

Abstract
In previous work, we designed peptides that showed potent inhibition of Newcastle disease virus (NDV) and infectious bronchitis virus (IBV) infections in chicken embryos. In this study, we demonstrate that peptides modified with cholesterol or 3 U of polyethylene glycol (PEG3) conjugated to the peptides' N termini showed even more promising antiviral activities when tested in animal models. Both cholesterol- and cholesterol-PEG3-tagged peptides were able to protect chicken embryos from infection with different serotypes of NDV and IBV when administered 12 h prior to virus inoculation. In comparison, the untagged peptides required intervention closer to the time of viral inoculation to achieve a similar level of protection. Intramuscular injection of cholesterol-tagged peptide at 1.6 mg/kg 1 day before virus infection and then three times at 3-day intervals after viral inoculation protected 70% of the chickens from NDV infection. We further demonstrate that the cholesterol-tagged peptide has an in vivo half-life greater than that of untagged peptides. It also has the potential to cross the blood-brain barrier to enter the avian central nervous system (CNS). Finally, we show that the cholesterol-tagged peptide could play a role before the viral fusion peptide's insertion into the host cell and thereby target an earlier stage of fusion glycoprotein activation. Our findings are of importance for the further development of antivirals with broad-spectrum protective effects.
INTRODUCTION
Antiviral agents can be achieved by targeting either viral life cycles or host response factors. The fusion process has been considered a significant antiviral target and received wide attention. Class I envelope viruses have similar mechanisms of membrane fusion, in which a key step is the formation of a six-helix bundle of three heptad repeat 1 (HR1) and HR2 trimers of the fusion glycoproteins (1, 2, 3). The current paradigm of the mechanism of HR-derived peptide action proposes that HR peptides bind to the postulated extended intermediate state after the fusion peptide has been inserted into the target membrane and prevent the transition to the postfusion conformation (3, 4, 5). The successful use of enfuvirtide against HIV has stimulated considerable efforts to develop more potent peptides (6, 7, 8, 9). Despite their high antiviral activity, HR-mimetic peptides are unsuitable as therapeutics because of their short half-lives. A favorite approach to improve peptide pharmacokinetics is to conjugate peptides to lipids, which prolongs their half-life in the circulation. The most typical derivatization involves long-chain fatty acids (10). Derivatization with cholesterol, however, has not been explored extensively. Recently, a few reports have revealed a different approach to increasing the antiviral potency of a peptide by targeting it to the cell compartment where fusion occurs, through the introduction of a cholesterol group membrane anchor (11, 12, 13, 14, 15).
Paramyxoviruses and coronaviruses feature a molecular mechanism similar to that of other class I envelope viruses (2, 3). The neurologic Newcastle disease virus (NDV) of the family Paramyxoviridae and the infectious bronchitis virus (IBV) of the family Coronaviridae are the two viral pathogens with the greatest economic impact on the poultry industry worldwide. These viruses can cause complex coinfections that hinder disease diagnosis and prevention (16). Our previous studies have demonstrated that newly designed peptides based on the HR region of the fusion glycoproteins from different viruses have more potent antiviral activity than their mother HR peptides. These peptides can efficiently inhibit NDV and IBV infections. We also assessed possible targets for these peptides. Our data strengthened the notion that HR2 is an attractive site for therapeutic intervention (5).
In the present study, two modification strategies involving cholesterol and small spacers made of 3 U of polyethylene glycol (PEG3), a well-known solubilizing moiety, were designed and exploited for their potential to enhance the peptides' antiviral activity. This refinement is based on a cholesterol tag at the N terminus of the fusion-inhibitory peptide. It gives the peptide a longer half-life and greater antiviral activity in vitro and in vivo than the unmodified peptide has.
MATERIALS AND METHODS
Synthesis and modification of peptides. (i) Peptide synthesis.
An untagged peptide (AU, referred to as N2 in a previous report [5]) was synthesized by 9-fluorenylmethoxy carbonyl/tert-butyl group chemistry with the peptide synthesizer SYMPHONY. The Wang resin was mixed with a 3-fold molar excess of protecting amino acids, a 10-fold molar excess of N,N-dimethylpyridin-4-amine and N,N′-dicyclohexylcarbodiimide in dimethylformamide (DMF). The amino acids were activated with a 3-fold molar excess of 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate (Novabiochem) and a 10-fold molar excess of N-methyl morpholine in DMF for 30 min. At the end of the synthesis, the peptide-resin mixture was washed with DMF (10 ml/g) once and with methanol (10 ml/g) and DMF (10 ml/g) twice and then lyophilized after removal of the Val protecting amino acid.
(ii) Peptide modification.
Cholesterol-tagged peptide (CAU) was prepared by chemoselective conjugation of the peptide AU and the cholesterol derivative of cholesteryl chloroformate (Sigma). In particular, the peptide precursor AU-resin mixture was dissolved in a DMF-dimethyl sulfoxide solution (2:1, vol/vol) before being mixed with a 3-fold molar excess of cholesterol. N,N-Diisopropylethylamine was added to the mixture to regulate the pH at 8 to 9. Dichloromethane was then added, and the solution was left stirring at 40°C for 120 min. At the end of the synthesis, the dry peptide-Wang resin mixtures were treated individually with a combination of 94% trifluoroacetic acid, 2.5% water, 2.5% 1,2-ethanedithiol, and 1% thioanisole for 120 min. The reaction was monitored by liquid chromatography(LC)-mass spectrometry (MS) with a Shimadzu platform and a Kromasil 100-5 C18 column (4.6 by 250 mm, 5 μm) by using as eluents 0.1% trifluoroacetic acid in water (buffer A) and 0.1% trifluoroacetic in 100% acetonitrile (buffer B) and a linear gradient of 10 to 50% (vol/vol) buffer B in 16 min at a flow rate of 1 ml/min. After 1 h of incubation, the reaction was complete. Additional incubation was conducted with a mixture of trifluoroacetic acid, 5% phenol, 2% triisopropylsilane, and 5% water for 1.5 h at room temperature. The final pellets were dried, resuspended in H2O and 20% acetonitrile, and lyophilized. For a cholesterol-PEG3-tagged peptide (CPAU), synthesis and modification were conducted by a process similar to the one described above. A PEG3 spacer was introduced between the cholesterol moiety and the N-terminal peptide.
Plaque formation inhibition assay.
Primary chicken embryo fibroblasts (CEFs) grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal calf serum (FCS) were allowed to attach overnight. NDV strain F48E9 a multiplicity of infection of 0.01 was incubated for 2 h at 37°C. Virus inoculations were removed and replaced with DMEM supplemented with 2% FCS. The cells were further incubated for 36 h at 37°C. Consistent and uniform plaques were observed and counted under an Olympus microscope, and images were captured with the DP Controller software. To assess the plaque formation-inhibiting effects of the peptides, the cells were incubated with a mixture of virus and increasing concentrations of the peptides (6, 12, 25, 50, and 100 nM) at 37°C for 2 h. The inoculums were replaced with DMEM supplemented with 2% FCS and incubated for another 36 h. Plaques were counted, and the percentage of plaque inhibition (i.e., the reduction rate) was calculated against the infected cells without peptide treatment, which was set at 0%. Experiments were performed in triplicate. The results are expressed as the averages of triplicates ± the standard deviations. All experiments were conducted in parallel with each peptide.
