

Abstract
Leptomycin B (LMB) is a highly specific inhibitor of CRM1, a cellular karyopherin-β that transports nuclear export signal-containing proteins from the nucleus to the cytoplasm. Previous work has shown that LMB blocks herpes simplex virus 1 (HSV-1) replication in Vero cells and that certain mutations in viral immediate early protein ICP27 can confer LMB resistance. However, little is known of the molecular mechanisms involved. Here we report that HSV-2, a close relative of HSV-1, is naturally resistant to LMB. To see whether the ICP27 gene determines this phenotypic difference, we generated an HSV-1 mutant that expresses the HSV-2 ICP27 instead of the HSV-1 protein. This recombinant was fully sensitive to LMB, indicating that one or more other viral genes must be important in determining HSV-2's LMB-resistant phenotype. In additional work, we report several findings that shed light on how HSV-1 ICP27 mutations can confer LMB resistance. First, we show that LMB treatment of HSV-1-infected cells leads to suppression of late viral protein synthesis and a block to progeny virion release. Second, we identify a novel type of ICP27 mutation that can confer LMB resistance, that being the addition of a 100-residue amino-terminal affinity purification tag. Third, by studying infections where both LMB-sensitive and LMB-resistant forms of ICP27 are present, we show that HSV-1's sensitivity to LMB is dominant to its resistance. Together, our results suggest a model in which the N-terminal portion of ICP27 mediates a nonessential activity that interferes with HSV-1 replication when CRM1 is inactive. We suggest that LMB resistance mutations weaken or abrogate this activity.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) is a widely distributed human alphaherpesvirus that is capable of causing serious and in some cases life-threatening infections. The HSV-1 infectious cycle involves both nuclear and cytoplasmic processes, and thus, productive viral replication requires that viral and cellular protein and RNA components travel efficiently between the nucleus and cytoplasm. Such movement is largely mediated by dedicated cellular transporters, one important class of which consists of the karyopherin-β family of proteins (1). Among this family, CRM1 is perhaps the most well studied member. It is an essential protein that exports ribosomal subunits and various protein cargos from the nucleus to the cytoplasm (2–4). Transport by CRM1 is dependent on the presence of a specific nuclear export signal (NES) on the cargo protein. This signal consists of an ∼15-residue consensus sequence that is rich in leucine and other hydrophobic residues. The NES binds to a hydrophobic cleft on the surface of nucleus-localized CRM1 (5, 6). Following binding, CRM1 escorts the cargo through the nuclear pore to the cytoplasm, whereupon it is released. One of the first leucine-rich NESs described was that of the human immunodeficiency virus type 1 (HIV-1) Rev protein (7). Rev uses CRM1 to facilitate the nuclear export of unspliced and partially spliced HIV-1 transcripts. Since then, numerous other cellular and viral proteins have been shown to possess similar, most often leucine-rich NESs that allow them access to the CRM1 export pathway.
The study of CRM1 has been greatly facilitated by the use of a highly specific inhibitor, leptomycin B (LMB) (8). LMB is a polyketide metabolite produced by a Streptomyces sp. and was originally identified as an antifungal agent. It binds to the hydrophobic cleft of CRM1 and becomes covalently attached to cysteine residue 528 (9), thus preventing the binding of bona fide NESs (5, 6). Several years ago, Murata et al. showed that the growth of HSV-1 strain KOS is inhibited by LMB (10), indicating that HSV-1 productive replication requires CRM1. Although the specific effects of LMB on HSV-1 replication have not been studied extensively, work from Soliman and Silverstein showed that LMB inhibits the steady-state accumulation of several viral late (L) proteins and their mRNAs (11). LMB also enhances the cytoplasmic localization of the viral immediate early (IE) proteins ICP0 and ICP4 (12). Thus, CRM1 appears to play a role in HSV-1 gene expression and/or viral protein transport.
In their original study, Murata et al. isolated an LMB-resistant mutant of HSV-1 (10). This virus was obtained after repeated passages of HSV-1 strain KOS in Vero cells in the presence of LMB. Marker rescue and sequencing experiments identified the resistance mutation as a methionine-to-threonine change at codon 50 (M50T) of the viral IE ICP27 protein. We subsequently verified this finding in strain KOS1.1 (a subclone of KOS) and additionally showed that another ICP27 mutant, dAc, is also LMB resistant (12). dAc has an in-frame deletion of ICP27 codons 21 to 63. These studies implicate a specific N-terminal region of ICP27, centered on residues 21 to 63, in the sensitivity of HSV-1 to LMB.
ICP27 is an essential, multifunctional IE protein that stimulates the expression of a subset of viral delayed-early (DE) and L genes. The L genes, which predominantly encode structural proteins, are subdivided into two types on the basis of the degree to which their expression is dependent on viral DNA replication: leaky L genes are expressed at low levels in the absence of viral DNA synthesis, whereas true L genes stringently require viral DNA synthesis. ICP27 likely uses multiple mechanisms to stimulate DE/L gene expression, but at least part of its stimulatory effects involves its ability to mediate the transport of unspliced viral mRNAs from the nucleus to the cytoplasm. Consistent with this role in mRNA export, ICP27 is an RNA-binding protein (13, 14) that continually shuttles between the nucleus and cytoplasm (15–18). Interestingly, ICP27 possesses a leucine-rich NES-like sequence near its N terminus (residues 6 to 19) that is required for both its shuttling capability and viral mRNA export function (17, 19). However, despite the similarity of this sequence to CRM1-dependent NESs, multiple studies show that the nuclear export of ICP27 is insensitive to LMB (12, 20, 21). This finding is consistent with a variety of data that indicate that ICP27's mRNA export function depends on its physical interaction with NXF1 (21–24), a non-karyopherin-β transporter that is responsible for bulk mRNA export in mammalian cells (25). ICP27's NES-like sequence is required for NXF1 interaction (22).
Given that there is little evidence that ICP27 interacts with CRM1, it is unclear how mutations in ICP27 confer LMB resistance to HSV-1. Here, we have further investigated this question and present several findings that lead us to a model to explain the linkage between ICP27 and CRM1. In the course of our studies, we also made the surprising discovery that the closely related human virus HSV-2 is naturally resistant to LMB, indicating that its replication is CRM1 independent. It is conceivable that this type-specific difference is relevant to one or more of the clinical and biological differences that distinguish HSV-1 and HSV-2.
MATERIALS AND METHODS
Cells, viruses, and infections.
Most infections were carried out in Vero cells, obtained from the American Type Culture Collection (ATCC), or in V27 cells, a derivative of Vero cells that expresses ICP27 upon HSV-1 infection (26). Life-extended human foreskin fibroblasts (HFFs) (27), obtained from Wade Bresnahan, were also used. The cells were propagated in Dulbecco modified Eagle medium containing 50 units/ml penicillin, 50 μg/ml streptomycin, and 5% (Vero and V27 cells) or 10% (HFFs) heat-inactivated fetal calf serum. The medium for V27 cells also contained 300 μg of G418 per ml. Strain KOS1.1 (28) was the primary wild-type (WT) HSV-1 strain used in this study. Additional WT HSV-1 strains F, 17syn+, and Patton were obtained from Jim Lokensgard, Alistair McGregor, and Ian Mohr, respectively. HSV-2 strain G was obtained from the ATCC, and HSV-2 strains HG52 and MS were obtained from Jin-Young Han. The LMB-resistant mutant M50T has been described previously (12). The derivation of HSV-1 mutants NTAP-27 and K2F1 is described below. LMB was obtained from LC Labs (Woburn, MA) and when used was added at 1 h postinfection (hpi), following viral adsorption. We noted that the relative potency of LMB varied slightly between different stock preparations. However, within a given experiment, all samples received LMB from the same stock. In some experiments, 400 μg/ml phosphonoacetic acid (PAA; Sigma-Aldrich) was used to inhibit viral DNA replication; it was added at 1 hpi. For all virus yield assays except that shown in Fig. 4A, cells were infected at a multiplicity of infection (MOI) of 0.01 and incubated for 2 days. LMB was used at a concentration of 30 ng/ml and was supplemented by another 30 ng/ml at 24 hpi. Viral infections were terminated by addition of an equal volume of sterile nonfat milk, followed by freezing the cultures at −70°C. In the viral yield experiment whose results are shown in Fig. 4A, cells were infected at an MOI of 10 and LMB was used at a concentration of 30 ng/ml. At 18 hpi, infected cells and supernatant were collected separately and frozen in a 1:1 mixture of medium and sterile nonfat milk. In all viral yield experiments, progeny virus was released by three cycles of freeze-thawing and viral yields were determined by plaque assay of the lysates on Vero cells.
Fig 4.
