes. QM7 and HeLa cells were transfected with pcDNA (data not shown) or pcDNA-VP3, as described in Materials and Methods. At 24 h p.i., cells were processed for indirect immunofluorescence analysis with anti-VP3 (1/500). (A) VP3 distribution in QM7 cells. In HeLa cells, double immune staining was performed with anti-EEA1. The insets show the colocalization of VP3 (green signal) and EEA1 (red signal) at the juxtanuclear region. (B) pcDNA-VP3-transfected HeLa or QM7 cells were incubated with LysoTracker or DQ-BSA, as described in the text. The cells were then processed for indirect immunofluorescence analysis. The insets show the lack of colocalization of LysoTracker (red signal) or DQ-BSA (red signal) with VP3 (green signal) at the perinuclear region. The nuclei were stained with Hoechst stain (blue). Images are representative of three independent experiments. Scale bars represent 10 μm.
IBDV VP3 associates with the cytoplasmic side of the Golgi apparatus of infected cells.
As shown in Fig. 2a, e, i, m, q, and u, VP3 exhibited a punctate distribution with a marked localization in the juxtanuclear region, reminiscent of a Golgi localization pattern. Therefore, we next examined whether VP3 colocalized with Golgi-associated proteins by double-staining analysis in mammalian and avian IBDV-infected cells. As shown in Fig. 6Aa to h, we found that in both cell types, VP3 associates with GM130 (red signal), which is a peripheral membrane protein exposed to the cytoplasmic side of the cis-Golgi complex (40, 41). Colocalization of VP3 with another Golgi-associated protein, CTR433, a cis-medial-cisterna marker (25), was also examined. As shown in Fig. 6Bi to p, VP3 was strongly associated with CTR433 at the juxtanuclear region. However, overlap between VP3 and the two analyzed Golgi marker signals was never observed, indicating that the VP3-containing structures remain in close proximity to the Golgi apparatus upon IBDV infection in both cell types.
Fig 6.
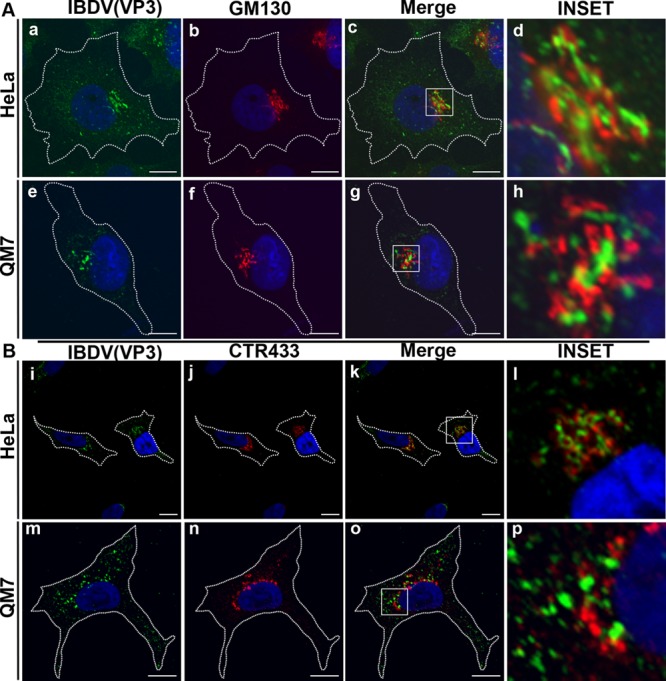
Juxtanuclear localization of IBDV VP3. Infected HeLa and QM7 cells were processed for indirect immunofluorescence analysis at 36 h p.i. with anti-VP3 (1:500) and the corresponding Alexa 488-conjugated secondary antibodies. Double labeling was performed with anti-GM130 (A) or anti-CTR433 (B) and the corresponding Cy3-conjugated secondary antibodies. The insets show the strong association of VP3 (green signal) and the Golgi markers (red signal) at the perinuclear region. The nuclei were stained with Hoechst stain (blue). Images are representative of three independent experiments. Scale bars represent 10 μm.
To further characterize the association of VP3, we analyzed the Golgi-resident glycolipid glycosyltransferases SialT2 (GM3α2,8-sialyltransferase or GD3 synthase) and GalNAcT (GM3/GD3 β1,4-N-acetylgalactosaminyltransferase). These enzymes concentrate at the proximal or cis- and trans-Golgi networks, respectively (42). The N-terminal domains of SialT2 and GalNAcT fused to YFP (43) were transfected into HeLa or QM7 cells. Transfected cells were then infected, and VP3 expression was detected by immunostaining. As shown in Fig. 7Aa to h, a strong association of the VP3 signal with the YFP fluorescence from both SialT2 and GalNAcT glycosyltransferases in HeLa cells was observed. A similar observation was made with avian cells (data not shown). In all cases, we were able to detect a well-defined association of both structures, in which some VP3 structures appeared completely surrounded by SialT2- or GalNAcT-labeled cisternae, nevertheless without overlapping of the specific fluorescent signals. We next addressed whether VP3 was associated with Golgi-derived membranes remaining on the outer side of the organelle or if it was located between the Golgi stacks. To address this issue and to establish the spatial relationship of the viral VP3 protein with the Golgi apparatus, 3D surface reconstruction imaging was employed. By using the 3D visualization and surface modeling software program ImageJ, we created images that display the 3D structure of portions of Golgi apparatus and the spatial location of the VP3 signal. Figure 7Ba to d shows the 2D projection image obtained from the z-stack of a single cell employed for the reconstruction, while Fig. 7Be to h shows different views of the 3D reconstruction image (the complete movie is shown in Movie S1 in the supplemental material). When the 3D image was rotated to achieve a side view, it became clear that the VP3 protein is located outside the Golgi stacks. We then asked if the VP3-labeled structures corresponded to the VP3 associated with endosomes. Double immune staining of VP3 and EEA1 in YFP-SialT2-transfected IBDV-infected cells was performed, and we observed that VP3 structures colocalizing with EEA1 were associated with the Golgi stacks (Fig. 7C, white arrows). Taken together, these results suggest that VP3-containing endosomal structures associate with the Golgi stacks in infected cells.
