

Abstract
Influenza A viruses, including H1N1 and H5N1 subtypes, pose a serious threat to public health. Neuraminidase (NA)-related immunity contributes to protection against influenza virus infection. Antibodies to the N1 subtype provide protection against homologous and heterologous H1N1 as well as H5N1 virus challenge. Since neither the strain-specific nor conserved epitopes of N1 have been identified, we generated a panel of mouse monoclonal antibodies (MAbs) that exhibit different reactivity spectra with H1N1 and H5N1 viruses and used these MAbs to map N1 antigenic domains. We identified 12 amino acids essential for MAb binding to the NA of a recent seasonal H1N1 virus, A/Brisbane/59/2007. Of these, residues 248, 249, 250, 341, and 343 are recognized by strain-specific group A MAbs, while residues 273, 338, and 339 are within conserved epitope(s), which allows cross-reactive group B MAbs to bind the NAs of seasonal H1N1 and the 1918 and 2009 pandemic (09pdm) H1N1 as well as H5N1 viruses. A single dose of group B MAbs administered prophylactically fully protected mice against lethal challenge with seasonal and 09pdm H1N1 viruses and resulted in significant protection against the highly pathogenic wild-type H5N1 virus. Another three N1 residues (at positions 396, 397, and 456) are essential for binding of cross-reactive group E MAbs, which differ from group B MAbs in that they do not bind 09pdm H1N1 viruses. The identification of conserved N1 epitopes reveals the molecular basis for NA-mediated immunity between H1N1 and H5N1 viruses and demonstrates the potential for developing broadly protective NA-specific antibody treatments for influenza.
INTRODUCTION
Neuraminidase (NA) is one of the two major glycoproteins on the surface of influenza virus. The primary biological role of NA is to cleave terminal sialic acid residues that serve as receptors for the hemagglutinin (HA), promoting the release of progeny virions from host cells (1). This enzymatic activity contributes to the transmission of influenza virus (2) and facilitates influenza virus infection by removing decoy receptors on mucins, cilia, and the cellular glycocalyx (3). Inhibition of NA enzyme activity by either drugs or NA-specific antibodies limits the spread of influenza virus, thus reducing viral load and disease symptoms.
Influenza A viruses are differentiated by HA and NA subtypes. Seventeen influenza HA subtypes (H1 to H17) and 10 NA subtypes (N1 to N10) have been identified (4), but only H1N1, H2N2, and H3N2 viruses have caused pandemics and subsequent seasonal epidemics in humans. The NA of the 1918 pandemic (18pdm) H1N1 virus enhances virus replication in mouse lungs and human airway cells (5) and therefore may have contributed to the extraordinary number of deaths during this pandemic. NA plays a role in the transmissibility of the 2009 pandemic (09pdm) H1N1 (2, 6) and host adaptation of H5N1 virus (7), highlighting its importance in the emergence of pandemic viruses.
Although antibodies against NA do not prevent attachment and entry of influenza virus into cells, they sharply limit virus spread (8) and thereby contribute to immunity against influenza virus (9, 10). A mouse monoclonal antibody (MAb) specific for H5N1 viral NA has therapeutic benefit against H5N1 infection in mice and ferrets (11). Studies in mice demonstrate that while antibodies specific for NA of the N2 subtype provide the greatest protection to the homologous H3N2 virus, they also provide substantial immunity against heterologous equine influenza viruses that share the same subtype (12, 13). Similar broad reactivity has been demonstrated for N1-specific antibodies. Polyclonal antiserum with specificity for the NA of 09pdm H1N1 virus has measurable inhibition of H5N1 NA activity (14). Moreover, heterologous protection has been attributed to NA antibodies in several studies. The NA of the seasonal H1N1 virus induces cross-reactive antibodies that reduce the lethality of 09pdm H1N1 virus (15), and immunization with a DNA vaccine expressing seasonal H1N1 NA (16) or virus-like particles containing 09pdm H1N1 NA (17) provides significant protection against lethal H5N1 challenge in mice. Similar NA-associated protection against H5N1 has been observed in ferrets immunized with recombinant 18pdm H1N1 NA or seasonal trivalent inactivated vaccine (18).
Despite the significant role of N1 in the pathogenesis and immunity of H1N1 and H5N1 viruses, there is surprisingly little information regarding its antigenic domains. Antibodies against two conserved NA peptides consisting of residues 222 to 230 (N2 numbering) and the 12 residues at the NA terminus, have been generated and explored for NA detection and quantification (19). In addition, antigenic epitopes of NA subtypes N2 and N9 have been identified (20–26). However, these do not provide sufficient information for understanding N1 antigenic determinants. To address this and, in particular, to identify conserved epitopes corresponding to N1-related heterologous immunity, we mapped antigenic domains of the NA of a recent seasonal H1N1 virus, A/Brisbane/59/2007 (BR/07), using a panel of N1-specific MAbs and tested the ability of cross-reactive antibodies to protect mice against homologous and heterologous H1N1 and H5N1 virus challenge.
MATERIALS AND METHODS
Viruses and plasmids.
Reassortant H6N1 viruses, H6N1BR/07, H6N1CA/09, and H6N1VN/04, which contain the HA gene of H6N2 virus A/turkey/Massachusetts/3740/1965 and the NA gene of seasonal H1N1 BR/07, 09pdm H1N1 A/California/07/2009 (CA/09), or H5N1 virus A/Vietnam/1203/2004 (VN/04), were rescued using reverse genetics (27). Reassortant viruses, wild-type (wt) viruses (Table 1), and an attenuated VN/04 virus (ΔVN/04, a reassortant virus containing the HA [polybasic residues in the cleavage motif deleted] and NA genes of VN/04 and other genes of A/Puerto Rico/8/1934) were grown in 10-day-old embryonated chicken eggs. Viruses were inactivated with β-propiolactone (Sigma-Aldrich, St. Louis, MO) and purified by sucrose gradient centrifugation if necessary. The pCAGGS-NIH/09 and pCAGGS-BM/18 plasmids, which contain the NA genes of seasonal H1N1 A/Bethesda/NIH50/2009 (NIH/09) and 18pdm H1N1 A/Brevig Mission/1/1918 (BM/18) viruses, respectively, were constructed as reported previously (17).
Table 1.
Wild-type influenza A viruses used in the present study
| Virus name (abbreviation)a | Subtype |
|---|---|
| A/Texas/36/1991 (TX/91) | H1N1 |
| A/New Caledonia/20/1999 (NC/99) | H1N1 |
| A/Solomon Islands/3/2006 (SI/06) | H1N1 |
| A/Brisbane/59/2007 (BR/07) | H1N1 |
| A/Chongqing/150/2007 | H1N1 |
| A/Guangdong/51/2008 | H1N1 |
| A/Bethesda/NIH50/2009 (NIH/09) | H1N1 |
| A/California/07/2009 (CA/09) | H1N1 |
| A/duck/Eastern China/103/03 (Y103) | H1N1 |
| A/duck/Eastern China/233/03 (Y233) | H3N1 |
| A/Vietnam/1203/2004 (VN/04) | H5N1 |
| A/duck/Eastern China/1/2008 | H6N1 |
| A/turkey/Ontario/6188/68 | H8N4 |
| A/duck/Eastern China/01/05 | H8N4 |
| A/duck/eastern China/008/2008 | H5N5 |
| A/duck/eastern China/031/2009 | H5N5 |
| A/shearwater/Australia/1/72 | H6N5 |
| A/duck/Yangzhou/013/2008 | H6N5 |
| A/duck/Ukraine/1/63 | H3N8 |
| A/duck/Eastern China/18/05 | H3N8 |
| A/duck/Jiangsu/K1203/2010 | H5N8 |
| A/duck/Eastern China/163/02 | H6N8 |
| A/duck/Eastern China/36/02 | H3N2 |
| A/chicken/Shanghai/14/2001 | H9N2 |
| A/duck/China/397/03 | H10N3 |
| A/duck/China/376/04 | H4N6 |
| A/chicken/Germany/n/49 | H10N7 |
| A/duck/Memphis/546/74 | H11N9 |
Viruses in bold were tested in IFAs.