To assess the effects of peptides at the possible stage of NDV infection, four different methods of treatment were tested with 12 nM peptide on cell monolayers. (i) For virus pretreatment, the virus was first incubated with peptides for 1 h at 37°C and then inoculated onto the cells. (ii) For cell pretreatment, the cells were incubated with peptides for 30 min at 4°C and then washed thoroughly with phosphate-buffered saline (PBS). The cells were then infected with NDV. (iii) For cotreatment, the cells were incubated with peptides in the presence of virus for 1 h at 37°C. (iv) For posttreatment, the cell monolayers were infected with virus for 45 min at 37°C. The peptides were then added, and the mixture was incubated for an additional 30 min at 37°C. The monolayers were incubated for 36 h at 37°C in DMEM supplemented with 2% FCS.
Biodistribution analysis.
Four- to 5-week-old specific-pathogen-free (SPF) chickens were injected intramuscularly with 1.6 mg/kg of peptides dissolved in sterile ultrapure water. At the indicated times, blood was obtained by cardiac puncture and collected in chilled Na-heparin tubes. Plasma was separated by centrifugation, snap-frozen, and stored at −80°C until analysis. Brain tissue was also collected and frozen. Before analysis, the plasma or tissue homogenates were treated with 3 volumes of acetonitrile and 10% formic acid to precipitate proteins. After centrifugation, clarified supernatant was analyzed by LC-MS with a C18 reverse-phase high-performance LC column and an acetonitrile-water gradient system as described above. The peaks were analyzed by MS with electrospray ionization in the multiple-reaction-monitoring mode.
Circular dichroism (CD) spectrum analysis.
Peptides were dissolved in PBS (pH 7.4) at a concentration of 30 μM. The wavelength dependence of molar ellipticity (θ) was monitored from 195 to 245 nm at 25°C by an average of eight scans in a spectropolarimeter (J-710; JASCO) with a thermoelectric temperature controller and cuvettes with a 0.2-cm path length. The buffers were filtered in a vacuum pump system with 0.2-μm (pore size) membrane filters. Routine calibration of the machine was performed with d-10-camphorsulfonic acid (60 mg 100 ml−1) according to the equation [Q] = 100 Q cnl−1, where Q is the ellipticity (millidegrees), c is the peptide concentration (millimolar), n is the number of residues, and l is the path length (centimeters). Data analyses and acquisition were performed with the spectrum manager software provided by the equipment manufacturer. On average, three scans were taken at a scanning rate of 200 nm min−1. The results are expressed as the mean residue ellipticity, [Q] (10−3 degrees cm2 dmol−1). A CD spectral analysis was performed to study the secondary structural changes in single peptides and any combination of two peptides (peptide mixtures) at an equimolar concentration in PBS. In these studies, single peptides were prepared at a concentration of 30 μM and peptide mixtures were prepared at equimolar concentrations in a constant volume (for example, for each peptide mixture, the final concentration of each peptide was 15 μM). The thermal denaturation temperature was measured at 222 nm by recording the CD signal over a temperature range of 20 to 100°C at a scan rate of 5°C/min.
Assays for F activation, receptor retention, and receptor release.
Monolayers of HeLa cells transiently expressing the NDV fusion glycoprotein (F) and attachment glycoprotein (HN) were washed three times and incubated with 1% red blood cell (RBC) suspensions at pH 7.5 for 30 min at 4°C. After rinsing to remove unbound RBCs, 1 μM peptide was added. The plates were placed into a 37°C incubator for a prespecified time period. Zanamivir at a concentration of 10 mM was then added, and the plates were incubated at 37°C until the end of the time period (60 min). The plates were rocked, and the liquid phase was collected in V-bottom tubes to measure the released RBCs. The monolayers were incubated at 4°C with 200 ml of RBC lysis solution. The lysis of unfused RBCs with NH4Cl removed the RBCs whose membranes had not fused with HN/F-coexpressing cells. The liquid phase was collected in V-bottom 96-well plates for measurement of irreversibly bound RBCs. The cells were then lysed in 200 ml 0.2% Triton X-100/PBS and then transferred to flat-bottom 96-well plates for quantification of the pool of fused RBCs. The quantity of RBCs in each of the above three compartments was determined by measurement of absorption at 410 nm.
Virus yield reduction assay in chicken embryos.
To determine the virus-inhibiting effects of the two modified peptides in chicken embryos, 104 PFU of three strains of NDV (F48E9, Guangxi, and ND/03) and 50 PFU of three strains of IBV (H52, Conne, and H120) were used. Each peptide was injected into the allantoic cavity of a 9-day-old SPF chicken egg before, after, or at the time of virus inoculation. After peptide injection and virus inoculation, the eggs were incubated at 37°C for 140 h. Observations of viable signs were done every 6 h. Dead embryos, when found, were immediately stored at 4°C for later examination of pathological changes. The viral titer present in the allantoic fluid was measured when the embryo was still alive at the endpoint (140 h postinfection [p.i.]). Hemorrhage is a typical symptom of NDV infection, and the pathology was rated as none (−), mild (+), moderate (++), or severe (+++) according to the pathological degree of hemorrhage. Embryo dwarfism is a typical symptom of IBV infection, and the pathology was rated as none (−), mild (about 5/6, +), moderate (4/5, ++), or severe (2/3, +++) according to the pathological difference in length between infected and uninfected embryos.
For canine distemper virus (CDV) (strain A75-17), 100 PFU of virus was injected into the chorioallantoic membranes (CAMs) of 7-day-old SPF chicken eggs. After 6 days of additional incubation, pale spots and protuberances on the CAMs were observed under an Olympus microscope. In the cotreatment protocol, a mixture of the CDV with peptides at various concentrations was injected into the CAMs and incubated at 37°C for 6 days. The size and shape of the pathological change were then measured and analyzed.
For Marek's disease virus MDV (strain RB1B), 103 PFU of CEF-associated virus was injected into the yolk sacs of 6- to 7-day-old SPF chicken eggs. After 9 days of additional incubation, the surviving embryos were chilled overnight at 4°C and inspected for lesion formation. In the cotreatment protocol, a mixture of MDV with various peptides at a concentration of 0.25 mM in 60 μl was injected into the yolk sacs and incubated at 37°C for 9 days. The CAMs at day 9 postincubation were fixed in 10% buffered formalin. Lesions (pox) were observed and counted under an Olympus microscope, and images were captured with DP Controller software. The ratio of lesion counts to the no-peptide sample control is presented as the percentage of infection (by arithmetic conversion of the mean percent lesion formation).
All experiments were conducted in parallel and triplicate with 5 eggs per condition for each peptide and scramble peptide.
In vivo antiviral assay.