Fig 7.

IBDV VP3 associates with Golgi stacks. (A) pYFP-SialT2- and pYFP-GalNAcT-transfected HeLa cells were infected and processed for indirect immunofluorescence analysis with anti-VP3 (1:500) and the corresponding Cy3-conjugated secondary antibodies. The insets show a well-defined association of both structures, in which VP3 appears completely surrounded by SialT2- or GalNAcT-labeled structures. The nuclei were stained with Hoechst stain (blue). (B) z-stack fluorescence images were captured from a single cell, as described in Materials and Methods. (a to d) 2D images showing brightest-point projections of 10 images collected at 0.39-μm steps in the z axis. To better appreciate the green and red signal distributions, a close-up of the entire cell was done, showing only the perinuclear region where the signals are located. The nuclei were stained with Hoechst stain (blue). (e to h) Different rotation planes of the same data, displayed as 3D topographical reconstructions of VP3 and YFP-SialT2. (C) YFP-SialT2-transfected, IBDV-infected HeLa cells were processed for double immune staining with rat anti-VP3 and the corresponding Cy3-conjugated secondary antibody followed by anti-EEA1 and the corresponding Cascade Blue-conjugated secondary antibody. (Top) Image of a complete cell. (Bottom) Images of an optical zoom done with the cell in the top panel. White arrows indicate VP3-positive and EEA1-positive vesicles in close proximity to the Golgi stacks (green signal). Images are representative of three independent experiments. Scale bars represent 10 μm.
Golgi disruption with nocodazole does not affect the IBDV life cycle.
Since we observed such a close association of VP3 with the Golgi apparatus, we next investigated the role of this organelle in both VP3 localization and the viral replication cycle by depolymerizing the microtubules (MTs) with nocodazole (ND). MTs participate in maintaining the Golgi structure, and MT depolymerization is known to result in the reorganization of the Golgi complex into characteristic ministacks, which appear as punctate structures throughout the cell cytoplasm (44). To determine whether VP3 localization and viral replication require a normal Golgi apparatus, ND was added to infected HeLa or QM7 cells at 12 h p.i., and cells were maintained in medium with the drug for an additional 24-h period to avoid the rebuilding of the cytoskeleton network. Afterwards, supernatants were collected to determine virus titers. In addition, cells were processed for Western blotting to detect viral VP3 levels and for IF to analyze VP3 subcellular distribution. To confirm the formation of the ND-induced Golgi ministacks in treated cells, we employed the YFP-GalNAcT and YFP-SialT2 Golgi complex markers (Fig. 8Aa and e). As shown in Fig. 8Ab and f, a marked change in the VP3 pattern upon ND treatment was observed, depicting a punctate distribution scattered throughout the cytoplasm of infected cells. Surprisingly, in the large majority of the YFP-GalNAcT- and YFP-SialT2-expressing infected cells, we observed a strong association of VP3 and the Golgi ministacks forming cup-shaped structures (Fig. 8Ac, d, g, and h), where the Golgi ministacks resemble a cup (green signal) and VP3 is the content. Interestingly, Western blot analysis of nontreated and treated infected cells revealed that Golgi disruption does not significantly affect VP3 synthesis, as shown in Fig. 8B, with similar intensity levels of VP3 in both cell lines. Moreover, infective progeny yields were also unaffected by ND treatment (Fig. 8C). Taken together, these results indicate that although a marked change in the VP3 subcellular distribution was observed upon cell treatment with ND, the IBDV replication cycle was not critically affected by the dispersion of the Golgi apparatus.
Fig 8.

Nocodazole treatment affects IBDV VP3 subcellular distribution. pYFP-SialT2- and pYFP-GalNAcT-transfected and infected HeLa cells were treated with 2 mM nocodazole (ND) or left in control medium. At 36 h p.i., cells were washed and processed for indirect immunofluorescence analysis. (A) Anti-VP3 (1:500) and the corresponding Cy3-conjugated secondary antibodies were employed. Only ND-treated HeLa cells are shown. The nuclei were stained with Hoechst stain (blue). The insets show a well-defined association of both structures. Scale bars represent 10 μm. (B) To determine the levels of VP3, cells were analyzed by Western blotting, as described in Materials and Methods. Complete VP3/actin values are 1.36 ± 0.02, 1.32 ± 0.04, 1.37 ± 0.06, and 1.41 ± 0.05. (C) Supernatants were collected and viral infective particles were titrated as described in the text. Images and data were obtained from three independent experiments.
