

Abstract
The host restriction factors TRIM5α and TRIMCyp potently inhibit retrovirus infection by binding to the incoming retrovirus capsid. TRIM5 proteins are dimeric, and their association with the viral capsid appears to be enhanced by avidity effects owing to formation of higher-order oligomeric complexes. We examined the stoichiometric requirement for TRIM5 functional recognition by quantifying the efficiencies of restriction of HIV-1 and murine leukemia virus (MLV) particles containing various proportions of restriction-sensitive and -insensitive CA subunits. Both TRIMCyp and TRIM5α inhibited infection of retrovirus particles containing as little as 25% of the restriction-sensitive CA protein. Accordingly, we also observed efficient binding of TRIMCyp in vitro to capsid assemblies containing as little as one-fourth wild-type CA protein. Paradoxically, the ability of HIV-1 particles to abrogate TRIMCyp restriction in trans was more strongly dependent on the fraction of wild-type CA than was restriction of infection. Collectively, our results indicate that TRIM5 restriction factors bind to retroviral capsids in a highly cooperative manner and suggest that TRIM5 can engage a capsid lattice containing a minimum of three or fewer recognizable subunits per hexamer. Our study supports a model in which localized binding of TRIM5 to the viral capsid nucleates rapid polymerization of a TRIM5 lattice on the capsid surface.
INTRODUCTION
The host range of retroviruses is governed by dominant-acting host factors that inhibit replication in a virus-specific and host species-specific manner. Members of the TRIM5 family of proteins prevent infection by targeting the viral capsid, leading to disruption of the capsid lattice and accelerated uncoating in target cells (1–7). TRIM5 proteins, including the rhesus macaque TRIM5α protein and the owl monkey TRIMCyp protein, block HIV-1 infection by recognizing the outer surface of the viral capsid (8, 9). TRIM5 proteins contain RING, B-box, and coiled-coil domains and, in the case of TRIM5α, a B30.2/SPRY domain (10). In contrast, TRIMCyp proteins contain a replacement of the B30.2/SPRY domain with a cyclophilin A (CypA) protein domain. Accordingly, restriction by owl monkey TRIMCyp, which recognizes the cyclophilin A-binding loop in CA, can be inhibited by addition of cyclosporine (CsA) or by alterations of the CypA-binding loop (11, 12). The human TRIM5α protein, which does not efficiently restrict HIV-1, potently inhibits infection by N-tropic murine leukemia virus (N-MLV) (13–16), but not B-tropic MLV (B-MLV), in a manner akin to that of the murine restriction factor Fv1.
TRIM5 restriction factor binding to the incoming viral capsid results in an early block in the virus life cycle, normally prior to reverse transcription (17, 18). Restriction is rapid and irreversible, as inferred from time-of-addition experiments employing CsA to prevent restriction by TRIMCyp (19). The TRIM5-imposed block to reverse transcription is linked to cellular proteasome activity: treatment of target cells with chemical inhibitors of proteasome activity relieves the block to reverse transcription, but the virus is then inhibited for nuclear entry (20). The mechanism by which TRIM5 proteins trigger a proteasome-dependent block to reverse transcription is unclear, but it likely involves ubiquitylation via the protein's RING domain (21). Engagement of the incoming viral capsids also results in proteasome-dependent degradation of the TRIM5 restriction factor, thus further implicating recruitment of proteasomes to the restricted retroviral capsid (22).
Assembled retroviral capsids consist of a hexameric lattice of the CA protein, and binding of TRIM5 proteins requires the assembled lattice, suggesting that they recognize a regular array of CA subunits. In cells in which proteasome activity is inhibited, TRIM5 proteins have been observed to form a coat surrounding the viral core, suggesting that the restriction factor may completely enclose the capsid (23). Although restriction by TRIM5α appears to be dependent on the RING and B-box domains, a recombinant fragment of TRIM5α consisting of only the coiled-coil and B30.2/SPRY domains was sufficient, at high concentrations, to inflict structural damage to the capsid, including fragmentation of tubular CA assemblies and viral cores in vitro (24). TRIM5 proteins are dimeric (6), and mutations that perturb dimerization result in a loss of restriction (25). TRIM5-capsid interactions are also enhanced by higher-order oligomer interactions mediated by the B-box domain (26), suggesting that binding occurs in a cooperative fashion. Collectively, these observations argue that the repeating hexameric lattice of the viral capsid is essential for efficient binding and coating of the viral capsid by TRIM5 proteins. However, it is also possible that local binding and recruitment of proteasome activity are sufficient for potent restriction.
In the present study, we examined the capsid recognition requirements for TRIM5 restriction by testing the sensitivity of viruses containing various proportions of sensitive and resistant CA proteins. We observed that restriction is distinctly nonlinear with respect to the quantity of restriction-sensitive CA, with as little as 25% restriction-sensitive CA sufficing for potent restriction. Our results suggest that local binding of TRIM5 to the viral capsid, followed by polymerization of a TRIM5 lattice on the capsid surface and recruitment of active proteasomes, results in efficient blockade of infection.
MATERIALS AND METHODS
Cells and viruses.
CrFK, CrFK-TRIMCyp, and CrFK-huTRIM5α cells were the generous gift of Greg Towers. Owl monkey kidney (OMK) and HeLa lines were purchased from the American Type Culture Collection. 293T cells expressing myc-tagged TRIMCyp have been reported previously (5, 22). All cells were cultured at 37°C in 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) supplemented with fetal bovine serum (10%), penicillin, and streptomycin.
Virus stocks were generated by transfection of 293T cells. HIV-1 reporter particles were produced from HIV-GFP, an Env-defective HIV-1 proviral construct encoding green fluorescent protein (GFP) in place of Nef, or a corresponding gag mutant encoding a substitution of valine for glycine at position 89 of CA (G89V). Particles were pseudotyped by vesicular stomatitis virus glycoprotein (VSV-G) via cotransfection of the proviral constructs (20 μg) with pHCMV-G (5 μg) (27). GFP-transducing MLV vector particles were produced by cotransfection of pCIG-N and pCIG-B (encoding N-tropic and B-tropic MLV Gag-Pol, respectively; 20 μg in total) (28) with pBABE-EGFP (15 μg) (transducing vector) and pHCMV-G (5 μg). HIV-1 particles used in abrogation-of-restriction assays were produced from the R9ΔE and corresponding G89V mutant proviral constructs (29). HIV-1 stocks were assayed by p24 enzyme-linked immunosorbent assay (ELISA) to normalize inoculation doses, and MLV stocks were normalized by the reverse transcriptase activities of the virus stocks.
