

Abstract
The mammalian type I interferon (IFN) response is a primary barrier for virus infection and is essential for complete innate and adaptive immunity. Both IFN production and IFN-mediated antiviral signaling are the result of differential cellular gene expression, a process that is tightly controlled at transcriptional and translational levels. To determine the potential for microRNA (miRNA)-mediated regulation of the antiviral response, small-RNA profiling was used to analyze the miRNA content of human A549 cells at steady state and following infection with the Cantell strain of Sendai virus, a potent inducer of IFN and cellular antiviral responses. While the miRNA content of the cells was largely unaltered by infection, specific changes in miRNA abundance were identified during Sendai virus infection. One miRNA, miR-203, was found to accumulate in infected cells and in response to IFN treatment. Results indicate that miR-203 is an IFN-inducible miRNA that can negatively regulate a number of cellular mRNAs, including an IFN-stimulated gene target, IFIT1/ISG56, by destabilizing its mRNA transcript.
INTRODUCTION
Virus infection of mammalian cells induces immediate and robust changes in cellular gene expression. Detection of virus infection by cellular signaling machinery triggers the transcription of antiviral genes including primary antiviral cytokines in the type I interferon (IFN) family as well as diverse effectors of the antiviral state (1). These cytokines and antiviral genes also drive further gene expression to amplify and regulate a primary cellular antiviral response that not only serves as a barrier to virus replication but also functions to educate the innate and adaptive immune systems. Inappropriate activation of these antiviral programs can lead to cytotoxicity and cell death; accordingly, diverse feedback inhibitors and other signal attenuators have evolved that serve to modulate the intensity and duration of IFN signaling and antiviral responses.
One of the primary mediators of antiviral gene expression is the IFN-JAK-STAT signaling system that directly links IFN production to target gene expression. IFN receptor engagement induces the assembly of the heterotrimeric transcription factor interferon-stimulated gene factor 3 (ISGF3) from preexisting latent reservoirs of STAT1, STAT2, and interferon regulatory factor 9 (IRF9) (2, 3). ISGF3 translocates to the nucleus, binds to target sites in interferon-stimulated gene (ISG) promoters, and activates the transcription of a large number of antiviral effector genes.
In addition to activation by virus infections of the expression of protein-coding mRNAs, it has become widely recognized that virus infections can also regulate the expression of noncoding RNAs, including microRNAs (miRNAs) involved in RNA interference pathways (4–6). MicroRNAs are generated from primary RNA polymerase II transcripts that are processed in the nucleus to create precursor miRNA hairpins. The precursor hairpins are further processed in the cytoplasm to create a mature 17- to 24-bp miRNA duplex that is incorporated into the RNA-induced silencing complex. MicroRNAs function to regulate the level of protein production by base pairing with short seed regions typically within the 3′ untranslated region (UTR) of target mRNAs (7–11). Recognition of mRNA targets by miRNAs can reduce protein expression either by inhibiting target mRNA translation or by promoting target mRNA degradation. Mounting evidence indicates that mRNA destabilization is a predominant means of miRNA-mediated translational repression (12–17).
Antiviral responses mediated by RNA interference are well documented in organisms such as flies, worms, and plants (18). In these cases, double-stranded viral genomic RNA or RNA replication intermediates are used to generate small interfering RNAs (siRNAs) that directly target the viral genome or mRNAs for efficient degradation (19). While there is scant evidence for a similar mechanism occurring naturally during the mammalian antiviral response (20), intact RNA interference pathways are required for optimal murine antiviral responses during vesicular stomatitis virus infection (21). The requirement of RNA interference machinery for mounting an antiviral response suggests an evolutionarily conserved role for the RNA interference pathway during virus infections. Further support of this concept has derived from studies that describe individual miRNAs that are regulated by virus infections or that control the response to virus infection. For example, influenza virus, vesicular stomatitis virus (VSV), Kaposi's sarcoma-associated herpesvirus (KSHV), and pathogenic bacteria induce miRNAs miR-132 and miR-146a, resulting in attenuation of cell signaling proteins mitogen-activated protein kinase 3 (MAPK3), p300, and IRAK1 to modulate antiviral signaling (6, 22–24). The well-characterized hepatocyte miRNA, miR-122, is used by hepatitis C virus (HCV) for efficient replication (25), but IFN signaling can decrease miR-122 abundance to limit virus replication (26). IFN has also been implicated in regulation of hepatocyte miRNAs that were reported to directly target the HCV genome in order to prevent its replication (27, 28). Diverse virus infections, including porcine reproductive and respiratory syndrome virus, respiratory syncytial virus, rabies virus, Epstein-Barr virus, influenza virus, human immunodeficiency virus 1, primate foamy virus, and hepatitis C virus, have been found to alter the cellular miRNA content during infection (29) and regulate the cellular antiviral response (6, 30, 31) or virus replication (27, 28, 32–35). These examples provide and reinforce strong correlative links between antiviral signaling and miRNA regulation.