Immunization of mice and fusion and screening of hybridomas.
All animal experiments were performed under protocols approved by the CBER Animal Care and Use Committee. BALB/c mice (6 weeks old; The Jackson Laboratory) were infected intranasally with BR/07 virus twice at an interval of 4 weeks and boosted intravenously with inactivated, purified BR/07 on day 3 before the fusion. Splenocytes from immunized mice were fused with Sp2/0 cells. Hybridomas were initially screened with enzyme-linked immunosorbent assays (ELISAs) using HA-mismatched H6N1BR/07 virus-coated plates. Positive clones were further screened in cell-based ELISAs with NA expressed on human embryonic kidney 293 (HEK293) cells that were transfected with pCAGGS-NIH/09 plasmid. Hybridomas secreting NA-specific antibodies were subcloned twice by limiting dilution. Ascitic fluid was generated for each hybridoma in BALB/c mice (Antibody and Immunoassay Consultants, Rockville, MD), and representative MAbs were purified using protein G columns (GE Healthcare, Uppsala, Sweden).
IFAs.
An immunofluorescence assay (IFA) was performed as described previously (28). Madin-Darby canine kidney (MDCK) cells growing in 96-well plates were infected with each virus at a multiplicity of infection of 1, followed by incubation at 35°C in 5% CO2 for 48 h. Cells were fixed with acetone and ethanol (3:2, vol/vol) and incubated with each MAb, followed by incubation with fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG (Sigma-Aldrich, St. Louis, MO). Cells were then examined under a Leica fluorescence microscope (DMI3000B).
Site-directed mutagenesis.
Nucleotide changes corresponding to a panel of 19 single mutations (K150A, D199N, N200A, K221N, N248D, K249E, W296A, D311E, N336G, D341N, N365I, R366S, K369A, G370K, D398E, G401K, S403K, L430R, and T435R) were introduced into the NIH/09 virus NA gene in the pCAGGS-NIH/09 plasmid with a QuikChange Multi Site-Directed Mutagenesis Kit (Stratagene, La Jolla, CA). The mutations were designed based on alignment of H1N1 and H5N1 NA sequences, the MAb reactivity pattern, and previously mapped epitopes on N2 and N9 (21–24). Mutant plasmids were sequenced to verify the presence of introduced mutations and the absence of additional, unwanted mutations.
Cell-based ELISA.
NA was expressed on HEK293 cells by transfection with wt or mutant plasmids using Lipofectamine 2000 reagent (Invitrogen, Grand Island, NY). Expression of NA containing the single mutation D199N, W296A, G370K, G401K, or S403K was poor, as confirmed with hyperimmune mouse serum against BR/07 virus (with a hemagglutination inhibition titer of ≥640), and therefore only the remaining 14 mutant NAs were examined in the subsequent cell-based ELISAs. The transfected cells were fixed with glutaraldehyde and blocked with 3% bovine serum albumin (BSA) in phosphate-buffered saline (PBS). Cells were then incubated with cell culture supernatant of hybridomas or purified MAbs, followed by incubation with horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Sigma-Aldrich). The signal was developed using o-phenylenediamine dihydrochloride (OPD) as the substrate. The reaction was stopped with sulfuric acid, and the optical density at 490 nm (OD490) was read. Hyperimmune mouse serum was used as a positive control. In assays to identify epitopes, the signals generated by MAb binding to mutant NAs were normalized to the signal generated by the positive control and therefore expressed as relative binding. A decrease of >20% in relative binding compared to wt NA was arbitrarily considered significant for the purpose of epitope identification.
ELLA.
The inhibition of NA enzyme activity by MAbs was measured with an enzyme-linked lectin assay (ELLA) in 96-well plate format as described previously (29). Briefly, serial dilutions of MAbs were mixed with a predetermined amount of virus diluted with 1% BSA in PBS containing Tween 20 (PBST). The mixture was transferred to 96-well plates coated with fetuin (Sigma-Aldrich) and incubated overnight at 37°C. Plates were washed with PBST, followed by the addition of peanut agglutinin conjugated to HRP (Sigma-Aldrich). Plates were incubated at room temperature for 2 h in the dark and then washed with PBST before the addition of OPD substrate. The reaction was stopped, and OD490 values were read. The median inhibition concentration (IC50) was calculated as the inverse dilution of antibody that resulted in 50% inhibition of NA activity.
Plaque size reduction assay.
Confluent MDCK cells in six-well plates were inoculated with virus dilutions. After 15 min at 4°C, cells were incubated at 37°C for 45 min and then overlaid with agar supplemented with MAbs. Cells inoculated with virus and overlaid with agar without MAbs were set up as controls. On day 3 after virus inoculation, cells were stained with crystal violet solution to visualize plaques.
Selection of MAb escape mutant viruses and identification of NA mutations.
MAb escape variants of BR/07 were selected as previously reported (30). Briefly, ascitic fluid containing MAbs was mixed with 106 PFU of cloned, wt BR/07 virus. After incubation at room temperature, the mixture was inoculated into eggs. Successful virus growth was confirmed by a hemagglutination assay, and the potential escape variants were further cloned in a plaque assay in the presence of MAb. Three plaques with sizes larger than those formed by the parental wt virus were picked for each variant and propagated in eggs. Allantoic fluid was collected, and the NA gene of each variant was amplified by reverse transcription-PCR (RT-PCR) (31). PCR products were sequenced at the core facility, Center for Biologics Evaluation and Research (CBER), FDA. Sequences were analyzed to identify nucleotide and amino acid changes.
Mouse challenge experiments.
The cross-reactive group B MAbs 1H5 and 3H10 were administered intraperitoneally at 15 mg/kg of body weight to groups of nine DBA/2 mice (7 weeks old; The Jackson Laboratory) 12 h before intranasal challenge with 10 median mouse lethal doses (MLD50) of seasonal H1N1 BR/07, 09pdm H1N1 CA/09-X179A, or attenuated H5N1 ΔVN/04 virus. On day 3 postchallenge (p.c.), four mice from each group were euthanized, and the lungs were collected for virus titration in MDCK cells. Body weights and mortality of the remaining mice were monitored for up to 14 days. Mice that lost more than 25% of body weight were euthanized. To examine the prophylactic efficacy of the same set of MAbs against challenge with wild-type (wt), highly pathogenic H5N1 virus, MAbs were administered at the same dose (15 mg/kg) to groups of 15 BALB/c mice (7 weeks old; The Jackson Laboratory) before challenge with 1 MLD50 or 20 MLD50 of wt VN/04 virus. Animal monitoring (n = 10) and virus titrations (n = 5) were performed as for H1N1 viruses. Mice that received the strain-specific group A MAb 3A2 and PBS served as controls for both H1N1 and H5N1 challenges. The wt H5N1 virus challenge was conducted in enhanced biosafety level 3 (BSL3+/ABSL3+) facilities under a protocol approved by the University of Maryland Animal Care and Use Committee.
RESULTS
Breadth of reactivity of N1-specific MAbs.