Four- to 5-week-old SPF chickens were obtained from a commercial source and kept in an isolation space (caged) with a 0.3-μm air inlet filter system. Virus challenge was done via the intranasal and intraocular (eye drop) routes with different dilutions of NDV in a 100-μl volume. For peptide treatment, groups of five chickens were treated with 1.6 mg/kg peptide in a 100-μl volume (equal to 0.5 mg or 1.25 mM in 100 μl per animal) by intramuscular injection. Different time periods of peptide treatments were scheduled: 1 or 2 days before infection, day of infection or 1 or 2 days after infection (i.e., day −2, −1, 0, 1, or 2, respectively). The experiments were then repeated once on day 6 p.i. or three times at 3-day intervals. The infected animals were observed daily for clinical signs (neurological signs and diet conditions). Animals were isolated to avoid chickens pecking at each other, which usually happens when severe neurological symptoms are observed. All of the experiments were completed in an isolated space under biosafety level 2 conditions.
LDH assay for toxicity analysis.
Peptide cytotoxicity was assessed with a lactate dehydrogenase (LDH) assay that was done according to the manufacturer's instructions by means of a cytotoxicity detection kit (Roche).
RESULTS
Peptide was modified with cholesterol or cholesterol and PEG3.
In our previous study, an untagged peptide (AU) was designed on the basis of the HR sequences of NDV and IBV. In general, peptides have a short half-life in vivo and various modification strategies have been investigated (7, 11, 14, 15). In the present study, we first modified the AU peptide by conjugating a cholesterol group to it, and the N-terminally tagged peptide was designated CAU. With a substitution reaction, in which cholesterol and the first amino acid of the peptide interacted, the chloride ion (Cl) of cholesterol chloroformate was replaced. The theoretical molecular mass was 4,322.19 Da (3,909.58 [molecular mass of AU] + 449.11 [molecular mass of cholesteryl chloroformate] − 36.5 [molecular mass of HCl]), and the observed mass was 4,322.2 Da. The synthetic scheme for the cholesterol-tagged peptide is shown in Fig. 1. The reaction was assessed by LC-MS, and the purity was >80%. The amino acid sequences of the AU peptides and scrambled peptides, which have the same sequences at the a, b, c, f, and g sites and similar features at the d and e sites, are shown in Fig. 2. We also modified the AU peptide with cholesterol and PEG3. The modified peptide was named CPAU, in which the cholesterol group and the AU peptide were linked by PEG3 (not shown here).
Fig 1.
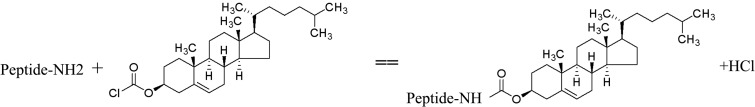
Scheme used to synthesize the cholesterol-tagged peptide described in this report.
Fig 2.

Sequence alignment of AU and scrambled peptides. The “a” site amino acids of peptides are underlined.
Cholesterol-tagged and untagged peptides have similar effects on plaque formation.
To compare the in vitro antiviral effects of the CAU and AU peptides, CEF cells were infected with NDV in the presence or absence of each peptide at various concentrations. Both CAU and AU displayed efficient fusion inhibition activities on the same order of magnitude, with 50% inhibitory concentrations (IC50s) of 8.1 and 10.2 nM, respectively (Fig. 3A). The CAU and AU peptides at a concentration of 12 nM (Fig. 3B) were further tested to address the possible difference in their inhibitory activities with four different treatments. In general, CAU showed higher antiviral activity than the AU peptide in all of the assays. Furthermore, plaque formation was almost completely inhibited by 12 nM CAU and was inhibited by 75% in the presence of AU in a cell pretreatment assay. It was also observed that plaque formation was reduced approximately 70% in the presence of CAU and 14% in the presence of AU in a virus pretreatment assay. These results demonstrate the potential antiviral activity of the CAU peptide.
Fig 3.
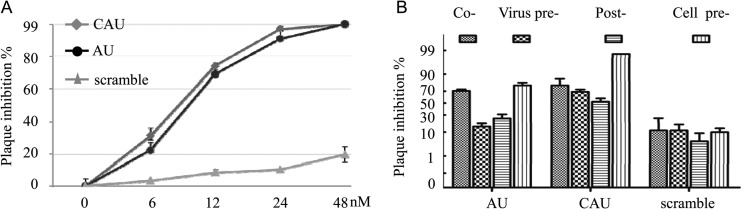
Effects of peptides at various concentrations on plaque formation. (A) CEF cells were exposed to active peptides at concentrations of 6, 12, 24, and 48 nM. Both CAU and AU displayed efficient fusion inhibition activity on the same orders of magnitude at the concentrations tested. (B) CEF cells were exposed to CAU at a concentration of 12 nM in four different modes. The results shown are representative of three independent experiments. P < 0.01 (t test).
Cholesterol-tagged peptide extended half-life and penetrated of the central nervous system (CNS).
To determine whether or not modification with cholesterol enhances the in vivo half-life of AU, the biodistribution of AU and CAU in plasma and brain tissue was examined (Fig. 4). A peak value of 170 nM in the plasma was observed at 4 h postinjection. The peptide then quickly decreased to an undetectable level at 8 h postinjection when the animals were injected intramuscularly with peptide AU at 1.6 mg/kg. However, we observed an increase in the plasma peptide concentration to a peak value of 820 nM at 8 h postinjection of CAU. Importantly, the concentration dropped slowly to 210 nM at 24 h and remained at approximately 120 nM after 48 h. These plasma peptide concentrations were considerably higher than the in vitro IC50 of 8.1 nM and IC90 of 13 nM. We then sought to determine the biodistribution of the peptides that enter the CNS—the target tissue of NDV. The CAU peptide was detected in the chicken brain, where its concentration reached 42 nM at 8 h postinjection. The upward tendency slowed at 24 h postinjection, and the concentration had increased to 300 nM at 48 h. The CNS distribution of the CAU peptide is particularly relevant to the course of NDV infection. The peptide AU was not found to penetrate the CNS.
Fig 4.
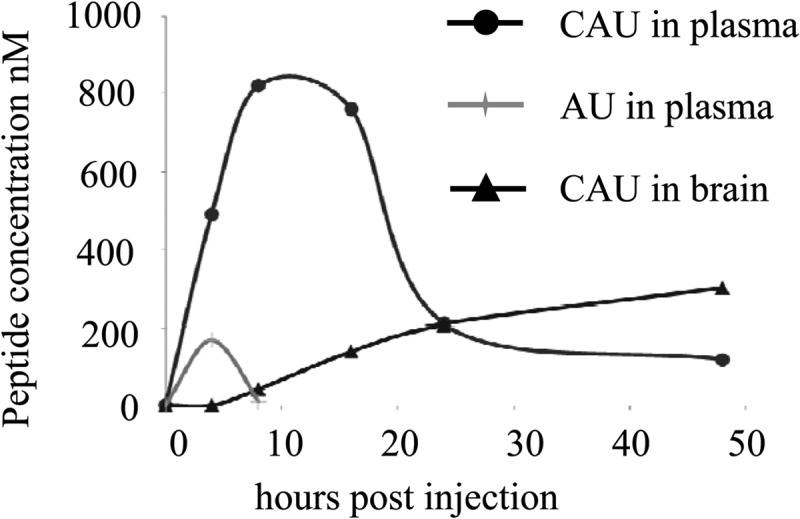
Biodistribution of cholesterol-tagged CAU peptide. Four- to 5-week-old chickens were injected with 1.6 mg/kg of CAU or AU peptide. At 10, 20, 30, 40, and 50 h postinjection (x axis), the animals (two for each data point) were sacrificed and the plasma and brain peptide concentrations were quantified.