To explore the importance of miRNA regulation during the cellular antiviral response, small-RNA profiling by next-generation sequencing was carried out to characterize the miRNA content of human cells at steady state and following Sendai virus infection. Differentially expressed miRNAs were identified that responded to virus infection, including miR-203, which is demonstrated to be an IFN-inducible miRNA capable of targeting the IFN-stimulated antiviral mediator IFIT1/ISG56. These findings provide evidence for diverse miRNA regulation by a negative-strand RNA virus infection and characterize an example of a complete miRNA regulatory circuit in the IFN antiviral response.
MATERIALS AND METHODS
Cell culture and viruses.
A549 cells (ATCC) were maintained in Ham's F12 medium with Kaign's modification (F12K; Gibco) supplemented with 10% cosmic calf serum (CCS; HyClone), 500 units/ml penicillin, and 500 μg/ml streptomycin. Vero cells were maintained in Dulbecco's modified eagle medium (DMEM) supplemented with 10% CCS, 500 units/ml penicillin, and 500 μg/ml streptomycin.
Sendai virus (Cantell strain) was grown in embryonated chicken eggs, and titers were determined on Vero cells. The A/Udorn/72 and A/WSN/33 strains of influenza virus (gift of R. A. Lamb, Northwestern University) were propagated, and titers were determined on MDCK cells. Virus infections were performed in serum-free medium (SFM) supplemented with 1% bovine serum albumin (BSA). At 2 h postinfection, the inoculation medium was replaced with medium containing 2% CCS, and cells were washed with SFM prior to RNA purification.
Cell treatments and transfection.
Recombinant human IFN-α (Hoffman LaRoche) was added to F12K medium at a concentration of 1,000 units/ml. Cells were treated for 10 h prior to RNA purification.
MicroRNA nontargeting control mimic and inhibitor or miR-203-specific mimic and inhibitor (Dharmacon) were transfected into A549 cells at a concentration of 25 nM using Lipofectamine 2000 (Invitrogen) according to the manufacturer's recommended RNA interference (RNAi) transfection protocol.
For cycloheximide (CHX) treatments, A549 cells were treated with 100 μg/ml CHX in F12K medium for 1 h prior to IFN treatment or Sendai virus infection. Cells were maintained in F12K medium containing CHX for the duration of the experiment.
RNA purification, reverse transcription, real-time PCR, and miRNA analysis.
Total RNA was purified from cells using an miRNeasy RNA isolation kit (Qiagen) and size fractionated with an RNeasy MinElute Cleanup Kit (Qiagen). For mRNA analysis, high-molecular-weight RNA was reverse transcribed using oligo(dT) primers and Superscript III reverse transcriptase (Invitrogen). PCR was performed using SYBR green detection and primers specific for the following: IFN-β, 5′-CATTACCTGAAGGCCAAGGA-3′ (forward) and 3′-CAATTGTCCAGTCCCAGAGG-3′ (reverse); ISG15, 5′-AATGCGACGAACCTCTGAAC-3′ (forward) and 5′-GAAGGTCAGCCAGAACAGGT-3′ (reverse); IFIT1/ISG56, 5′-GCAGCCAAGTTTTACCGAAG-3′ (forward) and 5′-AGCCCTATCTGGTGATGCAG-3′ (reverse); actin, 5′-GGCATCCTCACCCTGAAGTA-3′ (forward) and 5′-AGGTGTGGTGCCAGATTTTC-3′ (reverse); glyceraldehyde-3-phosphate dehydrogenase (GAPDH), 5′-ACAGTCAGCCGCATCTTCTT-3′ (forward) and 5′-ACGACCAAATCCGTTGACTC-3′ (reverse).
Relative mRNA abundance was determined by normalizing the mRNA of interest to GAPDH. All real-time PCR data are presented as 2−ΔΔCT (where CT is threshold cycle) (36).