Brefeldin A treatment of infected avian cells affects virus assembly.
To gain further insight into the functional role of the Golgi apparatus in the IBDV infection cycle, we tested the effect of brefeldin A (BFA). This drug causes profound alterations of the Golgi apparatus, resulting in the transfer of Golgi enzymes to the lumen of the ER (45, 46). Additionally, dispersed tubulovesicular clusters of membranes are left in the cytoplasm, termed Golgi remnants, which contain matrix proteins such GM130 (47). BFA was added to infected HeLa or QM7 cells at 24 h p.i., and the drug was maintained in the medium for an additional 12-h period to avoid the rebuilding of the Golgi network. These experimental conditions did not induce cell toxicity, as measured by trypan blue exclusion (data not shown). Supernatants were collected to determine the titer of extracellular virus, and the cells were processed by Western blotting to analyze VP3 levels and to quantitate intracellular virus titers. To corroborate BFA-induced Golgi dispersion, we employed antibodies against the matrix protein GM130, since this protein (together with other Golgi matrix proteins) is found in dispersed, punctate structures after BFA treatment (48) (Fig. 9Aa, e, i, and m). As expected, a strong change in the VP3 pattern upon BFA treatment was observed, showing a punctate distribution scattered throughout the cytoplasm of infected cells (Fig. 9Ab, f, j, and n). Levels of VP3 expression were analyzed by immunoblotting. Interestingly, we found a differential behavior of VP3 accumulation in treated HeLa and QM7 cells. As shown in Fig. 9B, a significant reduction of the VP3 expression level was observed in BFA-treated HeLa cells. However, when we quantitated intra- and extracellular virus titers, we found no differences in the formation and release, respectively, of infective particles in nontreated and treated HeLa cells in spite of the reduced VP3 accumulation levels (Fig. 9C, upper panel). Given the peculiarity of these results, we analyzed viral RNA levels in nontreated and treated HeLa cells and found a perfect correlation between RNA and VP3 levels (Fig. 9D). In contrast, QM7 cells treated with BFA showed no significant differences in VP3 accumulation levels compared with the levels in untreated control cells. However, we observed a moderate but consistent reduction in the amount of released infective virus in BFA-treated QM7 cells (Fig. 9C). Thus, we next aimed to determine if the reduction of virus release was due to impairment of a virus egress mechanism or a negative effect on the assembly of infective viral particles. To address this issue, we titrated intracellular infective viral particles in control and BFA-treated cells. Interestingly, we observed a reduction of intracellular virus titers in BFA-treated QM7 cells (Fig. 9B, lower panel). Thus, these results indicate that in avian QM7 cells, BFA treatment partially alters the assembly of infective viral particles, likely due to the negative impact of BFA on the functionality of the Golgi apparatus.
Fig 9.

Brefeldin A treatment partially affects virus assembly. Infected HeLa and QM7 cells were treated with 5 μg/ml brefeldin A (BFA) or left in control medium. At 36 h p.i., cells were washed and processed for indirect immunofluorescence analysis, as described in the text. (A) Double-staining IF analyses were performed by incubating the cells with anti-VP3 (1:500) and anti-GM130 antibodies followed by the corresponding Alexa 488- or Cy3-conjugated secondary antibodies. The nuclei were stained with Hoechst stain (blue). The insets show the association of both structures with a completely different cellular distribution upon BFA treatment. Scale bars represent 10 μm. (B) To determine the levels of VP3, cells were analyzed by Western blotting, as described in Materials and Methods. Complete VP3/tubulin values are 1.46 ± 0.08, 1.30 ± 0.02, 0.78 ± 0.04, and 1.47 ± 0.17. (C) Intracellular and extracellular viral infective particles were titrated from a cellular extract or supernatants, respectively, of control and BFA-treated infected HeLa and QM7 cells, as described in the text. Images and data were obtained from three independent experiments. (D) qRT-PCR analyses of HeLa cell extracts were performed as described in Materials and Methods, in an experiment similar to the one depicted in panel B. Relative quantification (RQ) of viral RNA compared to the control GAPDH (glyceraldehyde-3-phosphate dehydrogenase) gene is shown.
DISCUSSION
While several viral families constitute the dsRNA virus group, our study constitutes the first attempt to analyze how birnaviruses manage to establish their factories in infected cells without having a transcriptional core as the other viruses of this group do. Only members of the Reoviridae family, such as orthoreoviruses and rotavirus, have well-characterized replication and assembly sites. Reoviruses are nonenveloped viruses with a genome encapsidated by two protein shells, an outer capsid and an inner core shell. After internalization, the outer capsid is lost, and the core is delivered into the cytoplasm, where it imports substrates from the cytosol to synthesize and then export viral mRNAs representing the replication complex. Viral mRNAs transcribed in the cytoplasm make viral proteins that eventually form large perinuclear inclusions, called virus factories, that function as sites of further virus replication and assembly (49). Orthoreovirus factories are intimately associated with the microtubule network (50), and rotavirus factories are composed of electron-dense viroplasm often in proximity to membranes derived from the ER (51).
IBDV viroplasm localizes at an endocytic compartment.
