

Abstract
Poxviruses, including vaccinia virus (VV) and canarypox virus (ALVAC), do not indiscriminately infect all cell types of the primary human leukocytes (PHLs) that they encounter but instead demonstrate an extremely strong bias toward infection of monocytes and monocyte lineage cells. We studied the specific molecular events that determine the VV tropism for major PHL subsets including monocytes, B cells, neutrophils, NK cells, and T cells. We found that VV exhibited an extremely strong bias of cell surface protein-dependent binding to monocytes, B cells, and activated T cells to a similar degree and to neutrophils to a much lesser extent. Resting T cells and resting NK cells exhibited only trace amounts of VV binding. Activated T cells, however, became permissive to VV binding, infection, and replication, while activated NK cells still resisted VV binding. VV binding strongly colocalized with lipid rafts on the surfaces of all VV binding-susceptible PHL subsets, even when lipid rafts were relocated to cell uropods upon cell polarization. Immunosera raised against detergent-resistant membranes (DRMs) from monocytes or activated T cells, but not resting T cells, effectively cross-blocked VV binding to and infection of PHL subsets. CD29 and CD98, two lipid raft-associated membrane proteins that had been found to be important for VV entry into HeLa cells, had no effect on VV binding to and infection of primary activated T cells. Our data indicate that PHL subsets express VV protein receptors enriched in lipid rafts and that receptors are cross-presented on all susceptible PHLs.
INTRODUCTION
In general, all viruses must bind to their receptors on the surface of target cells to initiate infection. Virus-receptor interactions determine the cell type, organ specificity, and host range and therefore constitute an interspecies barrier. Poxviruses are a family of large, complex, enveloped DNA viruses that show species specificities (1, 2); for example, variola virus is a strict human-specific pathogen that causes smallpox in humans only (1), and myxoma virus is a rabbit-specific poxvirus that causes a lethal disease (myxomatosis) in rabbits only (1, 2). However, the molecular basis underlying the strict species barrier for poxviruses remains mysterious. In particular, no specific cellular receptor for any poxvirus has yet been identified. Poxviruses infect a wide variety of cell lines in culture, leading to the presumptions either that specific receptors for these viruses may not be required or that conserved and ubiquitous receptors may be widely distributed on the surface of diverse cell types (1). These conjectures may have impeded attempts to identify cellular receptor(s) that mediate poxvirus binding and infection. However, recent reports from our group and others have shown that vaccinia virus (VV), the prototypical member of the poxvirus family, and canarypox virus (ALVAC) do not indiscriminately infect all cell types of the primary human hematopoietic cells that they encounter but instead demonstrate an extremely strong preference for infection of monocytes among peripheral blood mononuclear cells (PBMCs) and monocyte lineage cells in the bone marrow (3, 4). Significantly, expression of VV receptor(s) can be induced de novo on primary human T cells upon T cell activation (3). As a consequence, activated T cells become susceptible to VV binding, infection, and replication, in contrast to resting T cells that are nonpermissive to VV binding (3, 4). These receptors are likely proteins because inhibitors of transcription (actinomycin D), protein synthesis (cycloheximide), and intracellular protein transport (brefeldin A) significantly reduce VV binding to activated primary human T cells and also because treatment of primary human monocytes or activated T cells with trypsin or pronase completely diminishes VV binding and infection (3).
Poxviruses not only bind to and infect monocytes but also use these cells to initiate a systematic infection. A recent study using high doses of variola virus, the most virulent member of the poxvirus family, to infect Cynomolgus macaques in an attempt to develop an animal model of smallpox has demonstrated that variola virus is disseminated by means of monocytic cell-associated viremia (5), suggesting that monocytes play a significant role in the initiation of systematic infection. Monocytes may use putative viral receptors to grab infectious variola virus particles and then disseminate them to uninfected cells and tissues, resulting in a generalized infection. However, the specific molecular events that determine poxvirus bias toward monocyte binding and infection remain unclear. In the present study, we investigated the susceptibility of major subsets of primary human leukocytes (PHLs) to VV binding and infection. Our data demonstrate that PHL subsets express and share protein VV receptors that are enriched in lipid rafts on the cytoplasmic membrane and that VV receptors are induced de novo on certain but not all PHL subsets.
MATERIALS AND METHODS
Antibodies and flow cytometric analysis.
The following anti-human monoclonal antibodies (MAbs) or polyclonal Abs (pAbs) conjugated with fluorochrome were purchased from BD PharMingen (San Diego, CA): anti-CD3APC, anti-CD4PerCP, anti-CD8PE, anti-CD14APC, anti-CD16PE, anti-CD19PE, anti-CD56PE, and matched-isotype control Abs conjugated with fluorescein isothiocyanate (FITC), phycoerythrin (PE), peridinin chlorophyll protein (PerCP), or allophycocyanin (APC). Anti-human neutrophil lipocalin (HNL) and anti-human CD66bPE Abs were purchased from Novus Biologicals (Littleton, CO) and Biolegend (San Diego, CA), respectively. Rabbit pAbs against full-length human integrin beta-1 (CD29) and rabbit pAbs against human amino acid transporter SLC3A2 (CD98) were purchased from Abnova (Taipei, Taiwan) and Thermo Fisher Scientific (Pittsburgh, PA), respectively. Isolated PHL subsets including monocytes, B cells, T cells, neutrophils, and NK cells were subjected to VV binding and surface staining with different combinations of Abs, followed by flow cytometric analysis (fluorescence-activated cell sorter [FACS]) using a BD FACSCalibur (BD Biosciences, San Diego, CA). The data were analyzed using FlowJo software (TreeStar, San Carlos, CA). Appropriate isotype controls were used at the same protein concentration as the test Abs, and control staining was performed during every FACS.
Preparation of human primary leukocyte subsets.
Whole-blood samples were collected from healthy donors. Written consent was obtained from each participant, and all investigational protocols were approved by Institutional Review Boards for Human Research at the Indiana University School of Medicine (Indianapolis, IN). PBMCs were separated from blood samples by gradient centrifugation on Ficoll-Hypaque (Amersham Pharmacia Biotech AB, Uppsala, Sweden). Monocytes, B cells, and NK cells were then purified by negative isolations using Ab-conjugated magnetic beads in the Monocyte, B cell, and NK cell negative isolation kits (Dynal, Oslo, Norway). The resulting cell preparations contained more than 95% desired cell types, assessed by CD14, CD3, CD4, CD8, CD19, or CD56 staining and FACS. Resting T cells were isolated from the PBMCs using the Pan T Cell isolation kit II (Miltenyi Biotec, Auburn, CA), which yielded >95% purity of CD3+ T cells. CD3+ T cells were activated by incubating with anti-CD3/anti-CD28 Abs-coated magnetic beads (Life Technologies, Carlsbad, CA) and allowed to culture in RPMI 1640 medium supplemented with 10% fetal bovine serum (FBS) and 2 mM l-glutamine (complete RPMI 1640 medium). Neutrophils were isolated from whole blood from healthy donors by density gradient separation in Lympholyte-Poly solution (Cedarlane Labs, Hornby, ON) to isolate polymorphonuclear cells, followed by treatment with a hypotonic solution to lyse red blood cells. Neutrophil purity was >98% as determined by FACS analysis of HNL+CD66b+ cells.