For analysis of miRNAs, TaqMan miRNA assays (Applied Biosystems) were used. Low-molecular-weight RNA was reverse transcribed using an miRNA-specific primer and Multiscribe reverse transcriptase (Applied Biosystems.) PCR was performed with an miRNA-specific probe according to the manufacturer's instructions (Applied Biosystems). Relative miRNA abundance was determined by normalizing the miRNA of interest to U6 small nuclear RNA using the 2−ΔΔCT method as above. The following TaqMan miRNA assays were used: hsa-miR-16, hsa-miR-125b, hsa-miR-147b, mmu-miR-187, hsa-miR-203, hsa-miR-376, hsa-miR-449b, hsa-miR-483-3p, and hsa-miR-652.
Library preparation and bioinformatics analysis.
To generate a library for SOLiD small-RNA sequencing, A549 cells were either mock infected or infected with Sendai virus (5 PFU/cell). RNA was purified from cells at 10 h postinfection and size fractionated. One microgram of low-molecular-weight RNA was used to generate the cDNA libraries using the SOLiD Small RNA Expression Kit (Applied Biosystems) according to the manufacturer's instructions. Emulsion PCR and SOLiD sequencing using the SOLiD4 platform (Applied Biosystems) were performed at the Center for Genetic Medicine at Northwestern University.
Sequence tags were analyzed using the BioScope software package (Applied Biosystems). The sequence tags were aligned to the human genome with high stringency, allowing for zero mismatches between the sequence tag and the genome. Sequence tags that mapped to the human genome were further characterized to determine their identity by BLAST. Sequence tags from the virus-infected library that could not be mapped to the human genome were then mapped to the Sendai virus genome.
Immunoblotting.
A549 cells were washed in ice cold phosphate-buffered saline before being lysed in whole-cell extract buffer containing 50 nM Tris, pH 8.0, 280 nM NaCl, 0.5% Igepal, 0.2 mM EDTA, 2 mM EGTA, 10% glycerol, 1 mM dithiothreitol (DTT), 0.1 mM sodium vanadate, and protease inhibitor mixture. Five micrograms of total protein was separated by SDS-PAGE, and protein was then transferred to nitrocellulose membrane, blocked in 5% nonfat milk in Tris-buffered saline plus Tween 20 (TBST), and detected by specific antibodies for IFIT1/ISG56 (Thermo Pierce)-, GAPDH (Santa Cruz Biotechnologies)-, and MDA5-specific antisera. Antibody detection was visualized by chemiluminescence (PerkinElmer Life Sciences). Densitometry analysis was performed using Vision Works software (UVP, LLC, Upland, CA).
Gene expression profiling.
Gene expression profiling and analysis were performed as described by Buggele et al. (6). A549 cells were left untreated, treated with 1,000 units/ml IFN, infected with Sendai virus (5 PFU/cell), and treated or infected in the presence of 50 nM miR-203 mimic. RNA was purified from cells 10 h after treatment and hybridized to a whole-genome microarray (Illumina bead array) (37, 38). MicroRNA seed matches were determined with the miRWalk algorithm (39), and gene ontology analysis was performed utilizing InnateDB and DAVID (40–43).
Accession numbers.
Nucleotide sequences of the sequence tags mapped to the human genome were deposited in the NCBI Gene Expression Omnibus (GEO) database under accession number GSE43966. Microarray data were deposited in the GEO database under accession numbers GSE47865 and GSE47866.
RESULTS
MicroRNA profile of Sendai virus-infected cells.