In this report, we have revealed, for the first time, an important issue in the life cycle of birnaviruses: the vesicular nature of the cellular compartment employed by IBDV for localization of the replication complexes. It was recently shown that RNP complexes act as efficient transcription units of IBDV (6). Our data strongly suggest that IBDV replication occurs on endocytic membranes. At 36 h p.i., infected HeLa and QM7 cells labeled with anti-VP3, anti-VP1, or anti-dsRNA showed a perinuclear punctate pattern, concentrated mostly on one side of the nucleus, compatible with a vesicular pattern. When we analyzed the nature of these vesicles, we found that they correspond to an endocytic compartment bearing features of early and late endosomes (i.e., labeled with EEA1 and Rab5 as well as LAMP-1 and LAMP-2) (Fig. 1). Our results suggest that VP3 localizes to modified membranous compartments belonging to the endocytic pathway, as is the case for members of the Togaviridae family. Togavirus factories are organized around endosomes and lysosomes, which appears to be unique to this family (52). In infected cells, modified endosomes and lysosomes constitute special vesicular structures, known as cytopathic vacuoles (CPVs), which serve as RNA replication sites. Viral nonstructural proteins and a threadlike RNP containing viral RNA localize to the cytoplasmic face of the CPVs (35). Indeed, the possibility that IBDV, like togaviruses, associates with the cytoplasmic face of the endosomal compartment is supported by our findings that recombinant VP3 is targeted to endocytic structures when overexpressed in noninfected cells.
Role for the Golgi apparatus in assembly of IBDV.
By using imaging techniques and a set of well-established markers, we have shown a close association of VP3 with Golgi apparatus stacks in infected cells (Fig. 6 and 7A). The question of whether VP3 would be located inside the stacks of the Golgi apparatus was solved by 3D surface reconstruction imaging, which clearly showed that VP3 is not inside the stacks but associated with them (Fig. 7Ba to h). To date, the involvement of the Golgi apparatus in virus assembly has been described for transmissible gastroenteritis coronavirus (TGEV) (53), the prototype member of the Bunyaviridae family (54), and rubella virus (RUBV) (55). Studies of RUBV assembly revealed two novel aspects of virion assembly: (i) the polymorphism of virus particles detected inside the Golgi complex which strongly suggested the existence of a structural maturation process that takes place in the Golgi stacks and (ii) the association between Golgi stacks and CPVs containing the viral replication complex, which indicates a potential physical connection between the sites of viral replication and virion assembly (55). Based on the physical association between VP3-containing vesicles and the Golgi stacks suggested by the Golgi ministacks and VP3 forming cup-shaped structures observed after ND treatment, it is tempting to hypothesize that IBDV could enact a scenario similar to the one described for RUBV. Nevertheless, further studies are necessary to fully address this possibility.
Nocodazole and BFA treatment of infected cells.
ND is an antimitotic agent that disrupts microtubules by binding to β-tubulin, inhibiting microtubule dynamics and, as a consequence, causing fragmentation of the Golgi complex. Golgi disruption by ND did not significantly affect either VP3 synthesis (Fig. 8B) or infective progeny yields (Fig. 8C) in both cell types analyzed. This result is consistent with the fact that dispersed Golgi ministacks remain functional after ND treatment (56) and points to a functional role of the Golgi complex in the IBDV life cycle. Most importantly, we found a marked change in the VP3 subcellular pattern, depicting a punctate distribution scattered throughout the cytoplasm of infected cells with a strong association of Golgi ministacks and VP3, forming cup-shaped structures (Fig. 8Ac, d, g, and h). This observation constitutes strong evidence of a physical association between VP3-containing vesicles and Golgi stacks, reinforcing the idea that the Golgi stacks must have a protagonist role in the IBDV life cycle. On the other hand, BFA was demonstrated to have dramatic effects on the structure and function of intracellular organelles, particularly the Golgi apparatus, which fuses with the endoplasmic reticulum (ER), leading to a complete block of protein transport out of the fused ER-Golgi system (46). Intriguingly, in HeLa cells, we observed a marked reduction of VP3 and viral RNA levels in BFA-treated cells (0.78 versus 1.46) (Fig. 9B), but no differences in intra- and extracellular viral titers were observed. Regarding this observation, an excess of VP3 in untreated infected HeLa cells (control situation) would be a probable explanation. Supporting this idea is the fact that in the control situation, the level of production of infectious viral particles in HeLa cells (4.3 × 102 PFU/ml) is always markedly lower than that in QM7 cells (5.4 × 103 PFU/ml), even with similar levels of VP3 (1.46 in HeLa cells versus 1.30 in QM7 cells). This “unbalanced situation” was a constant observation in our experiments, which we attribute to the fact that even though HeLa cells are susceptible to IBDV infection, IBDV does not naturally infect cells originating from humans, as avian cells (such as QM7 cells) are the natural target for efficient replication. Interestingly, we found a reduction in intracellular virus titers in BFA-treated compared to control cells in the avian model (Fig. 9C), suggesting a role for the Golgi apparatus in the assembly step of the IBDV life cycle.
Is there a relationship between virus internalization and replication on endocytic membranes?