Cholesterol-tagged peptide interacts with HR2 in the intermediate stage of glycoprotein F conformation change.
CD spectroscopy indicated that the CAU, AU, and scrambled peptides were well folded and formed a typical α-helical structure with double minima at 208 and 222 nm in a PBS solution. CPAU, however, kept an unfolded structure (Fig. 5A). If no interaction takes place between the two peptides, then each peptide will keep its original structure. The spectrum will exhibit a distinct value based on the average of the two constituent peptides. On the other hand, if two peptides interact with each other, a structural change will take place, with a great difference in the spectrum. Figure 5A also shows the CD results of the mixed peptides composed of AU-targeted HR2 and the peptides mentioned above (AU and CAU). The significant CD difference between the individual and mixed peptides indicates that the peptides interacted with each other to produce a conformation change. The thermal stability of the mixed peptides was assessed by CD by monitoring ellipticity at 222 nm as a function of temperature (Fig. 5B). It is interesting that although the tendency of the AU-HR2 peptide mixture to form coiled-coil structures was more obvious than that in the CAU-HR2 mixture, the latter was much more resistant to thermal denaturation than the parent AU-HR2 mixture. In conclusion, these results indicate that CAU yields a more stable interaction with target HR2 than the previously described peptide AU, thereby supporting the idea that the HR2 region of the viral F glycoprotein is a specific target of CAU.
Fig 5.

CD spectroscopy results. CD results of single or mixed peptides at equimolar concentrations are shown. (A) CD spectroscopy indicated that the CAU, AU, and scrambled peptides formed a typical helical structure, whereas CPAU kept an unfolded structure. Mixed peptides demonstrated an obvious tendency to form helical structures, as opposed to each individual peptide solution. (B) the CAU-HR2 mixture was much more resistant to thermal denaturation than the parent AU-HR2 mixture was.
Cholesterol-tagged peptide targeted to glycoprotein F prior to membrane insertion.
To investigate the exact target of CAU in the process of NDV entry into the host cell, we used an assay that was developed to distinguish the different states of F glycoprotein activation, such as HN-receptor association, HN-F protein interaction, fusion peptide membrane insertion, F protein conformation change, and membrane fusion (15). In an evaluation of reversible binding, zanamivir was used to interfere with the HN-receptor interaction. Our results (Fig. 6A) reveal that the CAU, AU, and scrambled peptides inhibited RBC release by 50, 40, and 27% at 30 min after treatment with zanamivir, respectively. Similar inhibition levels of 60, 50, and 55% were observed at 60 min. Most of the RBCs, however, were released by zanamivir in the absence of peptide, suggesting that the binding is HN dependent and lacks fusion peptide insertion into a membrane.
Fig 6.

Cholesterol-tagged peptide interacts with F glycoprotein. Monolayers of HeLa cells coexpressing HN and F were allowed to bind to receptor-bearing RBCs at 4°C. Upon transfer to 37°C, medium containing AU, CAU, scrambled peptide, or no peptide was added. Zanamivir was added to block HN-receptor interaction at the indicated time points, and RBCs that were reversibly bound by HN-receptor interaction (A, P ≤ 0.05), irreversibly bound (B, P ≤ 0.05), or fused (C) were quantified. The ordinate values are means ± standard deviations of triplicate samples.
The monolayers (after the zanamivir-treated liquid was collected) were incubated at 4°C with an RBC lysis solution and then treated with NH4Cl to remove the RBCs whose membranes were not fused with HN/F-coexpressing cells. As shown in Fig. 6B, our results demonstrate that for CAU, there was a progressive increase in the quantity of the RBCs that were not detached by zanamivir. Specifically, 53% of the RBCs were irreversibly bound in the presence of CAU at 30 min after zanamivir treatment. At that time, however, only 33 and 16% of the RBCs were irreversibly bound in the presence of the AU and scrambled peptides, respectively. These results demonstrate that the N-terminally cholesterol-tagged peptide CAU played a role in the early stage of membrane fusion and interfered with HN-F association. We hypothesize that this is the basis of the peptide's efficacy.
Cells lysed in Triton X-100–PBS were transferred to plates for quantification of the pool of fused RBCs. As expected, only minimal fusion (less than 10%) was measured before fusion peptide insertion (15 and 30 min) for all the peptides with and without treatments (Fig. 6C).
Two peptide modifications efficiently protected chicken embryos from NDV, IBV, and MDV infections.
To investigate whether the modified CAU and CPAU peptides have the ability to inhibit virus infection, we evaluated their antiviral effects in a model of chicken embryos (5, 17, 18) infected with different serotypes of NDV, IBV, CDV, and MDV (Table 1).
Table 1.
Effects of the peptides on chicken embryo infectivitya
| Virus (no. of PFU), strain, and peptide | Time (h) postinfection | Pathological change | Protective effect (%) |
|---|---|---|---|
| NDV (50) | |||
| F48E9 | |||
| AU | −6/0 | Hemo (−) | 100 |
| CAU | −12/−6/0 | Hemo (−) | 93 |
| CPAU | −12/−6/0 | Hemo (−) | 93 |
| CPAU | −18b | Hemo (+) | 20 |
| Scramble | −18/−12/−6/0/6 | Hemo (+++) | 100 |
| AU | −6/0 | Hemo (−) | 100 |
| CAU | −12/−6/0 | Hemo (−) | 87 |
| Guangxi | |||
| CPAU | −12/−6/0 | Hemo (−) | 100 |
| Scramble | −18/−12/−6/0/6 | Hemo (+++) | 100 |
| AU | −6/0 | Hemo (−) | 87 |
| CAU | −12/−6/0 | Hemo (−) | 100 |
| ND/03 | |||
| CPAU | −12/−6/0 | Hemo (−) | 100 |
| CPAU | −18b | Hemo (+) | 27 |
| Scramble | −18/−12/−6/0/6 | Hemo (+++) | 100 |
| IBV (104) | |||
| H52 | |||
| AU | −6/0 | Dwar (−) | 100 |
| CAU | −12/−6/0 | Dwar (−) | 87 |
| CPAU | −18/−12/−6/0 | Dwar (−) | 100 |
| CPAU | 6 | Dwar (+) | 93 |
| Scramble | −18/−12/−6/0/6 | Dwar (+++) | 100 |
| AU | −6/0 | Dwar (−) | 100 |
| CAU | −12/−6/0 | Dwar (−) | 93 |
| Conne | |||
| CPAU | −18/−12/−6/0/6 | Dwar (−) | 87 |
| Scramble | −18/−12/−6/0/6 | Dwar (+++) | 100 |
| AU | −6/0 | Dwar (−) | 87 |
| CAU | −12/−6/0 | Dwar (−) | 93 |
| H120 | |||
| CPAU | −18/−12/−6/0 | Dwar (−) | 100 |
| CPAU | 6 | Dwar (++) | 93 |
| Scramble | −18/−12/−6/0/6 | Dwar (+++) | 100 |
Antiviral efficiency includes the postinfection time of peptide treatment, pathological changes caused by the virus in infected embryos, and the corresponding protective effect (percent). Severity of pathology is rated as mild (+) to severe (+++). Hemorrhage (Hemo) is the typical symptom of NDV infection, and embryo dwarfism (Dwar) is the typical symptom of IBV infection. Each experiment was done three times with 5 eggs every time, for a total up to 15 eggs.
