

Abstract
p53 is a critical factor in the cellular response to a broad range of stress factors through its ability to regulate various cellular pathways. In this study, tandem affinity purification of transiently expressed herpes simplex virus 1 (HSV-1) regulatory protein ICP22 coupled with mass spectrometry-based proteomics technology and subsequent analyses showed that ICP22 interacted with p53 in HSV-1-infected cells. In p53−/− cells, replication of wild-type HSV-1 was reduced compared to that in parental p53+/+ cells, indicating that p53 had a positive effect on HSV-1 replication. In contrast, the levels of viral replication of an ICP22-null mutant virus were similar in both p53−/− and p53+/+ cells. At 2 h postinfection, the level of expression of ICP27, an essential viral regulatory protein, in p53−/− cells infected with wild-type HSV-1 or the ICP22-null mutant virus was lower than in p53+/+ cells. In contrast, at 18 h postinfection, the level of expression of ICP0, a critical viral regulatory protein, in p53−/− cells infected with the ICP22-null mutant virus was higher than in p53+/+ cells, although the levels of ICP0 expression in p53−/− and p53+/+ cells infected with wild-type HSV-1 were almost identical. These results suggested that p53 overall promoted HSV-1 replication and that p53 played both positive and negative roles in HSV-1 replication: upregulating ICP27 expression very early in infection and downregulating ICP0 expression later in infection, which was antagonized by ICP22.
INTRODUCTION
Herpes simplex virus 1 (HSV-1) virions have three morphologically distinct structures: the nucleocapsid, an icosahedral capsid containing the 152-kbp double-stranded DNA viral genome encoding at least 84 viral proteins; the tegument, a proteinaceous layer surrounding the nucleocapsid; and the envelope, a lipoprotein membrane with a host cell-derived lipid bilayer enclosing the nucleocapsid and tegument (1). After fusion of the virion envelope and host cell membrane, tegument proteins are released into the cytoplasm and function to establish an environment for effective initiation of very early viral infection (1). Nucleocapsids then reach the cell's nucleopores, enabling entry of the HSV-1 genome into the nucleus and initiation of viral gene transcription (1). There are three major classes of HSV-1 genes, designated immediate early (IE), early (E), and late (L) genes, with gene expression coordinately regulated and sequentially ordered in a cascade fashion (1). IE genes are expressed first and are primarily activated by a multiprotein enhanceosome complex containing VP16 (2), one of the tegument proteins. Several IE gene products, including ICP0, ICP4, ICP22, Us1.5, and ICP27, are required for proper expression of the IE, E, and/or L genes (1).
In the present study, we focused on IE protein ICP22, which is encoded by the Us1 gene. ICP22 is highly modified at the posttranslational level, including phosphorylation mediated by viral protein kinases UL13 and Us3 (3) and nucleotidylylation mediated by cellular casein kinase II (4, 5). The Us1 gene encodes both full-length ICP22 and Us1.5, an amino terminal truncated form of ICP22 (6). Most of the known functions of ICP22 map to the domain overlapping Us1.5 (7), suggesting that the reported functions of ICP22 may involve a combination of functions of the two proteins, although Us1.5 appears to be expressed much less than ICP22 in infected cells (6). Us1 gene products ICP22 and Us1.5 have been suggested to be critical for viral replication and pathogenicity, based on studies showing that recombinant viruses lacking the Us1 gene are significantly impaired (2 to 3 logs) in growth in cell cultures in a cell type-dependent manner, pathogenicity in mouse models, establishment of latency, and reactivation from latency (8). Although the mechanism by which ICP22 acts in viral replication and pathogenicity remains unknown at present, it has been suggested that ICP22 upregulates transcription of a subset of viral genes based on the following observations. (i) Null mutations in the Us1 gene reduce accumulation of both mRNAs and proteins of the ICP0 IE gene and a subset of L genes, including the UL26, UL26.5, UL38, UL41, and Us11 genes (7, 9, 10). (ii) ICP22 forms an in vitro complex with the HSV-1 transcriptional machinery, including TFIID, ICP4, and ICP27 (11, 12). (iii) ICP22 is specifically recruited to discrete nuclear domains containing host cell RNA polymerase II (Pol II) and ICP4 in infected cells (12). It has also been reported that (i) HSV-1 infection induces dramatic changes in the phosphorylation status of the carboxyl-terminal domain (CTD) of Pol II (13), which is critical for regulation of Pol II activity (14), and ICP22 is required for phosphorylation of Pol II in HSV-1-infected cells (15), (ii) ICP22 can form a complex with cyclin-dependent kinase 9 (cdk9) in vitro (16), and the Pol II CTD is phosphorylated in vitro by a complex containing cdk9 from HSV-1-infected cells in a ICP22- and HSV-1-encoded protein kinase Us3-dependent manner (16), (iii) both knockdown of cdk9 and a specific inhibitor of cdk9 downregulate expression of the subset of L genes regulated by ICP22 in infected cells (17), and (iv) cdk9 is recruited to nuclear domains containing Pol II in an ICP22-dependent manner in infected cells (17). These observations suggested that ICP22 recruits cdk9 to sites of viral transcription for phosphorylation of the Pol II CTD, which leads to upregulation of the subset of L genes regulated by ICP22.
Although the role of ICP22 in regulating expression of a subset of L genes has been gradually elucidated, the mechanism by which ICP22 regulates expression of ICP0 remains unknown. Moreover, the recent consensus that HSV-1 regulatory proteins, such as ICP0 and ICP27, are multifunctional proteins regulating many aspects of cellular and viral functions (18–20) prompted us to hypothesize that there may be additional cellular targets for ICP22 that need to be identified for a further understanding of ICP22. In this study, we screened cellular proteins interacting with ICP22 by tandem affinity purification of transiently expressed ICP22 and mass spectrometry-based proteomics technology. Of the putative ICP22-interacting cellular proteins identified, we focused on p53, a novel ICP22 interacting protein.
p53 is a key cellular transcription factor that plays a central role in cellular responses to a broad range of stress factors through its regulation of a variety of cellular pathways, such as apoptosis, cell cycle, cellular senescence, DNA repair, autophagy, and innate immune control (21, 22). Based on previous studies, p53 appears to have both positive and negative effects on various viral infections. Since viral infection is a type of cell stress, p53's effects can reduce replication of some viruses, and, therefore, these viruses have acquired mechanisms to counteract p53 in infected cells (23–25). In contrast, some viruses, including human cytomegalovirus, Epstein-Barr virus (EBV), and human immunodeficiency virus, appear to utilize p53 for efficient viral replication (26–28). HSV-1 infection has been reported to induce stabilization of p53 in a cell type-dependent manner (29), and p53 is recruited into the replication compartments where viral DNA replication takes place in infected cells (30). The role of p53 in HSV-1 infection is of interest, because stabilization of p53 is one of the key steps in its activation in response to cellular stress factors (21). However, the biological significance of p53 in HSV-1 replication remains to be elucidated.
In the present study, we investigated the role(s) of p53 in HSV-1 replication using p53+/+ and p53−/− cells. The results of this study suggested that p53 played both positive and negative roles in HSV-1 replication and that the negative action of p53 was antagonized by ICP22.
MATERIALS AND METHODS
Cells and viruses.
Vero, rabbit skin, HEL, 293T, and Plat-GP cells and the HSV-1F wild-type strain were described previously (31–33). HCT116 p53+/+ and HCT116 p53−/− cells (generous gifts from B. Vogelstein) (34) were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal calf serum. A recombinant virus YK711 expressing HSV-1 glycoprotein B (gB) fused to an MEF (Myc epitope–tobacco etch virus [TEV] protease cleavage site–Flag epitope) tag with Myc and Flag epitopes and a TEV protease cleavage site (MEF-gB) was described previously (35).
Plasmids.
To construct pcDNA-MEF-ICP22, an expression plasmid for ICP22 fused to an MEF tag, the Us1 open reading frame (ORF) without a start codon was amplified by PCR from pBC1013 (36) and cloned into pcDNA-MEF (37) (Fig. 1A). The transfer plasmid pBS-ICP22-Kan, for generating recombinant virus YK422 (ΔICP22-repair) in which the ICP22-null mutation was repaired, was constructed by cloning the EcoRV-NotI fragment of pcDNA-MEF-ICP22 containing the Us1 ORF into pBluescript II KS(+) (Stratagene) to yield pBS-ICP22. A kanamycin resistance cassette was then amplified by PCR from pEP-KanS using primers 5′-GCCTGCAGGTGTCCGGCGGAACCTGGGGAGGATGACGACGATAAGTAGGG-3′ and 5′-GCCTGCAGCAAACGGCACCACGTGCGCGCAACCAATTAACCAATTCTGATTAG-3′ and inserted into the PstI site of pBS-ICP22. For generating a fusion protein of glutathione S-transferase (GST) and the C-terminal domain of ICP22 (GST-ICP22283–420), pGEX-ICP22C was constructed by cloning ICP22 gene codons 283 to 420, amplified by PCR from pBS-ICP22, into pGEX-4T-1 (GE Healthcare) in frame with GST. pMXs-p53, a retrovirus vector expressing human p53, was constructed by amplifying the p53 ORF by PCR from cDNA synthesized from the total RNA of HEL cells and cloning it into pMXs-puro (31). Total RNA was isolated from HEL cells with a High Pure RNA isolation kit (Roche), and cDNA was synthesized from the isolated RNA with a Transcriptor first-strand cDNA synthesis kit (Roche) according to the manufacturer's instructions. To construct pcDNA-VP16, an expression plasmid for VP16, the VP16 ORF was amplified by PCR from pBC1007 (36) and inserted into pcDNA5/FRT (Invitrogen). pcDNA-p53, another expression plasmid for p53, was constructed by cloning the p53 ORF, amplified by PCR as described above, into pcDNA5/FRT. pICP27-luc, a reporter plasmid for luciferase assays, was constructed by amplifying the ICP27 promoter region (nucleotides [nt] −896 to +78, relative to the ICP27 transcription initiation site) by PCR from pBC1007 and inserting it into pGL3-Basic (Promega). pSRα-luc, another reporter plasmid, was constructed by cloning the HindIII-EcoRI fragment of pME18S (38) containing the SRα promoter region into pGL3-Basic.
Fig 1.