It has been demonstrated that chicken heat shock protein 90 (Hsp90) and the α4β1 integrin are components of the cellular receptor complex mediating IBDV infection (29, 57). We and others have indeed analyzed the internalization of IBDV, and our preliminary results indicate that IBDV is endocytosed and that a functional endocytic pathway is critical for viral infection (17). Birnaviruses present characteristic peptides associated with the virus particles generated during the processing of the C terminus of the pVP2 capsid precursor protein by the VP4 protease. One of those peptides, pep46, contains a membrane-active domain that is suggested to deform biological membranes, leading to the formation of pores. However, the diameter of the pore (<10 nm) is smaller than that of the virus particle (70 nm), so the authors of that study suggested that an exchange of small molecules between endosomal ghosts and the cytoplasm allows the initial transcription of the genome (19). Related to our findings, it is tempting to hypothesize that the virus is endocytosed by the host cell and builds up its replication factory associated with the endosomal membrane. As explained above, when recombinant VP3 was overexpressed in noninfected cells, VP3 was targeted to endosomes, indicating a marked tropism of this protein for endocytic membranous compartments. Taking advantage of the normal endosomal maturation pathway, which occurs with movement of endocytic vesicles toward the perinuclear region by microtubule-mediated transport, IBDV would reach the perinuclear region, where it likely completes the morphogenetic process associated with the Golgi stacks, as do other viruses (58–60). Since birnaviruses lack the typical T=2 core, where viral RNA metabolism of all dsRNA viruses takes place, it is conceivable that the RNPs instead would be extruded from the pierced endosomes targeting the surrounding membrane of this organelle. Subsequently, these endosome-associated RNPs could traffic along microtubules to reach the perinuclear region close to the Golgi complex, establishing the replication factory on the cytosolic surface of endocytic membranes, a possibility which appears to be employed by many positive ssRNA viruses (61–64). Based on our studies and studies by others, we propose a model, depicted in Fig. 10, in which IBDV RNPs reach the cellular endocytic compartment to initiate viral replication and complete the assembly step associated with the Golgi apparatus.
Fig 10.

Proposed model for IBDV RNP cellular localization. Hsp90 and α4β1 integrin are components of the cellular receptor complex mediating IBDV infection (29, 57). Virions are internalized into the target cells by employing the endocytic pathway (17). Inside the endosome, the trimeric structure of VP2 in the viral capsid destabilizes (67), allowing the exposure of pep 46, the capsid-associated peptide. Pep 46 is able to deform biological membranes, leading to the formation of pores on the endosomal membrane (19). We propose that IBDV RNPs would be extruded from the pierced endosomes associating with the limiting membrane of this organelle, through the VP3 membrane-targeting ability, where viral genome replication occurs. Afterwards, the RNPs associated with the endocytic vesicles could traffic along microtubules to reach the perinuclear region, establishing physical contact with the Golgi complex, where viral assembly takes place. BFA treatment affects specific steps in both the endocytic and secretory pathways (68).
What is the mechanism involved in membranous targeting of IBDV VP3?
VP3 is a scaffolding protein that accomplishes several tasks during viral replication and morphogenesis, with a highly hydrophilic C-terminal tail region rich in charged amino acids and proline residues. This C-terminal tail was predicted to be well exposed and consequently able to bind proteins and nucleic acids. The VP3-associated activities have been located in its C-terminal region, which participates in VP3 oligomerization and RNA polymerase RNA-dependent (RpRd) VP1 recruitment into the capsid during viral morphogenesis (11). In addition, VP3 interacts with the viral dsRNA, forming RNP complexes reminiscent of those of the nucleoproteins of certain dsDNA viruses (6). The amino acid sequence of the VP3 polypeptide from the IBDV Soroa strain was scanned for conserved domains that could potentially be responsible for its location at vesicles when expressed alone or in the context of viral infection. We employed different software programs to scan for conserved domains involved in membrane targeting, but none of them showed a positive match (data not shown). Additionally, the possibility that VP3 contains a posttranslational modification site for myristoylation, prenylation, or palmitoylation was also explored, since these modifications are involved in protein targeting to membranes. However, none of these putative sites were found in the VP3 nucleotide sequence. Finally, the presence of polybasic clusters in the protein sequence of VP3 was analyzed by using BioEdit (65). The interaction of small clusters of positively charged amino acids on proteins with negatively charged lipids in membranes constitutes an important issue in the field of protein-membrane interactions (66). Interestingly, we found that the C-terminal region of VP3 contains several positively charged amino acid residues, which could be involved in membrane interactions. However, even though this is a very attractive idea, we cannot rule out the possibility that a cellular component could be involved either in the association of RNPs with the endosome or in the endosome targeting of VP3 when expressed alone. Further studies are required to adequately address this issue. Indeed, we are currently performing point mutations of some of these residues in order to assess the importance of the positive-charge clusters in the association of the VP3 protein with a membranous compartment.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by grants PICT 2008 0192 and SeCTyP 06/J361 to María I. Colombo and by a grant from the Spanish Ministry of Economy and Competitiveness, AGL2011-24758, to José F. Rodríguez.
We thank María Roqué and Diego Lijavetzky for extremely valuable and generous help with the qRT-PCR technique and Alejandra Medero, Marcelo Furlán, and Graciela Gutierrez for valuable technical assistance.
Footnotes
Published ahead of print 5 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.03152-12.