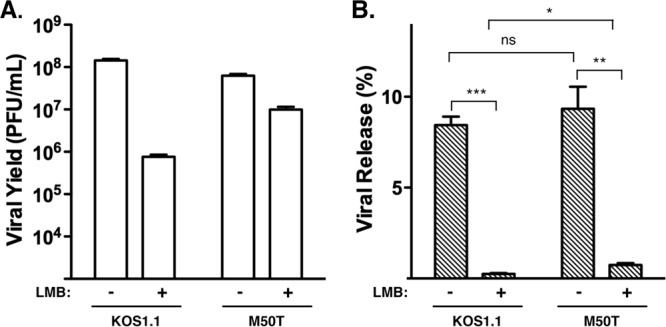
Effect of LMB on the efficiency of viral release. Vero cells were infected in triplicate at an MOI of 10 in the absence or presence of 30 ng/ml LMB. At 18 hpi, cell-associated and medium fractions were harvested separately and titers were determined on Vero cells to determine the amount of infectious progeny in each. (A) Total titers. Total titers were determined by summing the titers from the cell-associated and supernatant fractions. Means are shown, with error bars denoting SEMs. (B) Efficiency of viral release. To determine the efficiency of viral release, supernatant titers were divided by the total titers. Mean values (in percent) are shown; error bars denote SEMs. Statistical analysis was performed using the Student t test. P values: *, P < 0.05; **, P < 0.01; ***, P ≤ 0.001; ns, not significant (P > 0.05).
Derivation of NTAP-27 and K2F1.
NTAP-27 is a strain of HSV-1 that expresses an ICP27 molecule having an N-terminal tandem affinity purification (TAP) tag. It was engineered in the following way. First, we generated a TAP-encoding DNA fragment that could be cloned into the ICP27 gene. To do this, PCR was carried out on plasmid pNTAP-A (Stratagene, Inc.) using AgeI restriction site-containing primers GGAGCCACCGGTATGAAGCGACGATGGAAA and CCCGCTACCGGTGCCCAGCTTGCAGCCGCC (underlined sequences denote AgeI sites). The resulting product was digested with AgeI and ligated to pBS27 (29) that had been linearized with AgeI and treated with calf intestinal alkaline phosphatase (AgeI cuts in the 5′ untranslated region [UTR] of the ICP27 gene, 65 nucleotides upstream of the start codon). A plasmid, pNTAP27, that had the TAP insert in the sense orientation with respect to the ICP27 gene was isolated. Translation of this gene is predicted to give rise to an altered ICP27 that has a 100-residue N-terminal extension. The extension consists of a 78-residue TAP tag, comprised of calmodulin- and streptavidin-binding domains, followed by a 22-residue linker peptide derived from the 5′ UTR (TGAPPPEAISDTPAPTAADSPV). A plasmid-based complementation assay (30) showed that pNTAP27 efficiently complements the growth of an ICP27 deletion mutant in Vero cells (data not shown), indicating that the modified gene could be introduced into the HSV-1 genome by marker rescue of an ICP27-deficient strain. To facilitate marker rescue, the modified gene was engineered into a new plasmid so as to extend the flanking HSV-1 sequences. To do this, the TAP-ICP27 gene was isolated from pNTAP27 as a 2.7-kb BamHI-SstI fragment and cloned in pPs27pd1 (26) in place of the WT BamHI-SstI fragment. This plasmid was designated pPsNTAP27. Lastly, marker rescue (26) was used to engineer NTAP-27. To do this, pPsNTAP27 was digested with PstI and then cotransfected with d27lacZ DNA into V27 cells. The culture was incubated for several days until an extensive cytopathic effect developed, at which time a small stock was prepared. A plaque emanating from this stock was isolated on Vero cells, further purified, and designated NTAP-27. Sequencing confirmed that NTAP-27 had the expected sequence at the 5′ end of the ICP27 gene (the TAP tag through codon 69 of ICP27). The same marker transfer procedure was carried out a second time to derive an independent isolate of NTAP-27, which was designated NTAP-27b.
To engineer an HSV-1 strain that encodes HSV-2 ICP27 in place of the HSV-1 gene, we first cloned the ICP27 gene from HSV-2 strain HG52. To do this, the gene was PCR amplified from HSV-2 strain HG52 DNA using primers TCGTGCACAGAGCCGATCCGG and TGGTTGACCAGGTCCGCGAAG. The resulting 2,467-bp product included the regions 439 bp upstream and 492 bp downstream of the ICP27-coding region, encompassing the native promoter and polyadenylation site. This PCR fragment was cloned in pCR2.1-TOPO (Invitrogen), resulting in plasmid pT2-27. Sequencing of the entire ICP27-coding region revealed a single change relative to the published HG52 sequence (31). This was an AAC at codon 161 (encoding asparagine) instead of the expected AAA (encoding lysine). However, this was not a PCR-generated artifact, as sequencing of a PCR fragment derived from HG52 DNA revealed that this same alteration was present in our HG52 stock. To introduce the HSV-2 ICP27 gene into the HSV-1 genome, we first attached flanking HSV-1 DNA sequences to the HSV-2 ICP27 gene. To do this, primers GATCACCGGTCTCTTCCGACACGCGCCCCCTCGGAG and GATCGATCAATATTTGCCGTGCACATATAAGGGGGCGATA were used to PCR amplify the HSV-2 ICP27 ORF from a pT2-27 template. These primers have 5′ AgeI and SspI restriction sites (underlined), respectively, to facilitate ligation to HSV-1 sequences. The resulting product was digested with AgeI and SspI and then cloned into a pPs27pd1 (32) vector fragment that had most of the ICP27 open reading frame (ORF) removed by complete AgeI digestion and partial SspI digestion. This resulted in isolation of plasmid pPs27-T2, which had a hybrid ICP27 ORF composed of HSV-2 sequences from codons 1 to 504 and HSV-1 sequences from codons 505 to 512. However, since the last 8 amino acids of HSV-1 and HSV-2 ICP27 are identical, the encoded protein corresponds exactly to HSV-2 ICP27. The sequence of the HSV-2 insert in pPs27-T2 was confirmed by DNA sequencing. To insert the modified ICP27 gene into an HSV-1 recombinant, pPs27-T2 was digested with NcoI and cotransfected with infectious d27lacZ DNA into V27 cells. Plaques were purified on Vero cells and grown into stocks. Two genetically independent isolates, K2F1 and K2F1b, were isolated from different transfections. Their genomic structures in the ICP27 gene region were confirmed by Southern blotting (see Fig. 5B) using pBS27 as a probe.
Fig 5.

NTAP-27 is LMB resistant. (A) Generation of NTAP-27. To engineer the NTAP-27 ICP27 gene, an ∼300-bp DNA sequence (black bar) encoding tandem calmodulin- and streptavidin-binding domains was cloned at an AgeI site (Ag) in the 5′ UTR of the ICP27 gene. Translation of the modified gene leads to expression of an ICP27 molecule having a 100-residue N-terminal extension composed of a 70-residue TAP tag (crosshatched bar) followed by 30 residues derived from the 5′ UTR (narrow bar). The mutated gene was engineered into a recombinant virus, NTAP-27. (B) Immunoblot analysis. Vero cells were infected as shown, and proteins harvested at 6 hpi were analyzed by immunoblotting using ICP27 monoclonal antibody H1113 or H1119. (C) Growth of NTAP-27 in Vero cells. Vero cells were infected at an MOI of 0.01, and cultures were harvested at various time points. Viral yields were determined by plaque assay of the infected cell lysates on Vero cells. Each point represents the mean of triplicate infections; error bars denote the SEMs. (D and E) Viral plaque assays. (D) Titers of viral stocks were determined in the absence of LMB (leftmost bar in each set of bars) or in the presence of 3, 5, and 10 ng/ml LMB (triangles). The limit of detection was 1 × 103 PFU/ml; asterisks indicate that the titer was below this value. (E) Viral growth analysis. Vero cells were infected at an MOI of 0.01 PFU per cell in the absence or presence of 30 ng/ml LMB, and cultures were incubated for 2 days. Viral yields were determined by plaque assay of the infected cell lysates on Vero cells.
Protein analysis.
Immunoblotting and analysis of viral protein synthesis by metabolic pulse-labeling with [35S]methionine were performed as described previously (30, 33). Several antibodies were used for immunoblotting. To detect ICP27, mouse monoclonal antibodies H1113 (Virusys) and H1119 (Rumbaugh-Goodwin Institute) were used. Mouse monoclonal antibodies were also used to detect Aly/REF (11G5; EMD-Millipore), beta-actin (AC-15; Abcam), ICP8 (H1115; Rumbaugh-Goodwin Institute), ICP5 (3B6; Abcam), glycoprotein B (gB; HA-056; Virusys), and glycoprotein C (gC; H1104; Rumbaugh-Goodwin Institute). Rabbit polyclonal antisera were used to detect viral proteins VP22 (34), VP16 (35), and US11 (36). These reagents were kind gifts from G. Elliott, Steve Weinheimer, and J. J. Diaz, respectively.