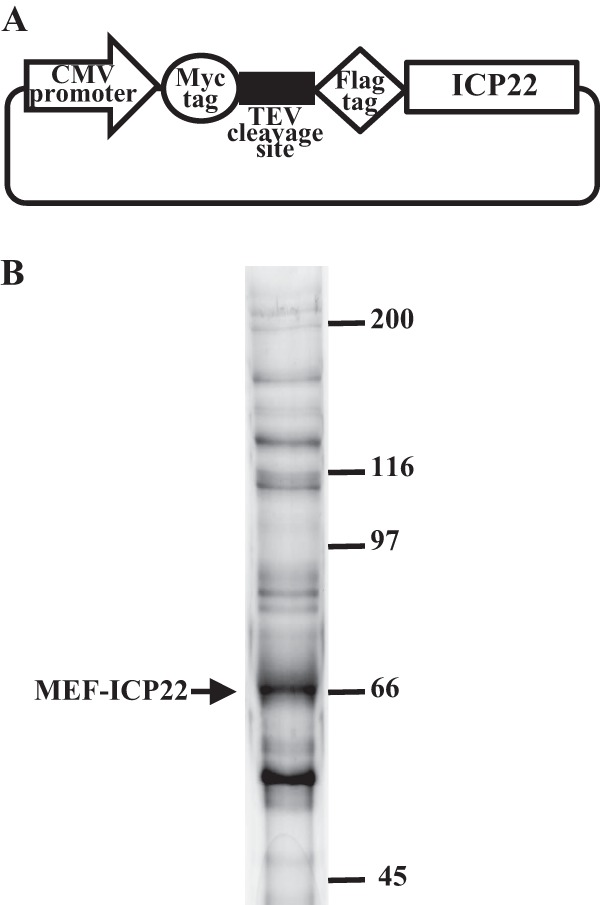
(A) Schematic of expression plasmid pcDNA-MEF-ICP22 encoding ICP22 tagged with MEF. (B) 293T cells were transfected with plasmid pcDNA-MEF-ICP22 described for panel A, harvested, and immunoprecipitated with anti-Myc and anti-Flag antibodies. Immunoprecipitates were separated in a denaturing gel and silver stained. The arrow marks MEF-ICP22. Molecular mass markers are indicated on the right.
Identification of proteins that interact with ICP22.
293T cells were transfected with pcDNA-MEF-ICP22 using polyethylenimine as described previously (39), harvested at 48 h posttransfection, and lysed in 0.1% NP-40 buffer (50 mM Tris-HCl [pH 8.0], 120 mM NaCl, 50 mM NaF, 0.1% NP-40) containing a protease inhibitor cocktail (Nacalai Tesque). After centrifugation, the supernatants were immunoprecipitated with an anti-Myc monoclonal antibody, and the immunoprecipitates were incubated with AcTEV protease (Invitrogen). After another centrifugation, the supernatants were immunoprecipitated with an anti-Flag monoclonal antibody, and the immunoprecipitates were washed three times with wash buffer (50 mM Tris-HCl [pH 8.0], 120 mM NaCl, 50 mM NaF). Flag elution buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, 0.5 mg Flag peptide/ml) was added, and the immunoprecipitates were rotated for 2 h at 4°C. A tenth of the eluted protein solution was analyzed by electrophoresis in a denaturing gel, followed by silver staining (Fig. 1B); the remaining 90% of the eluted protein solution was trypsinized and analyzed by nano liquid chromatography tandem mass spectrometry (nanoLC-MS/MS) as described previously (35). For this analysis, we used Q-STAR Elite (AB SCIEX) coupled with Dina (KYA Technologies). The MS/MS signals were then analyzed against the human proteins in the RefSeq database (National Center for Biotechnology Information; 38,963 sequences as of 5 July 2010) by using the Mascot algorithm (version 2.2.04; Matrix Science) with the following parameters: variable modifications, oxidation (Met), protein N-terminal acetylation, pyroglutamination (Gln), phosphorylation (Ser, Thr, and Tyr); maximum missed cleavages, 2; peptide mass tolerance, 200 ppm; and MS/MS tolerance, 0.5 Da. Protein identification was based on the criterion of having at least one MS/MS data signal with a Mascot score that exceeded the threshold (P < 0.05).
Mutagenesis of viral genomes in Escherichia coli and generation of recombinant HSV-1.
To generate recombinant virus YK423 expressing ICP22 fused to an MEF tag (MEF-ICP22), a two-step Red-mediated mutagenesis procedure was carried out using E. coli GS1783 containing pYEbac102 (33), a full-length infectious HSV-1F clone, as described previously (40), except with primers 5′-TGCTCGTCGGGCGGGGGGAAGCCACTGTGGTCCTCCGGGACGTTTTCTGGATGGAGCAAAAGCTCATTTC-3′ and 5′-GGACGCCGCGCTTTTACACAAGGCGCAAAAGCGCCTGGGGAAATGTCGGCATCTTTGTCATCGTCGTCCT-3′ (Fig. 2). An ICP22-null mutant virus, YK421 (ΔICP22), in which the entire Us1 ORF was deleted (Fig. 2), was generated as described above, except primers 5′-CGGGGGGAAGCCACTGTGGTCCTCCGGGACGTTTTCTGGGTCCGGTCGCCCCGACCCCCAGGATGACGACGATAAGTAGGG-3′ and 5′-TTTTATTTGGGGACATACAAGGGGGTCGGGGCGACCGGACCCAGAAAACGTCCCGGAGGACAACCAATTAACCAATTCTGATTAG-3′ were used and rabbit skin cells were cotransfected with pYEbac102 carrying the ICP22-null mutation and pcDNA-MEF-ICP22, an ICP22 expression plasmid. This protocol was used because transfection of rabbit skin cells with only pYEbac102 carrying the ICP22-null mutation produced no reconstituted ICP22 mutant virus (data not shown). Recombinant virus YK422 (ΔICP22-repair), in which the ICP22-null mutation was repaired (Fig. 2), was generated as described above, except with pBS-ICP22-Kan and primers 5′-GCGGGGGGAAGCCACTGTGGTCCTCCGGGACGTTTTCTGGATGGCCGACATTTCCCCAGGCGC-3′ and 5′-CTTTTATTTGGGGACATACAAGGGGGTCGGGGCGACCGGACTCACGGCCGGAGAAACGTGT-3′.
Fig 2.
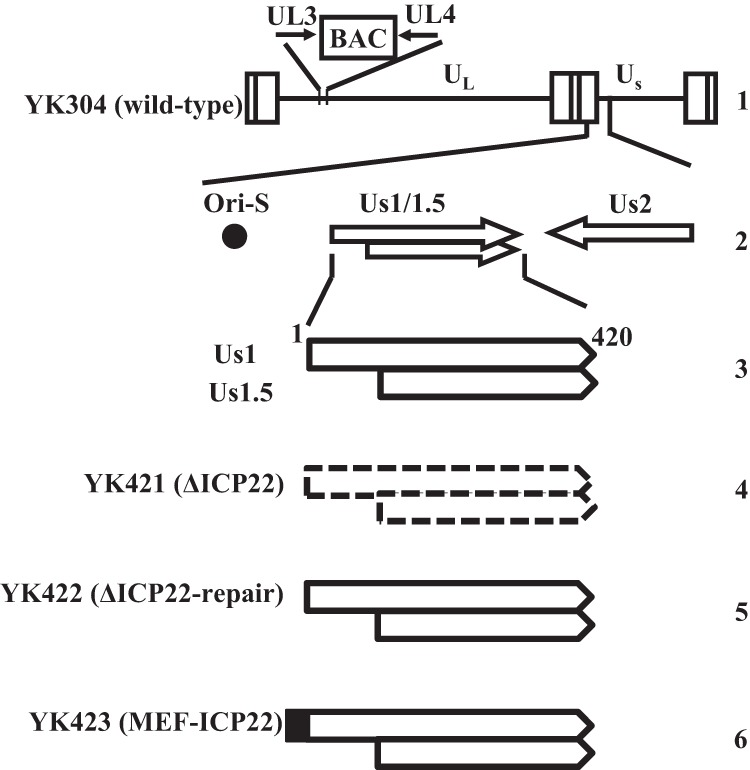
Schematic of the genome structure of wild-type virus YK304 and the relevant domains of the recombinant viruses in this study. Line 1, linear representation of the YK304 genome with a bacmid (BAC) in the intergenic region between UL3 and UL4; line 2, domains encoding the viral replication origin S (Ori-S) and the Us1 and Us2 open reading frames; line 3, domains of ICP22 and Us1.5; lines 4 to 6, schematics of the recombinant viruses in this study.
Antibodies.
The commercial antibodies used in this study were mouse monoclonal antibodies to Flag (M2; Sigma), Myc (PL14; MBL), p53 (DO-1; BioLegend), α-tubulin (DM1A; Sigma), ICP27 (8.F.137B; Abcam), and ICP0 (1112; Goodwin Institute) and rabbit polyclonal antibodies to Flag (DDDDK; MBL) and laminB1 (ab16048; Abcam). To generate a rabbit polyclonal antibody to ICP22, a rabbit was immunized, according to the standard protocol at MBL, with GST-ICP22283–420 that had been expressed in E. coli and purified as described previously (41). Serum from the immunized rabbit was used as the anti-ICP22 polyclonal antibody.
Antibody analyses.
Immunoprecipitation, immunoblotting, and immunofluorescence were performed as described previously (32, 36).
Establishment of HCT116 p53−/− cells expressing p53 exogenously.
Plat-GP cells, a 293T-derived murine leukemia virus-based packaging cell line, were cotransfected with pMxs-p53-puro or pMxs-puro in combination with pMDG encoding vesicular stomatitis virus envelope G protein. Supernatants were harvested 2 days posttransfection. HCT116 p53−/− and HCT116 p53+/+ cells were transduced by infection with retrovirus-containing supernatants of transfected Plat-GP cells and selected with 2 μg puromycin/ml in maintenance medium. Single colonies of HCT116 p53−/− transduced with pMXs-p53 were isolated and screened by immunoblotting with an anti-p53 antibody, which led to isolation of HCT116 p53−/− p53+ cells. Puromycin-resistant HCT116 p53−/− and HCT116 p53+/+ cells transduced by recombinant retrovirus derived from pMxs-puro in the presence of puromycin were designated HCT116 p53−/−-empty and HCT116 p53+/+-empty cells, respectively.
Quantitative reverse transcription (RT)-PCR.