REFERENCES
- 1. Delmas B, Kibenge F, Leong J, Mundt E, Vakharia V, Wu J. 2005. Birnaviridae, p 561–569 In Fauquet CM, Mayo MA, Maniloff J, Desselberger U, Ball LA. (ed), Virus taxonomy. Eighth report of the International Committee on Taxonomy of Viruses Elsevier Academic Press, San Diego, CA [Google Scholar]
- 2. Lombardo E, Maraver A, Espinosa I, Fernandez-Arias A, Rodriguez JF. 2000. VP5, the nonstructural polypeptide of infectious bursal disease virus, accumulates within the host plasma membrane and induces cell lysis. Virology 277:345–357 [DOI] [PubMed] [Google Scholar]
- 3. Da Costa B, Chevalier C, Henry C, Huet JC, Petit S, Lepault J, Boot H, Delmas B. 2002. The capsid of infectious bursal disease virus contains several small peptides arising from the maturation process of pVP2. J. Virol. 76:2393–2402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Luque D, Rivas G, Alfonso C, Carrascosa JL, Rodriguez JF, Caston JR. 2009. Infectious bursal disease virus is an icosahedral polyploid dsRNA virus. Proc. Natl. Acad. Sci. U. S. A. 106:2148–2152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. von Einem UI, Gorbalenya AE, Schirrmeier H, Behrens SE, Letzel T, Mundt E. 2004. VP1 of infectious bursal disease virus is an RNA-dependent RNA polymerase. J. Gen. Virol. 85:2221–2229 [DOI] [PubMed] [Google Scholar]
- 6. Luque D, Saugar I, Rejas MT, Carrascosa JL, Rodriguez JF, Caston JR. 2009. Infectious bursal disease virus: ribonucleoprotein complexes of a double-stranded RNA virus. J. Mol. Biol. 386:891–901 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ahlquist P. 2005. Virus evolution: fitting lifestyles to a T. Curr. Biol. 15:R465–R467. 10.1016/j.cub.2005.06.016 [DOI] [PubMed] [Google Scholar]
- 8. Schwartz M, Chen J, Janda M, Sullivan M, den Boon J, Ahlquist P. 2002. A positive-strand RNA virus replication complex parallels form and function of retrovirus capsids. Mol. Cell 9:505–514 [DOI] [PubMed] [Google Scholar]
- 9. Chevalier C, Lepault J, Da Costa B, Delmas B. 2004. The last C-terminal residue of VP3, glutamic acid 257, controls capsid assembly of infectious bursal disease virus. J. Virol. 78:3296–3303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Saugar I, Luque D, Ona A, Rodriguez JF, Carrascosa JL, Trus BL, Caston JR. 2005. Structural polymorphism of the major capsid protein of a double-stranded RNA virus: an amphipathic alpha helix as a molecular switch. Structure 13:1007–1017 [DOI] [PubMed] [Google Scholar]
- 11. Garriga D, Navarro A, Querol-Audi J, Abaitua F, Rodríguez JF, Verdaguer N. 2007. Activation mechanism of a noncanonical RNA-dependent RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 104:20540–20545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mackenzie J. 2005. Wrapping things up about virus RNA replication. Traffic 6:967–977 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Salonen A, Ahola T, Kaariainen L. 2005. Viral RNA replication in association with cellular membranes. Curr. Top. Microbiol. Immunol. 285:139–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hagiwara Y, Komoda K, Yamanaka T, Tamai A, Meshi T, Funada R, Tsuchiya T, Naito S, Ishikawa M. 2003. Subcellular localization of host and viral proteins associated with tobamovirus RNA replication. EMBO J. 22:344–353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kopek BG, Perkins G, Miller DJ, Ellisman MH, Ahlquist P. 2007. Three-dimensional analysis of a viral RNA replication complex reveals a virus-induced mini-organelle. PLoS Biol. 5:e220. 10.1371/journal.pbio.0050220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Novoa RR, Calderita G, Arranz R, Fontana J, Granzow H, Risco C. 2005. Virus factories: associations of cell organelles for viral replication and morphogenesis. Biol. Cell 97:147–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Giménez MC, Rodríguez JF, Colombo MI, Delgui LR. 2012. Study of the internalization pathway involved in infectious bursal disease virus infection. Biocell 36(Suppl):78 http://www.cricyt.edu.ar/biocell [Google Scholar]
- 18. Yip CW, Hon CC, Zeng F, Leung FC. 2012. Cell culture-adapted IBDV uses endocytosis for entry in DF-1 chicken embryonic fibroblasts. Virus Res. 165:9–16 [DOI] [PubMed] [Google Scholar]
- 19. Galloux M, Libersou S, Morellet N, Bouaziz S, Da Costa B, Ouldali M, Lepault J, Delmas B. 2007. Infectious bursal disease virus, a non-enveloped virus, possesses a capsid-associated peptide that deforms and perforates biological membranes. J. Biol. Chem. 282:20774–20784 [DOI] [PubMed] [Google Scholar]
- 20. Lombardo E, Maraver A, Caston JR, Rivera J, Fernandez-Arias A, Serrano A, Carrascosa JL, Rodriguez JF. 1999. VP1, the putative RNA-dependent RNA polymerase of infectious bursal disease virus, forms complexes with the capsid protein VP3, leading to efficient encapsidation into virus-like particles. J. Virol. 73:6973–6983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fernandez-Arias A, Risco C, Martinez S, Albar JP, Rodriguez JF. 1998. Expression of ORF A1 of infectious bursal disease virus results in the formation of virus-like particles. J. Gen. Virol. 79:1047–1054 [DOI] [PubMed] [Google Scholar]