Quantitative analysis of CA proportions in virions.
Virions were pelleted through 20% sucrose, dissolved in Laemmli buffer, and subjected to SDS-PAGE on Bio-Rad Criterion gels. Proteins were visualized by staining with colloidal Coomassie blue, and bands corresponding to CA proteins were subsequently excised and digested with trypsin. Peptides specific to both variants of either the MLV (N- and B-tropic) or HIV-1 (WT and G89V) CA proteins were analyzed by selected reaction monitoring liquid chromatography-tandem mass spectrometry (SRM LC-MS/MS), using a ThermoScientific Vantage triple-quadrupole tandem mass spectrometer coupled with a Waters NanoAcquity autosampler/UPLC system. For the HIV CA assays, the trypsin-derived peptides LHPVHAGPIAPGQMR (wild-type [WT]) and LHPVHAVPIAPGQMR (V89 mutant) were assayed for both unmodified and oxidized methionine. For the MLV CA assays, the N-MLV- and B-MLV-specific peptides, i.e., LKEAYR and LKGAYR, respectively, and PDWDYTTQR and PDWDYTTTEGR, respectively, were similarly monitored. Parent and transition m/z values for each peptide analyzed by SRM are listed in Table 1.
Table 1.
Transitions employed in SRM analysis of HIV-1 and MLV CA proteins
| Virus | CA peptide sequence | m/z transitions |
|---|---|---|
| HIV-1 (WT) | LHPVHAGPIAPGQMR | 546.9681–604.2872 (y5), 675.3243 (y6), 885.4611 (y8), 984.5295 (y9), 1,055.5666 (y10) |
| HIV-1 (G89V)a | LHPVHAVPIAPGQMR | 541.6364–588.2922 (y5), 659.3294 (y6), 869.4662 (y8), 968.5346 (y9), 1,039.5717 (y10) |
| N-MLV | LKEAYR | 390.2241–338.1823 (y2), 409.2194 (y3), 538.262 (y4), 666.357 (y5) |
| PDWDYTTQR | 591.2647–505.2729 (y4), 668.3362 (y5), 783.3632 (y6), 969.4425 (y7), 1,084.469 (y8) | |
| B-MLV | LKGAYR | 354.2136–466.2409 (y4), 594.3358 (y5), 297.6715 (+2 y5) |
| PDWDYTTTEGR | 670.7913–563.2784 (y5), 827.3894 (y7), 942.4164 (y8), 1,128.496 (y9) |
The mutated amino acid is underlined in the peptide sequence.
For each experiment, replicate injections of a known 50-50 mixture of the cognate peptides (generated by SDS-PAGE analysis of equal quantities of N-MLV and B-MLV particles and excision of CA bands, with digestion with trypsin) were included to calibrate the sensitivity of the assay for each peptide. The SRM data reported were the summed areas under the curves of the extracted ion chromatograms for each set of transitions after normalization using the ratios generated from the known 50-50 mixture. Selection of peptide collision energies, management of SRM method building, and data analysis were performed using Skyline (University of Washington). Three to six transitions were monitored for each peptide, using a scan width of 0.2 m/z and a 50-ms scan time for each transition. A vented-column configuration with a 60-min liquid chromatography gradient was employed: mobile phases were 0.1% formic acid in water (A) and 0.1% formic acid in acetonitrile (B). The trapping column contained 5 cm of 3-μm Jupiter C18 reverse-phase medium (Phenomenex) pressure packed into a fused-silica capillary (360-μm outer diameter [OD] and 100-μm inner diameter [ID]), and the analytical column contained 20 cm of either 3-μm Aqua or 3-μm Jupiter C18 reverse-phase medium (Phenomenex) pressure packed into a fused-silica capillary (360-μm OD and 100-μm ID) with a laser-pulled tip for electrospray ionization at 400 nl/min. Electrospray voltage was applied via a liquid junction, using a micro-cross junction placed between the trapping and analytical columns. An open fused-silica vent line (360-μm OD and 150-μm ID) was also connected to the micro-cross junction, with the vent line flow path being controlled via a secondary switching valve. This enabled the sample flow path to load peptides onto the trapping column and flow directly to waste during the loading phase of an analytical run, after which the vent line was closed and the high-pressure liquid chromatography (HPLC) gradient started as peptides eluted from the trapping column directly onto the analytical column.
Infection assays.
Target cells (20,000) were seeded into 48-well plates, cultured overnight, and inoculated with normalized quantities (0.3 ml) of viruses in the presence of Polybrene (8 μg/ml). Two days later, cells were detached with trypsin, fixed in 4% paraformaldehyde, and analyzed by flow cytometry for GFP expression. The extent of infection was determined as the percentage of GFP-positive (GFP+) cells in each culture. Abrogation of restriction was assayed with OMK cells as previously described (29).
Assay of TRIMCyp binding to CA tubes.
Recombinant A14C/E45C and A14C/E45C/G89V CA proteins were expressed in Escherichia coli and purified under reducing conditions by anion-exchange chromatography, to >98% purity (30). Protein concentrations were determined by spectrophotometry. CA proteins were coassembled (10-mg/ml total protein concentration; 50-μl volume) into tubular structures at predetermined ratios by dialysis overnight into buffer A (50 mM Tris-HCl [pH 8.0], 1 M NaCl, 20 mM 2-mercaptoethanol). The tubular complexes were subsequently allowed to form disulfide cross-links by dialysis into buffer A lacking 2-mercaptoethanol. To prepare cell extracts containing TRIMCyp, cells were collected from eight confluent 10-cm dishes of cultured 293T cells stably expressing myc-tagged TRIMCyp, resuspended in 2.5 ml of buffer (50 mM Tris-HCl [pH 7.5], 5 mM MgCl2, 0.5 mM EDTA, 1 mM dithiothreitol [DTT], mammalian protease inhibitor [Roche]), and lysed by 10 passes with a ball-bearing homogenizer (18-μm gap), as previously described (5). Lysates were clarified by centrifugation at 13,000 × g for 12 min at 4°C. The supernatants were flash-frozen in aliquots and stored at −80°C. Binding reactions were performed as previously reported (1), using 5 μl of the assembled CA tubes and 200 μl of cell extract. Following incubation for 1 h at room temperature, reaction mixtures were layered onto 1-ml 70% sucrose cushions and centrifuged at 45,000 rpm (90,700 × g) for 1 h at 4°C in a Beckman TLA-55 rotor. Pellets were dissolved in 50 μl of 1× SDS-PAGE sample buffer, and proteins were subjected to SDS-PAGE and immunoblotting with antibodies to the myc epitope tag, CypA, and CA. Blots were scanned with a Li-Cor Odyssey instrument at a 169-μm resolution, at medium quality and a laser intensity setting of 1, at which the bands of interest were not saturated. Integrated band intensities were determined with the instrument software, and an average background value for each band was determined by the top/bottom method, with a border width of 3, and subtracted automatically using the instrument software.