Total RNA was isolated from infected cells, and cDNAs were synthesized from the isolated RNA as described above. The amount of cDNA of specific genes was quantitated using the Universal ProbeLibrary (Roche) with TaqMan Master (Roche) and the LightCycler 1.5 system (Roche) according to the manufacturer's instructions. Gene-specific primers and universal probes were designed using ProbeFinder software (Roche). The primer sequences and probes for ICP27 were 5′-TCCGACAGCGATCTGGAC-3′, 5′-TCCGACGAGGAACACTCC-3′, and Universal ProbeLibrary probe 56, for ICP0 were 5′-ACCACCATGACGACGACTC-3′, 5′-AGCCCCGTCTCGAACAGT-3′, and Universal ProbeLibrary probe 56, and for 18S rRNA were 5′-GCAATTATTCCCCATGAACG-3′, 5′-GGGACTTAATCAACGCAAGC-3′, and ProbeLibrary probe 48. The amounts of ICP27 and ICP0 expression were normalized to the amount of 18S rRNA expression. The relative amount of each gene expression was calculated using the comparative cycle threshold (2−ΔΔCt) method (42).
Luciferase assay.
HCT116 p53−/− cells in a 48-well plate were transfected using polyethylenimine with 125 ng of either reporter plasmid pICP27-luc (ICP27 promoter) or pSRα-luc (SRα promoter) encoding the firefly luciferase gene driven by the indicated promoter. Cells in all wells were also transfected with 12.5 ng of the internal control plasmid pRL-CMV encoding the Renilla luciferase gene driven by the cytomegalovirus promoter (Promega). One-quarter of the pICP27-luc-transfected cells and one-quarter of the pSRα-luc-transfected cells were also transfected with 30 ng pcDNA-VP16 and 240 ng pcDNA5/FRT (empty); one-quarter of each were transfected with 30 ng pcDNA-VP16 and 240 ng of pcDNA-p53, one-quarter of each were transfected with 30 ng pcDNA5/FRT (empty) and 240 ng pcDNA-p53, and one-quarter of each were also transfected with 270 ng pcDNA5/FRT (empty) as described above. pcDNA5/FRT (empty) was used to adjust the DNA concentration so the transfection mixture in each well contained the same amount of DNA. Firefly and Renilla luciferase activities were independently assayed using the dual-luciferase reporter assay system (Promega) according to the manufacturer's instructions. Relative promoter activity was calculated as firefly luciferase activity/Renilla luciferase activity.
RESULTS
Identification of cell proteins that interact with ICP22.
To identify host cell proteins that interact with ICP22, we used a tandem affinity purification approach coupled with mass spectrometry-based proteomics technology. These experiments identified 329 cell proteins that coimmunoprecipitated with transiently expressed MEF-tagged ICP22 (MEF-ICP22) (Fig. 1B). Although it was necessary to confirm by further experiments whether these identified cell proteins specifically interacted with ICP22 in infected cells, they included cell proteins known to interact with ICP22, e.g., p78, Hsc70, EAP, and subunits of casein kinase II (5, 12, 43, 44). Of these proteins, we focused on p53, since it has been reported that p53 functions in regulation of a variety of cellular processes, as described above.
Characterization of recombinant viruses generated in this study.
To investigate the interaction between ICP22 and p53, and its biological significance in infected cells, we constructed the ICP22-null mutant virus YK421 (ΔICP22), its repaired virus YK422 (ΔICP22-repair), and recombinant virus YK423 (MEF-ICP22) expressing MEF-ICP22 (Fig. 2). As reported previously (10), the polyclonal antibody to ICP22 generated in this study reacted with multiple bands of ICP22 (Mr of approximately 67 to 72) in HEL cells infected with wild-type HSV-1F or YK422 (ΔICP22-repair) but not with mock-infected or YK421 (ΔICP22)-infected HEL cells (Fig. 3A). In agreement with previous characterization of other ICP22-null mutant viruses (8, 10), HEL cells infected with YK421 (ΔICP22) produced less ICP0 protein than cells infected with wild-type HSV-1F or YK422 (ΔICP22-repair), whereas cells infected with YK421 (ΔICP22) produced ICP27 protein at a level similar to cells infected with wild-type HSV-1F or YK422 (ΔICP22-repair) (Fig. 3A). In addition, YK421 (ΔICP22) replicated in HEL cells much less efficiently than wild-type HSV-1F or YK422 (ΔICP22-repair) (Fig. 3B), whereas growth of YK421 (ΔICP22) in Vero cells was comparable to that of wild-type HSV-1F and YK422 (ΔICP22-repair) (Fig. 3C).
Fig 3.
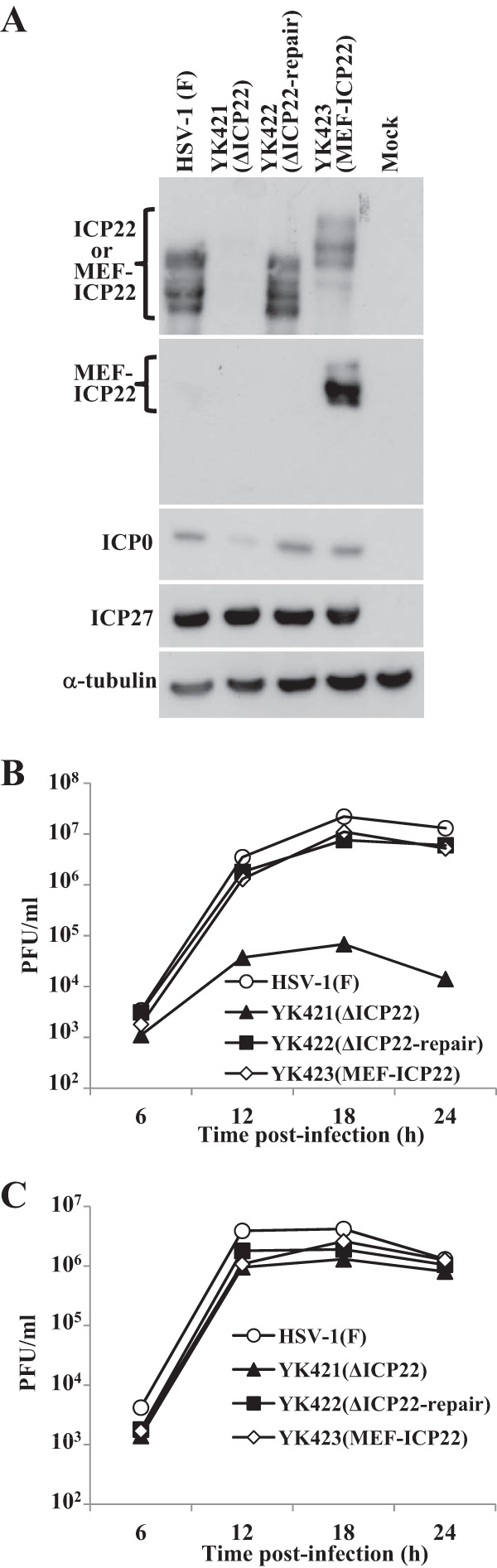
Characterization of the recombinant viruses constructed for this study. (A) HEL cells mock-infected or infected with wild-type HSV-1F, YK421 (ΔICP22), YK422 (ΔICP22-repair), or YK423 (MEF-ICP22) at an MOI of 5 for 18 h were analyzed by immunoblotting with antibody to ICP22, Flag epitope, ICP0, ICP27, or α-tubulin. HEL (B) and Vero (C) cells were infected with HSV-1F, YK421 (ΔICP22), YK422 (ΔICP22-repair), or YK423 (MEF-ICP22) at an MOI of 5. Total virus from the cell culture supernatants and infected cells was harvested at the indicated times and assayed on Vero cells.
Anti-Flag antibody and anti-ICP22 antibody both reacted with multiple bands of MEF-ICP22 in HEL cells infected with YK423 (MEF-ICP22) (Fig. 3A). The YK432 MEF-ICP22 bands migrated in a denaturing gel more slowly than the corresponding wild-type ICP22 bands due to the MEF tag on ICP22 in YK432 (Fig. 3A). The MEF tag on ICP22 appeared to have no effect on the function(s) of ICP22 in infected cells, since HEL cells infected with YK423 (MEF-ICP22) produced ICP0 at a level similar to that in cells infected with wild-type HSV-1F, and the growth curve of YK423 (MEF-ICP22) in HEL cells was comparable to that of wild-type HSV-1F (Fig. 3B).
Interaction of ICP22 with p53 in HSV-1-infected cells.
To confirm the interaction of ICP22 with p53 in infected cells, two series of experiments were carried out. In the first series of experiments, HEL cells infected with YK423 (MEF-ICP22), YK711 (MEF-gB), or wild-type HSV-1F were lysed and immunoprecipitated with anti-Flag antibody, and the immunoprecipitates were analyzed by immunoblotting with antibodies to p53, laminB1, and Flag. As shown in Fig. 4A, anti-Flag antibody coprecipitated p53 from lysates of cells infected with YK423 (MEF-ICP22) but did not from lysates of wild-type HSV-1F-infected cells. Furthermore, anti-Flag antibody coprecipitated p53 but not laminB1 from lysates of cells infected with YK423 (MEF-ICP22) but did not from lysates of cells infected with YK711 (MEF-gB) (Fig. 4B). We noted that as the MEF tag was fused at the amino terminus of ICP22, anti-Flag antibody reacted with MEF-ICP22 but was not able to react with Us1.5 since it is an amino terminal truncated form of ICP22 (Fig. 2), which suggested that full-length ICP22 formed a stable complex with p53 in infected cells. In the second series of experiments, HEL cells infected with YK423 (MEF-ICP22) or wild-type HSV-1F were analyzed by immunofluorescence with antibodies to the p53 and Flag epitopes. As reported previously (12, 30), ICP22 and p53 were predominantly localized to the nucleus and were in part colocalized (Fig. 4C). These results indicated that ICP22 interacted with p53 in infected cells.
Fig 4.