VV preparation, binding, and infection.
EGFP-VV and vA5L-YFP, two recombinant viruses of the Western Reserve (WR) VV strain, were obtained from J. W. Yewdell and B. Moss (NIH, Bethesda, MD). EGFP-VV contains a chimeric gene encoding the influenza virus nucleoprotein, the ovalbumin SIINFEKL peptide, and enhanced green fluorescence protein (EGFP) regulated by the P7.5 early-late promoter (6). EGFP expression was used in this study to monitor VV infection. The vA5L-YFP virions were constructed with VV core protein A5L fused with yellow fluorescence protein (YFP) and can be directly visualized by fluorescence microscopy or FACS (7). Viral stocks including EGFP-VV and vA5L-YFP were grown and titrated in primary chicken embryo fibroblasts (Charles River Laboratories, Wilmington, MA) or the monkey kidney cell line CV-1 (ATCC, Manassas, VA) in Dulbecco's minimal essential medium (DMEM) supplemented with 10% FBS and 2 mM l-glutamine. VV intracellular mature virions (IMV) were purified by 24 to 40% sucrose gradient as previously described (8), and the viral titers were determined by a plaque assay in CV-1 cells (8).
To determine the susceptibility of individual PHL subsets to VV binding, vA5L-YFP virions were incubated with isolated monocytes, B cells, resting T cells, activated T cells, neutrophils, or resting or activated NK cells at a multiplicity of infection (MOI) of 10 at 4°C for 30 min in complete RPMI 1640 medium, conditions that permit virus binding but not entry (3, 8, 9). After extensive washing with ice-cold phosphate-buffered saline (PBS), cells were fixed with 1% paraformaldehyde (PFA) and then subjected to confocal microscopy analysis and FACS to determine VV binding at single-cell and whole-cell, respectively, population levels. TA3 cells (a mouse B cell hybridoma cell line) that were previously shown not to bind with VV were used as a negative control of VV binding (8, 10).
VV-EGFP was used to monitor the susceptibility of individual PHL subsets to VV infection as previously described (4). Infection intensity was calculated by the percentage of EGFP-positive cells or the median fluorescence intensity (MFI).
Confocal microscopy.
Cells were incubated with cholera toxin subunit B (CTB) conjugated with Alexa Fluor 647 (Life Technologies, Carlsbad, CA) at 4°C for 20 min to stain ganglioside M1 (GM1) lipid rafts. For CTB patching, cells were treated with 1:100 diluted goat anti-CTB pAbs (Millipore, Darmstadt, Germany) in 2% FBS–PBS for 30 min on ice and then incubated at 37°C for 20 min as previously described (11). After washing, cells were then incubated with 10 to 30 PFU/cell of vA5L-YFP for 30 min under the binding conditions described above. Cells were fixed with 2% PFA, permeabilized with 0.1% saponin, and then incubated with phalloidin conjugated with Alexa Fluor 546 (Life Technologies, Carlsbad, CA) to stain F-actin. Cells were then adhered to poly-l-lysine-coated coverslips and mounted onto glass slides using the ProLong Gold Antifade reagent (Life Technologies, Carlsbad, CA) containing 4′,6-diamidino-2-phenylindole (DAPI) dye for fluorescent staining of DNA content and nuclei. Cells were analyzed using an Olympus FV1000-MPE confocal/multiphoton microscope fitted with a 60× objective. For each field, a z-series of images was collected and the number of virions per cell was determined on a three-dimensional (3D) image of cells constructed using the open source software FluoRender 2.7 (University of Utah) and ImageJ 1.44p (NIH, Bethesda, MD).
Polarization of primary human leukocyte subsets.
Individual PHL subsets were treated with various agents to induce membrane polarization and lipid raft relocation. Briefly, isolated monocytes were incubated with 100 ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) (BioVision, Milpitas, CA) for 24 h (12), B cells were incubated with 100 ng/ml SDF-1 (Biolegend, San Diego, CA) over rhICAM-2 (fc)-coated coverslips for 30 min (13), activated T cells were adhered to anti-CD44-coated coverslips for 30 min (14, 15), and neutrophils were treated with 10 nM the bacterial peptide fNLPNTL (Bachem, Torrence, CA) for 5 min (16). After treatment, all cell types were fixed with 2% PFA and subjected to VV binding and the fluorescence microscopy assay.
Immunosera raised against cell membrane extracts or whole cells.
All animal experimentation was conducted following the NIH guidelines for housing and care of laboratory animals and performed in accordance with Indiana University Institutional regulation after review and approval by the institutional Animal Care and Use Committee at Indiana University (approved IACUC protocol number, MD-0000003309-R). Female BALB/c mice, 6 to 8 weeks of age, from the Jackson Laboratories (Bar Harbor, ME) were subjected to immunization through intraperitoneal (i.p.) injections. Mice were divided into nine groups with 3 mice per group and then subjected to immunization with (i) detergent-resistant membranes (DRMs), (ii) crude membrane extracts (CMEs), or (iii) whole cells. These immunogens were prepared from 40 × 106 cells, either monocytes, resting T cells, or activated T cells from the same blood donors. DRMs and CMEs were prepared from each type of these cells as previously described (17). Briefly, 40 × 106 cells were lysed with 1% Triton X-100–PBS plus 1× protease inhibitor cocktail (Fisher Scientific, Pittsburgh, PA) at 4°C for 1 h. Lysates were clarified with centrifugation at 1,000 × g for 10 min, and the resulting supernatants were mixed with sucrose to make 45% sucrose solutions, which were added to the bottom of an ultracentrifuge tube. Equal volumes of 35% and 5% sucrose–PBS were sequentially added to the tube to make discontinuous gradients. The tube was centrifuged at 166,000 × g at 4°C for 18 h in an Optima LE-80K ultracentrifuge (Beckman Coulter, Brea, CA). The light-scattering band near ∼20% sucrose (DRMs) was harvested, diluted in PBS containing 1× protease inhibitor cocktail, and then centrifuged at 166,000 × g at 4°C for 2 h. The DRM pellet was homogenized in PBS using a Dounce homogenizer. For CME preparation, 40 × 106 cells of each subset in PBS with 1× protease inhibitor cocktail were disrupted with a Dounce homogenizer. After clarification, the resulting supernatant was centrifuged at 120,000 × g, and the pellet was resuspended in PBS. Primary immunizations were followed by two immunization boosts on day 14 and day 28. Two weeks after the last immunization boost, animals were anesthetized with isoflurane and sacrificed by bleeding. Sera were collected from mice in each group.
An enzyme-linked immunosorbent assay (ELISA) was developed to titrate Abs against human CD55, a glycosylphosphatidylinositol (GPI) protein anchored in cell lipid rafts (18), to evaluate the immunization efficacy. Briefly, microplates were coated with recombinant CD55 protein at 0.1 μg/ml (R&D Systems, Minneapolis, MN). After washing and blocking with 5% FBS–PBS, plates were incubated with serially diluted immunosera, followed by addition of anti-mouse IgG MAb conjugated with horseradish peroxidase (HRP). Pooled preimmunization mouse sera were used as Ab-negative controls. Absorption was read at a wavelength of 450 nm by a plate spectrophotometer (BioTek, Winooski, VT).