RESULTS
HSV-1 and HSV-2 differ markedly in their sensitivity to LMB.
In their original study of HSV-1's sensitivity to LMB, Murata et al. utilized strain KOS (10). In our follow-up work (12), we used KOS1.1, a derivative of KOS (28). To investigate whether LMB sensitivity is a general property of HSV-1, we examined strains F, 17syn+, and Patton. In a viral plaque assay (Fig. 1A), strains F and 17syn+ were comparable to KOS1.1 in their degree of LMB sensitivity, while strain Patton was somewhat less sensitive. However, we noted that Patton plaques were dimorphic in the presence of LMB, with some plaques being small and others being quite large (not shown). This suggests that our Patton stock may harbor LMB-resistant variants, although we did not investigate this further. We also examined M50T, an LMB-resistant derivative of KOS1.1 that has a methionine-to-threonine change at residue 50 of ICP27 (12). As expected, the M50T mutant was resistant to LMB in the plaque assay. As another test of LMB sensitivity, we used a viral yield analysis (12) in which Vero cells were infected at an MOI of 0.01 PFU/cell and incubated for 2 days in the absence or presence of 30 ng/ml LMB (Fig. 1B). KOS1.1, F, and 17syn+ all showed marked sensitivity to LMB in this assay, while Patton was also sensitive but less so. From these data, we conclude that sensitivity to LMB is a general property of HSV-1.
Fig 1.

HSV-1 and HSV-2 are differentially sensitive to LMB. (A and B) Sensitivity of HSV-1 strains to LMB. (A) Plaque assay. The titers of WT strains of HSV-1 and the M50T mutant were determined on Vero cells in the absence of LMB (leftmost bar in each set of bars) or in the presence of 10, 20 and 30 ng/ml LMB (triangles). (B) Yield assay. Vero cells were infected at an MOI of 0.01 in the absence or presence of 30 ng/ml LMB. Progeny virus was harvested after 2 days, and viral yields were determined by plaque assay on Vero cells. The bars represent the means of triplicate infections, and error bars denote SEMs. (C to E) Sensitivity of HSV-2 strains to LMB. (C) Plaque assay. Details are as described in the legend to panel A, except that LMB was used at 2 and 4 ng/ml. (D) LMB increases the size of HSV-2 plaques. Aliquots of diluted HSV-1 KOS1.1 or HSV-2 HG52 stocks were plated on Vero cells, and viral plaque assays were carried out in the absence or presence of 3 ng/ml LMB. Monolayers were fixed and Giemsa stained 2 days later; digital images of representative areas of the stained monolayers are shown. (E) Yield assay. Analysis of viral growth was carried out as described in the legend to panel B. (F) LMB sensitivity of HSV-1 in life-extended HFFs. Analysis of viral growth in Vero and life-extended HFFs was carried out as described in the legend to panel B.
We next asked whether HSV-2, a closely related alphaherpesvirus, is similarly LMB sensitive. Surprisingly, HSV-2 strains HG52 and G were completely resistant to LMB in the plaque assay (Fig. 1C). A third HSV-2 strain, MS, was similarly resistant (not shown). Interestingly, we noted that HSV-2 plaques were reproducibly slightly larger in the presence of LMB (Fig. 1D and data not shown). The HSV-2 strains were also tested in the low-MOI growth assay (Fig. 1E). Strain G was completely resistant, whereas strains HG52 and MS were inhibited by less than 10-fold. This was in striking contrast to the findings for HSV-1 KOS1.1, which was inhibited by more than 1,000-fold. We conclude that HSV-2 is naturally resistant to LMB. Thus, unlike HSV-1, HSV-2 does not stringently require CRM1 to replicate in Vero cells.
To see if HSV-1's sensitivity to LMB was observed in another cell type, we used the low-MOI growth assay to examine the LMB sensitivity of KOS1.1 and the M50T mutant in life-extended HFFs (27) (Fig. 1F). As a control, Vero cell infections were carried out in parallel. In the Vero cells, the results were as before, with KOS1.1 exhibiting high LMB sensitivity (a >10,000-fold reduction in yield) and the M50T mutant showing partial resistance (an ∼70-fold reduction). In life-extended HFFs, however, the results were substantially different. KOS1.1 showed only a modest ∼10-fold inhibition by LMB, indicating that this strain is less sensitive to LMB in these cells. Additionally, the sensitivity of the M50T mutant was comparable to that of KOS1.1, suggesting that the N-terminal region of ICP27 does not modulate LMB sensitivity in life-extended HFFs. This experiment demonstrates that the sensitivity of HSV-1 to LMB depends on the type of cell infected.
HSV-2's resistance to LMB involves a viral gene other than the ICP27 gene.
Since HSV-1 LMB resistance mutations map to the ICP27 gene, we hypothesized that HSV-2 is resistant because its ICP27 gene encodes an LMB-resistant form of the protein. Supporting this conjecture, the sequence divergence between HSV-2 and HSV-1 ICP27 is highest (35%) in the N-terminal half of the protein, where HSV-1 LMB resistance mutations map (it should be noted, however, that HSV-2 ICP27 is similar to the type 1 protein in having a methionine at the position equivalent to residue 50, which is residue 51 in the HSV-2 protein). To test our hypothesis, we engineered K2F1, an HSV-1 recombinant that encodes HSV-2 ICP27 in place of the HSV-1 protein (Fig. 2A). To ensure that its phenotype can be ascribed to its HSV-2 ICP27 gene and not to any secondary mutations, we also engineered a second isolate, designated K2F1b. Southern blotting confirmed that both isolates have the expected genomic structure at their ICP27 gene loci (Fig. 2B). Additionally, viral growth analysis showed that both isolates replicate similarly to KOS1.1 in Vero cells, demonstrating that HSV-2 ICP27 can functionally substitute for HSV-1 ICP27 in the context of a Vero cell infection (D. Park and S. Rice, unpublished data).
Fig 2.

An HSV-1 recombinant encoding HSV-2 ICP27 is LMB sensitive. (A) Construction of K2F1 and K2F1b. The top bar shows a schematic of the HSV-1 genome (RL, repeat long; UL, unique long; RS, repeat short; US, unique short). Dotted lines indicate a magnified view of the 5.9-kb PstI fragment having the ICP27 ORF (white bar), with the ICP27 gene promoter (arrow) and polyadenylation site (pA) shown. Below is shown the modified gene, which has a replacement of an AgeI to SspI fragment with the corresponding DNA from HSV-2 strain HG52 (crosshatched bar). Relevant restriction sites are abbreviated as follows: Ag, AgeI; P, PstI; S, SspI; and X, XhoI. The HSV-1 probe used for Southern blotting is indicated. (B) Southern blot analysis. Two isolates each of K2F1 and K2F1b were plaque purified and analyzed by Southern blotting using the probe shown in panel A. Numbers to the left of the gel denote the positions, in kilobase pairs, of DNA size standards. (C) K2F1 and K2F1b are sensitive to LMB. The titers of viral strains were determined in the absence of LMB (leftmost bar in each set of bars) or in the presence of 3, 5, and 10 ng/ml LMB (triangles). The limit of detection was 1 × 103 PFU/ml; asterisks indicate that the titer was below this value. (D) Viral growth analysis. Vero cells were infected at an MOI of 0.01 PFU per cell in the absence or presence of 30 ng/ml LMB, and cultures were incubated for 2 days, at which time viral yields were determined by plaque assay on Vero cells.
Based on our hypothesis, we predicted that K2F1 and K2F1b would be LMB resistant. However, when tested in the viral plaque assay, both were indistinguishable from KOS1.1 in their high degree of LMB sensitivity (Fig. 2C). They were also similarly LMB sensitive in the low-MOI growth assay (Fig. 2D). Thus, we conclude that the HSV-2 ICP27 gene does not encode an LMB-resistant form of ICP27, at least not when expressed from the context of the HSV-1 genome. This result also shows that the type-specific difference in LMB sensitivity between HSV-1 and HSV-2 must depend on at least one other viral gene besides the ICP27 gene.
LMB suppresses late HSV-1 protein synthesis and inhibits virus release.