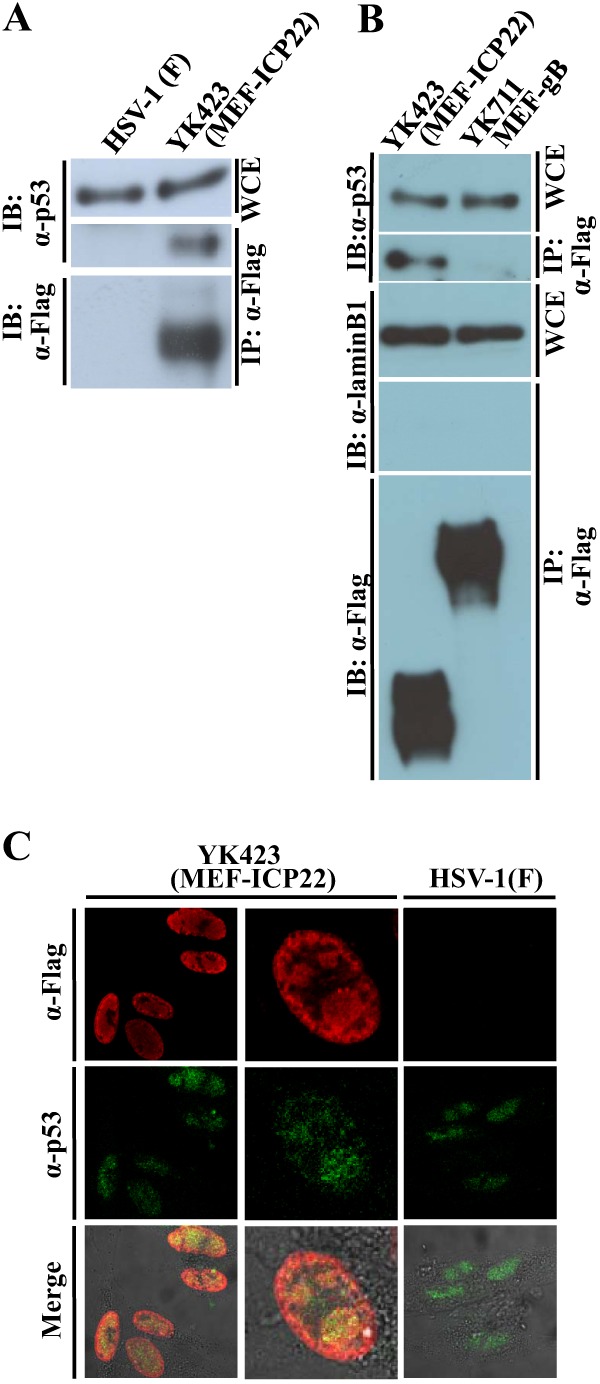
Interaction of ICP22 with p53. (A) HEL cells infected with wild-type HSV-1F (lane 1) or YK423 (MEF-ICP22) (lane 2) at an MOI of 5 for 9 h were harvested, immunoprecipitated (IP) with anti-Flag antibody (α-Flag), and analyzed by immunoblotting (IB) with anti-p53 antibody (α-p53) or anti-Flag antibody. WCE, whole-cell extract. (B) HEL cells infected with wild-type HSV-1F (lane 1) or YK711 (MEF-gB) (lane 2) at an MOI of 5 for 9 h were harvested, immunoprecipitated (IP) with anti-Flag antibody (α-Flag), and analyzed by immunoblotting (IB) with anti-p53 antibody (α-p53), anti-laminB1 (α-laminB1), or anti-Flag antibody. WCE, whole-cell extract. (C) HEL cells infected with YK423 (MEF-ICP22) or wild-type HSV-1F at an MOI of 10 for 9 h were fixed, permeabilized, stained with anti-Flag and anti-p53 antibodies, and examined by confocal microscopy. Upper and middle columns and the lower column show protein fluorescence and simultaneous acquisition of protein fluorescence and digital interference contrast, respectively.
Effect of a p53-null mutation on replication of HSV-1 wild-type and ICP22-null mutant viruses.
To investigate the role(s) of p53 in HSV-1 replication, we examined the effect of a p53-null mutation in HCT116 cells on progeny virus production of wild-type HSV-1F, YK421 (ΔICP22), and YK422 (ΔICP22-repair). As shown in Fig. 5, growth of wild-type HSV-1F and YK422 (ΔICP22-repair) in HCT116 p53−/− cells was less than that in HCT116 p53+/+ cells. The progeny virus titers of wild-type HSV-1F and YK422 (ΔICP22-repair) in HCT116 p53−/− cells 48 h postinfection were 22.0- and 22.8-fold lower than in HCT116 p53+/+ cells, respectively. In contrast, growth of YK421 (ΔICP22) in HCT116 p53−/− cells was similar to that in HCT116 p53+/+ cells, although the YK421 (ΔICP22) progeny virus yield was less than that of wild-type HSV-1F and YK422 (ΔICP22-repair) in HCT116 p53−/− and HCT116 p53+/+ cells (Fig. 5), as was also observed with HEL cells (Fig. 3B).
Fig 5.
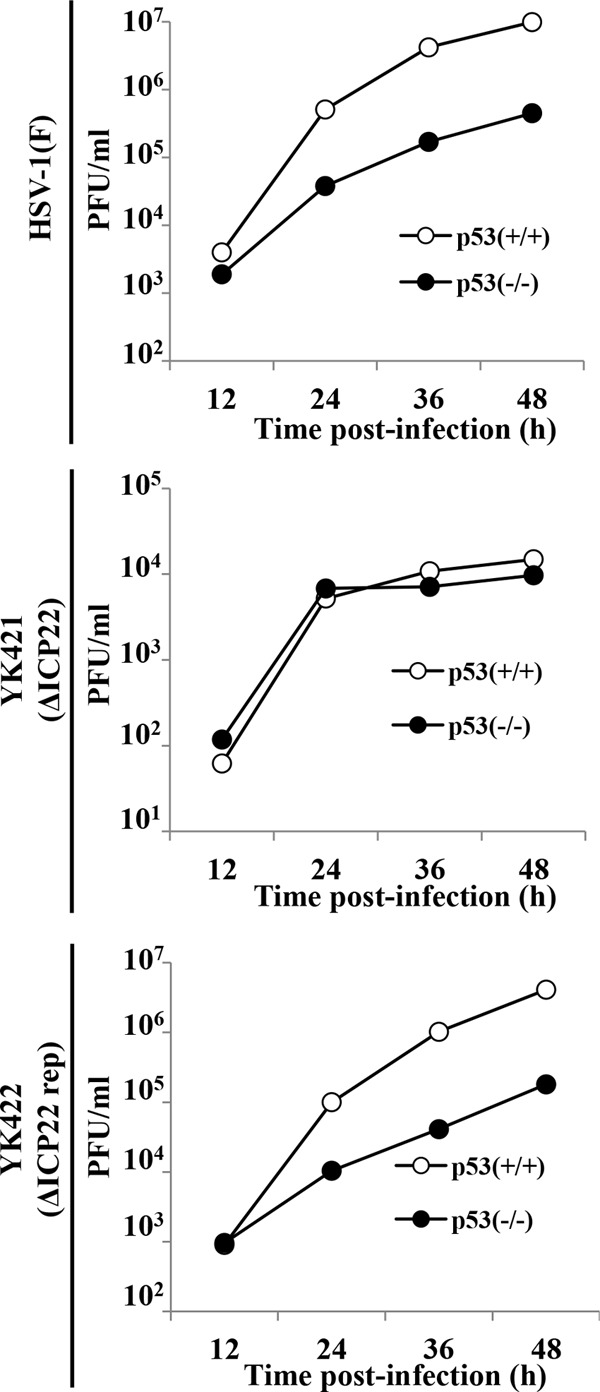
Effect of the p53-null mutation on growth of wild-type HSV-1F, YK421 (ΔICP22), and YK422 (ΔICP22-repair). HCT116 p53+/+ and HCT116 p53−/− cells were infected with wild-type HSV-1F, YK421 (ΔICP22), or YK422 (ΔICP22-repair) at an MOI of 0.01. Total virus from the cell culture supernatants and infected cells was harvested at the indicated times and assayed on Vero cells.
p53 is referred to as the major “guardian of the genome,” based on its pivotal roles for genomic stability (45), raising the question of whether the results of HSV-1 infection observed in HCT116 p53−/− cells were due to the absence of p53 or to some other protein product(s) that might be missing or aberrant in these cells. To address this question, we generated HCT116 p53−/− p53+ cells, in which p53 was reintroduced by transduction with a retrovirus expressing p53, and control HCT116 p53−/−-empty and HCT116 p53+/+-empty cells, in which HCT116 p53−/− and HCT116 p53+/+ cells were transduced by the empty retrovirus, respectively (Fig. 6A). As observed with HCT116 p53−/− and HCT116 p53+/+ cells (Fig. 5), progeny virus yields of wild-type HSV-1F and YK422 (ΔICP22-repair) in HCT116 p53−/−-empty cells infected at a multiplicity of infection (MOI) of 0.01 for 48 h were significantly lower than those in HCT116 p53+/+-empty cells (Fig. 6B). Reintroduction of p53 into HCT116 p53−/− cells significantly increased progeny virus yields of wild-type HSV-1F and YK422 (ΔICP22-repair), and the progeny virus yields of these viruses in HCT116 p53−/− p53+ cells were comparable to those in HCT116 p53+/+-empty cells (Fig. 6B). In contrast and in agreement with the observation that the p53-null mutation in HCT116 cells had no effect on replication of YK421 (ΔICP22) (Fig. 5), the YK421 (ΔICP22) progeny virus yield in HCT116 p53−/− p53+ cells was similar to that in HCT116 p53+/+-empty cells (Fig. 6B). These results suggested that the loss of p53 was responsible for the reduction in progeny virus yield of wild-type HSV-1F in HCT116 p53−/− cells.
Fig 6.