About 20 to 27% of the NDV-infected animals treated with CPAU at −18 h p.i. survived for 140 h.
As for therapeutic effect on NDV strain F48E9, our results showed that when used for treatment 6 h after infection (6 h p.i.), the AU, CAU, and CPAU peptides did not reduce the virus-induced pathological change in chicken embryos, as indicated by embryo hemorrhage. The severity of tissue damage was similar to that in embryos treated with scrambled peptides (severe hemorrhage, +++). When injected into eggs 12 h before virus inoculation (−12 and −6 h p.i.), however, both CAU and CPAU protected the embryos from NDV infection. The protective effect exceeded 93%. Under this treatment, all of the embryos survived the period of infection (140 h p.i.) without the pathological change of hemorrhage (−). When CAU or CPAU was given at the time of inoculation (0 h p.i.) by mixing with the virus, the same results were observed (−). The allantoic fluid of embryonic eggs contained a negligible virus titer. When CPAU treatment was given earlier before infection (−18 h p.i.), only 20% of the infected embryos survived for >140 h and the infected embryos showed mild hemorrhage (+). These data indicate that CAU and CPAU could protect chicken embryos from NDV infection. While AU could provide protection, even 100% protection, it only acted in a narrower time window (−6 to 0 h p.i.). Under the same experimental conditions, no embryos survived and all had severe hemorrhage (+++) in the scrambled-peptide treatment group.
For IBV strain H52 infection, virus-induced embryo dwarfism was used as an indicator. The scrambled peptide did not have any antiviral activity, and severe pathological dwarfism (+++) was observed in infected embryos. The embryo infected with IBV in the presence of the peptide CPAU did not display any pathological dwarfism (−) when the peptide was given 18 h prior to virus inoculation (−18, −12, −6, or 0 h p.i.). CPAU also efficiently reduced dwarfism when given at 6 h p.i., giving 93% protection, and only mild dwarfism (+) was observed. The CAU peptide showed a similar effect; 87% of the embryos did not show any pathological dwarfism (−) when the CAU peptide was given at −12, −6, or 0 h p.i. AU provided similar protection to 100% of the embryos when administered at −6 or 0 h p.i. Even though both CPAU and CAU showed efficient antiviral activity, CPAU had a longer-lasting effect than CAU.
We further tested the antiviral activities of CAU and CPAU against different serotypes of NDV (such as the Guangxi and ND/03 strains) and IBV (such as the Conne and H120 strains). Our results showed that they were also effective against those different serotypes (Table 1), but they showed no effect against CDV, another paramyxovirus, even at a higher concentration (1.25 mM). In MDV-infected embryos, the CAU and CPAU peptides reduced the CAM embryo lesions by 33 and 62% at a concentration of 0.25 mM, respectively (data not shown).
Cholesterol-tagged peptides have potent antiviral activity in vivo.
We next evaluated the prophylactic and therapeutic abilities of CAU and CPAU in chickens infected with NDV. On the basis of the results of chicken embryo experiments, the peptide dose was determined as 1.6 mg/kg. SPF chickens were infected with either 0.2 PFU (Fig. 7A) or 20 PFU (Fig. 7B) of NDV in 100 μl, and the peptides were given to the chickens two times, at day 0 and day 6 p.i. Our results showed that all of the infected chickens survived over the period of the experiment (40 days) when treated with CAU, while the survival time was about 10 to 16 days when they were treated with scrambled peptides. The surviving animals in the CAU treatment group had a higher serum hemagglutinin (HA) inhibition (HI) titer (212) than those in the scrambled peptide treatment group (28 to 29). CPAU was less effective under this experimental condition, in which only 40% of the chickens infected with 0.2 PFU of virus survived past day 40 p.i., with HI titers of 212, and all of the chickens infected with 20 PFU died around day 20 p.i., with HI titers of 29 to 210. The untagged AU peptide was effective in treating the animals infected with 0.2 PFU of virus, 100% of which survived at day 40 p.i. with HI titers of 212, whereas only 20% of the animals infected with 20 PFU of NDV survived when treated with the AU peptide, and 80% of the infected animals died at about day 20 p.i., corresponding to HI titers of 212 and 29 to 210, respectively.
Fig 7.
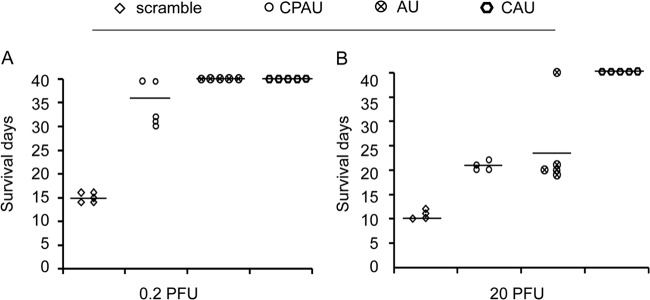
In vivo antiviral efficacies of peptides (lower viral dose). A 1.6-mg/kg dose of various peptide was administered to chickens; meanwhile, the chickens were challenged by intranasal point eye inoculation with 100 μl of a solution containing different dilutions of NDV and peptide administration was then repeated on day 6 p.i. (A) Treatment with 0.2 PFU of NDV (total of five animals per group). (B) Treatment with 20 PFU of NDV (total of five animals per group).
To examine whether CAU can protect chickens from a higher-dose NDV challenge, we infected chickens with 200 PFU of NDV and treated them with the CAU peptide (dose of 1.6 mg/kg) on different schedules. Five groups of chickens (five per group) were given the peptide CAU twice. The first dose was given 1 or 2 days before infection, the day of infection, or 1 or 2 days after infection, and the second was given at day 6 p.i. (Fig. 8A). Most of the infected chickens treated with the peptide CAU on five different dosing schedules survived approximately 3 to 5 days longer than those treated with scrambled peptides. In another treatment regimen test, the CAU peptide was administered as described above for the first dose and then given three more times at 3-day intervals to each of the five groups (10 chickens per group). Surprisingly, treatment started at day 1 before the virus challenge resulted in 70% protection over day 30 p.i. with an HI titer of 212, and treatment started 2 days after infection resulted in a survival rate of 50% over day 30 p.i (Fig. 8B). In addition, for those treated with CAU at the time of virus inoculation, only 20% of the infected animals survived about 5 days longer than those treated with scrambled peptides.
Fig 8.