Assay of HIV-1 reverse transcription in target cells.
Virus stocks were incubated with DNase I (20 μg/ml with 10 mM MgCl2) for 1 h at 37°C to remove plasmid DNA carried over from transfection. Viruses (100 ng p24) were inoculated onto CrFK-TRIMCyp cells (100,000 cells in 6-well dishes) for 8 h. Cells were detached, and DNA was purified (Qiagen DNeasy Blood & Tissue DNA extraction kit). HIV-1 DNA was quantified by real-time PCR using SYBR green chemistry and a Stratagene MX3000p instrument. The first strand-transfer products were amplified with primers 5′-AGCAGCTGCTTTTTGCCTGTACT-3′ and 5′-ACACAACAGACGGGCACACAC-3′, and the second strand-transfer products were amplified with primers 5′-AGCAGCTGCTTTTTGCCTGTACT-3′ and 5′-CCTGCGTCGAGAGAGCTCCTCTGG-3′. For each primer pair, a standard curve was generated with dilutions of plasmid DNA, and sample values were interpolated.
RESULTS
Owl monkey TRIMCyp recognizes and restricts HIV-1 by targeting CA. Mutation of Gly89 to Val in the CypA-binding loop of CA prevents binding of TRIMCyp, thereby rendering HIV-1 resistant to restriction (11, 12). We exploited this property of TRIMCyp restriction to examine the stoichiometric requirement for capsid recognition and restriction. Our approach involved generating virus particles containing various mixtures of wild-type and G89V mutant CA proteins by transfection of cells with specific mixtures of the corresponding proviral plasmids. The particles were harvested, and the p24 concentrations of the stocks were determined by antigen-capture ELISA. Titration of the particles on control CrFK cells lacking TRIMCyp revealed that all of the viruses were infectious, with the viruses containing G89V CA exhibiting minor decreases in the extent of infection (Fig. 1A). In contrast, on CrFK cells expressing owl monkey TRIMCyp, the wild-type virus was strongly restricted, while the G89V mutant was highly infectious (Fig. 1B). Titration curves for the mixed viruses resembled that for the wild type on TRIMCyp-expressing cells, suggesting that virus stocks containing as little as 25% wild-type CA are markedly inhibited by TRIMCyp. In additional studies, we observed that virus preparations generated from a 10% wild-type–90% G89V plasmid cotransfection exhibited a modest level of restriction by TRIMCyp, corresponding to an infection level intermediate between those of the 75% and 100% G89V viruses (data not shown). Analysis of the ratio of infection by the viruses on TRIMCyp-expressing versus control cells confirmed that all of the viruses containing capsids of mixed composition were restricted (Fig. 1C). These results suggested that efficient restriction requires only a minority of capsid subunits to be capable of binding TRIMCyp.
Fig 1.

TRIMCyp restricts HIV-1 particles containing mixtures of wild-type and G89V CA subunits. HIV-GFP reporter particles produced by transfection of the indicated ratios of wild-type and G89V mutant constructs were titrated onto the indicated cell lines, and the extent of infection was determined. (A) CrFK cells; (B) CrFK-TRIMCyp cells; (D) OMK cells; (E) OMK cells with CsA. (C) Percentages of infection of CrFK-TRIMCyp cells relative to control CrFK cells. (F) Percentages of infection of OMK cells relative to infection in the presence of CsA. The results are representative of three independent experiments.
The CrFK cell lines used for these experiments were engineered to express high levels of TRIMCyp and thus potently restricted wild-type HIV-1. To determine whether the observed infection results were due to TRIMCyp overexpression, we titrated the mixed capsid viruses on owl monkey kidney (OMK) cells, which express TRIMCyp endogenously. HIV-1 restriction in OMK cells is overcome under conditions with high virus inocula, indicating that the levels of the restriction factor are limited relative to those in engineered cell lines. We observed that restriction of HIV-1 in OMK cells was also distinctly nonlinear with respect to the levels of the wild-type CA protein, though the restriction of viruses containing 25% wild-type CA was not as potent as that observed in the CrFK-TRIMCyp cells (Fig. 1D). As expected, addition of the drug CsA, which prevents TRIMCyp binding to CA (11), rendered the cells permissive to all of the virus stocks, confirming that differences in infection resulted from restriction by TRIMCyp (Fig. 1E). Calculation of the extent of restriction revealed a nonlinear dependence of infection on the percentage of G89V mutant CA, though the steepness of the curve was less pronounced than that observed for the TRIMCyp-expressing CrFK cells (Fig. 1F). These results indicated that restriction of HIV-1 by TRIMCyp can occur with viruses containing a minority of the restriction-sensitive CA protein, even in cells that naturally express TRIMCyp.
A potential trivial explanation for the nonlinear relationship between the susceptibility of the mixed viruses to restriction and the CA ratios is that the particles contained compositions of CA that did not correspond to the input plasmid ratios used in the transfections. To rigorously test this, we quantified the proportions of wild-type and G89V mutant CA proteins by SRM LC-MS/MS of tryptic digests. The virus particles were pelleted and the proteins separated by SDS-PAGE. Protein bands were visualized by chemical staining of proteins in the gel, and the CA bands were excised, eluted, and cleaved with trypsin. Tryptic peptides corresponding to wild-type and G89V mutant CA in the mixtures were then quantified by SRM for unique peptides specific to the wild-type and mutant forms. As a control for peptide detection efficiency, samples containing equivalent quantities of the wild-type and G89V CA proteins were prepared by mixing lysates of equal quantities of pelleted wild-type and G89V particles (determined by ELISA) and then were analyzed in parallel by this approach. This sampling allowed the determination of a correction factor for differences in the detection efficiencies of the wild-type and mutant peptides. After applying this correction factor, we computed the ratio of wild-type to G89V CA in the samples. The results revealed a close correspondence between the input plasmid ratios and the ratios of the wild-type and G89V mutant CA proteins present in the virus particles (Fig. 2A). These results demonstrate that the input plasmid ratios used in the transfections accurately dictated the average CA protein ratios in the resulting virus particles.