Knockdown of CD29 and CD98 in HeLa cells and activated T cells.
Dharmacon Smartpool Accell small interfering RNA (siRNA) constructs against human CD29 and CD98 were purchased from Thermo Fisher Scientific (Pittsburgh, PA). These siRNA constructs were transfected into cells using the Amaxa Nucleofector system (Lonza, Basel, Switzerland) according to the manufacturer's instructions. Briefly, 150 to 300 nM each siRNA mixture was used per 5 × 106 activated T cells or per 1 × 106 HeLa cells. Transfected cells were allowed to culture for 48 h and then subjected to Western blotting or FACS using rabbit pAbs against human CD29 or CD98 to analyze knockdown of human CD29 or CD98. These cells were also subjected to VV binding and infection to determine the effects of CD29 and CD98 on VV binding and entry.
Statistical analysis.
Data were analyzed using Tukey's post hoc analysis of variance (ANOVA) test and the Student t test. P values of <0.05 were considered statistically significant.
RESULTS
VV differentially binds to primary human leukocyte subsets.
The majority of studies investigating the entry of VV into host cells have focused on single-enveloped VV IMV particles, since they are the most abundant (>98%) and maintain their membrane integrity after freezer storage (19, 20). The IMV particles of vA5L-YFP or EGFP-VV were therefore used in this study. Isolated monocytes, B cells, neutrophils, resting T cells, and NK cells were incubated with vA5L-YFP particles under binding conditions (4°C for 30 min) to study VV-binding profiles for these PHL subsets. At an MOI of 10, vA5L-YFP bound to 76% ± 10%, 71% ± 9%, 28% ± 2%, 3% ± 2%, and 2% ± 2% of monocytes, B cells, neutrophils, resting T cells, and NK cells, respectively (Fig. 1A and B). These values were the results (means ± standard deviations [SD]) from six healthy blood donors. VV binding to monocytes, B cells, and neutrophils was not affected by soluble heparan sulfate (HS) at 10 μg/ml (Fig. 1A and B), an optimal concentration that completely blocks VV nonspecific binding to HS glycosaminoglycan (GAG) side chains of cell surface proteoglycans of the BSC40 cell line (21). In contrast, HS at 10 μg/ml completely eliminated the trace amount of VV binding to resting T cells and NK cells (Fig. 1A and B), suggesting that the binding is GAG dependent. VV binding to monocytes, B cells, and neutrophils was markedly reduced by trypsin treatment (Fig. 1A and B). Next, we investigated whether activated T cells become sensitive to VV binding to confirm that VV receptor(s) can be induced de novo upon T cell activation (3). We found that VV binding was correlated to the degree of T cell activation (Fig. 1C and D). Similar to monocytes, B cells, and neutrophils, VV binding to activated T cells was markedly reduced by treatment with trypsin but not HS (Fig. 1E). In contrast, activated NK cells remained nonpermissive to VV binding (Fig. 1F). These results indicate that VV binding to monocytes, B cells, neutrophils, and activated T cells is mediated by protein VV receptors independent of HS GAGs and that these receptors are induced upon activation of T cells but not NK cells.
Fig 1.

VV binding and infection of PHL subsets. (A) Major PHL subsets including monocytes, B cells, neutrophils, resting T cells, and NK cells were treated or untreated with trypsin or HS and then subjected to vA5L-YFP binding at an MOI of 10. VV binding was measured by YFP intensity using FACS. (B) Pooled data represent means ± SD of VV binding (percentage of YFP-positive cells) to PHL subsets from 6 blood donors. (C) A representative sample's kinetics of VV binding to activated T cells. (D) Pooled data represent means ± SD (n = 6) of VV binding to activated T cells during 0- to 72-h activation periods. (E) VV binding to activated T cells on day 3 of activation was affected by treatment with trypsin but not with HS. (F) Both resting NK cells (CD69 negative) and interleukin-2 (IL-2)-activated NK cells (CD69 positive) were not permissive to VV binding. (G) Confocal microscope analysis of VV binding to monocytes versus resting T cells versus activated T cells at the single-cell level. (H) Pooled data represent means ± SD (n = 6) of VV binding to monocytes versus resting T cells versus activated T cells. (I) VV infection profiles of PHL subsets and effects of trypsin on VV infection of these cell types. (J) Pooled data represent means ± SD of VV infection (percentage of EGFP-positive cells) to PHL subsets from 6 blood donors. (K) Isolated monocytes were subjected to VV binding and then to surface staining with CD14 and CD16. Cells were then subjected to FACS to determine VV-binding efficacy of different monocyte subsets. Data represent VV binding to monocytes of 6 blood donors. Data were compared using Tukey's ANOVA assay. NS, not significant; *, P < 0.05; **, P < 0.01; HS, heparan sulfate; histogram in solid gray, virus binding control; histogram in dotted line, vA5L-YFP binding.
To visualize VV binding at the single-cell level, we incubated monocytes, resting T cells, activated T cells, and TA3 cells with vA5L-YFP particles under binding conditions and then used confocal microscopy to examine VV binding. At an MOI of 10, vA5L-YFP bound to monocytes at ∼39 virions per cell (mean from 100 cells counted) (Fig. 1G and H), whereas VV barely bound to resting T cells (0.05 virions per cell, mean from 100 cells counted), which shows no difference with the results of vA5L-YFP binding to TA3 cells, which were previously shown not to bind with VV (8, 10). After activation with anti-CD3/anti-CD28 MAbs-coated magnetic beads for 3 days, activated T cells became sensitive to VV binding with a binding degree similar to that of monocytes (Fig. 1G and H).
To determine whether VV binding was correlated to its infection tropism, we infected PHL subsets with EGFP-VV at an MOI of 10 for various durations. We confirmed that VV preferentially infected monocytes, as 65% ± 8% (n = 6) of these cells became EGFP positive 6 h postinfection, whereas 15% ± 5% (n = 6) of activated T cells became EGFP positive 24 h postinfection (Fig. 1I and J). In contrast, only 4% ± 2% (n = 6) of the B cells were infected, and neutrophils and resting T cells resisted VV infection, as only trace amounts of these cells were EGFP positive 24 h postinfection (Fig. 1I and J). VV infection of monocytes and activated T cells was significantly reduced by trypsin treatment but not HS treatment (Fig. 1I and J). Further analysis of monocyte subpopulations revealed that “classical” CD14highCD16− monocytes received the vast majority of bound virus in binding experiments and were sensitive to VV infection compared to “patrolling” CD14lowCD16+ monocytes (Fig. 1K). Two previous studies have demonstrated that VV-infected primary human monocytes do not produce either viral late-gene products or viral DNA copies, indicating that VV undergoes abortive infection in primary human monocytes (3, 22). Our data together with these results indicate that (i) monocytes are the most sensitive PHL subset to VV binding and infection, but the infection is abortive; (ii) B cells and neutrophils are sensitive to VV binding, albeit at different degrees, but nonpermissive to VV infection; (iii) NK cells (both resting and activated states) and resting T cells resist VV binding and infection; and (iv) activated T cells are the only cell type among PHLs to permit VV to complete the whole cycle of binding, infection, and replication.
VV strongly binds to lipid rafts on the surface of all susceptible PHL subsets.