The block that LMB imposes on HSV-1 replication is not well characterized. However, Soliman and Silverstein showed that LMB inhibits the accumulation of certain HSV-1 L mRNAs and their encoded proteins (11). To confirm and extend these findings, we used a metabolic labeling protocol to examine how LMB affects HSV-1 protein synthesis at various times postinfection. Thus, Vero cells were mock infected or infected with KOS1.1 or the M50T mutant in the absence or presence of 30 ng/ml LMB, and the cells were pulse-labeled with [35S]methionine at 4, 7, and 10 hpi. Protein synthesis was analyzed by SDS-PAGE and autoradiography (Fig. 3A). At 4 hpi, LMB had little apparent effect on viral protein synthesis, but at 7 hpi and especially 10 hpi, the synthesis of several infected cell-specific polypeptides (indicated by asterisks in Fig. 3A) was significantly reduced in the LMB-treated cells. These defects were partially suppressed in the M50T mutant infection. These results show that LMB inhibits HSV-1 late protein synthesis. Furthermore, this experiment demonstrates that the M50T mutant ICP27 allows more efficient viral protein synthesis when CRM1 is inhibited. In addition to inhibiting the synthesis of certain infected cell-specific proteins, we also noted that LMB increased the synthesis of others (indicated by upward-pointing arrows in Fig. 3A). It is possible that these correspond to viral IE or DE proteins, which are known to be upregulated when L-gene induction is blocked (37).
Fig 3.

LMB inhibits HSV-1 but not HSV-2 late protein expression. (A) Time course analysis of viral protein synthesis. Vero cells were mock infected or infected with KOS1.1 or the M50T mutant at an MOI of 10 in the absence or presence of 30 ng/ml LMB. Infected cells were pulse-labeled for 30 min with [35S]methionine at the time points shown. Viral protein synthesis was analyzed by SDS-PAGE and autoradiography. Asterisks and upward arrows on the right denote infected cell-specific proteins whose synthesis was decreased or increased, respectively, by LMB. (B) Comparison of the effects of LMB and PAA on HSV-1 protein synthesis. Infections were performed as described in the legend to panel A in the absence or presence of 30 ng/ml LMB or 400 μg/ml PAA. [35S]methionine pulse-labeling was carried out from 11.5 to 12 hpi, at which time total proteins were harvested. The labeled proteins were analyzed by SDS-PAGE and autoradiography. Subsequent immunoblotting analysis allowed the identification of viral and cellular proteins, as indicated. (C) Immunoblotting analysis. Protein samples from the gel shown in panel B were analyzed by immunoblotting to determine the abundance of specific cellular and viral proteins at 12 hpi. The ICP27 blot was performed with a mixture of H1113 and H1119 monoclonal antibodies. (D) HSV-2 late protein synthesis is minimally affected by LMB. Vero cells were infected with HSV-1 strain KOS1.1 or HSV-2 strain HG52 at an MOI of 5, and the cultures were pulse-labeled from 12 to 12.5 hpi and analyzed as described in the legend to panel A.
Inhibition of viral DNA replication by chemical agents such as PAA can also inhibit HSV-1 L-gene expression (38). To directly compare the effect of LMB to that of a viral DNA synthesis inhibitor, we carried out another metabolic labeling experiment wherein Vero cells were infected with KOS1.1 in the absence of any drug or in the presence of either LMB or PAA. The cells were pulse-labeled with [35S]methionine from 11.5 to 12 hpi, and the labeled proteins were analyzed by SDS-PAGE and autoradiography (Fig. 3B). As before, the addition of LMB to the KOS1.1-infected cells resulted in a striking inhibition of HSV-1 protein synthesis. The addition of PAA resulted in a very similar pattern, although there were some qualitative differences (e.g., in the synthesis of DE protein ICP8). These results indicate that the effects of LMB and PAA on viral protein synthesis are very similar and suggest that LMB may inhibit viral L-gene expression, at least in part by reducing viral DNA replication.
To examine how LMB and PAA affect the accumulation of specific viral proteins, we used the same protein samples, which were harvested at 12 hpi, for immunoblotting analysis (Fig. 3C). All infections, including the mock infection, showed similar levels of the cellular polypeptides Aly/REF and beta-actin, indicating comparable recovery and loading of the protein samples. Analysis of IE protein ICP27 and DE protein ICP8 indicated that neither LMB nor PAA had a major effect on the accumulation of these polypeptides. In contrast, both inhibitors similarly reduced the accumulation of several L proteins. Specifically, both agents caused a modest reduction in the levels of leaky L proteins ICP5, gB, VP22, and VP16. Severe reductions in the levels of true L proteins gC and US11 were seen. These results, which are consistent with the immunoblotting results of Soliman and Silverstein (11), indicate that LMB inhibits the accumulation of viral L proteins.
Because HSV-2 is resistant to LMB, we expected that its protein synthesis would not be inhibited by LMB. To test this, [35S]methionine pulse-labeling was used to compare protein synthesis in HSV-1 KOS1.1- and HSV-2 HG52-infected cells at 12 hpi in either the absence or presence of 30 ng/ml LMB (Fig. 3D). While HSV-1 protein synthesis was inhibited by LMB, as before, that of HSV-2 was minimally affected, if at all. These results highlight the marked difference in the LMB sensitivity of HSV-1 and HSV-2.
We previously found that the inhibitory effect of LMB on HSV-1 growth is more evident in infections with virus at low MOIs than in infections with virus at high MOIs (12). The outcome of infections with virus at low MOIs may be significantly influenced by the efficiency of virus release, i.e., the process by which progeny virions exit the infected cell into the extracellular environment or into neighboring cells. We therefore performed an experiment to investigate the effects of LMB on viral release. Thus, Vero cells were infected at an MOI of 10 PFU/cell with KOS1.1 or the M50T mutant either in the absence or in the presence of 30 ng/ml LMB. The experiment was carried out at a high rather than a low MOI, as we expected that viral release would be difficult to quantitate in an infection with virus at a low MOI, where released viruses could rapidly enter neighboring uninfected cells. At 18 hpi, cell-associated and supernatant fractions were harvested separately, and the titers were summed to determine the total viral yields (Fig. 4A). As expected, the M50T mutant replicated more efficiently than KOS1.1 in the presence of LMB, although the difference was not as great as that in the infection with virus at a low MOI (compare these results to those in Fig. 1B and E). To determine the efficiency of viral release, the supernatant titers were divided by the total titers (Fig. 4B). In the WT infection, 8.4% of the progeny virus was released at 18 hpi, but this fraction fell to 0.25% in the presence of LMB, a 34-fold decrease. These data indicate that, in addition to its suppressive effect on viral replication, LMB further inhibits the egress of viral progeny from the infected cell. The release defect was partially alleviated by the M50T mutation, as M50T mutant release was decreased only 13-fold by LMB (9.3%/0.74%). Statistical analysis indicated that the difference in release efficiency between the M50T mutant and WT in the presence of LMB is significant (P = 0.0125).
HSV-1 LMB resistance can be conferred by an N-terminal TAP moiety on ICP27.
To date, only two ICP27 mutations that confer LMB-resistant replication to HSV-1 have been described: point mutation M50T and the deletion of residues 21 to 63 in mutant dAc. In the course of a different project to identify ICP27's protein-binding partners, we identified a third ICP27 alteration that can confer LMB resistance. This finding arose when we engineered NTAP-27, a derivative of KOS1.1 that encodes an ICP27 molecule having a 100-residue N-terminal TAP tag (Fig. 5A). This moiety consists of tandem calmodulin- and streptavidin-binding domains and is intended to facilitate the biochemical isolation of ICP27 and interacting proteins by TAP chromatography (39). Analysis of NTAP-27-infected Vero cells by immunoblotting with ICP27-specific monoclonal antibody H1113 confirmed that NTAP-27 produces an ICP27 polypeptide of the expected larger size (Fig. 5B). Of note, this protein does not react with the monoclonal antibody H1119, which has been shown to bind to residues 1 to 11 of ICP27 (40). This suggests that H1119 recognizes this epitope only when it is exposed on the N terminus of the protein. Importantly, viral growth analysis showed that NTAP-27 replicates as efficiently as WT HSV-1 in Vero cells (Fig. 5C). This demonstrates that the TAP moiety does not interfere with ICP27's essential replication functions in Vero cells, making this strain potentially useful for identifying interacting proteins. However, in the course of analyzing NTAP-27, we were surprised to discover that it is LMB resistant. This could be seen in a viral plaque assay (Fig. 5D), where NTAP-27 resembled the M50T mutant in its LMB resistance. NTAP-27 was also resistant to LMB in the low-MOI growth assay (Fig. 5E). Although these results strongly suggest that the addition of the TAP moiety onto the N terminus of ICP27 is sufficient to confer LMB-resistant growth, it is conceivable that NTAP-27 had acquired another mutation that conferred LMB resistance. To rule this out, we engineered a second isolate of NTAP-27, designated NTAP-27b, and confirmed that it was similarly LMB resistant (data not shown). We therefore conclude that addition of a 100-residue TAP tag onto the N terminus of ICP27 is sufficient to confer LMB-resistant replication to HSV-1.