To identify antigenic domains of N1 and, in particular, to identify N1 epitopes that are conserved in H1N1 and H5N1 viruses, we generated mouse hybridomas secreting MAbs against the NA of BR/07 (H1N1), a component of the 2008-2009 and 2009-2010 seasonal influenza vaccines. Twenty-five N1-specific MAbs were selected for further characterization. These MAbs were placed into six different groups (A to F) based on their reactivity with the NAs of H1N1 and H5N1 viruses in ELISAs (Table 2).
Table 2.
Reactivities of N1 MAbs with the NA of H1N1 and H5N1 viruses
| MAb group | MAb | Isotype | Reactivity with N1 of the indicated virusa |
|||||||
|---|---|---|---|---|---|---|---|---|---|---|
| BR/07 | NIH/09 | TX/91 | NC/99 | SI/06 | BM/18 | CA/09 | VN/04 | |||
| A | 1C4 | IgG2b | + | + | − | − | − | − | − | − |
| 2D12 | IgG1 | + | + | − | − | − | − | − | − | |
| 3A2 | IgG2a | + | + | − | − | − | − | − | − | |
| 4D6 | IgG2b | + | + | − | − | − | − | − | − | |
| 4G2 | IgG1 | + | + | − | − | − | − | − | − | |
| B | 1H5 | IgG2b | + | + | + | + | + | + | + | + |
| 2D9 | IgG2b | + | + | + | + | + | + | + | + | |
| 2G6 | IgG2b | + | + | + | + | + | + | + | + | |
| 3H10 | IgG2b | + | + | + | + | + | + | + | + | |
| 4E9 | IgG2b | + | + | + | + | + | + | + | + | |
| C | 1C7 | IgG2a | + | + | − | + | ± | − | − | − |
| 2C3 | IgG2b | + | + | ± | + | + | − | − | − | |
| 2F4 | IgG2a | + | + | ± | + | + | − | − | − | |
| 3C2 | IgG1 | + | + | + | + | + | − | − | − | |
| 3G1 | IgG3 | + | ± | ± | ± | ± | − | − | − | |
| D | 2B4 | IgG2a | + | + | ± | + | + | + | − | − |
| 2B5 | IgG2a | + | + | + | + | + | ± | − | − | |
| 3C6 | IgG2a | + | + | + | + | + | + | − | − | |
| 3H4 | IgG2b | + | + | ± | + | + | + | − | − | |
| E | 1C9 | IgG2a | + | + | + | + | + | + | − | ± |
| 1H8 | IgG2b | + | + | + | + | + | + | − | ± | |
| 2D4 | IgG2b | + | + | + | + | + | + | − | + | |
| 3H3 | IgG2b | + | + | + | + | + | + | − | + | |
| 4B12 | IgG2b | + | + | + | + | + | + | − | + | |
| F | 4C4 | IgG2a | + | + | − | + | + | + | + | − |
Values were determined in ELISAs with H1N1 viruses A/Brisbane/59/2007 (BR/07), A/Texas/36/1991 (TX/91), A/New Caledonia/20/1999 (NC/99), A/Solomon Islands/3/2006 (SI/06), and A/California/07/2009 (CA/09)-X179A and the NA of A/Bethesda/NIH50/2009 (NIH/09), A/Brevig Mission/1/1918 (BM/18), and A/Vietnam/1203/2004 (VN/04) expressed on HEK293 cells. +, OD490 of >0.3; −, OD490 of <0.15; ±, OD490 value between 0.15 and 0.30.
Group A MAbs recognized only the NAs of BR/07 and NIH/09. The NA of NIH/09 shares 99% identity with that of BR/07 at both the nucleotide and amino acid levels, indicating that these MAbs are strain specific. In contrast, MAbs in groups B to F displayed various patterns of cross-reactivity. Specifically, group B MAbs were highly cross-reactive, binding to the NAs of all the tested seasonal H1N1 viruses, 18pdm H1N1 virus BM/18, and 09pdm H1N1 virus CA/09 as well as an H5N1 virus VN/04. Group E MAbs differed from those in group B in that they did not bind the NA of 09pdm H1N1, while the single group F MAb 4C4 was positive for the NAs of all tested viruses except those of H5N1 and H1N1 TX/91. MAbs in groups C and D exhibited narrower reactivity spectra than those in groups B, E, and F.
To determine whether the broad reactivity of group B MAbs extends to other NA subtypes (N2 to N9), an IFA was performed in MDCK cells infected with a broad spectrum of viruses (Table 1). All five of the MAbs in this group were tested. The IFA results revealed that group B MAbs recognized only N1, with reactivity to Eurasian human H1N1 and avian H6N1 viruses but not other NA subtypes (Fig. 1). Notably, group B MAbs did not react with two avian N1 viruses tested, A/Duck/Eastern China/103/03 (Y103, H1N1) and A/Duck/Eastern China/233/03 (Y233, H3N1). This is probably due to an amino acid mutation at position 338 in the NA, a position that we identified as crucial for epitope recognition by group B MAbs (see later results and discussion). We subsequently focused on characterizing group B MAbs since these MAbs had the broadest cross-reactivity with H1N1 and H5N1 virus NAs; other groups of MAbs, particularly those from group A, were included in relevant assays for comparison.
Fig 1.
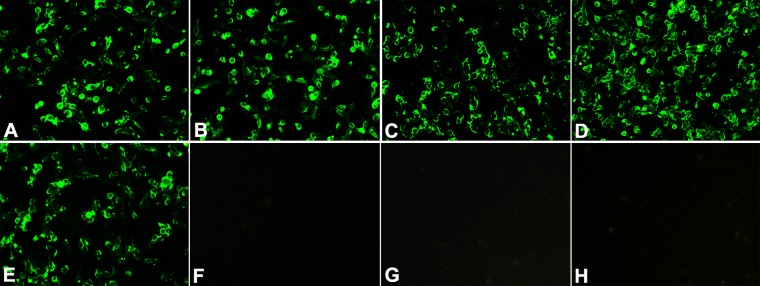
Reactivity of cross-reactive group B MAbs to N1 and other NA subtypes in IFAs. MDCK cells were infected with BR/07 (human H1N1) (A), CA/09 (human H1N1) (B), A/Chongqing/150/2007 (human H1N1) (C), A/Guangdong/51/2008 (human H1N1) (D), A/duck/Eastern China/1/2008 (avian H6N1) (E), A/duck/Eastern China/103/03 (Y103, avian H1N1) (F), A/duck/Eastern China/233/03 (Y233, avian H3N1) (G), A/turkey/Ontario/6188/68 (avian H8N4) (F), and other viruses as listed in Table 1. IFAs were performed with all group B MAbs, and the same reactivity patterns were observed. Shown are images generated with group B MAb 1H5. The remaining viruses tested (listed in Table 1) were negative for staining with group B MAbs, and therefore images are not shown.
Functional attributes of group B MAbs.
MAbs were tested for inhibition of NA activity by ELLA (29), which utilizes the large molecule fetuin as the substrate (Table 3). Group B MAbs inhibited BR/07 NA activity efficiently, with IC50s of 23.6 to 68.1 ng/ml, and moderately inhibited the activity of VN/04 NA (IC50, 1.41 to 2.42 μg/ml). The strain-specific group A MAbs inhibited only the homologous BR/07 NA, with IC50s of 10.4 and 7.7 ng/ml for MAbs 3A2 and 4G2, respectively. Surprisingly, despite reactivity in ELISA, none of the group B MAbs inhibited CA/09 NA even at 32 μg/ml, the highest concentration tested.
Table 3.