Lipid rafts (also called DRMs) play a critical role in VV entry into cell lines (23). VV particles colocalize with lipid rafts on HeLa cells (11), and VV uncoating, but not attachment, is inhibited by treatment of HeLa cells with methyl-β-cyclodextrin (mβCD), a drug that disrupts lipid rafts by depleting cellular cholesterol (23). To test whether VV-binding molecules are enriched in lipid rafts of primary human cells, we studied colocalization of VV binding with lipid rafts on the cell surface of all susceptible PHL subsets. Colocalization of VV with lipid rafts on PHL subsets including monocytes, B cells, neutrophils, and activated T cells was observed in both patched and unpatched states, while CXCR4 on monocytes, neutrophils, and activated T cells and CD19 on B cells did not colocalize with VV binding (Fig. 2A to D).
Fig 2.

VV binding to lipid rafts on the surface of PHL subsets. Isolated monocytes (A), B cells (B), activated T cells on day 3 of activation (C), and neutrophils (D) were patched or unpatched and then subjected to staining with CTB conjugated with Alexa Fluor 647 (red), anti-human CXCR4 Ab conjugated with Alexa Fluor 647 (red) for monocytes, activated T cells, and neutrophils, or anti-human CD19 Ab conjugated with Alexa Fluor 647 (red) for B cells. All cell types were then incubated with vA5L-YFP (green) at an MOI of 10 under binding conditions, fixed with 2% PFA, and adhered to poly-l-lysine-coated coverslips. Coverslips were mounted with mounting medium containing DAPI (blue) and analyzed by confocal microscopy. Scale bars represent 10 μM. The data represent the results of VV binding to lipid rafts on PHL subsets from 6 blood donors.
VV binds to lipid rafts enriched in uropods of polarized leukocytes.
To further analyze the association of VV binding with lipid rafts, we polarized monocytes, B cells, neutrophils, and activated T cells and then conducted lipid raft staining and VV binding. As previously reported, GM-CSF, SDF-1, bacterial peptide fNLPNTL, and anti-CD44-coated coverslips effectively induced polarization of monocytes (24), B cells (25), neutrophils (26), and activated T cells (15), respectively, as 80% ± 8% (n = 6) of monocytes, 65% ± 6% (n = 6) of B cells, 75% ± 11% (n = 6) of neutrophils, and 35% ± 4% (n = 6) of activated T cells displayed elongated phenotypes and uropod formation. In all polarized cell types, vA5L-YFP strongly colocalized with CTB-stained lipid rafts enriched in polarized cell uropods (Fig. 3A). In contrast, VV did not colocalize with F-actin molecules in lamellipodia in the leading edge of polarized cells (Fig. 3A). When monocytes were continually cultured for 7 days in GM-CSF-containing complete RPMI 1640 medium to differentiate into macrophages in vitro, these newly differentiated macrophages also maintained a polarized phenotype with bound VV strongly colocalized with lipid rafts in the uropods (Fig. 3B). These results indicate that the VV receptors are strongly associated with lipid rafts in PHL subsets in both ex vivo and polarized states.
Fig 3.

VV binding to lipid rafts enriched in uropods of polarized PHL subsets. (A) VV binding to lipid rafts enriched in uropods of polarized monocytes, B cells, activated T cells on day 3 of activation, and neutrophils. These cell types were treated with GM-CSF, SDF-1, anti-CD44-coated plates, and bacterial peptide fNLPNTL, respectively, to induce cell polarization. Polarized cells were subsequently fixed with 2% PFA and stained with CTB conjugated with Alexa Fluor 647 (red), phalloidin conjugated with Alexa Fluor 546 to stain actin filaments (pink), and DAPI (blue). Cells were then subjected to VV binding with vA5L-YFP (green) and confocal microscopy analysis. (B) VV binding to lipid rafts enriched in uropods of polarized macrophages that were differentiated from monocytes after 7 days of culture in vitro in the presence of GM-CSF. Scale bars represent 10 μM. The data represent the results of VV binding to these polarized cell types derived from 6 blood donors.
During PHL migration and/or polarization in vivo and in vitro, GM1-stained lipid rafts and raft components move to the uropod ends of cells (24–27). Many cell surface proteins also move in and out of lipid rafts during these processes to fulfill certain physiological roles. Thus, our polarization study not only provides another way to demonstrate colocalization of VV with lipid rafts but also presents a unique characteristic about the location of VV receptors during cell migration and polarization.
Immunosera raised against DRMs strongly block VV binding and infection.
Since VV receptors are strongly associated with lipid rafts in PHLs and are likely proteins, we hypothesized that DRMs from susceptible PHL subsets were able to induce Abs that would block VV binding. To this end, we immunized BALB/c mice with DRMs fractionated from monocytes, activated T cells, or resting T cells. Immunosera against DRMs from resting T cells would not block VV binding, as resting T cells do not express VV-binding receptors. Immunosera against whole cells or CMEs from monocytes, activated T cells, or resting T cells were also raised to be used for VV-blocking comparisons, as a previous study has reported that immunosera against whole monocytes or activated T cells effectively blocked VV binding to activated T cells (3). We found that all immunogens from all cell types effectively elicited Abs against CD55 protein (Fig. 4A). Because CD55 is associated with lipid rafts, DRMs from all cell types induced the highest titers of anti-CD55 Abs compared with immunogens of whole cells or CMEs (Fig. 4A). Immunosera raised against DRMs, whole cells, or CMEs from monocytes or activated T cells effectively cross-blocked VV binding to monocytes (Fig. 4B), B cells (Fig. 4C), and activated T cells (Fig. 4D). In contrast, immunosera raised against DRMs, whole cells, or CMEs from resting T cells did not affect VV binding to any of these cell types, which is similar to the results observed from preimmunization sera (Fig. 4B to D). Anti-DRM immunosera exhibited the strongest blockage activity, followed by immunosera against CMEs and then whole cells (Fig. 4B to D). Concordantly, infection of monocytes was drastically reduced when cells were pretreated with anti-DRM immunosera and to a lesser extent by anti-CME immunosera, but not by immunosera against DRMs or CMEs from resting T cells (Fig. 4E). Notably, anti-DRM immunosera did not affect endocytosis of latex beads (Fig. 4F), whereas cytochalasin D, a known endocytosis inhibitor, effectively blocked endocytosis of latex beads (Fig. 4F). Thus, immunosera raised against DRMs from monocytes or activated T cells effectively cross-blocked VV binding to and infection of VV-susceptible PHL subsets. The blocking activity is significantly higher than that mediated by immunosera raised against CMEs or whole cells. These results suggest that VV receptors are enriched in DRMs and these receptors are shared by the virus-susceptible subsets of PHLs.
Fig 4.