Fig 2.
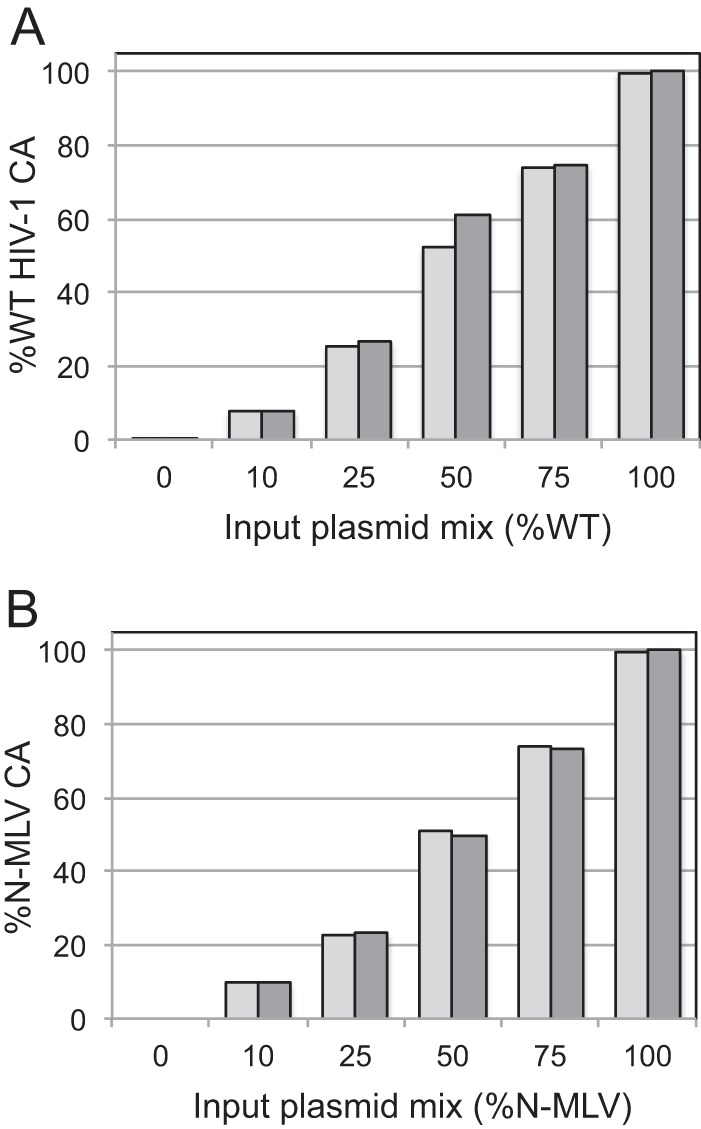
Quantitative analysis of the compositions of HIV-1 particles containing mixtures of CA proteins by selected reaction monitoring LC-MS/MS. The data shown are percentages of wild-type HIV-1 CA (A) and N-MLV CA (B) in the respective samples. The assays were performed in duplicate, and the values are presented as paired columns. Similar results were obtained in two independent experiments.
Previous studies have shown that murine leukemia virus particles containing mixed capsids are sensitive to endogenous restriction in murine and human cells (31, 32). N-tropic MLV is restricted by human TRIM5α, but B-tropic MLV is not, owing to sequence differences in CA. To examine the stoichiometric requirements for restriction by TRIM5α, we assayed the infection of MLV-GFP reporter particles containing mixtures of N- and B-tropic CA proteins on CrFK cells expressing human TRIM5α. As observed for TRIMCyp restriction of HIV-1, infection of TRIM5α-expressing cells by MLV was efficiently inhibited when 25% of the CA was derived from N-MLV (Fig. 3A). In contrast, all of the virus stocks were highly infectious on control CrFK cells, indicating that differences in infectivity were due to restriction by TRIM5α (Fig. 3B). Titration of the viruses on HeLa cells, a human cell line that endogenously expresses TRIM5α, yielded similar results (Fig. 3C). Proteomic analysis of the mixed MLV particles confirmed that the proportions of the N-MLV and B-MLV CA proteins were in concordance with the input plasmid ratios used to produce the virus stocks (Fig. 2B). We also tested MLV particles containing 10% N-MLV CA and observed infection levels on TRIM5α-expressing CrFK cells that were between those exhibited by viruses containing 25% and 100% N-MLV CA, with equivalent infection levels on control CrFK cells (data not shown). We concluded that, as with TRIMCyp restriction of HIV-1, MLV restriction by TRIM5α requires only a small fraction of the CA subunits to be capable of being recognized by the restriction factor.
Fig 3.
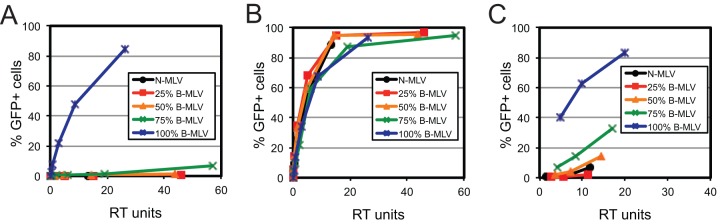
Human TRIM5α restricts MLV particles containing mixtures of N-tropic and B-tropic CA proteins. MLV vector particles were titrated onto CrFK cells expressing human TRIM5α (A), control CrFK cells (B), and HeLa cells (C). RT, reverse transcriptase.
The observation that viruses containing only 25% restriction-sensitive CA are efficiently inhibited by TRIM5 restriction factors implies that restriction factor binding requires only a small fraction of wild-type CA. Alternatively, localized binding of TRIM5α to the viral capsid may be sufficient for restriction. To examine the binding requirements for TRIMCyp, we produced tubular CA assemblies from mixtures of wild-type and G89V CA and quantified the extents of binding of TRIMCyp in cell lysates. Because wild-type CA assemblies are stable only under high-salt conditions, we utilized CA proteins with the A14C and E45C substitutions, which result in spontaneous formation of intersubunit disulfide bonds, thus stabilizing the assemblies by disulfide cross-linking (30). Cross-linked CA tubes are stable and thus can be employed in binding assays under conditions of physiologic ionic strength (33). Mixed CA complexes were generated by coincubation of 14C/45C and 14C/45C/89V CA proteins at various proportions under conditions that promote assembly. Binding of TRIMCyp was assessed by incubation with lysates of cells expressing hemagglutinin (HA)-tagged TRIMCyp, followed by pelleting of the complexes and quantitative immunoblotting analysis. As shown in Fig. 4, we observed comparable levels of TRIMCyp in pellets of reaction mixtures incubated with CA mixtures containing 100%, 75%, 50%, and 25% 14C/45C CA protein. However, binding of TRIMCyp was markedly reduced for the complexes containing 90% and 100% 14C/45C/G89V protein. For the same complexes, we also observed association of cellular CypA to an extent that was roughly proportional to the fraction of wild-type CA in the complexes, indicating that the CA complexes had the expected compositions. Collectively, these results indicate that TRIMCyp binds efficiently to CA assemblies containing as little as 25% wild-type CA.