Inhibition of enzyme activity by N1-specific MAbs
| MAb group | MAb | IC50 for the indicated virus (ng/ml)a |
||
|---|---|---|---|---|
| BR/07 | CA/09 | VN/04 | ||
| A | 3A2 | 10.4 | > | > |
| 4G2 | 7.7 | > | > | |
| B | 1H5 | 25.9 | > | 1,670 |
| 2D9 | 68.1 | > | 2,160 | |
| 2G6 | 31.2 | > | 1,410 | |
| 3H10 | 23.6 | > | 1,250 | |
| 4E9 | 27.0 | > | 2,420 | |
| C | 1C7 | 85.7 | > | > |
| 3C2 | 33.2 | > | > | |
| D | 2B5 | 28.2 | > | > |
| 3H4 | 63.4 | > | > | |
| E | 1H8 | 14.7 | > | > |
| 4B12 | 50.9 | > | 8,599 | |
| 2D4 | 196 | > | > | |
| F | 4C4 | 1,184 | > | > |
| CD6b | > | 44.0 | > | |
| Negative controlc | > | > | > | |
IC50, median inhibition concentration measured by ELLA of the reassortant H6N1 viruses containing the NA of each indicated virus (see Materials and Methods). >, greater than 32,000 ng/ml.
CD6 is a MAb raised against 09pdm H1N1 NA in our laboratory using a protocol similar to that for generating BR/07 NA MAbs.
Negative control, mouse MAb against influenza A virus nucleoprotein (ViroStat, Portland, ME).
A similar pattern of inhibition was evident in plaque size reduction assays using virus-infected MDCK cells (Fig. 2); group B MAbs significantly reduced the size of the homologous BR/07 as well as attenuated H5N1 VN/04 virus (ΔVN/04) plaques when applied at a concentration of 1 μg/ml in the overlaying agar. Group A MAbs reduced the size of only homologous BR/07 plaques. Consistent with the results obtained in ELLAs, none of the group B MAbs reduced the size of plaques formed by H6N1CA/09 virus (H6N1CA/09 virus was used as CA/09 does not form clear plaques in MDCK cells) at 1 μg/ml. However, at a higher concentration of 10 μg/ml, group B MAbs resulted in smaller plaques (Fig. 2, bottom row), with plaques formed in the presence of MAbs 1H5 and 3H10 having an average diameter of 0.97 and 1.18 mm, respectively, nearly half that of plaques formed in the presence of group A MAb 3A2 or in the absence of MAb (1.94 and 2.03 mm, respectively).
Fig 2.

Reduction of plaque size by cross-reactive group B MAbs. MDCK cells growing in six-well plates were inoculated with cloned, wt BR/07, attenuated VN/04 (ΔVN/04), or reassortant H6N1CA/09 virus and overlaid with agar supplemented with 1 μg/ml (the top three rows) or 10 μg/ml (the bottom row) of MAbs. Shown are images of plaques formed by these viruses in the presence of strain-specific group A MAb 3A2 and cross-reactive group B MAbs 1H5 and 3H10. The virus control shows plaques formed in the absence of MAb.
Identification of N1 residues crucial for MAb binding.
To map the epitope(s) recognized by N1-specific MAbs, mutant N1s bearing each of 14 single amino acid mutations were expressed in HEK293 cells, and MAb binding to each mutant NA was tested in a cell-based ELISA. None of the tested mutations showed a profound effect on binding of group B MAbs (data not shown). However, an N248D or K249E mutation abolished the binding of group A MAbs 3A2 and 4G2 (Fig. 3). In addition, a D341N mutation significantly affected the binding of 3A2 (>35% decrease in signal compared to wt N1) and abolished the binding of group C MAb 1C7.
Fig 3.

Effect of single amino acid mutations on MAb binding to N1. Binding of group A MAb 3A2 (A), group A MAb 4G2 (B), and group C MAb 1C7 (C) to mutant N1s was examined by cell-based ELISAs using HEK293 cells transfected with a panel of plasmids carrying each of 14 different single amino acid mutations. The signal generated with each MAb (1 μg/ml) was normalized to that of mouse hyperimmune serum and was expressed as relative binding.
To expand our search for key amino acids within group B epitope(s), we selected MAb escape variants of BR/07 in eggs. Variants selected with group B MAbs 1H5, 2D9, 2G6, and 3H10 all contained a single mutation in NA at position 339, changing from the parental T to A, I, N, or P (Table 4). This was not the only change selected by this MAb group. The variant selected by MAb 4E9 and a 3H10 variant had a V338M substitution in NA, while MAb 3H10 selected an additional variant carrying an N273D mutation. All three of these residues are located within loops on the perimeter of the NA head (Fig. 4).
Table 4.
Amino acid changes in NA of mutant BR/07 viruses selected with N1 MAbs
| MAb group | MAb for selection | Mutanta | Amino acid changeb |
|---|---|---|---|
| A | 3A2 | 3A2v1 | A250S |
| 4G2 | 4G2v1 | K249E | |
| 4G2v2 | A343E | ||
| B | 1H5 | 1H5v1 | T339A |
| 1H5v2 | T339I | ||
| 1H5v3 | T339N | ||
| 2D9 | 2D9v1 | T339I | |
| 2D9v2 | T339P | ||
| 2G6 | 2G6v1 | T339A | |
| 2G6v2 | T339I | ||
| 3H10 | 3H10v1 | N273D | |
| 3H10v2 | V338 M | ||
| 3H10v3 | T339N | ||
| 3H10v4 | T339P | ||
| 4E9 | 4E9v1 | V338 M | |
| C | 1C7 | 1C7v1 | D341N |
| 3C2 | 3C2v1 | T339A | |
| 3C2v2 | T339I | ||
| D | 2B5 | 2B5v1 | T339N |
| 3H4 | 3H4v1 | T397N | |
| E | 1H8 | 1H8v1 | T397A |
| 2D4 | 2D4v1 | I396T | |
| 2D4v2 | W456G | ||
| F | 4C4 | 4C4v1 | N309S |
MAb escape mutants of wt BR/07 were selected in eggs, followed by plaque purification.
Only a single mutation was detected in the NA of each escape variant.
Fig 4.

Location of 12 critical residues in N1 epitopes on an N1 dimer (Protein Data Bank code 3TI6). The image in the top panel was generated with PyMol software (Delano Scientific). Residues recognized by strain-specific group A MAbs and cross-reactive group B and E MAbs are highlighted in magenta, green, and yellow, respectively; residue 309 recognized by group F MAb 4C4 is at the bottom of the NA region and is shown in blue. Note that residues 339, 341, and 397 are shared by epitopes recognized by two or three groups of MAbs (Table 4 and Fig. 3). The bottom panel shows an alignment of the 12 residues in N1 viruses described in the present study.
Variants were also similarly selected with antibodies from groups A, C, D, E, and F. Three amino acid changes, K249E, A250S, and A343E, were identified in variants selected with group A antibodies. Group C MAbs selected variants with D341N, T339A, or T339I in the NA (Table 4). Notably, K249E and D341N mutations were independently identified in cell-based ELISAs with mutant NAs. Selection with group E MAbs resulted in escape variants containing mutations at residue 396, 397, or 456. A single variant with an N309S substitution, located on the bottom surface of the NA head region, was isolated after multiple rounds of selection with group F MAb 4C4, a weak inhibitor of NA activity. The NA activity of MAb escape mutant viruses was not substantially inhibited by the MAbs used for selection, with mutant viruses at least 10-fold less sensitive to inhibition by the selecting MAbs than wt BR/07 virus (Table 5), showing that the positions where amino acid mutations occurred are crucial for MAb binding/inhibition of NA.
Table 5.