Blockage of VV binding and infection of PHL subsets by immunosera. (A) Titers of anti-human CD55 Abs in immunosera raised against DRMs, CMEs, or whole cells from monocytes, resting T cells, or activated T cells were determined using an ELISA. Immunosera diluted at 1:10 with PBS were used to block VV binding to monocytes (B), B cells (C), and activated T cells (D). (E) Immunosera diluted at 1:10 with PBS were used to block VV infection of monocytes. (F) Effects of immunosera diluted at 1:10 with PBS and cytochalasin D on monocyte endocytosis of FITC-latex beads. The MFI of YFP (FITC for latex beads) or EGFP represented VV binding or infection intensity to PHL subsets of 6 blood donors. Data were compared using Tukey's ANOVA assay. Mono W, whole monocytes; Mono CME, monocyte crude membrane extracts (CMEs); Mono DRM, monocyte detergent-resistant membranes (DRMs); ActT W, whole activated T cells on day 3 of activation; ActT CME, activated T cell CMEs; ActT DRM, activated T cell DRMs; ResT W, whole resting T cells; ResT CME, resting T cell CMEs; ResT DRM, resting T cell DRMs; Pre, preimmunization sera. Statistical analysis was used to compare each experimental condition with medium (control). NS, not significant; *, P < 0.05; **, P < 0.01.
Immunosera depleted with VV-susceptible PHL subsets lose their blocking activity against VV binding.
To further confirm that monocytes, activated T cells, and B cells share VV receptors, we incubated anti-DRM immunosera with monocytes or activated T cells to deplete Abs in these immunosera. We found that anti-DRM immunosera depleted with either monocytes or activated T cells, but not resting T cells, profoundly reduced their activity in blocking VV binding to all cell types examined including monocytes (Fig. 5A), B cells (Fig. 5B), and activated T cells (Fig. 5C). PHL subsets from 6 healthy donors were used in the Ab depletion. In some cases, Ab-depleted anti-DRM immunosera totally lost their ability to block VV binding (data not shown). These results further indicate that VV receptors are proteins enriched in lipid rafts and that efforts to identify poxvirus receptors and to study interactions of individual poxvirus proteins with viral receptors should be focused on DRMs instead of soluble membrane proteins extracted from target cells by nonionic detergent lysis methods.
Fig 5.
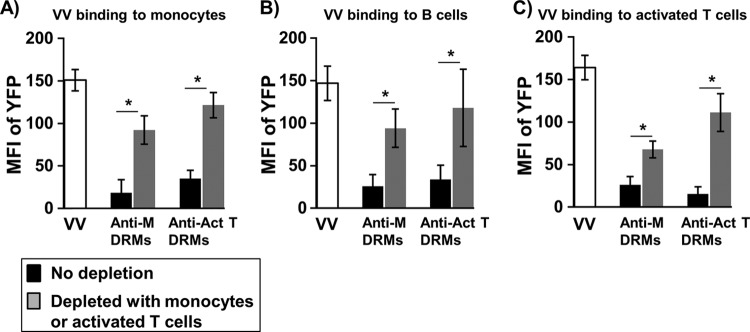
Immunosera depleted with VV-susceptible PHL subsets reduced their blocking activity against VV binding. Immunosera raised against DRMs from monocytes were diluted 1:10 in PBS and then treated with activated T cells (40 × 106), or immunosera raised against DRMs from activated T cells were treated with monocytes (40 × 106) to deplete Abs. Ab-depleted immunosera were then used to block VV binding to monocytes (A), B cells (B), and activated T cells (C). MFI of YFP data represent the immunoserum-mediated blockage of VV binding to PHL subsets of 6 blood donors. Data were compared using Tukey's ANOVA assay. Anti-M DRMs, immunosera raised against DRMs of monocytes and incubated with activated T cells to deplete Abs; Anti-Act T DRMs, immunosera raised against DRMs of activated T cells and incubated with monocytes to deplete Abs; *, P < 0.05.
These results add to the knowledge of characteristics of VV receptors on PHLs, where (i) receptors are mainly expressed on monocytes, B cells, and neutrophils among PHL subsets; (ii) receptors are induced de novo following T cell activation; (iii) receptors are upregulated on CD14highCD16− monocytes versus CD14lowCD16+ monocytes; and (iv) receptors are lipid raft associated. Using these criteria, a list of putative VV receptors was made according to mass spectrometry or transcriptome sequencing (RNA-seq) data available from previous studies (Table 1). These proteins were selected based on their presence in monocyte DRMs (28), the upregulation on CD14highCD16− monocytes versus CD14lowCD16+ monocytes (29), and the upregulation on activated T cells versus naive T cells (30).
Table 1.
Partial list of the lipid raft-associated proteins cross-presented on different PHL subsets
| Lipid raft-associated proteina | Known expression on: |
||||
|---|---|---|---|---|---|
| Monocytes | CD16− monocytes (upregulated) | Activated T cells | B cells | Neutrophils | |
| Alpha enolase | × | × | × | × | × |
| Annexin VI | × | × | × | × | × |
| ATP1A1 | × | × | × | × | |
| Carboxypeptidase M | × | × | |||
| Catenin, alpha 1 | × | × | × | × | |
| CAP1 | × | × | × | × | × |
| CCR5 | × | × | × | ||
| CD11b | × | × | × | × | × |
| CD18 | × | × | × | × | |
| CD1d | × | × | × | ||
| CD9 | × | × | × | × | × |
| CD29 | × | × | × | × | |
| CD33 | × | × | × | ||
| CD36 | × | × | |||
| CD44 | × | × | × | × | × |
| CD98 | × | × | × | × | |
| CXCR1 | × | × | × | × | |
| Flotillin 1 | × | × | × | × | × |
| Flotillin 2 | × | × | × | × | |
| Galectin-9 | × | × | |||
| IL-13Rα1 | × | × | × | × | × |
| Lamp2 | × | × | × | × | |
| LDLR | × | × | × | × | |
| Syntaxin 7 | × | × | × | × | |
Boldface indicates that the molecules were further investigated for their role in VV binding and entry.
Lipid raft-associated proteins CD29 and CD98 are not directly involved in VV binding.
The lipid raft-associated proteins CD29 and CD98 in HeLa cells and mouse embryonic fibroblasts (MEFs) play a critical role in VV entry into these cells, as knockdown or knockout of these proteins significantly reduces VV entry (31, 32). In addition, VV entry into GD25 cells (a mouse cell line that is deficient in CD29 expression) was less efficient than entry into GD25β1A cells (GD25 cells expressing human CD29) (31). We therefore wanted to determine whether these two lipid raft-associated proteins also play a role in VV binding to and infection of PHLs. We found that knockdown of either CD29 or CD98 in HeLa cells (Fig. 6A) or activated T cells (Fig. 6B) did not affect VV binding to these cells (Fig. 6C and D). However, knockdown of CD29 or CD98 in HeLa cells reduced VV infection, as the MFI of EGFP was significantly reduced in HeLa cells transfected with siRNA constructs against human CD29 or CD98 (Fig. 6C), which is consistent with previous reports as described above (31, 32). In contrast, knockdown of CD29 or CD98 in activated T cells had no effect on VV infection (Fig. 6D). In fact, CD29 expression on the surface of HeLa cells pre- or postknockdown of CD29 had no correlation with VV binding, as both the CD29-negative and CD29-positive population did not show any difference in VV binding (Fig. 6E). These results are in agreement with previous reports that CD29 and CD98 are important for VV infection in HeLa cells through mediating VV entry, but not attachment (31, 32). However, these two proteins have no effect on VV binding, entry, and infection of primary human T cells, although they are highly expressed on these cells.
Fig 6.