In vivo antiviral efficacies of peptides (higher viral dose). A CAU peptide dose of 1.6 mg/kg was administered to chickens 2 days or 1 day before infection (i.e., Day −2, Day −1), concurrently with infection (i.e., Day 0), and 1 or 2 days after infection (i.e., Day 1, Day 2). Meanwhile, the chickens were challenged by intranasal point eye inoculation with 100 μl of a solution containing 200 PFU of NDV. (A) Injections were repeated on day 6 p.i. (total of five animals per group). (B) Injections were repeated three times at 3-day intervals (total of five animals per group).
Cholesterol-tagged peptides do not display cell toxicity.
To investigate whether these peptides have toxic effects on cells, CEFs were cultured in medium containing different concentrations (0 μM, 0.2 μM, 1 μM, 5 μM, 25 μM, 50 μM, 250 μM, 0.5 mM, and 1.0 mM) of each peptide for 24 h. Cell viability was then assessed by LDH assay. There was no significant difference between untreated and peptide-treated cells at a concentration lower than 1.0 mM. In addition, in vivo tests showed that even at 5 mg/kg, no adverse effects, such as diet changes, status, or body weight alterations, occurred in the tested animals (data not shown).
DISCUSSION
Modified peptides can effectively inhibit existing virus serotypes of NDV and IBV infections.
In our previous reports, untagged AU peptide provided complete protection of chicken embryos against NDV, IBV, and MDV infections (5, 17). A series of systematic replacements with glutamic acid (E) or lysine (K) were made at solvent sites b, c, f, and g to design the peptide. Theoretically, these replacements should not change the local electrostatic balance. The important helix-forming residues at site a are highly conserved leucine (L) or isoleucine (I) residues, while the amino acids at sites d and e integrate the inherent features, such as polarity and hydrophobicity, of the corresponding regions from different viruses (5). In the present study, all of the AU and cholesterol- and/or PEG3-tagged AU (CAU and CPAU) peptides showed potent antiviral activity in the embryo experiment. When injected into eggs 12 h prior to virus inoculation, both CAU and CPAU protected the embryos from infection with three different serotypes of NDV or IBV, although the protection effects varied from 87 to 100% for different virus strains (Table 1). Notably, there are one to eight amino acid differences between the HR2 domains of the standard strain (F48E9) and the other strains. The interaction probability between the AU peptide and HR2 of other NDV strains was analyzed with the Coils software. An interaction probability of 90 to 100% was found (data not shown). We also found that mutation commonly occurred at b, c, f, and g sites of the HR2 region of different NDV serotypes (sequence alignment in Fig. 9). Moreover, the designed AU peptide is focused on mutations at d and e sites, which lends support to the suggestion that AU and its derivatives could be used to control outbreaks of existing virus serotypes.
Fig 9.

Alignment of HR2 domain sequences of different genotypes of NDV. The “a” site amino acids of peptides are underlined.
The cholesterol-tagged peptide CAU can penetrate the avian CNS.
Covalent modification with PEG, a nontoxic polymer commonly used in food, cosmetics, and pharmaceutical preparations for over 60 years, can profoundly influence the pharmacokinetic, pharmacologic, and toxicologic profiles of protein- and peptide-based therapeutics. The PEG3 spacer not only considerably improves peptide solubility in aqueous media but also enhances peptide activity by increasing its potency via a generic increase in affinity for membranes and/or via specific enrichment in the lipid rafts, where virus-cell fusion occurs (19). Recently, cholesterol and cholesterol-and-PEG double-modification strategies have been used to increase antiviral potency and dramatically extend peptide half-lives in mice (11, 15). Such techniques provided a basis for the development of longer-acting agents against virus infections. In our study, both CAU and CPAU completely inhibited NDV and IBV infections 12 h prior to virus inoculation. The longest-acting peptide was CPAU, which effectively inhibited IBV infection by >93% from 18 h preinfection to 6 h p.i. (Table 1). This result was not in agreement with our hypothesis, in which the design irrationality of double-modified CPAU leading to an unfolded secondary structure (Fig. 5) results in the inactivation of antiviral activity. Indeed, unlike CAU, CPAU only slightly reduced the mortality rate due to encephalitis in NDV-infected animals (Fig. 7 and 8). It is reasonable to speculate, on the basis of the present experimental results that PEG conjugation prevents cholesterol-modified peptides from crossing the poultry blood-brain barrier. In fact, NDV can infect the CNS to induce severe encephalitic lesions at approximately 10 days p.i (20, 21). Further investigation, including a series of pathology studies, however, is necessary to clarify these results.
Peptide antivirals against HIV (e.g., enfuvirtide) and the peptide AU designed by our group fail to cross the blood-brain barrier, which impairs the treatment of CNS diseases. The design of peptide CAU, which can cross into the CNS, could provide better therapeutics. We believe that the ability to cross into the CNS is essential for therapies against human and poultry diseases, including that due to highly pathogenic avian influenza virus infection. Further studies are needed to address the mechanism by which the cholesterol-tagged peptide crosses the blood-brain barrier. This mechanism is currently unknown and is of great interest. It has significant implications for the treatment of CNS diseases caused by enveloped viruses.
Novel N-terminal cholesterol modification strategy is effective.
Two major structural elements of a cell are the cytoskeleton and the lipid membranes. There have been reports of cholesterol's direct involvement in poultry blood-brain barrier formation (22). Cholesterol is involved in the life cycle of many viruses; cell membrane cholesterol mainly contributes to virus entry into cells, and viral envelope cholesterol is essential for virus infectivity (23, 24, 25, 26). The interaction of viruses with cholesterol during different stages of the replication cycle leads to another effective avenue in the design of antiviral strategies (27, 28). Recent studies have indicated that the removal of cellular cholesterol partially reduces viral binding, fusion, and infectivity, suggesting that cellular cholesterol may be required for optimal cell entry in the NDV infection cycle (29). Cholesterol modification has already been used to increase the antiviral potency of HR-mimetic peptides (11, 14, 15).
The formation of a six-helix bundle structure between fusion glycoprotein HR1 and HR2 is a key step in class I virus entry into a host cell, in which the a and d sites of HR1 form the center core and the a and d sites of HR2 and the HR1 e and g binding sites form a six-helix bundle in an antiparallel manner (1, 2, 3). A synthetic HR1-HR2 peptide can bind to endogenous HR2-HR1 in the intermediate state after the fusion peptide is inserted into the target membrane (3, 4, 5). If a specific orientation of the HR2-mimetic peptide on the cell surface is required to block the refolding of the fusion glycoprotein, it is likely that only the C-terminally tagged peptide would be properly oriented to maximize the interaction with HR1. Actually, C-terminal peptide tagging dramatically increases the peptide's antiviral potency and simultaneously increases its in vivo half-life (11, 15). It has been shown that N-terminal tagging of a peptide with cholesterol produces a 50-fold decrease in antiviral activity against HIV of the Retroviridae family (11). N-terminal tagging of the peptide, however, increases the inhibitor peptide's efficacy against human parainfluenza virus type 3 of the Paramyxoviridae family (12). The different effectiveness of tagging at the N terminus could be influenced by the kinetics of fusion glycoprotein activation (12). On the other hand, it has been reported that the thiol group of an extra cysteine residue (i.e., GSGC) via a chemoselective reaction is added between the HR-mimetic peptide and a bromoacetyl derivative of cholesterol (11, 12, 14, 15). The extra residue insertion cannot be excluded if it leads to a conformation change or functional effects on the target protein. Therefore, we reason that an N-terminal tagging strategy by direct substitution reaction would be suitable to modify the peptide AU.