Inhibition of NA activity of MAb escape mutants by selecting MAbs
| MAb group | MAb | IC50 (ng/ml) of MAb escape mutant bearing the indicated mutationa |
|||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| wt BR/07b | K249E | A250S | A343E | N273D | V338M | T339A | T339I | T339N | T339P | D341N | I396T | T397A | T397N | W456G | N309S | ||
| A | 3A2 | 10.4 | > | ||||||||||||||
| 4G2 | 7.7 | > | > | ||||||||||||||
| B | 1H5 | 25.9 | > | > | > | ||||||||||||
| 2G6 | 68.1 | > | > | ||||||||||||||
| 2D9 | 31.2 | > | > | ||||||||||||||
| 3H10 | 23.6 | > | > | > | > | ||||||||||||
| 4E9 | 27.0 | 1,269 | |||||||||||||||
| C | 1C7 | 85.7 | > | ||||||||||||||
| 3C2 | 33.2 | > | > | ||||||||||||||
| D | 2B5 | 28.2 | > | ||||||||||||||
| 3H4 | 63.4 | > | |||||||||||||||
| E | 1H8 | 14.7 | 200 | ||||||||||||||
| 2D4 | 196 | > | > | ||||||||||||||
| F | 4C4 | 1184 | ≫ | ||||||||||||||
IC50, median inhibition concentration measured with enzyme-linked lectin assay. Each mutant virus was tested only with the MAb used for its selection. >, no inhibition at 2 μg/ml, the highest MAb concentration tested; ≫, no inhibition at 16 μg/ml, the highest MAb concentration tested.
IC50 of wt BR/07 virus was included for comparison.
Taking these results together, we have identified 12 residues within antigenic epitopes of N1. These residues are shown on a model of N1 (Fig. 4, upper panel), together with an alignment of these residues in the NA of N1 viruses described in this study (Fig. 4, bottom panel). Of these, residues 273, 338, and 339 are essential for the binding/inhibition of NA by group B MAbs, while residues 396, 397, and 456 correspond to epitope(s) recognized by group E MAbs. Five residues, at positions 248, 249, 250, 341, and 343, are within the epitope(s) recognized by the strain-specific group A MAbs, while residue 309 is critical for the binding of group F MAb 4C4 to its corresponding epitope.
Although escape mutants could be selected by group B MAbs in this study, the epitope(s) recognized by these MAbs appears to be highly conserved in the NAs of H1N1 (seasonal and pandemic) viruses and to a lesser extent in H5N1 viruses. Among 10,630 human H1N1 NA sequences available in GenBank (as of 13 December 2012), N273 is universally conserved, while V338 and S/T339 (both S339 and T339 are recognized by group B MAbs) are present in ≥99% of these H1N1 viruses. In the NAs of 3,147 H5N1 viruses (GenBank; as of 13 December 2012), V338 is present in 83% of the sequences, while S/T339 is present in 93%.
Group B MAbs protect mice against both seasonal and pandemic H1N1 challenge.
Since group B antibodies had the broadest reactivity, we evaluated their ability to protect mice against lethal challenge with homologous H1N1 virus BR/07 and heterologous 09pdm H1N1 virus CA/09-X179A. DBA/2 mice, which are highly susceptible to most influenza A virus subtypes (32), were inoculated intranasally with 10 MLD50 of either BR/07 or CA/09-X179A 12 h after intraperitoneal administration of group A MAb 3A2, group B MAb 1H5, or a second group B MAb, 3H10, each at a dose of 15 mg/kg. All three of these MAbs fully protected mice from the lethal BR/07 challenge (Fig. 5A). Mice lost weight after the challenge but began to regain weight on day 4 or 5 postchallenge (p.c.) (Fig. 5B). Viral titration of mouse lungs collected on day 3 p.c. showed that mice treated with group A MAb 3A2 had an average titer ∼1,000-fold lower than mice in the PBS control group. Although all animals survived the challenge, interestingly, virus titers in the lungs of group B MAb (1H5 and 3H10)-treated mice were only slightly (2- and 4-fold) lower than those of the PBS control mice (Fig. 5C).
Fig 5.

Prophylactic efficacy of cross-reactive group B MAbs in mice against H1N1 and H5N1 challenge. (A to I) DBA/2 mice (9/group) and (J to L) BALB/c mice (15/group) were treated with group A MAb 3A2 and group B MAb 1H5 or 3H10 (15 mg/kg) intraperitoneally 12 h before virus challenge. Mice treated with PBS served as a control. DBA/2 mice were challenged intranasally with 10 MLD50 of seasonal H1N1 BR/07 (A to C), 09pdm H1N1 CA/09-X179A (D to F), or attenuated H5N1 ΔVN/04 virus (G to I). BALB/c mice were challenged with 20 MLD50 of wt VN/04 virus. Mortality (A, D, G, and J) and body weight (B, E, H, and K) were monitored daily for 14 days, and on day 3 postchallenge, animals (4/group for DBA/2 mice and 5/group for BALB/c mice) were euthanized, and lung viral titers (C, F, I, and L) determined in MDCK cells.
In the CA/09-X179A challenge experiment, administration of MAb 3A2 did not protect the mice against this heterologous virus infection. All mice died or had to be euthanized as did those in the PBS control group, confirming the strain specificity of this MAb. In contrast, group B MAbs 1H5 and 3H10 protected 80% and 100% of mice, respectively (Fig. 5D). Animals in both groups lost less than 20% of starting weight and began to regain weight at ∼1 week p.c. (Fig. 5E). Virus titers in the lungs of the 1H5- and 3H10-treated mice on day 3 p.c. were lower (18- and 240-fold, respectively) than those in 3A2- and the PBS-treated control groups (Fig. 5F). Protection against CA/09-X179A challenge by the cross-reactive MAbs 1H5 and 3H10 but not the strain-specific MAb 3A2 shows that the in vivo protection was mediated by a specific interaction between group B MAbs and the challenge virus. To further confirm the specificity of the protection provided by group B MAbs, we examined the protection of the group B MAb 3H10 against an H3N2 virus, A/Hong Kong/1/68-X31 (HK/68-X31). As shown in Fig. 6, while 3H10 fully protected mice against CA/09-X179A, it did not provide protection against HK/68-X31. These data demonstrate that protection against CA/09-X179A was due to the specific recognition of the H1N1 virus rather than a nonspecific effect.
Fig 6.

Group B MAbs protect mice against H1N1 but not H3N2 virus challenge. DBA/2 mice (5/group) were treated with group B MAb 3H10 (15 mg/kg) intraperitoneally 12 h before challenge with 10 MLD50 of 09pdm H1N1 CA/09-X179A or H3N2 A/Hong Kong/1/1968-X31 (HK/68-X31) virus. Mice treated with PBS and challenged with HK/68-X31 served as a control. Mortality (A) and body weight (B) were monitored daily for 14 days.
To evaluate the efficacy of group B MAbs and also understand why virus titers in the lungs harvested from group B MAb-treated mice were not reduced to a greater extent 3 days after challenge with BR/07 (Fig. 5C), we tested MAb 3H10 in a dose-dependent BR/07 challenge study. A single dose of 3H10 administered 12 h before challenge provided full protection to mice at ≥2 mg/kg and partial protection at a lower dose of 0.5 mg/kg (Fig. 7A and B). Again, the difference between virus titers in lungs harvested 3 days after challenge of the 3H10-treated mice and those treated with the control antibody was small. However, by day 6 p.c., the lung viral titers of mice treated with ≥2 mg/kg of 3H10 were 600- to 10,000-fold lower than those of mice treated with the control MAb (Fig. 7C). At 0.5 mg/kg, the reduction in virus titer on day 6 postchallenge was evident but low (15-fold), and there was no reduction in virus load when mice were treated with 0.1 mg/kg of 3H10, consistent with the lack of protection against death. These data demonstrate that the group B MAb did, indeed, impact BR/07 replication, although with different kinetics compared to the strain-specific MAb 3A02.
Fig 7.