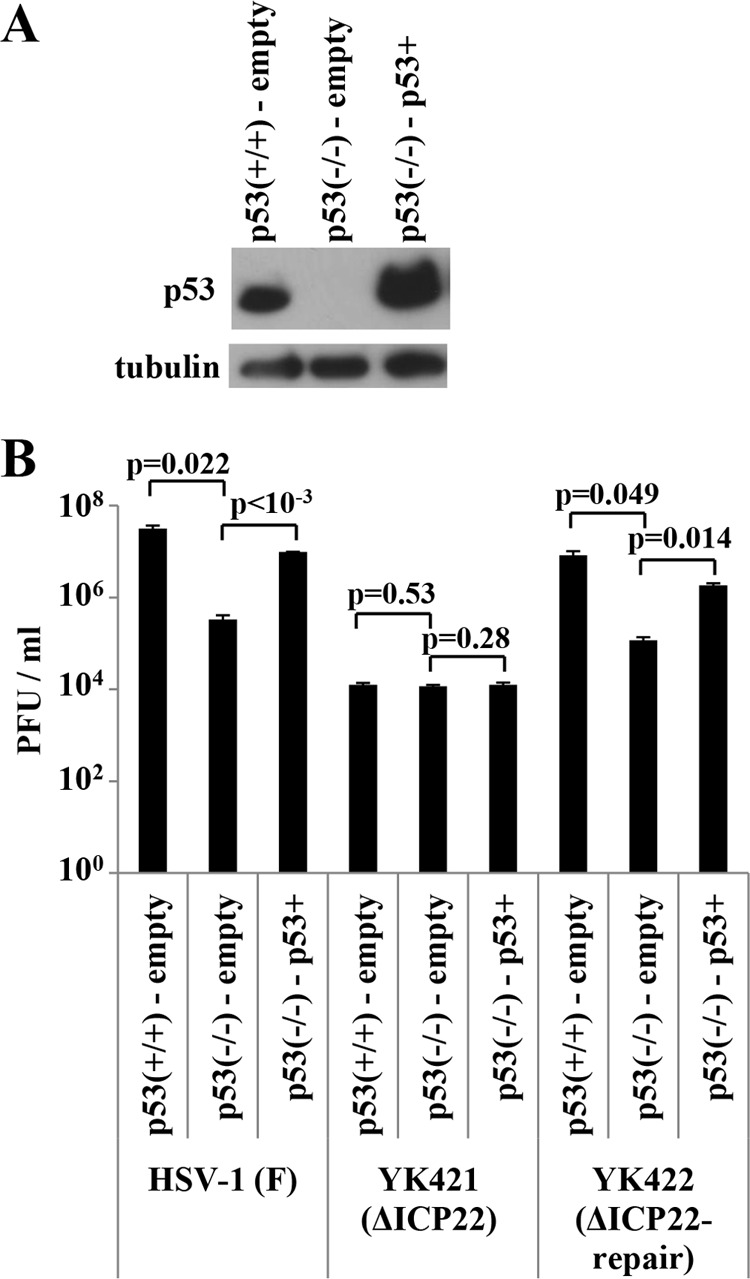
Effect of p53 reintroduction into HCT116 p53−/− cells on viral replication. (A) HCT116 p53+/+-empty, HCT116 p53−/−-empty, and HCT116 p53−/− p53+ cells were analyzed by immunoblotting with antibody to p53 or α-tubulin. (B) HCT116 p53+/+-empty, HCT116 p53−/−-empty, and HCT116 p53−/− p53+ cells were infected with wild-type HSV-1F, YK421 (ΔICP22), or YK422 (ΔICP22-repair) at an MOI of 0.01. Total virus from cell culture supernatants and infected cells was harvested at 48 h postinfection and assayed on Vero cells. P values were calculated by a two-tailed Student t test.
Taken together, these observations indicated that p53 was critical for replication of wild-type HSV-1 but not for replication of the ICP22-null mutant virus.
Effect of the p53-null mutation on expression of HSV-1 ICP27.
p53 is known to regulate expression of various downstream genes at both transcriptional and posttranscriptional levels (21, 46). Therefore, we hypothesized that p53 regulated expression of a specific HSV-1 gene(s). To test this hypothesis, we compared the expression levels of several HSV-1 proteins in HCT116 p53−/− and HCT116 p53+/+ cells infected with wild-type HSV-1F or YK421 (ΔICP22). Of the viral proteins tested, we found that the level of expression of an IE protein, ICP27, which plays an essential role in HSV-1 replication (20), at 2 h after wild-type HSV-1F infection in HCT116 p53−/− cells was reduced compared to that in HCT116 p53+/+ cells (Fig. 7A). Similar results were obtained with YK421 (ΔICP22) (Fig. 7A). In contrast, the level of expression of another IE protein, ICP22, at 2 h after wild-type HSV-1F infection was similar in HCT116 p53−/− cells and in HCT116 p53+/+ cells (Fig. 7B). At later times postinfection (8 and 18 h postinfection), the level of expression of ICP27 and ICP22 proteins was similar in HCT116 p53−/− and HCT116 p53+/+ cells (Fig. 7A and B). Furthermore, reintroduction of p53 into HCT116 p53−/− cells restored ICP27 protein expression at 2 h after wild-type HSV-1F infection to the level observed in HCT116 p53+/+-empty cells (Fig. 7C). We also compared the level of ICP27 mRNA expressed in HCT116 p53+/+ and HCT116 p53−/− cells infected with wild-type HSV-1F. In agreement with the observation obtained in Fig. 7, the level of ICP27 mRNA in HCT116 p53−/− cells infected with wild-type HSV-1F at 1.5 h postinfection was significantly lower than in infected HCT116 p53+/+ cells, whereas there was no significant difference in the level of ICP27 mRNA between HCT116 p53+/+ and HCT116 p53−/− cells at 8 h postinfection (Fig. 8). These results indicated that p53 was required for efficient expression of the ICP27 gene very early in infection (2 h postinfection), and this p53 regulatory effect was independent of ICP22.
Fig 7.
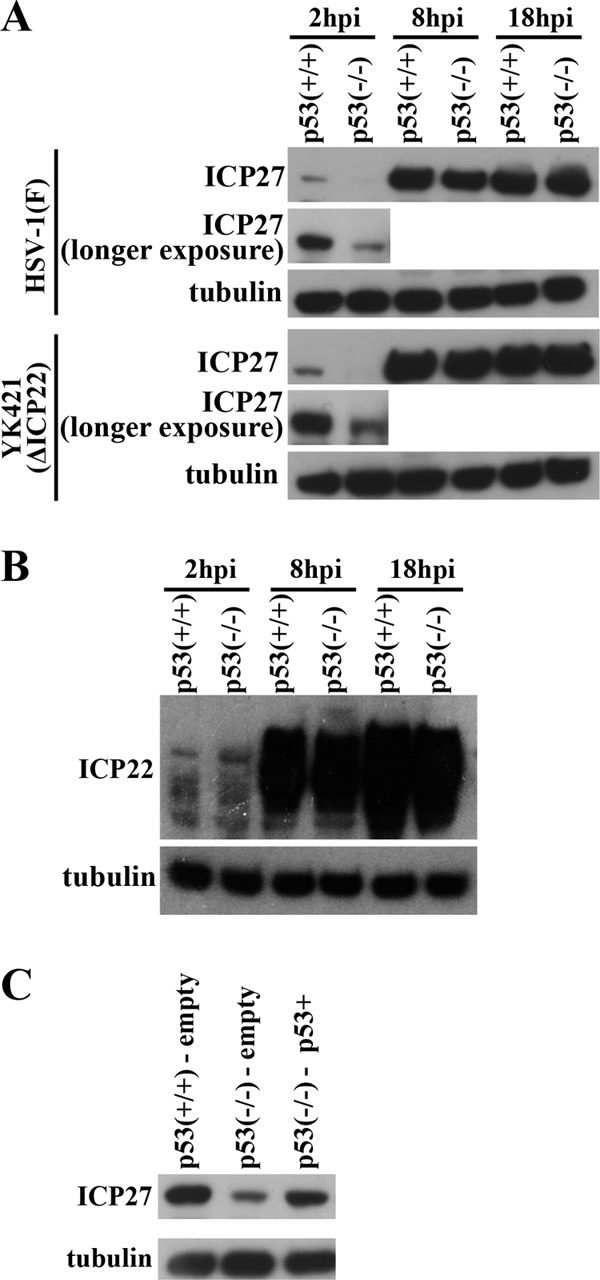
Effect of the p53-null mutation on expression of ICP27 protein in infected cells. (A) HCT116 p53+/+ and HCT116 p53−/− cells infected with wild-type HSV-1F or YK421 (ΔICP22) at an MOI of 5 were harvested at the indicated times, and the amounts of ICP27 and α-tubulin protein were analyzed by immunoblotting with antibody to ICP27 or α-tubulin. A longer exposure is shown for ICP27 at 2 h postinfection. (B) HCT116 p53+/+ and HCT116 p53−/− cells infected with wild-type HSV-1F at an MOI of 5 were harvested at the indicated times, and the amounts of ICP22 and α-tubulin protein were analyzed by immunoblotting with antibody to ICP22 and α-tubulin. (C) HCT116 p53+/+-empty, HCT116 p53−/−-empty, and HCT116 p53−/− p53+ cells infected with HSV-1F at an MOI of 5 for 2 h were harvested and analyzed by immunoblotting with antibody to ICP27 and α-tubulin.
Fig 8.
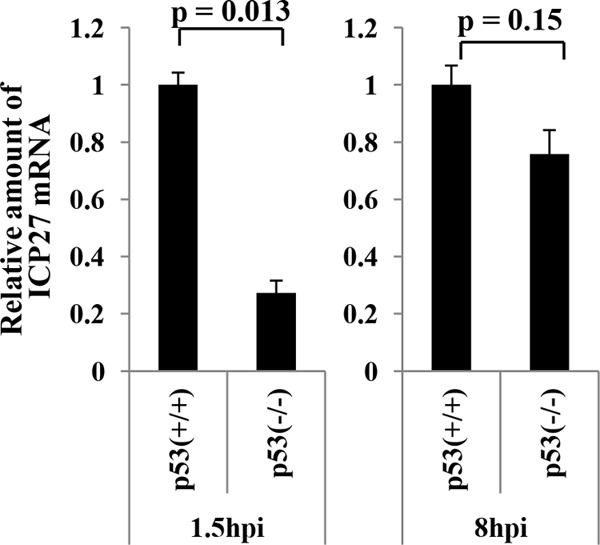
Effect of the p53-null mutation on expression of ICP27 mRNA in infected cells. HCT116 p53+/+ and HCT116 p53−/− cells infected with wild-type HSV-1F at an MOI of 5 were harvested at the indicated times, and the amounts of ICP27 mRNA were analyzed by quantitative RT-PCR. Each bar is the mean ± standard error of data from three independent experiments. The mean value for HCT116 p53−/− cells was calculated relative to that for HCT116 p53+/+ cells, which was normalized to 1. P values were calculated by a two-tailed Student t test.
Effect of p53 and/or VP16 on activation of the ICP27 promoter.
To examine effects of p53 on ICP27 promoter activity and on activation of the ICP27 promoter mediated by VP16, a primary component of the multienhancesome that activates IE genes very early in infection (2), we carried out luciferase reporter assays in HCT116 p53−/− cells. As reported previously, expression of VP16 by itself resulted in approximately 27-fold activation of the ICP27 promoter, whereas expression of p53 by itself had little effect (Fig. 9A). Interestingly, coexpression of p53 and VP16 significantly augmented activation of the ICP27 promoter, compared to that when VP16 was expressed alone, and resulted in 47-fold activation of the ICP27 promoter (Fig. 9A). In contrast, p53 by itself or p53 in combination with VP16 did not stimulate the unrelated SRα promoter (Fig. 9B). These results raised the possibility that p53 may have the ability to stimulate the ICP27 promoter in concert with VP16.
Fig 9.