- 22. Schonborn J, Oberstrass J, Breyel E, Tittgen J, Schumacher J, Lukacs N. 1991. Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res. 19:2993–3000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. 2006. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 80:5059–5064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kobayashi T, Stang E, Fang KS, de Moerloose P, Parton RG, Gruenberg J. 1998. A lipid associated with the antiphospholipid syndrome regulates endosome structure and function. Nature 392:193–197 [DOI] [PubMed] [Google Scholar]
- 25. Jasmin BJ, Cartaud J, Bornens M, Changeux JP. 1989. Golgi apparatus in chick skeletal muscle: changes in its distribution during end plate development and after denervation. Proc. Natl. Acad. Sci. U. S. A. 86:7218–7222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Manders EMM, Verbeek FJ, Aten JA. 1993. Measurement of co-localization of objects in dual-colour confocal images. J. Microsc. 169:375–382 [DOI] [PubMed] [Google Scholar]
- 27. Zinchuk V, Grossenbacher-Zinchuk O. 2009. Recent advances in quantitative colocalization analysis: focus on neuroscience. Prog. Histochem. Cytochem. 44:125–172 [DOI] [PubMed] [Google Scholar]
- 28. Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am. J. Epidemiol. 27:493–497 [Google Scholar]
- 29. Delgui L, Oña A, Gutierrez S, Luque D, Navarro A, Castón JR, Rodríguez JF. 2009. The capsid protein of infectious bursal disease virus contains a functional alpha 4 beta 1 integrin ligand motif. Virology 386:360–372 [DOI] [PubMed] [Google Scholar]
- 30. Nielsen E, Severin F, Backer JM, Hyman AA, Zerial M. 1999. Rab5 regulates motility of early endosomes on microtubules. Nat. Cell Biol. 1:376–382 [DOI] [PubMed] [Google Scholar]
- 31. Feng Y, Press B, Wandinger-Ness A. 1995. Rab 7: an important regulator of late endocytic membrane traffic. J. Cell Biol. 131:1435–1452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Matsuo H, Chevallier J, Mayran N, Le Blanc I, Ferguson C, Faure J, Blanc NS, Matile S, Dubochet J, Sadoul R, Parton RG, Vilbois F, Gruenberg J. 2004. Role of LBPA and Alix in multivesicular liposome formation and endosome organization. Science 303:531–534 [DOI] [PubMed] [Google Scholar]
- 33. Geuze HJ, Stoorvogel W, Strous GJ, Slot JW, Bleekemolen JE, Mellman I. 1988. Sorting of mannose 6-phosphate receptors and lysosomal membrane proteins in endocytic vesicles. J. Cell Biol. 107:2491–2501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Griffiths G, Hoflack B, Simons K, Mellman I, Kornfeld S. 1988. The mannose 6-phosphate receptor and the biogenesis of lysosomes. Cell 52:329–341 [DOI] [PubMed] [Google Scholar]
- 35. Lee JY, Marshall JA, Bowden DS. 1994. Characterization of rubella virus replication complexes using antibodies to double-stranded RNA. Virology 200:307–312 [DOI] [PubMed] [Google Scholar]
- 36. Targett-Adams P, Boulant S, McLauchlan J. 2008. Visualization of double-stranded RNA in cells supporting hepatitis C virus RNA replication. J. Virol. 82:2182–2195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Westaway EG, Khromykh AA, Mackenzie JM. 1999. Nascent flavivirus RNA colocalized in situ with double-stranded RNA in stable replication complexes. Virology 258:108–117 [DOI] [PubMed] [Google Scholar]
- 38. Pfeffer SR. 2001. Rab GTPases: specifying and deciphering organelle identity and function. Trends Cell Biol. 11:487–491 [DOI] [PubMed] [Google Scholar]
- 39. Gorvel JP, Chavrier P, Zerial M, Gruenberg J. 1991. rab5 controls early endosome fusion in vitro. Cell 64:915–925 [DOI] [PubMed] [Google Scholar]
- 40. Bloom GS, Brashear TA. 1989. A novel 58-kDa protein associates with the Golgi apparatus and microtubules. J. Biol. Chem. 264:16083–16092 [PubMed] [Google Scholar]
- 41. Gao YS, Alvarez C, Nelson DS, Sztul E. 1998. Molecular cloning, characterization, and dynamics of rat formiminotransferase cyclodeaminase, a Golgi-associated 58-kDa protein. J. Biol. Chem. 273:33825–33834 [DOI] [PubMed] [Google Scholar]
- 42. Maccioni HJ, Daniotti JL, Martina JA. 1999. Organization of ganglioside synthesis in the Golgi apparatus. Biochim. Biophys. Acta 1437:101–118 [DOI] [PubMed] [Google Scholar]
- 43. Uliana AS, Giraudo CG, Maccioni HJ. 2006. Cytoplasmic tails of SialT2 and GalNAcT impose their respective proximal and distal Golgi localization. Traffic 7:604–612 [DOI] [PubMed] [Google Scholar]
- 44. Thyberg J, Moskalewski S. 1985. Microtubules and the organization of the Golgi complex. Exp. Cell Res. 159:1–16 [DOI] [PubMed] [Google Scholar]