Dose-dependent prophylactic efficacy of group B MAbs against BR/07 (H1N1). DBA/2 mice (9/group) were treated with group B MAb 3H10 (15, 5, 2, 0.5, and 0.1 mg/kg) intraperitoneally 12 h before challenge with 10 MLD50 of BR/07. Mice treated with the 09pdm H1N1 NA-specific MAb CD6 (Table 3) at 15 mg/kg served as a control. Mortality (A) and weight (B) were monitored daily for 14 days. (C) lung viral titers were measured for 4 mice per group on days 3 and 6 postchallenge.
A single dose of group B MAb provides significant protection against highly pathogenic H5N1 infection.
We further assessed the ability of group B MAbs to protect against the attenuated H5N1 virus ΔVN/04 in DBA/2 mice and the highly pathogenic wt VN/04 virus in BALB/c mice. MAbs 1H5 and 3H10 fully protected DBA/2 mice from death caused by 10 MLD50 of ΔVN/04 virus (Fig. 5G) and clearly impacted virus replication, with lung virus titers of the 1H5- and 3H10-treated mice ∼100-fold lower than those in control mice (Fig. 5I). All BALB/c mice that received MAb 1H5 or 3H10 survived a low-dose (1 MLD50) wt VN/04 challenge (data not shown). When BALB/c mice were challenged with a relatively high dose of wt VN/04 (20 MLD50), a single dose of 1H5 and 3H10 given prophylactically significantly delayed the disease process, with body weights of the MAb-treated mice remaining stable until day 7 p.c. Substantial weight loss was observed thereafter, and the mice began to die by days 9 and 10. In contrast, mice in control groups (PBS- and group A MAb 3A2-treated mice) began to die on day 6 p.c., and all had died by day 9. It is remarkable that without additional group B MAb treatment, 20% of mice in each group survived (Fig. 5J and K). The delay in disease course corresponded with an early reduction in virus load: on day 3 p.c., the virus load in 1H5- and 3H10-treated mice was ∼100-fold lower than that in control animals (Fig. 5L).
DISCUSSION
Although the molecular structure of N1 has been defined (33–35), antigenic domains recognized by strain-specific and broadly reactive antibodies have not been mapped. Changes in sequence over time often point to amino acids that are recognized by antibodies; however, it is difficult to predict which antigenic domains are epidemiologically important because single mutations can have a large impact even on binding of polyclonal antiserum (27). Moreover, conserved epitopes on N1 that contribute to broad protection against heterologous viruses, including H5N1 (16, 17, 36), have not been identified.
We have used a relatively large panel of MAbs to identify the antigenic domains of N1. Differences in MAb binding to the NAs of seasonal and pandemic (18pdm and 09pdm) H1N1, as well as H5N1 viruses, highlight the presence of several epitopes on N1. Group A MAbs inhibited the NA activity of homologous virus in vitro most efficiently, reflecting the proximity of group A epitope(s) to the NA active site. The highly cross-reactive group B MAbs recognize epitope(s) containing residues 273, 338, and 339. Residues 339 and 341 are also shared by the epitope(s) corresponding to groups C and D MAbs, suggesting the presence of overlapping N1 epitopes, as has been observed for N2 (23).
In the epitope(s) recognized by group E MAbs, seasonal H1N1 and H5N1 viruses encode a T at position 397, while 09pdm H1N1 viruses have an N, explaining why group E MAbs recognize seasonal H1N1 and avian H5N1 viruses but not 09pdm H1N1 virus. The escape mutant selected with group F MAb 4C4 carries an amino acid change (N309S) on the bottom surface of the NA head, while changes detected in other groups of escape mutants are on the top surface. The weak inhibition of NA activity by 4C4 suggests that the corresponding epitope is located far from the NA active site. Group D MAbs 2B5 and 3H4 had similar reactivity patterns, binding NAs of seasonal and 18pdm H1N1 viruses but not the NA of either 09pdm H1N1 or H5N1. However, they selected variants with mutations at seemingly different positions on the tertiary structure (339 and 397, respectively), suggesting that the corresponding epitope(s) spans the loops containing these residues, as does the N9 epitope recognized by MAb NC41 (24). A clear picture of all residues within each N1 epitope will not be available until the crystal structures of N1-antibody complexes are solved.
The epitope(s) recognized by group B MAbs is likely highly conserved, particularly in H1N1 viruses, with three residues, N273, V338, and S/T339, critical for antibody binding/inhibition of NA. In the present study, group B MAbs 3H10 and 4E9 selected escape variants with the V338M mutation in NA. Consistent with this observation, the NAs of duck viruses Y103 (H1N1) and Y233 (H3N1) bear M residues at position 338, explaining why group B MAbs did not recognize these two viruses in IFAs. Viruses that possess either S339 or T339 are well recognized by group B MAbs. A small proportion of H1N1 and H5N1 viruses encode different residues at this position, e.g., A, F, L, P, V, or Y, but viruses bearing these substitutions have not become dominant in the population. Residue 339 is also critical for the binding of some group C and D antibodies (e.g., 3C2 and 2B5, respectively). Considering these findings and the fact that the group B epitope(s) is present in the 18pdm H1N1 virus as well as contemporary seasonal H1N1, 09pdm H1N1, and H5N1 viruses, it is plausible that residues within this epitope(s) are important for maintaining the structure and function of N1.
NA-specific immunity reduces the severity of clinical illness and virus shedding in influenza virus-infected persons (10, 37–39). This has been attributed in part to the inhibition of NA enzyme activity by NA-specific antibodies. In our study, the in vitro inhibition of NA activity of seasonal H1N1 and H5N1 viruses by group A and B MAbs was consistent with their protective efficacy in mice. Group B MAbs did not inhibit the NA activity of 09pdm H1N1 virus in the enzyme inhibition assay and only reduced 09pdm H1N1 virus plaque size at high concentrations. However, group B MAbs protected mice against lethal 09pdm H1N1 virus challenge. Although an alternative mechanism of protection, other than the direct inhibition of viral NA, is feasible, it is also possible that the in vitro assays used to measure enzyme inhibition do not reflect the antibodies' ability to inhibit other substrates that are relevant in vivo.
Recently, there have been several reports describing NA-associated serum cross-reactivity and heterologous protection among seasonal H1N1, 09pdm H1N1, and H5N1 viruses. In addition to the cross-reactivity/protection observed in the mouse model (15–17, 36), there is evidence that seasonal influenza vaccination increased antibody responses to the NA of 09pdm H1N1 in the elderly (40) and there is measurable NA-specific cross-reactivity between ferret serum against 09pdm H1N1 and H5N1 viruses (14). The presence of conserved group B, E, and F epitopes is likely the molecular basis for such cross-reactivity and heterologous protection. A single dose of the highly cross-reactive group B MAbs (e.g., 3H10) administered prophylactically provided full protection against lethal challenge with seasonal H1N1, 09pdm H1N1, or attenuated H5N1 virus in the highly susceptible DBA/2 mouse model. Group B MAbs also provided significant protection against highly pathogenic H5N1 virus challenge in BALB/c mice, reducing virus replication in lungs and delaying the disease process. Our findings add to the concept that immunization with both HA and NA could elicit broader immunity than immunization with HA alone (41–43). Conserved, protective N1 epitopes, together with conserved HA epitopes (44–47), are thus ideal targets for the development of universal influenza vaccines. Antibodies against these epitopes, administered alone or as a cocktail with broadly reactive HA antibodies, may be useful therapeutics to add to the currently limited arsenal to treat severe H1N1 and H5N1 infections.