The lipid raft-associated proteins CD29 and CD98 were not directly involved in VV binding to activated T cells. HeLa cells and primary human activated T cells were transfected with siRNA constructs against human CD29 and CD98, and knockdown was measured by Western blotting on HeLa cells (A) and surface expression on activated T cells (B). VV binding with vA5L-YFP and infection with VV-EGFP were performed on HeLa cells (C) and activated T cells (D) to analyze the effects on CD29 and CD98 on VV binding and infection. (E) A representative FACS plot of HeLa cell CD29 surface staining versus vA5L-YFP binding shows the relationship between CD29 expression and VV binding. NS, not significant; *, P < 0.05.
DISCUSSION
In the present study, we systemically studied the patterns of VV binding to and infection of major PHL subsets including monocytes, B cells, neutrophils, NK cells, and T cells. We found that VV exhibited an extremely strong bias toward binding to and infection of monocytes in PHLs (Fig. 1A, B, G, and J), and this result is in agreement with previous reports (3, 4, 33). VV also bound to primary B cells to a degree similar to that of monocytes and to neutrophils to a much lesser extent but barely infected these cells (Fig. 1A, B, I, and J). These results suggest that monocytes not only express VV receptors on the cell surface but also have all the pathways necessary for viral uptake, entry, transmembrane trafficking, and ultimately penetration to the cytoplasm, where the whole process of poxvirus replication takes place. However, VV infection of primary human monocytes is abortive because either viral late-gene products or viral DNA copies are not significantly generated in infected monocytes (3, 22). Primary B cells and neutrophils also express VV receptors on their surface, albeit at different degrees, but they likely lack cellular pathways for VV entry or other downstream events. It is also possible that VV may require more than one molecular species as receptors, and monocytes have all these molecular species, whereas B cells and neutrophils have only one of these molecular species. Many viruses such as poliovirus use a single molecular species as its receptor (34), whereas other viruses such as human immunodeficiency virus 1 (HIV-1) use more than one molecular species, including CD4 and CCR5 (34). Different outcomes of VV binding to monocytes versus B cells or neutrophils imply that these cell types can be used as cell models to dissect the molecular mechanisms of poxvirus binding, penetration, entry, and infection, eventually leading to a better understanding of poxvirus tropism and species specificities. These results may also provide a better understanding of why live VV-based vaccines were so effective against smallpox that we continue to use these vaccines today. This knowledge is requisite for the rational development of safer and more effective poxvirus-based vaccines against other infectious pathogens and tumors. Currently, over a dozen viral vaccines based on live poxvirus vectors are licensed in veterinary medicine (35). For humans, a combination of a poxvirus-based HIV-1 vaccine with HIV-1 envelope (Env) demonstrates a promising protective effect in the HIV-1 vaccine efficacy trial known as RV144 clinical trial (36). The success of these poxvirus-based vaccines greatly renews research interests in poxvirus biology and virology. Because VV replication is dependent on epidermal growth factor receptor (EGFR)/Ras pathway signaling, which is commonly active in epithelial cancers (37), VV has been developed as a promising oncolytic agent to kill tumor cells and has been engineered as a vehicle for the intravenous delivery and expression of antitumor siRNA and peptides (37–39). Therefore, characterization of VV binding and infection tropism will also advance the concept of using live viruses to treat cancer.
The VV envelope consists of approximately 25 surface membrane proteins, and several have been proposed as receptor-binding proteins (RBPs) (40). However, none of them have been validated as an RBP because they failed to pull down a specific ligand from soluble membrane proteins extracted from VV permissive cells by conventional detergent lysis methods, implying that VV receptors may exist not in soluble membrane extracts but in detergent-resistant lipid rafts. We found that VV colocalized with lipid rafts on the surface of all major PHL subsets (monocytes, B cells, and neutrophils) that are susceptible to VV binding (Fig. 2A to D). Activated T cells become sensitive to VV binding and infection because VV receptors are induced de novo upon T cell activation (3). The VV receptors newly induced on activated T cells were also found to colocalize with lipid rafts (Fig. 2C). Strikingly, these receptor molecules move together with lipid rafts, as VV binding is concentrated in lipid rafts even when they are relocated to uropods of polarized cells (Fig. 3A) and VV binding is dispersed on the cell surface when lipid rafts are dispersed by MβCD treatment (data not shown). Since VV receptors are strongly associated with lipid rafts, efforts to identify poxvirus receptors and to study interactions of individual poxvirus proteins with viral receptors should be focused on DRMs instead of soluble membrane proteins extracted from target cells by nonionic detergent lysis methods. If detergents are to be used in an attempt to isolate possible VV receptor proteins, ionic detergents should be preferred and lysates should not be cleared to avoid losing detergent-insoluble materials.
It is pertinent to note that lipid rafts of cell lines in culture versus PHLs may play different roles in VV binding and infection. In HeLa cells, membrane lipid rafts are important for VV penetration but not for VV binding, as MβCD treatment significantly inhibits VV uncoating without affecting virion attachment (23). In addition, HeLa cell surface CD29 and CD98, two lipid raft-associated proteins, are important for VV entry (31, 32), although not for VV binding (Fig. 6C and E). However, these two proteins have no effect on VV binding to and infection of primary human T cells, as knockdown of their expression on the surface of activated T cells does not affect viral binding and infection (Fig. 6) and pAbs against full-length human CD29 did not block VV binding to PHLs (data not shown). Furthermore, primary human NK cells express high levels of CD29 together with many other adhesion molecules (41), but these cells are resistant to VV binding and infection (3, 42) (Fig. 1A and B). These data indicate that VV receptors are strongly associated with lipid rafts, but not CD29 and CD98, on the cell surface of PHLs.
It has been reported that immunosera raised against whole monocytes or activated T cells effectively block VV binding to these cells (3). If VV receptors were enriched in lipid rafts, immunosera raised against DRMs would be more effective than immunosera raised against whole cells in blocking VV binding. In fact, anti-DRM immunosera significantly blocked VV binding to the highest degree, whereas immunosera raised against whole cells or CMEs also blocked VV binding but at much lesser degrees (Fig. 4B to D). Blockage of VV binding by these immunosera appears to be specific, as immunosera raised against either DRMs, CMEs, or whole cells of resting T cells did not affect VV binding (Fig. 4B to D). In addition, these immunosera lost their blocking activity if they were depleted against monocytes or activated T cells but not resting T cells (Fig. 5A to C). Importantly, immunosera raised against monocyte DRMs, CMEs, or whole cells were able to cross-block VV binding to B cells and activated T cells (Fig. 4B to D). Similarly, immunosera raised against activated T cell DRMs, CMEs, or whole cells were able to cross-block VV binding to B cells and monocytes (Fig. 4B to D). These data strongly suggest that monocytes, B cells, and activated T cells share one or more unique protein receptors for VV. Notably, the consequences of VV binding to these cell types are different. Activated T cells are permissive to VV binding, infection, and replication. In contrast, primary B cells and neutrophils are only sensitive to VV binding but not permissive to VV infection. VV binds to and enters monocytes to initiate virus infection, but the infection is abortive, as no viral late gene products have been detected (3) and viral DNA copies have not been found to increase in infected monocytes (22), which indicates that the state of certain cellular pathways in monocytes is not permissive for VV replication. However, monocytes bound with VV may help VV dissemination from initially infected sites to distant organs and tissues, as variola virus has been found to be disseminated by monocytic cell-associated viremia (5). It is possible that monocytes use putative viral receptors to grab infectious variola virus particles and then disseminate them to uninfected cells and tissues via filopodial extensions, a major mechanism that HIV-1 uses to disseminate virus infection from dendritic cells to other cell types (43). Cell-associated VV spread by filopodial extensions greatly reduces the time needed to infect neighboring cells in culture, and this process requires only VV early gene transcriptions that monocyte lineage cells are known to express during VV infection (44). It is also possible that VV binds to the uropods of phagocytes such as monocytes, macrophages, and neutrophils in PHLs to not only spread to tissues but also resist phagocytic activity. A recent report has demonstrated that Neisseria meningitidis binds to the uropods of migrating neutrophils to resist neutrophil phagocytic activity and to spread the bacteria during cell migration through epithelial cell layers (45). This report also showed that uropod-bound bacteria were resistant to phagocytosis, which occurs only at the pseudopod end (leading edge) (45). Our results also showed that VV would preferentially bind to the uropod ends of phagocytes, which may also provide protection from phagocytosis and assist in viral dissemination leading to generalized VV infections.