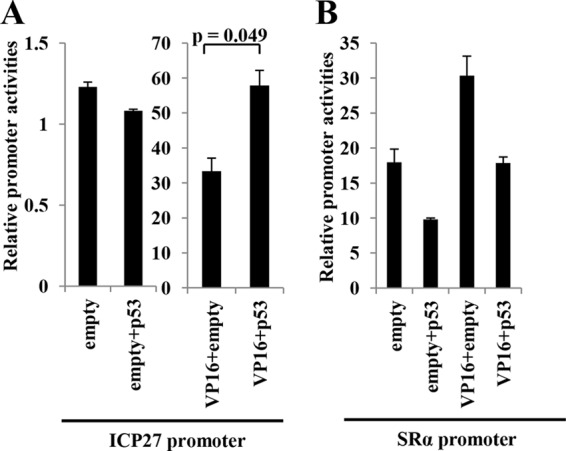
Effect of p53 by itself or p53 in combination with VP16 on the ICP27 promoter. HCT116 p53−/− cells were transfected with pRL-CMV and pICP27-luc (ICP27 promoter) (A) or pSRα-luc (SRα promoter) (B) in combination with pcDNA5/FRT (empty), pcDNA-VP16 (VP16), and/or pcDNA-p53 (p53). At 24 h after transfection, luciferase activity was assayed. Relative promoter activity was calculated as firefly luciferase activity/Renilla luciferase activity. The results were the means ± standard errors from the results of triplicate experiments. Data are representative of three independent experiments. P values were calculated by a two-tailed Student t test.
Effect of the p53-null mutation on expression of HSV-1 ICP0.
During comparison of the expression levels of viral proteins in HCT116 p53−/− and HCT116 p53+/+ cells infected with wild-type HSV-1F and YK421 (ΔICP22), we also obtained data showing that p53 appeared to affect expression of viral protein ICP0, a critical regulator of viral replication (18, 19), in cells infected with the ICP22-null mutant virus but not in wild-type virus-infected cells. As shown in Fig. 10A, HCT116 p53+/+ cells infected with YK421 (ΔICP22) produced less ICP0 than HCT116 p53+/+ cells infected with wild-type HSV-1F or YK422 (ΔICP22-repair) (Fig. 10A). These results are in agreement with the results obtained with HEL cells described above (Fig. 3A) and in a previous report (10). In contrast, HCT116 p53−/− cells infected with YK421 (ΔICP22) produced ICP0 at a level similar to HCT116 p53−/− cells infected with wild-type HSV-1F or YK422 (ΔICP22-repair) (Fig. 10A). The expression levels of the ICP27 protein were similar in HCT116 p53+/+ and HCT116 p53−/− cells infected with wild-type HSV-1F, YK421 (ΔICP22), or YK422 (ΔICP22-repair) (Fig. 10A). In addition, reintroduction of p53 into HCT116 p53−/− cells and then infection with YK421 (ΔICP22) reduced ICP0 protein expression at 18 h postinfection, and the level of ICP0 protein expression in HCT116 p53−/− p53+ was comparable to that in HCT116 p53+/+-empty cells (Fig. 10B). These results indicated that p53 downregulated expression of the ICP0 protein in ICP22-null mutant virus-infected cells late in infection (18 h postinfection) but not in wild-type virus-infected cells, suggesting that p53 downregulated expression of ICP0 protein in ICP22-null mutant virus-infected cells but that this effect was antagonized by ICP22 in wild-type virus-infected cells.
Fig 10.
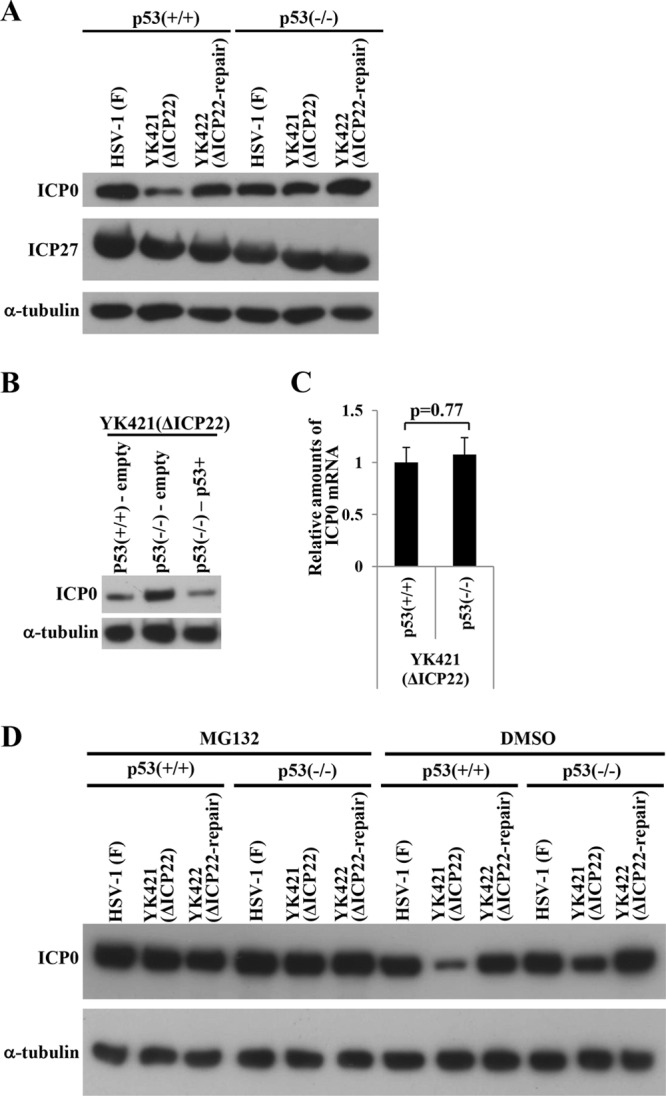
Effect of the p53-null mutation on ICP0 gene expression. (A) HCT116 p53+/+ and HCT116 p53−/− cells infected with wild-type HSV-1F, YK421 (ΔICP22), or YK422 (ΔICP22-repair) at an MOI of 5 for 18 h were harvested and ICP0, ICP27, and α-tubulin expression was analyzed by immunoblotting with antibody to ICP0, ICP27, or α-tubulin. (B) HCT116 p53+/+-empty, HCT116 p53−/−-empty, and HCT116 p53−/− p53+ cells infected with YK421 (ΔICP22) at an MOI of 5 for 18 h were harvested and analyzed by immunoblotting with antibody to ICP0 or α-tubulin. (C) HCT116 p53+/+ and HCT116 p53−/− cells infected with YK421 (ΔICP22) at an MOI of 5 for 18 h were harvested, and ICP0 mRNA was analyzed by quantitative RT-PCR. Each bar is the mean ± standard error of data from three independent experiments. The mean value for HCT116 p53−/− cells was calculated relative to that for HCT116 p53+/+ cells, which was normalized to 1. The P value was calculated by a two-tailed Student t test. (D) HCT116 p53+/+ and HCT116 p53−/− cells infected with wild-type HSV-1F, YK421 (ΔICP22), or YK422 (ΔICP22-repair) at an MOI of 5 were treated with 20 μM MG132 or DMSO at 4 h postinfection, and infection proceeded for an additional 14 h in the presence of MG132 or DMSO. Infected cells were then harvested and analyzed by immunoblotting with antibody to ICP0 or α-tubulin.
We also compared the amount of ICP0 mRNA expressed in YK421 (ΔICP22)-infected HCT116 p53+/+ and HCT116 p53−/− cells. As shown in Fig. 10C, there was no significant difference in the amount of ICP0 mRNA in YK421 (ΔICP22)-infected HCT116 p53−/− and HCT116 p53+/+ cells, suggesting that the regulatory effect of p53 was at the posttranscriptional level. We next examined the effect of the proteasomal inhibitor MG132 on ICP0 protein expression in YK421 (ΔICP22)-infected HCT116 p53+/+ and HCT116 p53−/− cells. In agreement with the results in Fig. 10A and B, ICP0 protein expression in YK421 (ΔICP22)-infected HCT116 p53+/+ cells treated with dimethyl sulfoxide (DMSO) was lower than that in YK421 (ΔICP22)-infected HCT116 p53−/− cells treated with DMSO (Fig. 10D). In contrast, MG132 treatment increased ICP0 protein expression, and the level of ICP0 protein expression in YK421 (ΔICP22)-infected HCT116 p53+/+ cells was equivalent to that in HCT116 p53−/− cells infected with YK421 (ΔICP22) and to HCT116 p53+/+ cells infected with wild-type HSV-1F (Fig. 10D). These results suggested that p53-mediated downregulation of ICP0 expression late in infection was due to proteasome degradation.
DISCUSSION
In the present study, tandem affinity purification of transiently expressed ICP22 coupled with mass spectrometry-based proteomics technology indicated a potential interaction between ICP22 and p53. This led us to investigate the interaction of ICP22 and p53 in HSV-1-infected cells and the role(s) of p53 in viral replication in the presence and absence of ICP22. The results of this study suggested that p53 overall promoted HSV-1 replication. Furthermore, we obtained data suggesting that p53 played both positive and negative roles in HSV-1 replication and that the negative effect of p53 was antagonized by ICP22. The salient features of our results that supported these conclusions are as follows.
(i) ICP22 interacted with p53 in HSV-1-infected cells. This conclusion was based on the observations that p53 coprecipitated with ICP22 tagged with MEF at its N terminus (MEF-ICP22) in the lysate of cells infected with recombinant virus YK423 expressing MEF-ICP22 and that p53 colocalized with MEF-ICP22 in the nucleus of cells infected with YK423 (MEF-ICP22). It appeared that the interaction of p53 with ICP22 did not require any other viral protein, since transiently expressed ICP22 in 293T cells coprecipitated p53. Although the results with YK423 (MEF-ICP22) clearly indicated that p53 interacted with ICP22 in infected cells, it is uncertain at present whether Us1.5, an N-terminal truncated form of ICP22, also interacted with p53 in infected cells.