In the present study, both CAU and AU demonstrated efficient fusion inhibition activity with the same IC50 but CAU showed antiviral activity superior to that of the AU peptide in different assays (Fig. 3). Furthermore, the affinity between AU and HR2 is stronger than that between CAU and HR2, although the latter complex is more stable than the former (Fig. 5). Finally, modification through the use of cholesterol at the N terminus of the peptide results in potent antiviral activity in the two types of in vivo models (Table 1; Fig. 7 and 8). Combined, the results indicate that these new chemical modifications are effective and can extend the in vivo half-life of the peptide.
Proposed mechanism by which the CAU peptide blocks NDV infection.
Our previous report (5) showed the interaction between peptide AU and HR2 by immunoprecipitation and glutathione S-transferase pulldown experiments. There was no evidence suggesting possible binding between HR1 and AU under the same assay conditions. The CD analysis further showed that equimolar mixtures of peptide AU and HR2 have more obvious α-helical structures than the HR1-HR2 complex. In addition, the CD spectrum clearly demonstrated that the AU-HR2 complex is more stable than the HR1-HR2 complex, which is a process of energy release (1, 4). These results support the possibility that AU blocks the formation of the six-helix bundle, thereby inhibiting virus infection by competitive combination with the endogenous HR2 region. Here, the CD results indicated that the CAU-HR2 mixture is much more resistant to thermal denaturation than the parent AU-HR2 mixture (Fig. 5), supporting the hypothesis that CAU blocked the formation of the six-helix bundle by competition for the HR2 region. Given that the cholesterol modification generally does not affect the peptide structure and its activity, it is reasonable to infer that HR2 is also the target of CAU antiviral action. It is reasonable to infer that even if CAU is inserted into the cell membrane first, CAU shows a tendency to associate with HR2 because of their high affinity.
Furthermore, it has been reported that cholesterol conjugation of antiviral peptides could be useful for viruses that fuse in the endosomal compartment, such as influenza virus, by localizing the peptides to the target cell membrane, allowing the peptides to trap viral HA glycoprotein in a transient intermediate state after fusion (30). These results provide proof of the concept that cholesterol-modified peptides can be distributed into intracellular spaces, as well as inserted into cellular membranes. Ingallinella et al. (11) concluded that when infection was initiated by addition of the retrovirus HIV, cholesterol-modified peptide is available for dominant negative interference with the formation of the six-helix bundle of fusion glycoprotein gp41 to drive the merging of the viral and cellular membranes. Porotto et al. (15) extended these earlier seminal findings by showing that a cholesterol-tagged peptide can interact with the F glycoprotein of paramyxovirus before the fusion peptide is inserted into the target cell membrane.
Our results indicate that in the presence of the CAU peptide, RBCs were retained to attach and bind to HN/F-expressing cells in an HN-independent manner before fusion peptide insertion (Fig. 6). This supports the notion (12, 15) that a cholesterol moiety group, as an appropriate type of lipid anchor on the cell surface, may localize the HR-mimetic peptide to the cell membrane and create a protective antiviral shield to target the earlier stage of the activation process of the F glycoprotein. Porotto et al. (15) further proposed that the uncleaved F glycoprotein must have undergone a conformational change to expose the HR1 and HR2 regions, and the cholesterol-tagged peptide then captures this transitional intermediate. This has broad-spectrum significance and is worthy of further exploration, although we cannot exclude the possibility that the cholesterol-tagged peptide CAU plays a role at two different stages of membrane fusion, including the HN-F association and the intermediate state of F glycoprotein conformation change.
General-utility perspective on cholesterol-tagged peptide CAU.
The present study focused on the molecular mechanisms of class I envelope virus entry based on newly designed peptides that have been developed to inhibit infection with enveloped viruses of three different families. This design can be used to explore approaches to expedite the development of relatively broad-spectrum antivirals or therapeutic drugs. Further research will explore antiviral peptides that have common leucine zipper regions with similar structures and functions and peptides rich in the α-helical structure. Extensive research will be conducted into viruses beyond the Paramyxoviridae, Coronaviridae, Retroviridae, and Herpesviridae families. When considering the time-effect study on the CAU peptide, it is notable that CAU is able to block viral infection at a postattachment entry step in vitro by 58% at a concentration of 12 nM (Fig. 3B). Furthermore, animals infected with 200 PFU of NDV and administered the peptide CAU on day 2 p.i. were protected and >50% survived to day 30 p.i. (Fig. 8B). These results provide proof of the concept of an antiviral strategy that is applicable to fusing viruses.
We also evaluated other administration methods, since intramuscular injection is not as convenient for poultry as for humans or other animals. We used an intranasal instillation method in the treatment with the peptide CAU. It was found that the infected animals lived only 2 to 3 days longer under the same experimental conditions. The effect of cholesterol modification on the mucosal barrier was not obvious. Finally, effective protection was achieved in the present study by administering the peptide CAU at a concentration of 1.6 mg/kg four times at 3-day intervals. Infected animals treated 1 day before a challenge exhibited 70% protection against disease for at least 30 days p.i. (Fig. 8B). This approach appeared to be more efficient than the existing techniques (14, 15), in which a cholesterol-tagged HR2 peptide was injected into mice daily at a concentration of 2 mg/kg for 10 to 14 days. A total of four peptide injections instead of a daily injection is a desirable scheme and provides a basis for the development of practical antiviral peptides at lower prices. This strategy of cholesterol modification and peptide injection may be applied to other peptide agents.
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (31101819) and by the Program for New Century Excellent Talents in University of China (NCET-11-0486).