Sensitivity of HSV-1 to LMB is mediated by a dominant function of ICP27.
LMB-resistant forms of HSV-1 ICP27 could confer resistance through two distinct types of mechanisms. First, the resistance mutation could abrogate or weaken an ICP27 activity that interferes with viral replication when CRM1 is inhibited. Alternatively, the resistance mutation could endow ICP27 with a new activity or could increase the strength of an existing activity. Such an activity could enhance replication under conditions where CRM1 is inhibited. These two types of mechanisms can potentially be distinguished by asking whether LMB sensitivity or LMB resistance is dominant when both sensitive and resistant forms of ICP27 are present in the same infected cell. Although this situation could arise in a coinfection, we previously showed that LMB resistance is most apparent in infections with virus at low MOIs (<1 PFU/cell) (12), as discussed above. This makes the coinfection approach difficult. We therefore took advantage of the V27 cell line, which is a derivative of Vero cells that possesses a stably transfected HSV-1 ICP27 gene (26). Although the ICP27 gene is not constitutively expressed, it is efficiently induced upon HSV-1 infection. Thus, Vero or V27 cells were infected at a low MOI with KOS1.1 or the M50T mutant, and progeny yields were determined after 2 days (Fig. 6A). Interestingly, WT HSV-1 was significantly more sensitive to LMB when the infections were carried out in V27 cells, suggesting that increased expression of WT ICP27 enhances HSV-1's sensitivity to LMB. In contrast, in the absence of LMB, KOS1.1 replicated comparably in the two cell lines. Importantly, the M50T mutant became significantly more sensitive to LMB when infections were carried out in V27 cells. Similar results were seen with NTAP-27 (Fig. 6B).
Fig 6.

Sensitivity of HSV-1 to LMB is enhanced in V27 cells. (A) The M50T mutant becomes LMB sensitive in V27 cells. Vero or V27 cells were infected at an MOI of 0.01 in the absence or presence of 30 ng/ml LMB, and the infected cells were incubated for 2 days. Viral yields were determined by plaque assay on Vero cells. The data show means of triplicate infections; error bars denote SEMs. Viral replication in the presence of LMB was compared between Vero and V27 cells using the Student t test. (B) NTAP-27 replication is LMB sensitive in V27 cells. Analysis was as described in the legend panel A, except that NTAP-27 was analyzed. (C) Comparable expression of LMB-sensitive and -resistant forms of ICP27 in V27 infections. Cultures parallel to the one whose results are shown in panel B were used for immunoblotting analysis. Total proteins were prepared at 1 day postinfection and analyzed using ICP27-specific antibody H1113. The top arrow shows the position of NTAP-27-encoded ICP27, whereas the bottom arrow denotes the position of WT ICP27. (D) HG52 remains comparably resistant to LMB in V27 cells. Analysis was as described in the legend to A. P values are labeled as follows: **, P < 0.01, ***, P ≤ 0.001; ns, not significant (P > 0.05).
The results described above suggest that the LMB-sensitive form of ICP27 is dominant to the LMB-resistant form. However, this interpretation is complicated by the possibility that the LMB-resistant and LMB-sensitive forms of ICP27 are expressed at different levels in the infected V27 cells. Fortunately, since the WT and NTAP-27 ICP27 molecules have distinct electrophoretic mobilities, we were able to use immunoblotting to compare their relative accumulation in a set of parallel infected cultures (Fig. 6C). The expression of both proteins was higher in infections in which virus replicated to higher titers, as expected due to the more efficient secondary spread of the infections with virus at low MOIs. Importantly, in the NTAP-27-infected V27 cells (Fig. 6C, lanes 7 and 8), there was comparable expression of KOS1.1- and NTAP-27-encoded ICP27. This shows that sensitivity to LMB is dominant in a situation where approximately equal levels of LMB-sensitive and LMB-resistant forms of ICP27 are present. Thus, our results strongly favor the first type of mechanism described above, in which LMB resistance mutations abrogate or weaken an ICP27 activity that interferes with replication when CRM1 is inhibited.
Since HSV-2 is similar to the M50T mutant and NTAP-27 in its LMB resistance, it was of interest to see whether its LMB sensitivity would be altered in V27 cells. We thus used the low-MOI yield assay to compare the LMB sensitivity of HSV-1 strain KOS1.1 and HSV-2 strain HG52 in Vero and V27 cells (Fig. 6D). Although HSV-1 was more sensitive to LMB in V27 cells, as expected, HSV-2 was not significantly different in its sensitivity. Thus, HSV-2 appears to be impervious to the LMB-sensitizing effect of WT HSV-1 ICP27. This suggests that the LMB resistance mechanism of HSV-2 is distinct from that utilized by LMB-resistant HSV-1 mutants.
DISCUSSION
HSV-1 and HSV-2 are differentially sensitive to LMB.
Using several WT strains of HSV-1, we have confirmed that the replication of this human pathogen is inhibited by LMB, a small organic molecule that covalently binds to and inactivates the cellular karyopherin-β CRM1. However, we were surprised to discover that a closely related virus, HSV-2, is inherently resistant to this inhibitor. Thus, HSV-2 differs from HSV-1 in that it does not require CRM1 for its efficient replication in Vero cells. In fact, we noted that HSV-2 plaques are somewhat larger when LMB is present, suggesting that CRM1 slightly impairs the replication and/or spread of HSV-2. The molecular basis of this enhancing effect is under investigation.
The markedly different requirements of HSV-1 and HSV-2 for CRM1 are unexpected, given that these two viruses are closely related genetically, having colinear genomes with a nearly perfect one-to-one gene correspondence (31). They also cause related diseases in humans. These similarities suggest that HSV-1's requirement for CRM1 does not involve a basic step in viral replication, as such a step would be expected to be conserved in HSV-2. Rather, the CRM1 dependence of HSV-1 may involve an ICP27-dependent process that is peripheral to the fundamental replication process, perhaps one involved in a virus-host cell interaction. In this regard, it should be noted that HSV-1 and HSV-2 differ in several clinical and biological properties. Perhaps most notably, they preferentially colonize different anatomical sites in the human host (oral/facial for HSV-1, genital for HSV-2) (41). Moreover, there is evidence that they clinically reactivate from these sites with distinct frequencies (42). Differences between HSV-1 and HSV-2 have also been reported in animal models of infection, where they show distinct properties with respect to neurovirulence (43), host immunological response (44), and latency (45, 46). It is conceivable that the type-specific difference in CRM1 dependence that we have identified is relevant to one or more of these clinical or biological differences.
Genetic basis of LMB resistance in HSV.
Given that LMB resistance mutations map to ICP27 in HSV-1, we hypothesized that HSV-2 is resistant by virtue of its ICP27 gene. However, when we replaced the HSV-1 ICP27 gene with that of HSV-2, the resulting recombinant remained fully sensitive to LMB. This demonstrates that the LMB resistance of HSV-2 must involve a gene (or genes) other than the ICP27 gene. However, at present it is not possible to say whether HSV-2 is LMB resistant because it possesses a gene that actively confers resistance or because it lacks a genetic determinant (presumably present in HSV-1) that sensitizes infected cells to LMB. One clue to the mechanism emerged when we found that HSV-2's resistance is unaffected by the presence of WT ICP27, unlike LMB-resistant HSV-1 mutants, which become more sensitive when WT ICP27 is present. This suggests that the resistance mechanism of HSV-2 is qualitatively different from that of HSV-1 LMB-resistant mutants.
To further understand HSV-2's resistance to LMB, it will be crucial to identify the non-ICP27 gene (or genes) in HSV-2 that determines this phenotype. Assuming that there is a single gene, it may be possibly to do so using a marker rescue approach. For example, defined segments of the HSV-2 genome could be cotransfected with infectious HSV-1 DNA into Vero cells, allowing intertypic recombinants to be generated. The cotransfected cultures could then be passaged in the presence of LMB to identify the HSV-2 sequence that can confer LMB resistance to HSV-1. Experiments along these lines have been initiated in our laboratory.
LMB inhibits late HSV-1 protein synthesis and virion release.
The remainder of our study focused on understanding how mutations in HSV-1 ICP27 can confer LMB resistance. Central to this issue is understanding the molecular role that CRM1 plays in the WT HSV-1 infection. One obvious possibility is that this karyopherin-β mediates the nuclear export of one or more viral proteins. Consistent with this, the HSV-1 proteins UL47 (47), γ34.5 (48), and US3 (49) have been found to have leucine-rich NESs that utilize CRM1 for nuclear export. However, the significance of this is unclear, as it is unknown whether the function of any of these proteins requires their transport out of the nucleus. Recently, an HSV-1 capsid protein, VP19C, was also shown to have a CRM1-dependent NES (50). Since HSV-1 nucleocapsids are assembled in the nucleus but mature in the cytoplasm, VP19C does need to travel from the nucleus to the cytoplasm. However, nuclear export of nucleocapsids proceeds via direct breach of the nuclear membrane via a process of envelopment and deenvelopment (51). Thus, at present there is no definitive evidence that HSV-1 requires CRM1 for export of viral proteins, raising the possibility that CRM1 plays another role in infection.