(ii) p53 promoted ICP27 expression in early infection in an ICP22-independent manner. ICP27 plays essential roles in HSV-1 replication by regulating all stages of mRNA biogenesis, including transcription, RNA processing, and export for translation (20). Therefore, the results of this study, that p53 was required for efficient expression of ICP27 in cells infected with wild-type HSV-1 and the ICP22-null mutant virus at 2 h postinfection, suggested that p53 played a positive role in HSV-1 replication by upregulating ICP27 expression in early infection, and this positive effect of p53 is independent of ICP22 binding. The consensus sequence of the p53 binding site for transcriptional regulation of its target genes has been identified as two inverted pentameric sequences with the pattern 5′-RRRC(A/T)(A/T)GYYY-3′ (where R can be A or G, and Y can be T or C) (47). However, we were not able to detect any match to this consensus sequence in a search of the sequence 3 kbp upstream and 1 kbp downstream of the ICP27 transcription initiation site (data not shown). Therefore, at present, the mechanism by which p53 upregulated ICP27 expression at the transcriptional level in early infection is unknown. Recently, it has been reported that lysine-specific demethylase 1 (LSD-1) and p300, which interact with p53 (48, 49), and VP16 are components of a multiprotein enhanceosome complex that mediates transcriptional activation of IE genes in very early infection (2). p53 may upregulate ICP27 gene expression by activating and/or stabilizing the multiprotein enhanceosome complex by interacting with the viral and cellular components of the enhanceosome complex in very early infection. In agreement with this hypothesis, our experiments showed that p53 significantly upregulated VP16-mediated activation of the ICP27 promoter in HCT116 p53−/− cells in a reporter gene assay, but p53 had no effect on VP16-mediated activation of an unrelated promoter (Fig. 9). It has also been reported that the requirement for VP16 for expression of individual IE genes varied in infected cells (50), and this may account for the effect of p53 on ICP27 expression but not on ICP22 expression.
(iii) p53 downregulated ICP0 expression in a proteasome-dependent manner in cells infected with the ICP22-null mutant virus but not in cells infected with the wild-type virus. ICP0 plays a critical role in viral replication, mainly by promoting the transition from IE to E and L gene expression and by blocking innate immunity responses mediated by interferons (18, 19). Therefore, the results of this study, that p53 reduced expression of ICP0 protein in ICP22-null mutant virus-infected cells and that this effect of p53 on ICP0 expression was not found in wild-type virus-infected cells, suggested that p53 played a negative role in replication of the ICP22-null mutant virus by downregulating ICP0 expression, but this negative effect of p53 was counteracted by ICP22 in wild-type virus-infected cells. The results that the effect of p53 on ICP0 expression was reduced by treatment with the proteasome inhibitor MG132 suggested that p53 promoted proteasome-mediated degradation of ICP0 in infected cells. ICP0 acts as an E3 ubiquitin ligase and targets specific proteins, such as promyelocytic leukemia protein (PML) and Sp100, for ubiquitination and proteasome-mediated degradation in infected cells (51, 52). The E3 ubiquitin ligase activity of ICP0 also autoubiquitinates ICP0, which leads to degradation of ICP0 (53). Interestingly, ICP0 interacts with ubiquitin-specific protease 7 (USP7), which removes ubiquitins from target proteins, and this interaction counteracts autoubiquitination-mediated ICP0 degradation, but USP7 is in turn subjected to ICP0-mediated degradation (54, 55). Furthermore, it has been reported that cellular E3 ubiquitin ligase SIAH-1 interacts with ICP0 and promotes proteasomal degradation of ICP0 in infected cells (56). Therefore, the stability of ICP0 in infected cells appears to be tightly regulated by multiple viral and cellular factors as described above and probably by a currently unknown factor(s). p53 may downregulate ICP0 protein expression by regulating the factor(s) involved in ICP0 stabilization as described above, and ICP22 may antagonize the effect(s) of p53 by interacting with it. Although SIAH-1 is a direct transcriptional target of p53 (57), our preliminary experiment indicated that the level of SIAH-1 mRNA and protein in HCT116 p53+/+ cells infected with YK421 (ΔICP22) and in HCT116 p53−/− cells infected with YK421 (ΔICP22) were similar (data not shown), which suggested that a factor(s) other than SIAH-1 was involved in p53-mediated downregulation of ICP0 expression in ICP22-null mutant virus-infected cells.
Taken together, these data raised a possibility that, in wild-type HSV-1-infected cells, the negative role of p53 in downregulating ICP0 expression was antagonized by ICP22, and only the positive role of p53 in upregulating ICP27 expression was effective, resulting in the reduction of viral replication in p53−/− cells. In contrast, in ICP22-null mutant virus-infected cells, both the positive and negative functions of p53 were effective, thereby diminishing the reduction of viral replication in p53−/− cells (Fig. 5). However, we cannot eliminate the possibility that the effects of p53 on HSV-1 replication described in this study, other than those on expression of ICP27 and ICP0 in infected cells, involved both types of p53 functions. The features of p53 in HSV-1 infection shown in this study seem to be in agreement with those of p53 in EBV infection. p53 has been reported to be required for efficient expression of BZLF1 (27), an EBV IE transcriptional regulator that plays a critical role in initiation of EBV lytic infection (58), but at later stages of lytic infection, BZLF1 mediates p53 degradation to counteract p53's negative effect(s) on viral replication (59). Therefore, viruses appear to have acquired multiple mechanisms to deal with p53's positive and negative effects in infected cells: they utilize one or more of p53's functions to regulate some cell components for efficient viral replication, but they also have mechanisms to antagonize p53-mediated cellular response(s) against viral infection. Viruses other than HSV-1 and EBV may also have acquired mechanisms to control p53's positive and negative effects on viral infection, since it has been reported that some other viruses regulate one of these p53 effects (60). Further studies will be needed to better understand the roles of p53 in HSV-1 replication in cells and in pathogenicity in vivo. Studies of the role of p53 in HSV-1 replication in vivo would be of particular interest, since p53, whose expression can be regulated by interferons, has been reported to contribute to immune responses for neutralization of pathogens (61).
ACKNOWLEDGMENTS
We thank Tomoko Ando and Shihoko Koyama for excellent technical assistance.
This study was supported by the Funding Program for Next Generation World-Leading Researchers and Grants for Scientific Research from the Japan Society for the Promotion of Science (JSPS), a contract research fund for the Program of Japan Initiative for Global Research Network on Infectious Diseases and Global COE Program “Center of Education and Research for the Advanced Genome-Based Medicine, for personalized medicine and the control of worldwide infectious diseases,” from the Ministry of Education, Culture, Science, Sports and Technology (MEXT) of Japan, and grants from the Takeda Science Foundation, the Naito Foundation, the Asahi Glass Foundation, and the Tokyo Biochemical Research Foundation. H.F. was supported by research fellowships from JSPS for Young Scientists.
Footnotes
Published ahead of print 19 June 2013
REFERENCES
- 1. Roizman B, Knipe DM, Whitley RJ. 2007. Herpes simplex viruses. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott-Raven Publishers, Philadelphia, PA [Google Scholar]
- 2. Kristie TM, Liang Y, Vogel JL. 2010. Control of alpha-herpesvirus IE gene expression by HCF-1 coupled chromatin modification activities. Biochim. Biophys. Acta 1799:257–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Purves FC, Roizman B. 1992. The UL13 gene of herpes simplex virus 1 encodes the functions for posttranslational processing associated with phosphorylation of the regulatory protein alpha 22. Proc. Natl. Acad. Sci. U. S. A. 89:7310–7314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blaho JA, Mitchell C, Roizman B. 1993. Guanylylation and adenylylation of the alpha regulatory proteins of herpes simplex virus require a viral beta or gamma function. J. Virol. 67:3891–3900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Mitchell C, Blaho JA, Roizman B. 1994. Casein kinase II specifically nucleotidylylates in vitro the amino acid sequence of the protein encoded by the alpha 22 gene of herpes simplex virus 1. Proc. Natl. Acad. Sci. U. S. A. 91:11864–11868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Carter KL, Roizman B. 1996. The promoter and transcriptional unit of a novel herpes simplex virus 1 alpha gene are contained in, and encode a protein in frame with, the open reading frame of the alpha 22 gene. J. Virol. 70:172–178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Ogle WO, Roizman B. 1999. Functional anatomy of herpes simplex virus 1 overlapping genes encoding infected-cell protein 22 and US1.5 protein. J. Virol. 73:4305–4315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sears AE, Halliburton IW, Meignier B, Silver S, Roizman B. 1985. Herpes simplex virus 1 mutant deleted in the alpha 22 gene: growth and gene expression in permissive and restrictive cells and establishment of latency in mice. J. Virol. 55:338–346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ng TI, Chang YE, Roizman B. 1997. Infected cell protein 22 of herpes simplex virus 1 regulates the expression of virion host shutoff gene U(L)41. Virology 234:226–234 [DOI] [PubMed] [Google Scholar]