Fig 4.
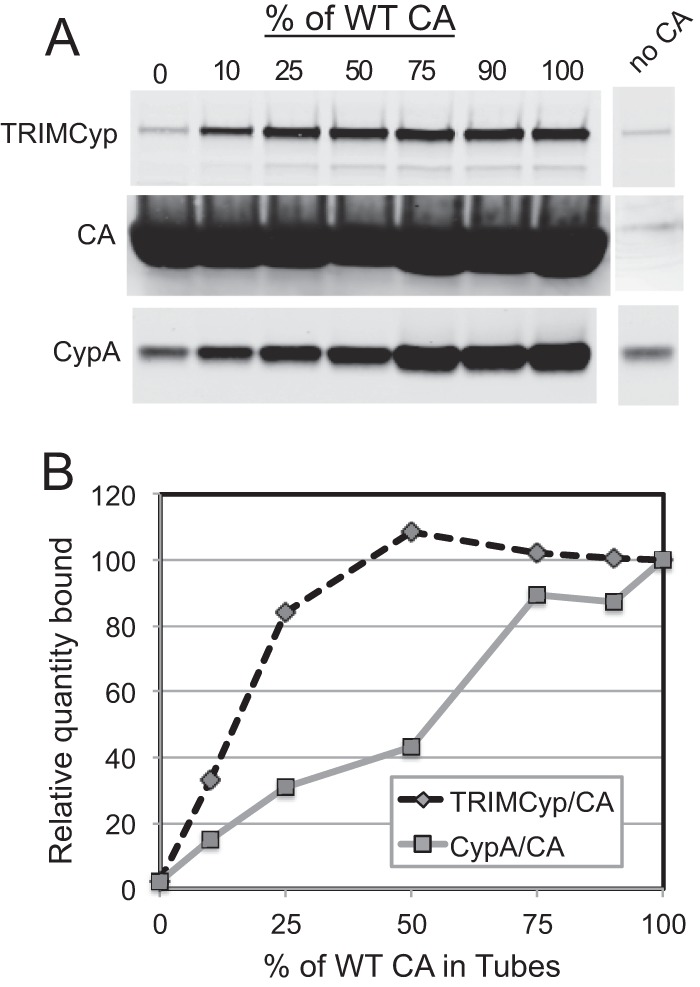
Binding of TRIMCyp to CA assemblies containing mixtures of wild-type and mutant CA in vitro. (A) Lysates of cells expressing myc-TRIMCyp were incubated with complexes assembled at the indicated ratios of wild-type and G89V mutant CA proteins, and the extent of binding was determined by quantitative immunoblotting. (B) Relative quantities of CypA, TRIMCyp, and CA in the pelleted complexes.
Restriction by TRIM5 proteins can be overcome at high virus inocula, and this phenomenon has been exploited as a tool to study the ability of incoming virions to interact with restriction factors (29, 34–36). To probe the interaction of cellular TRIMCyp with the incoming viral capsid, we tested the ability of mixed HIV-1 particles to prevent restriction of a wild-type HIV-GFP reporter virus in trans. Non-GFP-encoding virus particles containing various proportions of wild-type and G89V CA were titrated with a fixed quantity of HIV-GFP reporter virus on OMK cells, and the extent of infection by the reporter virus was quantified by flow cytometry. We observed that the ability of HIV-1 particles to overcome restriction depended strongly on the percentage of wild-type CA in the virions (Fig. 5). Thus, while a small fraction of wild-type CA is sufficient for binding TRIMCyp in vitro and to confer sensitivity to restriction in target cells, efficient abrogation of restriction in trans requires a majority of the CA subunits to be capable of being recognized by the restriction factor.
Fig 5.
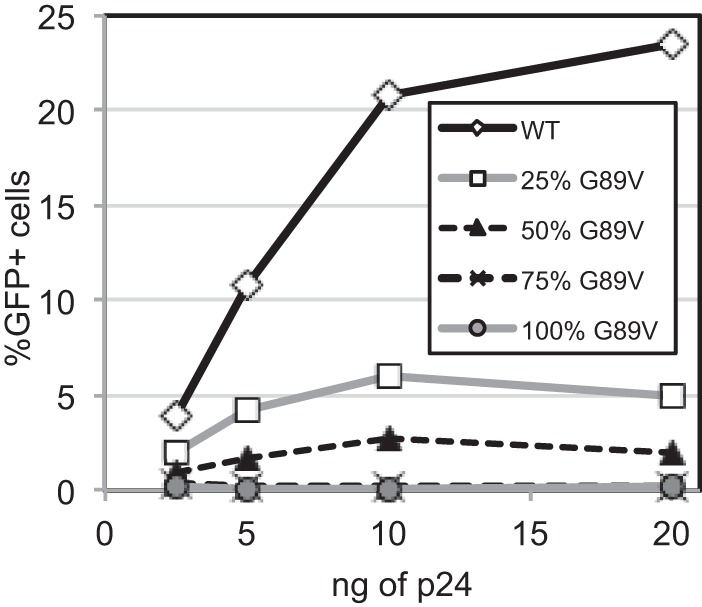
The ability of HIV-1 particles to abrogate TRIMCyp restriction in trans depends strongly on the subunit composition of the capsid. Non-GFP-encoding HIV-1 stocks were titrated onto OMK cultures with a fixed quantity of HIV-GFP reporter virus, and the extent of infection by the reporter virus was subsequently determined. The results shown are representative of three independent experiments.