To gain more insight into CRM1's function during HSV-1 infection, we examined the effects of LMB on viral protein synthesis. We found that at a middle to late stage of infection (7 hpi and thereafter), LMB suppresses the synthesis and accumulation of viral L proteins, indicating that CRM1 is needed for efficient viral gene expression. This is consistent with previous work showing that LMB inhibits the steady-state accumulation of several viral late proteins and their mRNAs (11). Although the mechanism by which CRM1 promotes HSV-1 gene expression is unknown, Soliman and Silverstein suggested that CRM1 enables nuclear export of a subset of the intronless viral mRNAs (11). Alternatively, as will be discussed further below, it is possible that HSV-1 gene expression does not require CRM1 but rather that a nonessential activity of ICP27 interferes with viral gene expression when CRM1 is inhibited. Finally, it is worth pointing out that the role of CRM1 in viral L-gene expression may be indirect, as late viral gene expression is contingent upon earlier events in viral replication, in particular, viral DNA synthesis (41). In this regard, we found that inhibition of viral DNA replication by PAA led to a viral gene expression defect that closely resembles that induced by LMB. Thus, future experiments should address whether CRM1 plays a role in viral DNA synthesis.
One additional finding of note is that we found that the M50T mutation in ICP27 alleviates the LMB-induced defects in viral protein synthesis. While this is perhaps not surprising, given that this mutant replicated more efficiently in the presence of LMB, it is formally possible that the M50T mutation promotes replication by enhancing a later step in viral replication, such as virus assembly, thus compensating for the reduced levels of viral gene products. Instead, our results suggest that mutations in ICP27 allow HSV-1 to overcome the viral gene expression defect that is imposed by LMB.
Because we had previously observed that the antiviral effect of LMB is greatest in infections with virus at low MOIs (12), we also investigated whether this drug inhibits the release of viral progeny from the infected cell. This was indeed found to be the case. Similar to the deficiency in viral protein synthesis, this defect was partially rescued by the M50T mutation, although M50T mutant viral release was still significantly inhibited by LMB treatment. The role of CRM1 in the process of viral release is presently unclear. Release of HSV-1 progeny occurs when cytoplasmic vesicles containing infectious enveloped virions travel via exocytosis to the cell surface, where they fuse with the plasma membrane (52). Although this process is not well understood, it likely involves the participation of HSV-1 glycoproteins (53), which are mostly encoded by L genes. Given this, it is possible that LMB's inhibitory effect on viral release is simply an indirect consequence of its ability to inhibit L gene expression.
A novel type of HSV-1 ICP27 mutation confers LMB resistance.
Prior to this work, only two HSV-1 ICP27 LMB-resistant mutants had been identified. Both have alterations in a specific N-terminal region of ICP27 (residues 21 to 63), implicating this portion of the protein in LMB sensitivity. In this study, we identified a new ICP27 alteration that can confer LMB resistance, that being the addition of a 100-residue N-terminal TAP tag. Although the NTAP-27 ICP27 molecule has residues 21 to 63 intact, the N-terminal tag is near these residues, at least in the primary sequence. Our findings thus underscore the importance of ICP27's N-terminal region in the LMB-resistant phenotype. Several functions have been ascribed to this part of the protein. First, the leucine-rich NES-like sequence, which maps to residues 6 to 19, is needed for efficient HSV-1 replication in Vero cells (19) and is critical for ICP27's interaction with NXF1 (17). All three LMB-resistant mutants have this sequence intact. Second, Hernandez and Sandri-Goldin have shown that the N and C termini of ICP27 interact in an intramolecular fashion (54) and that this interaction is important for several of ICP27's functions, including NXF1 interaction (55). It would be interesting to see whether the TAP moiety on the NTAP-27 ICP27 protein affects the association between N and C termini. It would also be worthwhile to see if other N-terminal additions to ICP27 (e.g., a green fluorescent protein moiety) lead to LMB resistance. Third, the N-terminal portion of ICP27 has been implicated in the ability of HSV-1 to stimulate several cellular signal transduction pathways (56–58). This activity and its possible relevance to LMB resistance are discussed further below.
A model to explain how ICP27 mutations confer LMB resistance in HSV-1.
Based on our findings, we propose a model to explain how ICP27 mutations confer LMB resistance (Fig. 7). The model proposes that the N-terminal region of ICP27 carries out an activity in Vero cells that actively interferes with viral replication when CRM1 is inhibited. This could be because the putative activity leads to a downstream event that requires CRM1, and in the absence of CRM1 function, a block to late viral gene expression ensues. This model is supported by two lines of evidence. The first and strongest is that LMB-resistant mutants such as the M50T mutant become LMB sensitive when infections are carried out in WT ICP27-expressing V27 cells. The most straightforward interpretation of this finding is that, in the absence of CRM1 function, WT ICP27 actively interferes with replication. If so, LMB-resistant mutations could abrogate or weaken this activity. An additional point is that the M50T mutant and other LMB-resistant mutants have no known phenotype in the absence of LMB. Therefore, it is likely that the ICP27 activity that is affected by the LMB resistance mutations is distinct from the essential replication functions that ICP27 mediates in Vero cells (such as enhancing viral mRNA export). The second line of evidence supporting the model stems from our discovery that addition of an N-terminal tag onto ICP27 can confer LMB resistance. This is consistent with the model because it is relatively easy to envision how a functionally unrelated moiety such as the TAP tag could interfere with an ICP27 activity. Conversely, it is difficult to envision how this moiety could increase an ICP27 activity or create a new one, as would be predicted by the alternate model.
Fig 7.
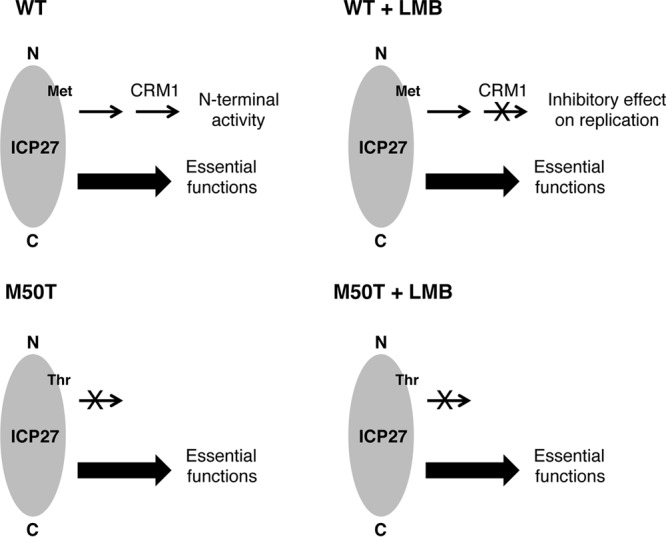
Model to explain how mutations in HSV-1 ICP27 can confer LMB resistance. The diagram shows representations of the WT and M50T mutant forms of ICP27, which have Met and Thr, respectively, at residue 50. See the text for further details.
If LMB-resistant mutations weaken or abrogate an ICP27 activity, what might that activity be? A possibility that is consistent with many of the existing data is that the mutations weaken ICP27's ability to induce cellular signaling pathways, including the mitogen-activated protein kinases p38 and Jun N-terminal protein kinase (JNK) (56, 58), and the NF-κB system (57). There are several reasons why this idea is appealing. First, as mentioned earlier, sequences in the N-terminal half of ICP27 are critical for induction of these pathways during HSV-1 infection (58, 59). Indeed, Hargett et al. found that the M50T mutant and dAc are unable to induce the p38, JNK, and NF-κB responses in CV-1 monkey kidney cells (59). Complicating the picture, however, Corcoran et al. reported that an independently derived HSV-1 M50T mutant is competent to initiate p38 signaling in HeLa cells (60). We are currently investigating whether HSV-1 LMB-resistant mutants are altered in their ability to activate the p38, JNK, and NF-κB pathways in Vero cells. Second, studies using chemical inhibitors of p38 and JNK suggest that activation of these pathways by HSV-1 is not absolutely essential for viral replication but rather modestly enhances progeny production (61, 62). This is consistent with our proposal that the ICP27 function is distinct from ICP27's essential replication activities. Third, CRM1 has been previously implicated in the regulation of both the p38 (63) and NF-κB (64) signaling cascades, consistent with the possibility that CRM1 plays a downstream role in ICP27's N-terminal activity (Fig. 7).