ACKNOWLEDGMENTS
We thank J. W. Yewdell and B. Moss at NIH (Bethesda, MD) for EGFP-VV and vA5L-YFP.
This work was supported in part by the Grand Challenges Explorations (GCE) Phase II grant through the Bill & Melinda Gates Foundation (OPP1035237 to Q.Y.), NIH 1R21AI104268 (Q.Y.), the Showalter Research Trust Fund (Q.Y.), NIH T32 AI060519 (D.B.), the National Natural Science Foundation of Zhejiang Province (Y2110608 to N.H.), and Research Facilities Improvement Program grant C06 RR015481-01 from the National Center for Research Resources, NIH, to Indiana University School of Medicine.
D.B., N.H., and Q.Y. designed and performed research, analyzed data, and wrote the paper. T.A., N.H., J.L., and S.H. contributed vital new reagents and assisted with animal immunization.
We declare that we have no conflicts of interest.
Footnotes
Published ahead of print 19 June 2013
REFERENCES
- 1. McFadden G. 2005. Poxvirus tropism. Nat. Rev. Microbiol. 3:201–213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Werden SJ, McFadden G. 2008. The role of cell signaling in poxvirus tropism: the case of the M-T5 host range protein of myxoma virus. Biochim. Biophys. Acta 1784:228–237 [DOI] [PubMed] [Google Scholar]
- 3. Chahroudi A, Chavan R, Kozyr N, Waller EK, Silvestri G, Feinberg MB. 2005. Vaccinia virus tropism for primary hematolymphoid cells is determined by restricted expression of a unique virus receptor. J. Virol. 79:10397–10407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yu Q, Jones B, Hu N, Chang H, Ahmad S, Liu J, Parrington M, Ostrowski M. 2006. Comparative analysis of tropism between canarypox (ALVAC) and vaccinia viruses reveals a more restricted and preferential tropism of ALVAC for human cells of the monocytic lineage. Vaccine 24:6376–6391 [DOI] [PubMed] [Google Scholar]
- 5. Jahrling PB, Hensley LE, Martinez MJ, Leduc JW, Rubins KH, Relman DA, Huggins JW. 2004. Exploring the potential of variola virus infection of cynomolgus macaques as a model for human smallpox. Proc. Natl. Acad. Sci. U. S. A. 101:15196–15200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mackett M, Smith GL, Moss B. 1984. General method for production and selection of infectious vaccinia virus recombinants expressing foreign genes. J. Virol. 49:857–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Katsafanas GC, Moss B. 2007. Colocalization of transcription and translation within cytoplasmic poxvirus factories coordinates viral expression and subjugates host functions. Cell Host Microbe 2:221–228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Law M, Smith GL. 2004. Studying the binding and entry of the intracellular and extracellular enveloped forms of vaccinia virus. Methods Mol. Biol. 269:187–204 [DOI] [PubMed] [Google Scholar]
- 9. Doms RW, Blumenthal R, Moss B. 1990. Fusion of intra- and extracellular forms of vaccinia virus with the cell membrane. J. Virol. 64:4884–4892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Chang W, Hsiao JC, Chung CS, Bair CH. 1995. Isolation of a monoclonal antibody which blocks vaccinia virus infection. J. Virol. 69:517–522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Janes PW, Ley SC, Magee AI. 1999. Aggregation of lipid rafts accompanies signaling via the T cell antigen receptor. J. Cell Biol. 147:447–461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ambarus CA, Krausz S, van Eijk M, Hamann J, Radstake TR, Reedquist KA, Tak PP, Baeten DL. 2012. Systematic validation of specific phenotypic markers for in vitro polarized human macrophages. J. Immunol. Methods 375:196–206 [DOI] [PubMed] [Google Scholar]
- 13. Vicente-Manzanares M, Rey M, Jones DR, Sancho D, Mellado M, Rodriguez-Frade JM, del Pozo MA, Yanez-Mo M, de Ana AM, Martinez AC, Merida I, Sanchez-Madrid F. 1999. Involvement of phosphatidylinositol 3-kinase in stromal cell-derived factor-1 alpha-induced lymphocyte polarization and chemotaxis. J. Immunol. 163:4001–4012 [PubMed] [Google Scholar]
- 14. Yeh JH, Sidhu SS, Chan AC. 2008. Regulation of a late phase of T cell polarity and effector functions by Crtam. Cell 132:846–859 [DOI] [PubMed] [Google Scholar]
- 15. Okamoto N, Nukada Y, Tezuka K, Ohashi K, Mizuno K, Tsuji T. 2004. AILIM/ICOS signaling induces T-cell migration/polarization of memory/effector T-cells. Int. Immunol. 16:1515–1522 [DOI] [PubMed] [Google Scholar]
- 16. Schmitt M, Cochrane CG. 1987. Cell-dependent chemiluminescence. Modulation of the N-formyl chemotactic peptide (FNLPNTL) mediated oxidative burst in human polymorphonuclear leukocytes (PMNL) by murine monoclonal antibody NMS-1. Free Radical Res. Commun. 2:359–368 [DOI] [PubMed] [Google Scholar]
- 17. Foster LJ, De Hoog CL, Mann M. 2003. Unbiased quantitative proteomics of lipid rafts reveals high specificity for signaling factors. Proc. Natl. Acad. Sci. U. S. A. 100:5813–5818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. de Gassart A, Geminard C, Fevrier B, Raposo G, Vidal M. 2003. Lipid raft-associated protein sorting in exosomes. Blood 102:4336–4344 [DOI] [PubMed] [Google Scholar]
- 19. Smith GL, Vanderplasschen A, Law M. 2002. The formation and function of extracellular enveloped vaccinia virus. J. Gen. Virol. 83:2915–2931 [DOI] [PubMed] [Google Scholar]