Restrictive TRIM5 proteins block reverse transcription in target cells, and inhibition of cellular proteasome activity relieves the block to reverse transcription without overcoming restriction. Therefore, we asked whether changing the proportion of wild-type to G89V CA alters the ability of TRIM5α to inhibit reverse transcription in target cells. Cultures of control CrFK cells and CrFK cells expressing TRIMCyp were inoculated with a fixed quantity of the mixed viruses (normalized by the amount of p24), and cells were harvested at 8 h postinfection for DNA purification and analysis. HIV-1 DNA was quantified by real-time PCR. The results demonstrated that the viruses containing mixed capsids were all inhibited for reverse transcription in TRIMCyp-expressing cells (Fig. 6), in agreement with their sensitivity to restriction. To control for potential contaminating plasmid DNA, parallel infections of CrFK cells with the viruses were performed in the presence of the drug efavirenz to prevent reverse transcription. This resulted in values of <1,000 copies, demonstrating that the PCR signals resulted from reverse transcription (data not shown). Based on these data, we concluded that a minimum level of restriction-sensitive CA is sufficient for HIV-1 to be susceptible to inhibition of reverse transcription by TRIMCyp.
Fig 6.
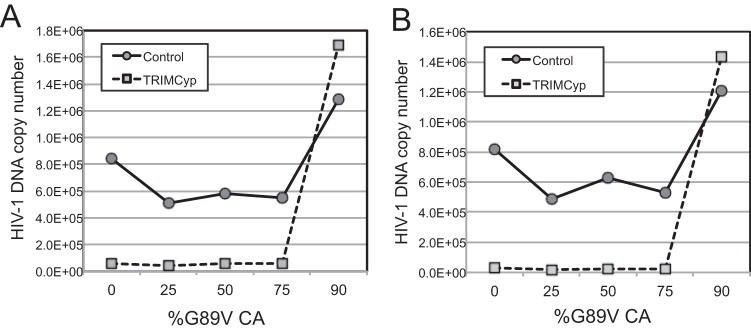
TRIMCyp inhibits reverse transcription of HIV-1 particles containing mixed capsids. CrFK and CrFK-TRIMCyp cells were inoculated with the viruses and cultured for 8 h. Cells were then harvested, and the levels of intracellular HIV-1 DNA were quantified by PCR. (A) Analysis of early reverse transcripts (minus-strand strong stop). (B) Quantitation of late reverse transcription products (second strand transfer).
DISCUSSION
It is well established that TRIM5 restriction factors inhibit infection by targeting the viral capsid lattice and perturbing capsid function, but the structural and mechanistic details of restriction are poorly understood. Specifically, the interaction of TRIM5 with the viral capsid and the resulting consequences of binding are poorly defined at the biochemical level. Recent studies have shown that TRIM5 can form a hexameric supralattice on the assembled CA surface, suggesting that the protein can form a cage-like structure around the viral capsid (37). TRIM5 restriction results in disassembly and/or degradation of the incoming retroviral capsid (1). Moreover, addition of a recombinant rhesus monkey TRIM5α protein containing a replacement of the RING domain from TRIM21 resulted in shortening of capsid assemblies in vitro (6), and a TRIM5α CC-SPRY fragment disassembled tubular CA assemblies into strips of CA hexamers (24). Collectively, these studies indicate that TRIM5 binding can result in structural disruption of the viral capsid lattice.
In the present study, we studied the binding to and functional recognition of the HIV-1 capsid by owl monkey TRIMCyp via an approach utilizing viruses containing mixtures of restriction-sensitive and -resistant CA proteins. HIV-1 particles containing as little as 25% wild-type CA were efficiently inhibited by TRIMCyp. We also observed that TRIMCyp can bind in vitro to assemblies of recombinant CA containing up to 75% nonrestricted G89V CA protein. The latter results suggest that TRIMCyp can initially interact with a few subunits of the assembled capsid and that this interaction is sufficient to promote further recruitment of TRIM5 molecules, which subsequently self-assemble on the capsid surface. We also observed similar results for TRIM5α restriction of MLV particles bearing mixtures of N- and B-tropic CA, consistent with previous reports showing that mixed capsids can be recognized by Fv1 and TRIM5α (31, 32). TRIM5 proteins form intracellular complexes known as cytoplasmic bodies, which can be observed as punctate structures by immunofluorescence microscopy (19, 38). While the role of cytoplasmic bodies in restriction is unclear, the existence of these complexes implies that TRIM5 is capable of self-assembly, a prediction that recently received experimental confirmation: Ganser-Pornillos and coworkers showed that the recombinant TRIM5-21R chimeric protein can form large hexameric lattices in vitro (37). Assembly of TRIM5 complexes may be facilitated by the reported ability of TRIM5 dimers to form higher-order oligomers owing to interactions of the B-box domain (26). In the present study, we observed efficient binding of TRIMCyp in vitro to CA assemblies containing up to 75% G89V CA subunits, further supporting a role of TRIM5 self-assembly in capsid binding and restriction. Thus, our results reinforce the conclusion that functional interaction with the viral capsid is enhanced by cooperative interactions between TRIM5 dimers (26).
Paradoxically, while virions containing only a minor fraction of wild-type CA were restricted by TRIMCyp, the ability of HIV-1 particles to abrogate TRIMCyp restriction in trans was strongly dependent on the proportion of wild-type CA. Thus, while restriction of an incoming capsid occurs upon recognition of only a fraction of CA subunits, saturation of the cellular pool of TRIMCyp appears to depend on the total quantity of restriction-sensitive CA entering the cell. While the basis for this apparent recognition difference is currently unclear, our group previously showed that cellular exposure to high doses of restriction-sensitive particles results in degradation of TRIM5 restriction factors by a proteasome-dependent mechanism (22), suggesting that degradation of TRIM5 may contribute to virus-dependent abrogation of restriction. We hypothesize that the binding of TRIM5 to mixed capsids in target cells occurs more slowly than binding to wild-type capsids, resulting in degradation of the bound TRIM5 and concomitant destruction of the viral capsid prior to maximal binding of the restriction factor. Hence, although cores with mixed capsids are efficiently restricted, they may not exhibit efficient abrogation activity owing to premature destruction triggered upon limited binding of the restriction factor.
Our proteomic analysis confirmed that the virus stocks contained CA proteins in proportion to the input plasmid ratios. Nonetheless, one potential confounding factor of our approach is the possibility that capsid assembly may be a nonrandom process and that mixtures of input plasmids do not accurately reflect the distribution of CA subunits in the assembled viral capsid. While we cannot formally exclude this possibility, we think it unlikely. The G89V substitution, which lies on the exterior face of the capsid, does not affect HIV-1 capsid assembly, nor do the CA substitutions that distinguish N- and B-tropic MLV. Moreover, we observed similar effects of capsid mixing for restriction of HIV-1 and MLV particles by TRIMCyp and human TRIM5α, respectively. The fact that similar patterns of restriction were observed for distinct retroviruses and different restriction factors suggests that the results reflect a fundamental aspect of the recognition of the capsid lattice by TRIM5 proteins.