A small-RNA deep-sequencing experiment was performed to profile cellular miRNAs and determine abundance changes induced by Sendai virus infection. A549 cells, a human alveolar epithelial cell line, were mock infected or infected with Sendai virus for 10 h, a time point previously determined to be suitable for both inducible mRNA and miRNA expression (6). Total RNA was purified and size fractionated to yield low-molecular-weight RNA (<200 nucleotides). The low-molecular-weight RNA was used to construct a library for SOLiD sequencing (Applied Biosystems). Both infected and uninfected cell libraries yielded in excess of 108 sequence tags. These sequence tags were mapped to the human genome with high stringency; no mismatches were allowed between the genome sequence and the sequence tags. This resulted in ∼3.5 × 107 unique mappable sequences per condition (Table 1). The mappable sequences were subjected to BLAST alignment (44) to determine their locus of origin, resulting in ∼1.5 × 106 miRNA sequence tags in each library. This procedure identified 778 miRNAs in the mock-infected library and 800 miRNAs in the Sendai virus-infected cell library (see Table S1 in the supplemental material). In addition to miRNAs, snRNA and snoRNA classes were identified. Fragmented large RNA classes including rRNA and mRNA were also identified. Sequence tags from the virus-infected cell library that could not be mapped to the human genome could be mapped to the Sendai virus genome. These small Sendai virus-derived RNAs displayed a size distribution of between 18 and 24 nucleotides, and genome-derived RNAs mapped uniquely to the 5′ end, similar to the reported RIG-I ligand generated during Sendai virus infection (45). Of the miRNAs identified, only 343 miRNAs in the mock library and 351 miRNAs in the Sendai virus library were identified by more than 100 sequence tags. To identify if any gross changes in miRNA abundances were the result of Sendai virus infection, the number of miRNAs identified in each library were compared. The mock library contained 19 exclusive miRNAs, and the Sendai library contained 27 exclusive miRNAs while both libraries contained a common 324 miRNAs (Fig. 1A). This analysis indicated that the vast majority of miRNAs identified by more than 100 sequence tags were common between libraries, consistent with the interpretation that miRNA expression is overall largely similar before and after infection. In addition, the 25 most abundant miRNAs comprised greater than 87% of all miRNAs in either library. Comparing normalized miRNA sequence tag abundances (miRNA sequence tags per 1,000 mappable sequence tags) between the two conditions revealed that the vast majority of miRNAs in A549 cells do not display greater than a 2-fold change during infection (Fig. 1B). Nonetheless, 52 miRNAs that increased in abundance by ≥1.5-fold and 33 miRNAs that decreased by ≥1.5-fold following infection were identified (see Table S2 in the supplemental material). Together, these data suggest that infection does not cause a bulk change in the miRNAs present within the cell; however, it does cause changes in abundance of specific miRNAs.
Table 1.
Sequencing library statistics
| Sequence tag groupa | No. of sequence tags by conditionb |
|
|---|---|---|
| Mock infection | Sendai virus infection | |
| Total | 97,542,060c | 108,423,000c |
| Mappable | 32,188,879 | 35,247,578 |
| microRNA | 18,012,238 | 11,917,137 |
| miscRNA | 183,009 | 411,399 |
| mtRNA | 483,546 | 389,294 |
| rRNA | 1,297,566 | 1,898,927 |
| snoRNA | 1,143,405 | 626,386 |
| snRNA | 188,817 | 127,418 |
| Other RNA | 11,294,960 | 19,877,017 |
RNA sequence tag classification identified through small-RNA deep sequencing. miscRNA, miscellaneous RNA; mtRNA, mitochondrial RNA.
A549 cells were either mock infected or infected with Sendai virus (5 PFU/cell) for 10 h.
Number of sequence tags that correspond to a particular RNA feature identified in the library.
Fig 1.

Small-RNA profile of uninfected and Sendai virus-infected A549 cells. A549 cells were mock infected or infected with Sendai virus (Cantell strain; 5 PFU/cell). RNA was purified 10 h later and size fractionated to yield RNA <200 nucleotides in length, and cDNA libraries were prepared for SOLiD sequencing. (A) Venn diagram illustrates the number of miRNAs identified (by greater than 100 sequence tags) that are either unique or common to mock-infected and Sendai virus-infected libraries. (B) Scatter plot indicates the abundance of each identified miRNA in the mock-infected or Sendai virus-infected library with greater than 100 sequence tags. Blue dots represent miRNAs with less than 1.5-fold change, and red dots represent miRNAs with greater than 1.5-fold change.
Cellular miRNAs respond to Sendai virus infection.
To verify the sequencing data, independent RNA samples were generated from mock-infected or Sendai virus-infected cells and subjected to individual miRNA analysis by reverse transcription-quantitative PCR (RT-qPCR) using TaqMan miRNA assays (Applied Biosystems). A group of miRNAs was selected for validation based on two criteria, initial abundance and differential expression induced by infection. These miRNAs are represented by more than 100 sequence tags in either library and also exhibit greater than a 1.5-fold change in response to Sendai virus infection. Several miRNAs were analyzed that displayed either positive (miR-125b, miR-147b, miR-203, miR-376, miR-449b, and miR-483-3p) or negative (miR-187 and miR-652) changes in abundance (Fig. 2). A high correlation between sequencing and independent RT-qPCR detection measurement was observed for all the tested miRNAs, confirming their reproducible differential expression. These findings verify that changes in miRNA abundance identified by small-RNA sequencing accurately reflect the miRNA content of uninfected and infected cells.