Possible therapeutic relevance.
Finally, although the development of LMB as an antiviral was not a focus of our work, it is worth mentioning that LMB derivatives and other CRM1 inhibitors are currently being developed as therapeutic anticancer agents (65, 66), consistent with the observation that CRM1-dependent nuclear export is often increased in cancer cells (67). On the basis of our findings, it is conceivable that some of these compounds could have efficacy against HSV-1 infections, although our results suggest that they may not be useful against HSV-2. However, it is important to point out that the sensitivity of HSV-1 to LMB was much lower in HFF cells than in Vero cells, and in these cells, ICP27 does not appear to modulate LMB sensitivity (Fig. 1F). Therefore, it will be important to assess the LMB sensitivity of HSV-1 and HSV-2 in additional cellular contexts. Furthermore, as our experiments have been done with laboratory-adapted strains, it would be worthwhile to examine the effect of CRM1 inhibitors on clinical isolates of HSV-1 and HSV-2.
ACKNOWLEDGMENTS
We thank Jamie Borten for designing and constructing the TAP-tagged ICP27 gene and Zhi Pan for cloning the HSV-2 ICP27 gene. We also acknowledge Lindsey Foran for excellent technical support and Leslie Schiff for many insightful conversations. Finally, we thank our colleagues Jin-Young Han, Jim Lokensgard, Alistair McGregor, and Ian Mohr for providing HSV-1 and HSV-2 strains.
This research was supported by a grant from the NIH (R01-AI42737).
Footnotes
Published ahead of print 5 June 2013
REFERENCES
- 1. Xu D, Farmer A, Chook YM. 2010. Recognition of nuclear targeting signals by karyopherin-beta proteins. Curr. Opin. Struct. Biol. 20:782–790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fornerod M, Ohno M, Yoshida M, Mattaj IW. 1997. CRM1 is an export receptor for leucine-rich nuclear export signals. Cell 90:1051–1060 [DOI] [PubMed] [Google Scholar]
- 3. Fukuda M, Asano S, Nakamura T, Adachi M, Yoshida M, Yanagida M, Nishida E. 1997. CRM1 is responsible for intracellular transport mediated by the nuclear export signal. Nature 390:308–311 [DOI] [PubMed] [Google Scholar]
- 4. Stade K, Ford CS, Guthrie C, Weis K. 1997. Exportin 1 (Crm1p) is an essential nuclear export factor. Cell 90:1041–1050 [DOI] [PubMed] [Google Scholar]
- 5. Dong X, Biswas A, Chook YM. 2009. Structural basis for assembly and disassembly of the CRM1 nuclear export complex. Nat. Struct. Mol. Biol. 16:558–560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Monecke T, Guttler T, Neumann P, Dickmanns A, Gorlich D, Ficner R. 2009. Crystal structure of the nuclear export receptor CRM1 in complex with Snurportin1 and RanGTP. Science 324:1087–1091 [DOI] [PubMed] [Google Scholar]
- 7. Cullen BR. 2003. Nuclear mRNA export: insights from virology. Trends Biochem. Sci. 28:419–424 [DOI] [PubMed] [Google Scholar]
- 8. Yoshida M, Horinouchi S. 1999. Trichostatin and leptomycin. Inhibition of histone deacetylation and signal-dependent nuclear export. Ann. N. Y. Acad. Sci. 886:23–36 [DOI] [PubMed] [Google Scholar]
- 9. Kudo N, Matsumori N, Taoka H, Fujiwara D, Schreiner EP, Wolff B, Yoshida M, Horinouchi S. 1999. Leptomycin B inactivates CRM1/exportin 1 by covalent modification at a cysteine residue in the central conserved region. Proc. Natl. Acad. Sci. U. S. A. 96:9112–9117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Murata T, Goshima F, Koshizuka T, Takakuwa H, Nishiyama Y. 2001. A single amino acid substitution in the ICP27 protein of herpes simplex virus type 1 is responsible for its resistance to leptomycin B. J. Virol. 75:1039–1043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Soliman TM, Silverstein SJ. 2000. Herpesvirus mRNAs are sorted for export via Crm1-dependent and -independent pathways. J. Virol. 74:2814–2825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lengyel J, Strain AK, Perkins KD, Rice SA. 2006. ICP27-dependent resistance of herpes simplex virus type 1 to leptomycin B is associated with enhanced nuclear localization of ICP4 and ICP0. Virology 352:368–379 [DOI] [PubMed] [Google Scholar]
- 13. Ingram A, Phelan A, Dunlop J, Clements JB. 1996. Immediate early protein IE63 of herpes simplex virus type 1 binds RNA directly. J. Gen. Virol. 77:1847–1851 [DOI] [PubMed] [Google Scholar]
- 14. Mears WE, Rice SA. 1996. The RGG box motif of the herpes simplex virus ICP27 protein mediates an RNA-binding activity and determines in vivo methylation. J. Virol. 70:7445–7453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Mears WE, Rice SA. 1998. The herpes simplex virus immediate-early protein ICP27 shuttles between nucleus and cytoplasm. Virology 242:128–137 [DOI] [PubMed] [Google Scholar]
- 16. Phelan A, Clements JB. 1997. Herpes simplex virus type 1 immediate early protein IE63 shuttles between nuclear compartments and the cytoplasm. J. Gen. Virol. 78:3327–3331 [DOI] [PubMed] [Google Scholar]
- 17. Sandri-Goldin RM. 1998. ICP27 mediates HSV RNA export by shuttling through a leucine-rich nuclear export signal and binding viral intronless RNAs through an RGG motif. Genes Dev. 12:868–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Soliman TM, Sandri-Goldin RM, Silverstein SJ. 1997. Shuttling of the herpes simplex virus type 1 regulatory protein ICP27 between the nucleus and cytoplasm mediates the expression of late proteins. J. Virol. 71:9188–9197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lengyel J, Guy C, Leong V, Borge S, Rice SA. 2002. Mapping of functional regions in the amino-terminal portion of the herpes simplex virus ICP27 regulatory protein: importance of the leucine-rich nuclear export signal and RGG box RNA-binding domain. J. Virol. 76:11866–11879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen IH, Sciabica KS, Sandri-Goldin RM. 2002. ICP27 interacts with the RNA export factor Aly/REF to direct herpes simplex virus type 1 intronless mRNAs to the TAP export pathway. J. Virol. 76:12877–12889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Koffa MD, Clements JB, Izaurralde E, Wadd S, Wilson SA, Mattaj IW, Kuersten S. 2001. Herpes simplex virus ICP27 protein provides viral mRNAs with access to the cellular mRNA export pathway. EMBO J. 20:5769–5778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chen IH, Li L, Silva L, Sandri-Goldin RM. 2005. ICP27 recruits Aly/REF but not TAP/NXF1 to herpes simplex virus type 1 transcription sites although TAP/NXF1 is required for ICP27 export. J. Virol. 79:3949–3961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Johnson LA, Li L, Sandri-Goldin RM. 2009. The cellular RNA export receptor TAP/NXF1 is required for ICP27-mediated export of herpes simplex virus 1 RNA, but the TREX complex adaptor protein Aly/REF appears to be dispensable. J. Virol. 83:6335–6346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Johnson LA, Sandri-Goldin RM. 2009. Efficient nuclear export of herpes simplex virus 1 transcripts requires both RNA binding by ICP27 and ICP27 interaction with TAP/NXF1. J. Virol. 83:1184–1192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rodriguez-Navarro S, Hurt E. 2011. Linking gene regulation to mRNA production and export. Curr. Opin. Cell Biol. 23:302–309 [DOI] [PubMed] [Google Scholar]
- 26. Rice SA, Knipe DM. 1990. Genetic evidence for two distinct transactivation functions of the herpes simplex virus alpha protein ICP27. J. Virol. 64:1704–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bresnahan WA, Hultman GE, Shenk T. 2000. Replication of wild-type and mutant human cytomegalovirus in life-extended human diploid fibroblasts. J. Virol. 74:10816–10818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hughes RG, Jr, Munyon WH. 1975. Temperature-sensitive mutants of herpes simplex virus type 1 defective in lysis but not in transformation. J. Virol. 16:275–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Rice SA, Lam V, Knipe DM. 1993. The acidic amino-terminal region of herpes simplex virus type 1 alpha protein ICP27 is required for an essential lytic function. J. Virol. 67:1778–1787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Rice SA, Lam V. 1994. Amino acid substitution mutations in the herpes simplex virus ICP27 protein define an essential gene regulation function. J. Virol. 68:823–833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dolan A, Jamieson FE, Cunningham C, Barnett BC, McGeoch DJ. 1998. The genome sequence of herpes simplex virus type 2. J. Virol. 72:2010–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Rice SA, Su LS, Knipe DM. 1989. Herpes simplex virus alpha protein ICP27 possesses separable positive and negative regulatory activities. J. Virol. 63:3399–3407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Perkins KD, Gregonis J, Borge S, Rice SA. 2003. Transactivation of a viral target gene by herpes simplex virus ICP27 is posttranscriptional and does not require the endogenous promoter or polyadenylation site. J. Virol. 77:9872–9884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Elliott G, O'Hare P. 1997. Intercellular trafficking and protein delivery by a herpesvirus structural protein. Cell 88:223–233 [DOI] [PubMed] [Google Scholar]