Our results have implications regarding the pattern of subunits within the capsid that is recognized by TRIM5 restriction factors. Assuming random mixing of CA subunits during capsid assembly, the composition of hexamers in the lattice as a function of the CA ratio is determined by a binomial distribution (39). Accordingly, a capsid containing 25% wild-type CA and 75% mutant CA is predicted to contain hexamers in the proportions shown in Table 2. Thus, 82% of hexamers will contain one or more wild-type subunits, 47% will have two or more, and 17% will contain three or more. Based on these calculations, our observation that viruses containing only 25% restriction-sensitive CA were efficiently restricted suggests that a hexamer with a single recognizable subunit may be engaged by TRIM5. It is also possible that TRIM5 initially binds the capsid at the less frequent hexamers containing larger numbers of recognizable subunits. However, because at this proportion the frequency of hexamers with more than three recognizable subunits is very low, it appears likely that capsid hexamers with at least three recognizable subunits can be engaged efficiently by TRIM5. Nonetheless, because binding of TRIM5 appears to require the assembled capsid lattice (1, 37, 40), it is also possible that a more complex pattern composed of multiple hexamers represents the functional TRIM5 recognition target.
Table 2.
Capsid hexamer distribution for a random mixture composed of 25% wild-type and 75% mutant subunits, determined from the binomial distribution
| Hexamer type | Calculated fraction |
|---|---|
| 0 WT/6 mutant | 0.178 |
| 1 WT/5 mutant | 0.356 |
| 2 WT/4 mutant | 0.279 |
| 3 WT/3 mutant | 0.132 |
| 4 WT/2 mutant | 0.033 |
| 5 WT/1 mutant | 0.0044 |
| 6 WT/0 mutant | 0.00024 |
ACKNOWLEDGMENTS
We thank Greg Towers for providing the CrFK-TRIMCyp and CrFK-huTRIM5α cell lines. Efavirenz was obtained via the NIH AIDS Research and Reference Reagent Program, Division of AIDS, NIAID, NIH. Analysis of the CA levels in mixed virus particles was performed in the Mass Spectrometry Research Center at Vanderbilt University School of Medicine.
This work was supported by NIH grant R01 AI076121.
Footnotes
Published ahead of print 19 June 2013
REFERENCES
- 1. Stremlau M, Perron M, Lee M, Li Y, Song B, Javanbakht H, Diaz-Griffero F, Anderson DJ, Sundquist WI, Sodroski J. 2006. Specific recognition and accelerated uncoating of retroviral capsids by the TRIM5alpha restriction factor. Proc. Natl. Acad. Sci. U. S. A. 103:5514–5519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kim J, Tipper C, Sodroski J. 2011. Role of TRIM5alpha RING domain E3 ubiquitin ligase activity in capsid disassembly, reverse transcription blockade, and restriction of simian immunodeficiency virus. J. Virol. 85:8116–8132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Perron MJ, Stremlau M, Lee M, Javanbakht H, Song B, Sodroski J. 2007. The human TRIM5alpha restriction factor mediates accelerated uncoating of the N-tropic murine leukemia virus capsid. J. Virol. 81:2138–2148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kar AK, Diaz-Griffero F, Li Y, Li X, Sodroski J. 2008. Biochemical and biophysical characterization of a chimeric TRIM21-TRIM5alpha protein. J. Virol. 82:11669–11681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Black LR, Aiken C. 2010. TRIM5alpha disrupts the structure of assembled HIV-1 capsid complexes in vitro. J. Virol. 84:6564–6569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Langelier CR, Sandrin V, Eckert DM, Christensen DE, Chandrasekaran V, Alam SL, Aiken C, Olsen JC, Kar AK, Sodroski JG, Sundquist WI. 2008. Biochemical characterization of a recombinant TRIM5alpha protein that restricts human immunodeficiency virus type 1 replication. J. Virol. 82:11682–11694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Kutluay SB, Perez-Caballero D, Bieniasz PD. 2013. Fates of retroviral core components during unrestricted and TRIM5-restricted infection. PLoS Pathog. 9:e1003214. 10.1371/journal.ppat.1003214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hatziioannou T, Cowan S, Von Schwedler UK, Sundquist WI, Bieniasz PD. 2004. Species-specific tropism determinants in the human immunodeficiency virus type 1 capsid. J. Virol. 78:6005–6012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Owens CM, Song B, Perron MJ, Yang PC, Stremlau M, Sodroski J. 2004. Binding and susceptibility to postentry restriction factors in monkey cells are specified by distinct regions of the human immunodeficiency virus type 1 capsid. J. Virol. 78:5423–5437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, Riganelli D, Zanaria E, Messali S, Cainarca S, Guffanti A, Minucci S, Pelicci PG, Ballabio A. 2001. The tripartite motif family identifies cell compartments. EMBO J. 20:2140–2151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Towers GJ, Hatziioannou T, Cowan S, Goff SP, Luban J, Bieniasz PD. 2003. Cyclophilin A modulates the sensitivity of HIV-1 to host restriction factors. Nat. Med. 9:1138–1143 [DOI] [PubMed] [Google Scholar]
- 12. Sayah DM, Sokolskaja E, Berthoux L, Luban J. 2004. Cyclophilin A retrotransposition into TRIM5 explains owl monkey resistance to HIV-1. Nature 430:569–573 [DOI] [PubMed] [Google Scholar]
- 13. Hatziioannou T, Perez-Caballero D, Yang A, Cowan S, Bieniasz PD. 2004. Retrovirus resistance factors Ref1 and Lv1 are species-specific variants of TRIM5alpha. Proc. Natl. Acad. Sci. U. S. A. 101:10774–10779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Keckesova Z, Ylinen LM, Towers GJ. 2004. The human and African green monkey TRIM5alpha genes encode Ref1 and Lv1 retroviral restriction factor activities. Proc. Natl. Acad. Sci. U. S. A. 101:10780–10785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Perron MJ, Stremlau M, Song B, Ulm W, Mulligan RC, Sodroski J. 2004. TRIM5α mediates the postentry block to N-tropic murine leukemia viruses in human cells. Proc. Natl. Acad. Sci. U. S. A. 101:11827–11832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yap MW, Nisole S, Lynch C, Stoye JP. 2004. Trim5alpha protein restricts both HIV-1 and murine leukemia virus. Proc. Natl. Acad. Sci. U. S. A. 101:10786–10791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Cowan S, Hatziioannou T, Cunningham T, Muesing MA, Gottlinger HG, Bieniasz PD. 2002. Cellular inhibitors with Fv1-like activity restrict human and simian immunodeficiency virus tropism. Proc. Natl. Acad. Sci. U. S. A. 99:11914–11919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Stremlau M, Owens CM, Perron MJ, Kiessling M, Autissier P, Sodroski J. 2004. The cytoplasmic body component TRIM5alpha restricts HIV-1 infection in Old World monkeys. Nature 427:848–853 [DOI] [PubMed] [Google Scholar]