Fig 2.

Validation of changes in miRNA abundance induced by Sendai virus infection. For each indicated miRNA, normalized sequence tag abundance is plotted to the left of the dashed line. Individual miRNAs were also measured in freshly prepared RNA derived from A549 cells that were either mock infected or infected with Sendai virus (5 PFU/cell; 10 h) and then subjected to specific TaqMan microRNA RT-qPCR assays; results are plotted to the right of the dashed line. Statistical significance was determined by a two-tailed t test (*, P < 0.05; **, P < 0.01).
IFN-mediated accumulation of miR-203.
A prior investigation of miRNAs induced by influenza virus infection revealed remarkable virus-specific responses (6). To determine if the Sendai virus-inducible miRNAs, miR-203 and miR-449b, are sensitive to other RNA virus infections, A549 cells were either mock infected or inoculated with 5 PFU/cell of Sendai virus, influenza A/Udorn/72 virus, or influenza A/WSN/33 virus for 10 h prior to miRNA analysis. The level of a control miRNA, miR-16, did not change in response to either Sendai virus or influenza virus infection, and miR-449b was induced by Sendai virus (∼2.5-fold) and even more dramatically in response to either strain of influenza virus (∼13.5-fold) (Fig. 3A). These results are consistent with our previously reported observation that influenza virus infection can be a more potent inducer of miRNA expression than other RNA viruses (6). However, a distinct behavior was observed for miR-203, which increased by >15-fold in response to Sendai virus infection but only by ∼3-fold during influenza virus infections (Fig. 3A).
Fig 3.

IFN mediates regulation of miR-203. (A) A549 cells were mock infected or infected with either influenza virus A/Udorn/72 (Udorn), A/WSN/33 (WSN), or Sendai virus Cantell Strain (Sendai) (5 PFU/cell; 10 h). RNA was purified at 10 h postinfection and size fractionated to yield RNA of <200 nucleotides in length, and the abundances of miR-203, miR-449b, and miR-16 were analyzed using TaqMan miRNA RT-qPCR assays. (B) High-molecular-weight RNA was used to measure the abundance of IFN-β mRNA during Sendai virus and influenza virus infection by RT-qPCR as indicated. (C) A549 cells were left untreated (UNT) or treated with PBS or increasing concentrations of IFN-α (250 units/ml, 500 units/ml, 1,000 units/ml, or 5,000 units/ml). RNA was purified at 10 h posttreatment, and the abundance of miR-203 was analyzed by TaqMan miRNA assays. (D) A549 cells were left untreated or treated as indicated with PBS or IFN-α (1,000 units/ml) and mock infected or infected with Sendai virus (5 PFU/cell). RNA was purified at 10 h posttreatment, and the abundance of miR-203 was analyzed by TaqMan miRNA assays. (E) A549 cells were left untreated, treated with PBS or IFN-α (1,000 units/ml), or transfected with poly(I·C) (5 μg/ml). RNA was purified at 10 h posttreatment, and the abundance of miR-203 was analyzed by TaqMan miRNA assays. (F) Vero cells were left untreated or treated as indicated with PBS or IFN-α (1,000 units/ml) and then mock infected or infected with Sendai virus (5 PFU/cell). RNA was purified at 10 h posttreatment, and the abundance of miR-203 was analyzed by TaqMan miRNA assays. Statistical significance was determined by a two-tailed t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001; n.s., not significant).
One significant difference in the cellular response to influenza virus infection compared to Sendai virus infection is that Sendai virus is a more robust activator of the antiviral cytokine IFN-β. The reason for this difference is known: the Cantell strain used for these studies is well documented to induce IFN production due to the presence of defective interfering genomes that activate antiviral responses (46). The IFN-β mRNA is highly induced by Sendai virus infection of A549 cells but only weakly induced by the influenza viruses (Fig. 3B). To determine the effect of IFN signaling on miR-203 expression, a dose-response experiment was performed. A549 cells were left untreated, treated with phosphate-buffered saline (PBS), or treated with increasing concentrations of IFN-α (250, 500, 1,000, or 5,000 units/ml). RNA was purified 10 h after treatment, and the abundance of miR-203 was determined by RT-qPCR. MicroRNA-203 abundance increased as the concentration of IFN increased to 1,000 units/ml (Fig. 3C). This concentration proved saturating for miR-203 induction compared to treatment of 5,000 units/ml. Higher IFN concentrations induce miR-203 to accumulate to higher abundance, similar to the enhanced accumulation observed during Sendai virus infection.