- 35. Weinheimer SP, Boyd BA, Durham SK, Resnick JL, O'Boyle DR., II 1992. Deletion of the VP16 open reading frame of herpes simplex virus type 1. J. Virol. 66:258–269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Diaz JJ, Simonin D, Masse T, Deviller P, Kindbeiter K, Denoroy L, Madjar JJ. 1993. The herpes simplex virus type 1 US11 gene product is a phosphorylated protein found to be non-specifically associated with both ribosomal subunits. J. Gen. Virol. 74(Pt 3):397–406 [DOI] [PubMed] [Google Scholar]
- 37. Honess RW, Roizman B. 1974. Regulation of herpesvirus macromolecular synthesis. I. Cascade regulation of the synthesis of three groups of viral proteins. J. Virol. 14:8–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Holland LE, Anderson KP, Shipman C, Jr, Wagner EK. 1980. Viral DNA synthesis is required for the efficient expression of specific herpes simplex virus type 1 mRNA species. Virology 101:10–24 [DOI] [PubMed] [Google Scholar]
- 39. Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, Wilm M, Seraphin B. 2001. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods 24:218–229 [DOI] [PubMed] [Google Scholar]
- 40. Mears WE, Lam V, Rice SA. 1995. Identification of nuclear and nucleolar localization signals in the herpes simplex virus regulatory protein ICP27. J. Virol. 69:935–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Roizman B, Knipe DM, Whitley RJ. 2007. Herpes simplex viruses, p 2501–2601 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 42. Lafferty WE, Coombs RW, Benedetti J, Critchlow C, Corey L. 1987. Recurrences after oral and genital herpes simplex virus infection. Influence of site of infection and viral type. N. Engl. J. Med. 316:1444–1449 [DOI] [PubMed] [Google Scholar]
- 43. McKendall RR. 1980. Comparative neurovirulence and latency of HSV1 and HSV2 following footpad inoculation in mice. J. Med. Virol. 5:25–32 [DOI] [PubMed] [Google Scholar]
- 44. Zheng M, Conrady CD, Ward JM, Bryant-Hudson KM, Carr DJ. 2012. Comparison of the host immune response to herpes simplex virus 1 (HSV-1) and HSV-2 at two different mucosal sites. J. Virol. 86:7454–7458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Margolis TP, Imai Y, Yang L, Vallas V, Krause PR. 2007. Herpes simplex virus type 2 (HSV-2) establishes latent infection in a different population of ganglionic neurons than HSV-1: role of latency-associated transcripts. J. Virol. 81:1872–1878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yoshikawa T, Hill JM, Stanberry LR, Bourne N, Kurawadwala JF, Krause PR. 1996. The characteristic site-specific reactivation phenotypes of HSV-1 and HSV-2 depend upon the latency-associated transcript region. J. Exp. Med. 184:659–664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Williams P, Verhagen J, Elliott G. 2008. Characterization of a CRM1-dependent nuclear export signal in the C terminus of herpes simplex virus type 1 tegument protein UL47. J. Virol. 82:10946–10952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cheng G, Brett ME, He B. 2002. Signals that dictate nuclear, nucleolar, and cytoplasmic shuttling of the gamma(1)34.5 protein of herpes simplex virus type 1. J. Virol. 76:9434–9445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wang X, Patenode C, Roizman B. 2011. US3 protein kinase of HSV-1 cycles between the cytoplasm and nucleus and interacts with programmed cell death protein 4 (PDCD4) to block apoptosis. Proc. Natl. Acad. Sci. U. S. A. 108:14632–14636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhao L, Zheng C. 2012. The first identified nucleocytoplasmic shuttling herpesviral capsid protein: herpes simplex virus type 1 VP19C. PLoS One 7:e41825. 10.1371/journal.pone.0041825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Mettenleiter TC, Klupp BG, Granzow H. 2006. Herpesvirus assembly: a tale of two membranes. Curr. Opin. Microbiol. 9:423–429 [DOI] [PubMed] [Google Scholar]
- 52. Mettenleiter TC, Klupp BG, Granzow H. 2009. Herpesvirus assembly: an update. Virus Res. 143:222–234 [DOI] [PubMed] [Google Scholar]
- 53. Mingo RM, Han J, Newcomb WW, Brown JC. 2012. Replication of herpes simplex virus: egress of progeny virus at specialized cell membrane sites. J. Virol. 86:7084–7097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hernandez FP, Sandri-Goldin RM. 2010. Herpes simplex virus 1 regulatory protein ICP27 undergoes a head-to-tail intramolecular interaction. J. Virol. 84:4124–4135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hernandez FP, Sandri-Goldin RM. 2010. Head-to-tail intramolecular interaction of herpes simplex virus type 1 regulatory protein ICP27 is important for its interaction with cellular mRNA export receptor TAP/NXF1. mBio 1(5):e00268–10. 10.1128/mBio.00268-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gillis PA, Okagaki LH, Rice SA. 2009. Herpes simplex virus type 1 ICP27 induces p38 mitogen-activated protein kinase signaling and apoptosis in HeLa cells. J. Virol. 83:1767–1777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Gregory D, Hargett D, Holmes D, Money E, Bachenheimer SL. 2004. Efficient replication by herpes simplex virus type 1 involves activation of the IkappaB kinase-IkappaB-p65 pathway. J. Virol. 78:13582–13590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Hargett D, McLean T, Bachenheimer SL. 2005. Herpes simplex virus ICP27 activation of stress kinases JNK and p38. J. Virol. 79:8348–8360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Hargett D, Rice S, Bachenheimer SL. 2006. Herpes simplex virus type 1 ICP27-dependent activation of NF-kappaB. J. Virol. 80:10565–10578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Corcoran JA, Hsu WL, Smiley JR. 2006. Herpes simplex virus ICP27 is required for virus-induced stabilization of the ARE-containing IEX-1 mRNA encoded by the human IER3 gene. J. Virol. 80:9720–9729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Karaca G, Hargett D, McLean TI, Aguilar JS, Ghazal P, Wagner EK, Bachenheimer SL. 2004. Inhibition of the stress-activated kinase, p38, does not affect the virus transcriptional program of herpes simplex virus type 1. Virology 329:142–156 [DOI] [PubMed] [Google Scholar]
- 62. Zachos G, Koffa M, Preston CM, Clements JB, Conner J. 2001. Herpes simplex virus type 1 blocks the apoptotic host cell defense mechanisms that target Bcl-2 and manipulates activation of p38 mitogen-activated protein kinase to improve viral replication. J. Virol. 75:2710–2728 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 63. Ben-Levy R, Hooper S, Wilson R, Paterson HF, Marshall CJ. 1998. Nuclear export of the stress-activated protein kinase p38 mediated by its substrate MAPKAP kinase-2. Curr. Biol. 8:1049–1057 [DOI] [PubMed] [Google Scholar]
- 64. Rodriguez MS, Thompson J, Hay RT, Dargemont C. 1999. Nuclear retention of IkappaBalpha protects it from signal-induced degradation and inhibits nuclear factor kappaB transcriptional activation. J. Biol. Chem. 274:9108–9115 [DOI] [PubMed] [Google Scholar]
- 65. Etchin J, Sun Q, Kentsis A, Farmer A, Zhang ZC, Sanda T, Mansour MR, Barcelo C, McCauley D, Kauffman M, Shacham S, Christie AL, Kung AL, Rodig SJ, Chook YM, Look AT. 2013. Antileukemic activity of nuclear export inhibitors that spare normal hematopoietic cells. Leukemia 27:66–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Mutka SC, Yang WQ, Dong SD, Ward SL, Craig DA, Timmermans PB, Murli S. 2009. Identification of nuclear export inhibitors with potent anticancer activity in vivo. Cancer Res. 69:510–517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Turner JG, Dawson J, Sullivan DM. 2012. Nuclear export of proteins and drug resistance in cancer. Biochem. Pharmacol. 83:1021–1032 [DOI] [PMC free article] [PubMed] [Google Scholar]