ACKNOWLEDGMENTS
This work was supported in part by the Intramural Research Program of the NIH and the NIAID (J.K.T.) and by intramural FDA funds. L.Z. and X.L. were funded by the Major State Basic Research Development Program of China (973 Program grant number 2011CB505003). K.X., H.C., and D.R.P. were funded by contract HHSN266200700010C from the NIAID, NIH. L.K.C., K.Y., M.R., and I.S. were supported by training funds administered by the Oak Ridge Institute for Science and Education.
We thank Yuelong Shu from China CDC for providing some of the viruses used in IFA and are indebted to staff of the Division of Veterinary Services, CBER, FDA, for excellent animal care.
Footnotes
Published ahead of print 19 June 2013
REFERENCES
- 1. Palese P, Tobita K, Ueda M, Compans RW. 1974. Characterization of temperature sensitive influenza virus mutants defective in neuraminidase. Virology 61:397–410 [DOI] [PubMed] [Google Scholar]
- 2. Lakdawala SS, Lamirande EW, Suguitan AL, Jr, Wang W, Santos CP, Vogel L, Matsuoka Y, Lindsley WG, Jin H, Subbarao K. 2011. Eurasian-origin gene segments contribute to the transmissibility, aerosol release, and morphology of the 2009 pandemic H1N1 influenza virus. PLoS Pathog. 7:e1002443. 10.1371/journal.ppat.1002443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Matrosovich MN, Matrosovich TY, Gray T, Roberts NA, Klenk HD. 2004. Neuraminidase is important for the initiation of influenza virus infection in human airway epithelium. J. Virol. 78:12665–12667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Tong S, Li Y, Rivailler P, Conrardy C, Castillo DA, Chen LM, Recuenco S, Ellison JA, Davis CT, York IA, Turmelle AS, Moran D, Rogers S, Shi M, Tao Y, Weil MR, Tang K, Rowe LA, Sammons S, Xu X, Frace M, Lindblade KA, Cox NJ, Anderson LJ, Rupprecht CE, Donis RO. 2012. A distinct lineage of influenza A virus from bats. Proc. Natl. Acad. Sci. U. S. A. 109:4269–4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Pappas C, Aguilar PV, Basler CF, Solorzano A, Zeng H, Perrone LA, Palese P, Garcia-Sastre A, Katz JM, Tumpey TM. 2008. Single gene reassortants identify a critical role for PB1, HA, and NA in the high virulence of the 1918 pandemic influenza virus. Proc. Natl. Acad. Sci. U. S. A. 105:3064–3069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ma W, Liu Q, Bawa B, Qiao C, Qi W, Shen H, Chen Y, Ma J, Li X, Webby RJ, Garcia-Sastre A, Richt JA. 2012. The neuraminidase and matrix genes of the 2009 pandemic influenza H1N1 virus cooperate functionally to facilitate efficient replication and transmissibility in pigs. J. Gen. Virol. 93:1261–1268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ilyushina NA, Bovin NV, Webster RG. 2012. Decreased neuraminidase activity is important for the adaptation of H5N1 influenza virus to human airway epithelium. J. Virol. 86:4724–4733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kilbourne ED, Laver WG, Schulman JL, Webster RG. 1968. Antiviral activity of antiserum specific for an influenza virus neuraminidase. J. Virol. 2:281–288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Clements ML, Betts RF, Tierney EL, Murphy BR. 1986. Serum and nasal wash antibodies associated with resistance to experimental challenge with influenza A wild-type virus. J. Clin. Microbiol. 24:157–160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Murphy BR, Kasel JA, Chanock RM. 1972. Association of serum anti-neuraminidase antibody with resistance to influenza in man. N. Engl. J. Med. 286:1329–1332 [DOI] [PubMed] [Google Scholar]
- 11. Shoji Y, Chichester JA, Palmer GA, Farrance CE, Stevens R, Stewart M, Goldschmidt L, Deyde V, Gubareva L, Klimov A, Mett V, Yusibov V. 2011. An influenza N1 neuraminidase-specific monoclonal antibody with broad neuraminidase inhibition activity against H5N1 HPAI viruses. Hum. Vaccin. 7(Suppl):199–204 [DOI] [PubMed] [Google Scholar]
- 12. Schulman JL, Khakpour M, Kilbourne ED. 1968. Protective effects of specific immunity to viral neuraminidase on influenza virus infection of mice. J. Virol. 2:778–786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schulman JL, Kilbourne ED. 1969. The antigenic relationship of the neuraminidase of Hong Kong virus to that of other human strains of influenza A virus. Bull. World Health Organ. 41:425–428 [PMC free article] [PubMed] [Google Scholar]
- 14. Chen Z, Kim L, Subbarao K, Jin H. 2012. The 2009 pandemic H1N1 virus induces anti-neuraminidase (NA) antibodies that cross-react with the NA of H5N1 viruses in ferrets. Vaccine 30:2516–2522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Marcelin G, DuBois R, Rubrum A, Russell CJ, McElhaney JE, Webby RJ. 2011. A contributing role for anti-neuraminidase antibodies on immunity to pandemic H1N1 2009 influenza A virus. PLoS One 6:e26335. 10.1371/journal.pone.0026335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sandbulte MR, Jimenez GS, Boon AC, Smith LR, Treanor JJ, Webby RJ. 2007. Cross-reactive neuraminidase antibodies afford partial protection against H5N1 in mice and are present in unexposed humans. PLoS Med. 4:e59. 10.1371/journal.pmed.0040059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Easterbrook JD, Schwartzman LM, Gao J, Kash JC, Morens DM, Couzens L, Wan H, Eichelberger MC, Taubenberger JK. 2012. Protection against a lethal H5N1 influenza challenge by intranasal immunization with virus-like particles containing 2009 pandemic H1N1 neuraminidase in mice. Virology 432:39–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Rockman S, Brown LE, Barr IG, Gilbertson B, Lowther S, Kachurin A, Kachurina O, Klippel J, Bodle J, Pearse M, Middleton D. 2013. Neuraminidase-inhibiting antibody is a correlate of cross-protection against lethal H5N1 influenza virus in ferrets immunized with seasonal influenza vaccine. J. Virol. 87:3053–3061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gravel C, Li C, Wang J, Hashem AM, Jaentschke B, Xu KW, Lorbetskie B, Gingras G, Aubin Y, Van Domselaar G, Girard M, He R, Li X. 2010. Qualitative and quantitative analyses of virtually all subtypes of influenza A and B viral neuraminidases using antibodies targeting the universally conserved sequences. Vaccine 28:5774–5784 [DOI] [PubMed] [Google Scholar]
- 20. Air GM. 2012. Influenza neuraminidase. Influenza Other Respi. Viruses 6:245–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Air GM, Els MC, Brown LE, Laver WG, Webster RG. 1985. Location of antigenic sites on the three-dimensional structure of the influenza N2 virus neuraminidase. Virology 145:237–248 [DOI] [PubMed] [Google Scholar]
- 22. Gulati U, Hwang CC, Venkatramani L, Gulati S, Stray SJ, Lee JT, Laver WG, Bochkarev A, Zlotnick A, Air GM. 2002. Antibody epitopes on the neuraminidase of a recent H3N2 influenza virus (A/Memphis/31/98). J. Virol. 76:12274–12280 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Webster RG, Brown LE, Laver WG. 1984. Antigenic and biological characterization of influenza virus neuraminidase (N2) with monoclonal antibodies. Virology 135:30–42 [DOI] [PubMed] [Google Scholar]