- 19. Perez-Caballero D, Hatziioannou T, Zhang F, Cowan S, Bieniasz PD. 2005. Restriction of human immunodeficiency virus type 1 by TRIM-CypA occurs with rapid kinetics and independently of cytoplasmic bodies, ubiquitin, and proteasome activity. J. Virol. 79:15567–15572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu X, Anderson JL, Campbell EM, Joseph AM, Hope TJ. 2006. Proteasome inhibitors uncouple rhesus TRIM5alpha restriction of HIV-1 reverse transcription and infection. Proc. Natl. Acad. Sci. U. S. A. 103:7465–7470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Roa A, Hayashi F, Yang Y, Lienlaf M, Zhou J, Shi J, Watanabe S, Kigawa T, Yokoyama S, Aiken C, Diaz-Griffero F. 2012. RING domain mutations uncouple TRIM5alpha restriction of HIV-1 from inhibition of reverse transcription and acceleration of uncoating. J. Virol. 86:1717–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Rold CJ, Aiken C. 2008. Proteasomal degradation of TRIM5alpha during retrovirus restriction. PLoS Pathog. 4:e1000074. 10.1371/journal.ppat.1000074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Campbell EM, Perez O, Anderson JL, Hope TJ. 2008. Visualization of a proteasome-independent intermediate during restriction of HIV-1 by rhesus TRIM5alpha. J. Cell Biol. 180:549–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhao G, Ke D, Vu T, Ahn J, Shah VB, Yang R, Aiken C, Charlton LM, Gronenborn AM, Zhang P. 2011. Rhesus TRIM5alpha disrupts the HIV-1 capsid at the inter-hexamer interfaces. PLoS Pathog. 7:e1002009. 10.1371/journal.ppat.1002009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Javanbakht H, Yuan W, Yeung DF, Song B, Diaz-Griffero F, Li Y, Li X, Stremlau M, Sodroski J. 2006. Characterization of TRIM5alpha trimerization and its contribution to human immunodeficiency virus capsid binding. Virology 353:234–246 [DOI] [PubMed] [Google Scholar]
- 26. Diaz-Griffero F, Qin XR, Hayashi F, Kigawa T, Finzi A, Sarnak Z, Lienlaf M, Yokoyama S, Sodroski J. 2009. A B-box 2 surface patch important for TRIM5alpha self-association, capsid binding avidity, and retrovirus restriction. J. Virol. 83:10737–10751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yee JK, Friedmann T, Burns JC. 1994. Generation of high-titer pseudotyped retroviral with very broad host range. Methods Cell Biol. 43:99–112 [DOI] [PubMed] [Google Scholar]
- 28. Bock M, Bishop KN, Towers G, Stoye JP. 2000. Use of a transient assay for studying the genetic determinants of Fv1 restriction. J. Virol. 74:7422–7430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Forshey BM, Shi J, Aiken C. 2005. Structural requirements for recognition of the human immunodeficiency virus type 1 core during host restriction in owl monkey cells. J. Virol. 79:869–875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pornillos O, Ganser-Pornillos BK, Banumathi S, Hua Y, Yeager M. 2010. Disulfide bond stabilization of the hexameric capsomer of human immunodeficiency virus. J. Mol. Biol. 401:985–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Rein A, Kashmiri SV, Bassin RH, Gerwin BL, Duran-Troise G. 1976. Phenotypic mixing between N- and B-tropic murine leukemia viruses: infectious particles with dual sensitivity to Fv-1 restriction. Cell 7:373–379 [DOI] [PubMed] [Google Scholar]
- 32. Towers G, Bock M, Martin S, Takeuchi Y, Stoye JP, Danos O. 2000. A conserved mechanism of retrovirus restriction in mammals. Proc. Natl. Acad. Sci. U. S. A. 97:12295–12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Pertel T, Hausmann S, Morger D, Zuger S, Guerra J, Lascano J, Reinhard C, Santoni FA, Uchil PD, Chatel L, Bisiaux A, Albert ML, Strambio-De-Castillia C, Mothes W, Pizzato M, Grutter MG, Luban J. 2011. TRIM5 is an innate immune sensor for the retrovirus capsid lattice. Nature 472:361–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Shi J, Aiken C. 2006. Saturation of TRIM5 alpha-mediated restriction of HIV-1 infection depends on the stability of the incoming viral capsid. Virology 350:493–500 [DOI] [PubMed] [Google Scholar]
- 35. Jiang J, Ablan SD, Derebail S, Hercik K, Soheilian F, Thomas JA, Tang S, Hewlett I, Nagashima K, Gorelick RJ, Freed EO, Levin JG. 2011. The interdomain linker region of HIV-1 capsid protein is a critical determinant of proper core assembly and stability. Virology 421:253–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dodding MP, Bock M, Yap MW, Stoye JP. 2005. Capsid processing requirements for abrogation of Fv1 and Ref1 restriction. J. Virol. 79:10571–10577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ganser-Pornillos BK, Chandrasekaran V, Pornillos O, Sodroski JG, Sundquist WI, Yeager M. 2011. Hexagonal assembly of a restricting TRIM5alpha protein. Proc. Natl. Acad. Sci. U. S. A. 108:534–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Song B, Diaz-Griffero F, Park DH, Rogers T, Stremlau M, Sodroski J. 2005. TRIM5alpha association with cytoplasmic bodies is not required for antiretroviral activity. Virology 343:201–211 [DOI] [PubMed] [Google Scholar]
- 39. Werbeck ND, Schlee S, Reinstein J. 2008. Coupling and dynamics of subunits in the hexameric AAA+ chaperone ClpB. J. Mol. Biol. 378:178–190 [DOI] [PubMed] [Google Scholar]
- 40. Yang H, Ji X, Zhao G, Ning J, Zhao Q, Aiken C, Gronenborn AM, Zhang P, Xiong Y. 2012. Structural insight into HIV-1 capsid recognition by rhesus TRIM5alpha. Proc. Natl. Acad. Sci. U. S. A. 109:18372–18377 [DOI] [PMC free article] [PubMed] [Google Scholar]