- 20. Senkevich TG, Ward BM, Moss B. 2004. Vaccinia virus A28L gene encodes an essential protein component of the virion membrane with intramolecular disulfide bonds formed by the viral cytoplasmic redox pathway. J. Virol. 78:2348–2356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Chung CS, Hsiao JC, Chang YS, Chang W. 1998. A27L protein mediates vaccinia virus interaction with cell surface heparan sulfate. J. Virol. 72:1577–1585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hu N, Yu R, Shikuma C, Shiramizu B, Ostrwoski MA, Yu Q. 2009. Role of cell signaling in poxvirus-mediated foreign gene expression in mammalian cells. Vaccine 27:2994–3006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chung CS, Huang CY, Chang W. 2005. Vaccinia virus penetration requires cholesterol and results in specific viral envelope proteins associated with lipid rafts. J. Virol. 79:1623–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rajendran L, Beckmann J, Magenau A, Boneberg EM, Gaus K, Viola A, Giebel B, Illges H. 2009. Flotillins are involved in the polarization of primitive and mature hematopoietic cells. PLoS One 4:e8290. 10.1371/journal.pone.0008290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vicente-Manzanares M, Montoya MC, Mellado M, Frade JM, del Pozo MA, Nieto M, de Landazuri MO, Martinez AC, Sanchez-Madrid F. 1998. The chemokine SDF-1alpha triggers a chemotactic response and induces cell polarization in human B lymphocytes. Eur. J. Immunol. 28:2197–2207 [DOI] [PubMed] [Google Scholar]
- 26. Rossy J, Schlicht D, Engelhardt B, Niggli V. 2009. Flotillins interact with PSGL-1 in neutrophils and, upon stimulation, rapidly organize into membrane domains subsequently accumulating in the uropod. PLoS One 4:e5403. 10.1371/journal.pone.0005403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Gomez-Mouton C, Abad JL, Mira E, Lacalle RA, Gallardo E, Jimenez-Baranda S, Illa I, Bernad A, Manes S, Martinez AC. 2001. Segregation of leading-edge and uropod components into specific lipid rafts during T cell polarization. Proc. Natl. Acad. Sci. U. S. A. 98:9642–9647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Li N, Mak A, Richards DP, Naber C, Keller BO, Li L, Shaw AR. 2003. Monocyte lipid rafts contain proteins implicated in vesicular trafficking and phagosome formation. Proteomics 3:536–548 [DOI] [PubMed] [Google Scholar]
- 29. Ancuta P, Liu KY, Misra V, Wacleche VS, Gosselin A, Zhou X, Gabuzda D. 2009. Transcriptional profiling reveals developmental relationship and distinct biological functions of CD16+ and CD16− monocyte subsets. BMC Genomics 10:403. 10.1186/1471-2164-10-403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Birzele F, Fauti T, Stahl H, Lenter MC, Simon E, Knebel D, Weith A, Hildebrandt T, Mennerich D. 2011. Next-generation insights into regulatory T cells: expression profiling and FoxP3 occupancy in Human. Nucleic Acids Res. 39:7946–7960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Izmailyan R, Hsao JC, Chung CS, Chen CH, Hsu PW, Liao CL, Chang W. 2012. Integrin beta1 mediates vaccinia virus entry through activation of PI3K/Akt signaling. J. Virol. 86:6677–6687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schroeder N, Chung CS, Chen CH, Liao CL, Chang W. 2012. The lipid raft-associated protein CD98 is required for vaccinia virus endocytosis. J. Virol. 86:4868–4882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sanchez-Puig JM, Sanchez L, Roy G, Blasco R. 2004. Susceptibility of different leukocyte cell types to Vaccinia virus infection. Virol. J. 1:10. 10.1186/1743-422X-1-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Grove J, Marsh M. 2011. The cell biology of receptor-mediated virus entry. J. Cell Biol. 195:1071–1082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gerdts V, Mutwiri GK, Tikoo SK, Babiuk LA. 2006. Mucosal delivery of vaccines in domestic animals. Vet. Res. 37:487–510 [DOI] [PubMed] [Google Scholar]
- 36. Rerks-Ngarm S, Pitisuttithum P, Nitayaphan S, Kaewkungwal J, Chiu J, Paris R, Premsri N, Namwat C, de Souza M, Adams E, Benenson M, Gurunathan S, Tartaglia J, McNeil JG, Francis DP, Stablein D, Birx DL, Chunsuttiwat S, Khamboonruang C, Thongcharoen P, Robb ML, Michael NL, Kunasol P, Kim JH. 2009. Vaccination with ALVAC and AIDSVAX to prevent HIV-1 infection in Thailand. N. Engl. J. Med. 361:2209–2220 [DOI] [PubMed] [Google Scholar]
- 37. Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQ, Nieva J, Hwang TH, Moon A, Patt R, Pelusio A, Le Boeuf F, Burns J, Evgin L, De Silva N, Cvancic S, Robertson T, Je JE, Lee YS, Parato K, Diallo JS, Fenster A, Daneshmand M, Bell JC, Kirn DH. 2011. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature 477:99–102 [DOI] [PubMed] [Google Scholar]
- 38. Liu TC, Hwang T, Park BH, Bell J, Kirn DH. 2008. The targeted oncolytic poxvirus JX-594 demonstrates antitumoral, antivascular, and anti-HBV activities in patients with hepatocellular carcinoma. Mol. Ther. 16:1637–1642 [DOI] [PubMed] [Google Scholar]
- 39. Breitbach CJ, Arulanandam R, De Silva N, Thorne SH, Patt R, Daneshmand M, Moon A, Ilkow C, Burke J, Hwang TH, Heo J, Cho M, Chen H, Angarita FA, Addison C, McCart JA, Bell JC, Kirn DH. 2013. Oncolytic vaccinia virus disrupts tumor-associated vasculature in humans. Cancer Res. 73:1265–1275 [DOI] [PubMed] [Google Scholar]
- 40. Laliberte JP, Weisberg AS, Moss B. 2011. The membrane fusion step of vaccinia virus entry is cooperatively mediated by multiple viral proteins and host cell components. PLoS Pathog. 7:e1002446. 10.1371/journal.ppat.1002446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Maenpaa A, Jaaskelainen J, Carpen O, Patarroyo M, Timonen T. 1993. Expression of integrins and other adhesion molecules on NK cells; impact of IL-2 on short- and long-term cultures. Int. J. Cancer 53:850–855 [DOI] [PubMed] [Google Scholar]
- 42. Yu Q, Hu N, Ostrowski M. 2009. Poxvirus tropism for primary human leukocytes and hematopoietic cells. Methods Mol. Biol. 515:309–328 [DOI] [PubMed] [Google Scholar]
- 43. Aggarwal A, Iemma TL, Shih I, Newsome TP, McAllery S, Cunningham AL, Turville SG. 2012. Mobilization of HIV spread by diaphanous 2 dependent filopodia in infected dendritic cells. PLoS Pathog. 8:e1002762. 10.1371/journal.ppat.1002762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Doceul V, Hollinshead M, van der Linden L, Smith GL. 2010. Repulsion of superinfecting virions: a mechanism for rapid virus spread. Science 327:873–876 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Soderholm N, Vielfort K, Hultenby K, Aro H. 2011. Pathogenic Neisseria hitchhike on the uropod of human neutrophils. PLoS One 6:e24353. 10.1371/journal.pone.0024353 [DOI] [PMC free article] [PubMed] [Google Scholar]