Footnotes
Published ahead of print 26 June 2013
REFERENCES
- 1. Colman PM, Lawrence MC. 2003. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 4:309–319 [DOI] [PubMed] [Google Scholar]
- 2. Bosch BJ, van der Zee R, de Haan CA, Rottier PJ. 2003. The coronavirus spike protein is a class I virus fusion protein: structural and functional characterization of the fusion core complex. J. Virol. 77:8801–8811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wang XJ, Bai YD, Zhang GZ, Zhao JX, Wang M, Gao GF. 2005. Structure and function study of paramyxovirus fusion protein heptad repeat peptides. Arch. Biochem. Biophys. 436:316–322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eckert DM, Kim PS. 2001. Mechanisms of viral membrane fusion and its inhibition. Annu. Rev. Biochem. 70:777–810 [DOI] [PubMed] [Google Scholar]
- 5. Wang XJ, Li CG, Chi XJ, Wang M. 2011. Characterisation and evaluation of antiviral recombinant peptides based on the heptad repeat regions of NDV and IBV fusion glycoproteins. Virology 416:65–74 [DOI] [PubMed] [Google Scholar]
- 6. Kilby JM, Lalezari JP, Eron JJ, Carlson M, Cohen C, Arduino RC, Goodgame JC, Gallant JE, Volberding P, Murphy RL, Valentine F, Saag MS, Nelson EL, Sista PR, Dusek A. 2002. The safety, plasma pharmacokinetics, and antiviral activity of subcutaneous enfuvirtide (T-20), a peptide inhibitor of gp41-mediated virus fusion, in HIV-infected adults. AIDS Res. Hum. Retroviruses 18:685–693 [DOI] [PubMed] [Google Scholar]
- 7. Eggink D, Langedijk JP, Bonvin AM, Deng Y, Lu M, Berkhout B, Sanders RW. 2009. Detailed mechanistic insights into HIV-1 sensitivity to three generations fusion inhibitors. J. Biol. Chem. 284:26941–26950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Nishikawa H, Nakamura S, Kodama E, Ito S, Kajiwara K, Izumi K, Sakagami Y, Oishi S, Ohkubo T, Kobayashi Y, Otaka A, Fujii N, Matsuoka M. 2009. Electrostatically constrained alpha-helical peptide inhibits replication of HIV-1 resistant to enfuvirtide. Int. J. Biochem. Cell Biol. 4:891–899 [DOI] [PubMed] [Google Scholar]
- 9. Miyamoto F, Kodama EN. 2013. Development of small molecule HIV-1 fusion inhibitors: linking biology to chemistry. Curr. Pharm. Des. 19:1827–1834 [DOI] [PubMed] [Google Scholar]
- 10. Madsen K, Knudsen LB, Agersoe H, Nielsen PF, Thøgersen H, Wilken M, Johansen NL. 2007. Structure-activity and protraction relationship of long-acting glucagon-like peptide-1 derivatives: importance of fatty acid length, polarity, and bulkiness. J. Med. Chem. 50:6126–6132 [DOI] [PubMed] [Google Scholar]
- 11. Ingallinella P, Bianchi E, Ladwa NA, Wang YJ, Hrin R, Veneziano M, Bonelli F, Ketas TJ, Moore JP, Miller MD, Pessi A. 2009. Addition of a cholesterol group to an HIV-1 peptide fusion inhibitor dramatically increases its antiviral potency. Proc. Natl. Acad. Sci. U. S. A. 106:5801–5806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Porotto M, Yokoyama CC, Palermo LM, Mungall B, Aljofan M, Cortese R, Pessi A, Moscona A. 2010. Viral entry inhibitors targeted to the membrane site of action. J. Virol. 84:6760–6768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eade CR, Wood MP, Cole AM. 2012. Mechanisms and modifications of naturally occurring host defense peptides for anti-HIV microbicide development. Curr. HIV Res. 10:61–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pessi A, Langella A, Capitò E, Ghezzi S, Vicenzi E, Poli G, Ketas T, Mathieu C, Cortese R, Horvat B, Moscona A, Porotto M. 2012. A general strategy to endow natural fusion-protein-derived peptides with potent antiviral activity. PLoS One 7:e36833. 10.1371/journal.pone.0036833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Porotto M, Rockx B, Yokoyama CC, Talekar A, Devito I, Palermo LM, Liu J, Cortese R, Lu M, Feldmann H, Pessi A, Moscona A. 2010. Inhibition of Nipah virus infection in vivo: targeting an early stage of paramyxovirus fusion activation during viral entry. PLoS Pathog. 6:e1001168. 10.1371/journal.ppat.1001168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Roussan DA, Haddad R, Khawaldeh G. 2008. Molecular survey of avian respiratory pathogens in commercial broiler chicken flocks with respiratory diseases in Jordan. Poult. Sci. 87:444–448 [DOI] [PubMed] [Google Scholar]
- 17. Wang X, Chi X, Wang M. 2011. Structural characteristics and antiviral activity of multiple peptides derived from MDV glycoproteins B and H. Virol. J. 8:190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chi XJ, Zhao P, Li CG, Wang XJ. 2013. Interaction domain between glycoproteins gB and gH of Marek's disease virus and identification of antiviral peptide with dual functions. PLoS One 8:e54761. 10.1371/journal.pone.0054761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wonganan P, Croyle MA. 2010. PEGylated adenoviruses: from mice to monkeys. Viruses 2:468–502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wilczynski SP, Cook ML, Stevens JG. 1977. Newcastle disease as a model for paramyxovirus-induced neurologic syndromes. II. Detailed characterization of the encephalitis. Am. J. Pathol. 89:649–666 [PMC free article] [PubMed] [Google Scholar]
- 21. Ecco R, Susta L, Afonso CL, Miller PJ, Brown C. 2011. Neurological lesions in chickens experimentally infected with virulent Newcastle disease virus isolates. Avian Pathol. 40:145–152 [DOI] [PubMed] [Google Scholar]
- 22. Dietschy JM, Turley SD. 2004. Thematic review series: brain lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 45:1375–1397 [DOI] [PubMed] [Google Scholar]
- 23. Zhu L, Ding X, Tao J, Wang J, Zhao X, Zhu G. 2010. Critical role of cholesterol in bovine herpesvirus type 1 infection of MDBK cells. Vet. Microbiol. 144:51–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carette JE, Raaben M, Wong AC, Herbert AS, Obernosterer G, Mulherkar N, Kuehne AI, Kranzusch PJ, Griffin AM, Ruthel G, Dal Cin P, Dye JM, Whelan SP, Chandran K, Brummelkamp TR. 2011. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 477:340–343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Haspot F, Lavault A, Sinzger C, Laib Sampaio K, Stierhof YD, Pilet P, Bressolette-Bodin C, Halary F. 2012. Human cytomegalovirus entry into dendritic cells occurs via a macropinocytosis-like pathway in a pH-independent and cholesterol-dependent manner. PLoS One 7:e34795. 10.1371/journal.pone.0034795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Poh MK, Shui G, Xie X, Shi PY, Wenk MR, Gu F. 2012. U18666A, an intra-cellular cholesterol transport inhibitor, inhibits dengue virus entry and replication. Antiviral Res. 93:191–198 [DOI] [PubMed] [Google Scholar]
- 27. Pollock S, Nichita NB, Böhmer A, Radulescu C, Dwek RA, Zitzmann N. 2010. Polyunsaturated liposomes are antiviral against hepatitis B and C viruses and HIV by decreasing cholesterol levels in infected cells. Proc. Natl. Acad. Sci. U. S. A. 107:17176–17181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Favoreel HW, Van den Broeke C, Desplanques A, Deruelle M, Van Minnebruggen G, Nauwynck H, Glorieux S, Van Opdenbosch N, De Regge N. 2010. Alphaherpesvirus use and misuse of cellular actin and cholesterol. Vet. Microbiol. 143:2–7 [DOI] [PubMed] [Google Scholar]
- 29. Martín JJ, Holguera J, Sánchez-Felipe L, Villar E, Muñoz-Barroso I. 2012. Cholesterol dependence of Newcastle disease virus entry. Biochim. Biophys. Acta 1818:753–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lee KK, Pessi A, Gui L, Santoprete A, Talekar A, Moscona A, Porotto M. 2011. Capturing a fusion intermediate of influenza hemagglutinin with a cholesterol-conjugated peptide, a new antiviral strategy for influenza virus. J. Biol. Chem. 286:42141–42149 [DOI] [PMC free article] [PubMed] [Google Scholar]