- 45. Doms RW, Russ G, Yewdell JW. 1989. Brefeldin A redistributes resident and itinerant Golgi proteins to the endoplasmic reticulum. J. Cell Biol. 109:61–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lippincott-Schwartz J, Yuan LC, Bonifacino JS, Klausner RD. 1989. Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell 56:801–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Seemann J, Jokitalo E, Pypaert M, Warren G. 2000. Matrix proteins can generate the higher order architecture of the Golgi apparatus. Nature 407:1022–1026 [DOI] [PubMed] [Google Scholar]
- 48. Nakamura N, Rabouille C, Watson R, Nilsson T, Hui N, Slusarewicz P, Kreis TE, Warren G. 1995. Characterization of a cis-Golgi matrix protein, GM130. J. Cell Biol. 131:1715–1726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Netherton C, Moffat K, Brooks E, Wileman T. 2007. A guide to viral inclusions, membrane rearrangements, factories, and viroplasm produced during virus replication. Adv. Virus Res. 70:101–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Parker JS, Broering TJ, Kim J, Higgins DE, Nibert ML. 2002. Reovirus core protein mu2 determines the filamentous morphology of viral inclusion bodies by interacting with and stabilizing microtubules. J. Virol. 76:4483–4496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Altenburg BC, Graham DY, Estes MK. 1980. Ultrastructural study of rotavirus replication in cultured cells. J. Gen. Virol. 46:75–85 [DOI] [PubMed] [Google Scholar]
- 52. Magliano D, Marshall JA, Bowden DS, Vardaxis N, Meanger J, Lee JY. 1998. Rubella virus replication complexes are virus-modified lysosomes. Virology 240:57–63 [DOI] [PubMed] [Google Scholar]
- 53. Salanueva IJ, Carrascosa JL, Risco C. 1999. Structural maturation of the transmissible gastroenteritis coronavirus. J. Virol. 73:7952–7964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Salanueva IJ, Novoa RR, Cabezas P, Lopez-Iglesias C, Carrascosa JL, Elliott RM, Risco C. 2003. Polymorphism and structural maturation of bunyamwera virus in Golgi and post-Golgi compartments. J. Virol. 77:1368–1381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Risco C, Carrascosa JL, Frey TK. 2003. Structural maturation of rubella virus in the Golgi complex. Virology 312:261–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Pfeffer SR. 2007. Unsolved mysteries in membrane traffic. Annu. Rev. Biochem. 76:629–645 [DOI] [PubMed] [Google Scholar]
- 57. Lin TW, Lo CW, Lai SY, Fan RJ, Lo CJ, Chou YM, Thiruvengadam R, Wang AH, Wang MY. 2007. Chicken heat shock protein 90 is a component of the putative cellular receptor complex of infectious bursal disease virus. J. Virol. 81:8730–8741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Marsh M, Bron R. 1997. SFV infection in CHO cells: cell-type specific restrictions to productive virus entry at the cell surface. J. Cell Sci. 110(Part 1):95–103 [DOI] [PubMed] [Google Scholar]
- 59. Mercer J, Schelhaas M, Helenius A. 2010. Virus entry by endocytosis. Annu. Rev. Biochem. 79:803–833 [DOI] [PubMed] [Google Scholar]
- 60. Sodeik B. 2000. Mechanisms of viral transport in the cytoplasm. Trends Microbiol. 8:465–472 [DOI] [PubMed] [Google Scholar]
- 61. Bienz K, Egger D, Pfister T, Troxler M. 1992. Structural and functional characterization of the poliovirus replication complex. J. Virol. 66:2740–2747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Pedersen KW, van der Meer Y, Roos N, Snijder EJ. 1999. Open reading frame 1a-encoded subunits of the arterivirus replicase induce endoplasmic reticulum-derived double-membrane vesicles which carry the viral replication complex. J. Virol. 73:2016–2026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Restrepo-Hartwig MA, Ahlquist P. 1996. Brome mosaic virus helicase- and polymerase-like proteins colocalize on the endoplasmic reticulum at sites of viral RNA synthesis. J. Virol. 70:8908–8916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Schaad MC, Jensen PE, Carrington JC. 1997. Formation of plant RNA virus replication complexes on membranes: role of an endoplasmic reticulum-targeted viral protein. EMBO J. 16:4049–4059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. 2009. Jalview version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics 25:1189–1191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Kim J, Mosior M, Chung LA, Wu H, McLaughlin S. 1991. Binding of peptides with basic residues to membranes containing acidic phospholipids. Biophys. J. 60:135–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Garriga D, Querol-Audi J, Abaitua F, Saugar I, Pous J, Verdaguer N, Caston JR, Rodríguez JF. 2006. The 2.6-angstrom structure of infectious bursal disease virus-derived T=1 particles reveals new stabilizing elements of the virus capsid. J. Virol. 80:6895–6905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Klausner RD, Donaldson JG, Lippincott-Schwartz J. 1992. Brefeldin A: insights into the control of membrane traffic and organelle structure. J. Cell Biol. 116:1071–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.