- 10. Purves FC, Ogle WO, Roizman B. 1993. Processing of the herpes simplex virus regulatory protein alpha 22 mediated by the UL13 protein kinase determines the accumulation of a subset of alpha and gamma mRNAs and proteins in infected cells. Proc. Natl. Acad. Sci. U. S. A. 90:6701–6705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kalamvoki M, Roizman B. 2011. The histone acetyltransferase CLOCK is an essential component of the herpes simplex virus 1 transcriptome that includes TFIID, ICP4, ICP27, and ICP22. J. Virol. 85:9472–9477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Leopardi R, Ward PL, Ogle WO, Roizman B. 1997. Association of herpes simplex virus regulatory protein ICP22 with transcriptional complexes containing EAP, ICP4, RNA polymerase II, and viral DNA requires posttranslational modification by the U(L)13 protein kinase. J. Virol. 71:1133–1139 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Rice SA, Long MC, Lam V, Spencer CA. 1994. RNA polymerase II is aberrantly phosphorylated and localized to viral replication compartments following herpes simplex virus infection. J. Virol. 68:988–1001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hsin JP, Manley JL. 2012. The RNA polymerase II CTD coordinates transcription and RNA processing. Genes Dev. 26:2119–2137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rice SA, Long MC, Lam V, Schaffer PA, Spencer CA. 1995. Herpes simplex virus immediate-early protein ICP22 is required for viral modification of host RNA polymerase II and establishment of the normal viral transcription program. J. Virol. 69:5550–5559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Durand LO, Advani SJ, Poon AP, Roizman B. 2005. The carboxyl-terminal domain of RNA polymerase II is phosphorylated by a complex containing cdk9 and infected-cell protein 22 of herpes simplex virus 1. J. Virol. 79:6757–6762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Durand LO, Roizman B. 2008. Role of cdk9 in the optimization of expression of the genes regulated by ICP22 of herpes simplex virus 1. J. Virol. 82:10591–10599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hagglund R, Roizman B. 2004. Role of ICP0 in the strategy of conquest of the host cell by herpes simplex virus 1. J. Virol. 78:2169–2178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roizman B. 2011. The checkpoints of viral gene expression in productive and latent infection: the role of the HDAC/CoREST/LSD1/REST repressor complex. J. Virol. 85:7474–7482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sandri-Goldin RM. 2011. The many roles of the highly interactive HSV protein ICP27, a key regulator of infection. Future Microbiol. 6:1261–1277 [DOI] [PubMed] [Google Scholar]
- 21. Kruse JP, Gu W. 2009. Modes of p53 regulation. Cell 137:609–622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Menendez D, Shatz M, Resnick MA. 2013. Interactions between the tumor suppressor p53 and immune responses. Curr. Opin. Oncol. 25:85–92 [DOI] [PubMed] [Google Scholar]
- 23. Lepik D, Ilves I, Kristjuhan A, Maimets T, Ustav M. 1998. p53 protein is a suppressor of papillomavirus DNA amplificational replication. J. Virol. 72:6822–6831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Pampin M, Simonin Y, Blondel B, Percherancier Y, Chelbi-Alix MK. 2006. Cross talk between PML and p53 during poliovirus infection: implications for antiviral defense. J. Virol. 80:8582–8592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Staib C, Pesch J, Gerwig R, Gerber JK, Brehm U, Stangl A, Grummt F. 1996. p53 inhibits JC virus DNA replication in vivo and interacts with JC virus large T-antigen. Virology 219:237–246 [DOI] [PubMed] [Google Scholar]
- 26. Casavant NC, Luo MH, Rosenke K, Winegardner T, Zurawska A, Fortunato EA. 2006. Potential role for p53 in the permissive life cycle of human cytomegalovirus. J. Virol. 80:8390–8401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chang SS, Lo YC, Chua HH, Chiu HY, Tsai SC, Chen JY, Lo KW, Tsai CH. 2008. Critical role of p53 in histone deacetylase inhibitor-induced Epstein-Barr virus Zta expression. J. Virol. 82:7745–7751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Pauls E, Senserrich J, Clotet B, Este JA. 2006. Inhibition of HIV-1 replication by RNA interference of p53 expression. J. Leukoc. Biol. 80:659–667 [DOI] [PubMed] [Google Scholar]
- 29. Boutell C, Everett RD. 2004. Herpes simplex virus type 1 infection induces the stabilization of p53 in a USP7- and ATM-independent manner. J. Virol. 78:8068–8077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wilcock D, Lane DP. 1991. Localization of p53, retinoblastoma and host replication proteins at sites of viral replication in herpes-infected cells. Nature 349:429–431 [DOI] [PubMed] [Google Scholar]
- 31. Kato A, Arii J, Shiratori I, Akashi H, Arase H, Kawaguchi Y. 2009. Herpes simplex virus 1 protein kinase Us3 phosphorylates viral envelope glycoprotein B and regulates its expression on the cell surface. J. Virol. 83:250–261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sugimoto K, Uema M, Sagara H, Tanaka M, Sata T, Hashimoto Y, Kawaguchi Y. 2008. Simultaneous tracking of capsid, tegument, and envelope protein localization in living cells infected with triply fluorescent herpes simplex virus 1. J. Virol. 82:5198–5211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tanaka M, Kagawa H, Yamanashi Y, Sata T, Kawaguchi Y. 2003. Construction of an excisable bacterial artificial chromosome containing a full-length infectious clone of herpes simplex virus type 1: viruses reconstituted from the clone exhibit wild-type properties in vitro and in vivo. J. Virol. 77:1382–1391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. 1998. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 282:1497–1501 [DOI] [PubMed] [Google Scholar]
- 35. Arii J, Goto H, Suenaga T, Oyama M, Kozuka-Hata H, Imai T, Minowa A, Akashi H, Arase H, Kawaoka Y, Kawaguchi Y. 2010. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 467:859–862 [DOI] [PubMed] [Google Scholar]
- 36. Kawaguchi Y, Van Sant C, Roizman B. 1997. Herpes simplex virus 1 alpha regulatory protein ICP0 interacts with and stabilizes the cell cycle regulator cyclin D3. J. Virol. 71:7328–7336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tanaka M, Sata T, Kawaguchi Y. 2008. The product of the herpes simplex virus 1 UL7 gene interacts with a mitochondrial protein, adenine nucleotide translocator 2. Virol. J. 5:125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kitamura T, Hayashida K, Sakamaki K, Yokota T, Arai K, Miyajima A. 1991. Reconstitution of functional receptors for human granulocyte/macrophage colony-stimulating factor (GM-CSF): evidence that the protein encoded by the AIC2B cDNA is a subunit of the murine GM-CSF receptor. Proc. Natl. Acad. Sci. U. S. A. 88:5082–5086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Boussif O, Lezoualc'h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. 1995. A versatile vector for gene and oligonucleotide transfer into cells in culture and in vivo: polyethylenimine. Proc. Natl. Acad. Sci. U. S. A. 92:7297–7301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kato A, Tanaka M, Yamamoto M, Asai R, Sata T, Nishiyama Y, Kawaguchi Y. 2008. Identification of a physiological phosphorylation site of the herpes simplex virus 1-encoded protein kinase Us3 which regulates its optimal catalytic activity in vitro and influences its function in infected cells. J. Virol. 82:6172–6189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kawaguchi Y, Bruni R, Roizman B. 1997. Interaction of herpes simplex virus 1 alpha regulatory protein ICP0 with elongation factor 1delta: ICP0 affects translational machinery. J. Virol. 71:1019–1024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schmittgen TD, Livak KJ. 2008. Analyzing real-time PCR data by the comparative C(T) method. Nat. Protoc. 3:1101–1108 [DOI] [PubMed] [Google Scholar]
- 43. Bastian TW, Livingston CM, Weller SK, Rice SA. 2010. Herpes simplex virus type 1 immediate-early protein ICP22 is required for VICE domain formation during productive viral infection. J. Virol. 84:2384–2394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Bruni R, Roizman B. 1998. Herpes simplex virus 1 regulatory protein ICP22 interacts with a new cell cycle-regulated factor and accumulates in a cell cycle-dependent fashion in infected cells. J. Virol. 72:8525–8531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lane DP. 1992. Cancer p53, guardian of the genome. Nature 358:15–16 [DOI] [PubMed] [Google Scholar]
- 46. Puca R, Nardinocchi L, Givol D, D'Orazi G. 2010. Regulation of p53 activity by HIPK2: molecular mechanisms and therapeutical implications in human cancer cells. Oncogene 29:4378–4387 [DOI] [PubMed] [Google Scholar]
- 47. el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. 1992. Definition of a consensus binding site for p53. Nat. Genet. 1:45–49 [DOI] [PubMed] [Google Scholar]
- 48. Huang J, Sengupta R, Espejo AB, Lee MG, Dorsey JA, Richter M, Opravil S, Shiekhattar R, Bedford MT, Jenuwein T, Berger SL. 2007. p53 is regulated by the lysine demethylase LSD1. Nature 449:105–108 [DOI] [PubMed] [Google Scholar]
- 49. Lill NL, Grossman SR, Ginsberg D, DeCaprio J, Livingston DM. 1997. Binding and modulation of p53 by p300/CBP coactivators. Nature 387:823–827 [DOI] [PubMed] [Google Scholar]
- 50. Ace CI, McKee TA, Ryan JM, Cameron JM, Preston CM. 1989. Construction and characterization of a herpes simplex virus type 1 mutant unable to transinduce immediate-early gene expression. J. Virol. 63:2260–2269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Everett RD, Freemont P, Saitoh H, Dasso M, Orr A, Kathoria M, Parkinson J. 1998. The disruption of ND10 during herpes simplex virus infection correlates with the Vmw110- and proteasome-dependent loss of several PML isoforms. J. Virol. 72:6581–6591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Parkinson J, Everett RD. 2000. Alphaherpesvirus proteins related to herpes simplex virus type 1 ICP0 affect cellular structures and proteins. J. Virol. 74:10006–10017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Boutell C, Sadis S, Everett RD. 2002. Herpes simplex virus type 1 immediate-early protein ICP0 and is isolated RING finger domain act as ubiquitin E3 ligases in vitro. J. Virol. 76:841–850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Boutell C, Canning M, Orr A, Everett RD. 2005. Reciprocal activities between herpes simplex virus type 1 regulatory protein ICP0, a ubiquitin E3 ligase, and ubiquitin-specific protease USP7. J. Virol. 79:12342–12354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Canning M, Boutell C, Parkinson J, Everett RD. 2004. A RING finger ubiquitin ligase is protected from autocatalyzed ubiquitination and degradation by binding to ubiquitin-specific protease USP7. J. Biol. Chem. 279:38160–38168 [DOI] [PubMed] [Google Scholar]
- 56. Nagel CH, Albrecht N, Milovic-Holm K, Mariyanna L, Keyser B, Abel B, Weseloh B, Hofmann TG, Eibl MM, Hauber J. 2011. Herpes simplex virus immediate-early protein ICP0 is targeted by SIAH-1 for proteasomal degradation. J. Virol. 85:7644–7657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Fiucci G, Beaucourt S, Duflaut D, Lespagnol A, Stumptner-Cuvelette P, Geant A, Buchwalter G, Tuynder M, Susini L, Lassalle JM, Wasylyk C, Wasylyk B, Oren M, Amson R, Telerman A. 2004. Siah-1b is a direct transcriptional target of p53: identification of the functional p53 responsive element in the siah-1b promoter. Proc. Natl. Acad. Sci. U. S. A. 101:3510–3515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kieff ER, Rickinson AB. 2007. Epstein-Barr virus and its replication. In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed Lippincott-Raven Publishers, Philadelphia, PA [Google Scholar]
- 59. Sato Y, Shirata N, Kudoh A, Iwahori S, Nakayama S, Murata T, Isomura H, Nishiyama Y, Tsurumi T. 2009. Expression of Epstein-Barr virus BZLF1 immediate-early protein induces p53 degradation independent of MDM2, leading to repression of p53-mediated transcription. Virology 388:204–211 [DOI] [PubMed] [Google Scholar]
- 60. Sato Y, Tsurumi T. 17 December 2012. Genome guardian p53 and viral infections. Rev. Med. Virol. [Epub ahead of print.] 10.1002/rmv.1738 [DOI] [PubMed] [Google Scholar]
- 61. Takaoka A, Hayakawa S, Yanai H, Stoiber D, Negishi H, Kikuchi H, Sasaki S, Imai K, Shibue T, Honda K, Taniguchi T. 2003. Integration of interferon-alpha/beta signalling to p53 responses in tumour suppression and antiviral defence. Nature 424:516–523 [DOI] [PubMed] [Google Scholar]