- 24. Tulip WR, Varghese JN, Laver WG, Webster RG, Colman PM. 1992. Refined crystal structure of the influenza virus N9 neuraminidase-NC41 Fab complex. J. Mol. Biol. 227:122–148 [DOI] [PubMed] [Google Scholar]
- 25. Venkatramani L, Bochkareva E, Lee JT, Gulati U, Graeme Laver W, Bochkarev A, Air GM. 2006. An epidemiologically significant epitope of a 1998 human influenza virus neuraminidase forms a highly hydrated interface in the NA-antibody complex. J. Mol. Biol. 356:651–663 [DOI] [PubMed] [Google Scholar]
- 26. Colman PM, Varghese JN, Laver WG. 1983. Structure of the catalytic and antigenic sites in influenza virus neuraminidase. Nature 303:41–44 [DOI] [PubMed] [Google Scholar]
- 27. Sandbulte MR, Westgeest KB, Gao J, Xu X, Klimov AI, Russell CA, Burke DF, Smith DJ, Fouchier RA, Eichelberger MC. 2011. Discordant antigenic drift of neuraminidase and hemagglutinin in H1N1 and H3N2 influenza viruses. Proc. Natl. Acad. Sci. U. S. A. 108:20748–20753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Higgins AD, Shaw CJ, Johnson JG, Navarro A, Chapman NA, Ewers SD, Stockwell JW, Carpenter JM, Olivo PD, Miao LY. 2010. Monoclonal antibody kit for identification of the novel 2009 H1N1 influenza A virus. J. Clin. Microbiol. 48:2677–2682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lambre CR, Terzidis H, Greffard A, Webster RG. 1990. Measurement of anti-influenza neuraminidase antibody using a peroxidase-linked lectin and microtitre plates coated with natural substrates. J. Immunol. Methods 135:49–57 [DOI] [PubMed] [Google Scholar]
- 30. Gerhard W, Webster RG. 1978. Antigenic drift in influenza A viruses. I. Selection and characterization of antigenic variants of A/PR/8/34 (HON1) influenza virus with monoclonal antibodies. J. Exp. Med. 148:383–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hoffmann E, Stech J, Guan Y, Webster RG, Perez DR. 2001. Universal primer set for the full-length amplification of all influenza A viruses. Arch. Virol. 146:2275–2289 [DOI] [PubMed] [Google Scholar]
- 32. Boon AC, deBeauchamp J, Krauss S, Rubrum A, Webb AD, Webster RG, McElhaney J, Webby RJ. 2010. Cross-reactive neutralizing antibodies directed against pandemic H1N1 2009 virus are protective in a highly sensitive DBA/2 mouse influenza model. J. Virol. 84:7662–7667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Li Q, Qi J, Zhang W, Vavricka CJ, Shi Y, Wei J, Feng E, Shen J, Chen J, Liu D, He J, Yan J, Liu H, Jiang H, Teng M, Li X, Gao GF. 2010. The 2009 pandemic H1N1 neuraminidase N1 lacks the 150-cavity in its active site. Nat. Struct. Mol. Biol. 17:1266–1268 [DOI] [PubMed] [Google Scholar]
- 34. Xu X, Zhu X, Dwek RA, Stevens J, Wilson IA. 2008. Structural characterization of the 1918 influenza virus H1N1 neuraminidase. J. Virol. 82:10493–10501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Russell RJ, Haire LF, Stevens DJ, Collins PJ, Lin YP, Blackburn GM, Hay AJ, Gamblin SJ, Skehel JJ. 2006. The structure of H5N1 avian influenza neuraminidase suggests new opportunities for drug design. Nature 443:45–49 [DOI] [PubMed] [Google Scholar]
- 36. Quan FS, Kim MC, Lee BJ, Song JM, Compans RW, Kang SM. 2012. Influenza M1 VLPs containing neuraminidase induce heterosubtypic cross-protection. Virology 430:127–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Couch RB, Kasel JA, Gerin JL, Schulman JL, Kilbourne ED. 1974. Induction of partial immunity to influenza by a neuraminidase-specific vaccine. J. Infect. Dis. 129:411–420 [DOI] [PubMed] [Google Scholar]
- 38. Beutner KR, Chow T, Rubi E, Strussenberg J, Clement J, Ogra PL. 1979. Evaluation of a neuraminidase-specific influenza A virus vaccine in children: antibody responses and effects on two successive outbreaks of natural infection. J. Infect. Dis. 140:844–850 [DOI] [PubMed] [Google Scholar]
- 39. Ogra PL, Chow T, Beutner KR, Rubi E, Strussenberg J, DeMello S, Rizzone C. 1977. Clinical and immunologic evaluation of neuraminidase-specific influenza A virus vaccine in humans. J. Infect. Dis. 135:499–506 [DOI] [PubMed] [Google Scholar]
- 40. Marcelin G, Bland HM, Negovetich NJ, Sandbulte MR, Ellebedy AH, Webb AD, Griffin YS, DeBeauchamp JL, McElhaney JE, Webby RJ. 2010. Inactivated seasonal influenza vaccines increase serum antibodies to the neuraminidase of pandemic influenza A(H1N1) 2009 virus in an age-dependent manner. J. Infect. Dis. 202:1634–1638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Johansson BE. 1999. Immunization with influenza A virus hemagglutinin and neuraminidase produced in recombinant baculovirus results in a balanced and broadened immune response superior to conventional vaccine. Vaccine 17:2073–2080 [DOI] [PubMed] [Google Scholar]
- 42. Chen Z, Matsuo K, Asanuma H, Takahashi H, Iwasaki T, Suzuki Y, Aizawa C, Kurata T, Tamura S. 1999. Enhanced protection against a lethal influenza virus challenge by immunization with both hemagglutinin- and neuraminidase-expressing DNAs. Vaccine 17:653–659 [DOI] [PubMed] [Google Scholar]
- 43. Bosch BJ, Bodewes R, de Vries RP, Kreijtz JH, Bartelink W, van Amerongen G, Rimmelzwaan GF, de Haan CA, Osterhaus AD, Rottier PJ. 2010. Recombinant soluble, multimeric HA and NA exhibit distinctive types of protection against pandemic swine-origin 2009 A(H1N1) influenza virus infection in ferrets. J. Virol. 84:10366–10374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ekiert DC, Bhabha G, Elsliger MA, Friesen RH, Jongeneelen M, Throsby M, Goudsmit J, Wilson IA. 2009. Antibody recognition of a highly conserved influenza virus epitope. Science 324:246–251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sui J, Hwang WC, Perez S, Wei G, Aird D, Chen LM, Santelli E, Stec B, Cadwell G, Ali M, Wan H, Murakami A, Yammanuru A, Han T, Cox NJ, Bankston LA, Donis RO, Liddington RC, Marasco WA. 2009. Structural and functional bases for broad-spectrum neutralization of avian and human influenza A viruses. Nat. Struct. Mol. Biol. 16:265–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ekiert DC, Friesen RH, Bhabha G, Kwaks T, Jongeneelen M, Yu W, Ophorst C, Cox F, Korse HJ, Brandenburg B, Vogels R, Brakenhoff JP, Kompier R, Koldijk MH, Cornelissen LA, Poon LL, Peiris M, Koudstaal W, Wilson IA, Goudsmit J. 2011. A highly conserved neutralizing epitope on group 2 influenza A viruses. Science 333:843–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Corti D, Voss J, Gamblin SJ, Codoni G, Macagno A, Jarrossay D, Vachieri SG, Pinna D, Minola A, Vanzetta F, Silacci C, Fernandez-Rodriguez BM, Agatic G, Bianchi S, Giacchetto-Sasselli I, Calder L, Sallusto F, Collins P, Haire LF, Temperton N, Langedijk JP, Skehel JJ, Lanzavecchia A. 2011. A neutralizing antibody selected from plasma cells that binds to group 1 and group 2 influenza A hemagglutinins. Science 333:850–856 [DOI] [PubMed] [Google Scholar]