To compare miR-203 induction by Sendai virus infection with that of IFN treatment, A549 cells were treated with 1,000 units/ml IFN-α or infected with Sendai virus (5 PFU/cell). Ten hours later, total RNA was prepared and size fractionated, and the low-molecular-weight RNA was used to measure the abundance of miR-203. IFN-α stimulation increased miR-203 levels by 3.4-fold, and infection with Sendai virus increased miR-203 by 8.3-fold (Fig. 3D). These data indicate that miR-203 is inducible by direct IFN-α stimulation but that Sendai virus infection is a more potent miR-203 inducer than IFN-α treatment alone, possibly indicating that additional factors are generated during Sendai virus infection that further enhance miR-203 expression or stability.
To assess the role of RIG-I-like receptor (RLR) signaling in the activation of miR-203, A549 cells were untreated, treated with PBS, treated with 1,000 units/ml IFN-α, or transfected with the synthetic double-stranded RNA poly(I·C). RNA was purified 10 h after treatment, and the abundance of miR-203 was determined by RT-qPCR. RLR signaling induced miR-203 to levels statistically identical to those with IFN treatment alone (Fig. 3E). Together, the data indicate that while IFN mediates induction, robust activation of miR-203 requires synergism of multiple antiviral pathways activated during Sendai virus infection but not influenza virus infection, IFN stimulation, or RLR signaling alone.
To determine if type I IFN is required for miR-203 induction during Sendai virus infection, an experiment was performed using Vero cells, a cell line that does not induce IFN in response to infection due to genetic defects (47, 48). Vero cells were left untreated, treated with PBS, treated with 1,000 units/ml IFN-α, or infected with Sendai virus (5 PFU/cell). Similar to results in A549 cells, miR-203 increased 3.3-fold following IFN-α treatment. Unlike the A549 cells, miR-203 did not increase in response to Sendai virus-infection compared to a mock-infected sample (Fig. 3F). This result further corroborates the IFN-α inducibility of miR-203 and also is consistent with the notion that IFN production initiated by Sendai virus infection is required for miR-203 activation; however, it remains possible that there may be additional factors produced in A549 cells that are not produced in Vero cells that could contribute to the differential miR-203 expression.
Activation of miR-203 by immediate and delayed signaling pathways.
To further explore the kinetics of miR-203 induction, a time course experiment was performed. A549 cells were infected with Sendai virus (5 PFU/cell), RNA was purified at 2, 4, 10, and 24 h postinfection, and the abundance of miR-203 was measured. Elevated miR-203 was detected as early as 2 h postinfection, and the level continued to increase during the 24 h of infection despite loss of cells due to cytopathic effects. These results suggested that signaling pathway accumulation is required for continuous miR-203 expression throughout infection (Fig. 4A).
Fig 4.

Activation of miR-203 by immediate and delayed signaling pathways. (A) A549 cells were infected with Sendai virus (5 PFU/cell), and the abundance of miR-203 was measured at indicated times after Sendai virus infection by TaqMan miRNA RT-qPCR. (B) A549 cells were stimulated with IFN-α (1,000 units/ml) or infected with Sendai virus (SeV) (5 PFU/cell) for 4, 10, and 24 h in the presence (+) or absence (−) of cycloheximide (CHX 100 ng/ml; 1 h pretreatment and continuous thereafter). Abundances of miR-203 and miR-449b were determined by TaqMan microRNA assays at the indicated time points. Statistical significance was determined by a two-tailed t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001. M, mock; hpt, hours posttransfection.
To determine if the changes in miR-203 abundance result from a primary or secondary response to cell stimulation, a time course experiment was carried out in the presence of the protein synthesis inhibitor cycloheximide (CHX). A549 cells were treated with IFN-α (1,000 units/ml) or infected with Sendai virus (5 PFU/cell) in the presence or absence of CHX. RNA samples were prepared 4, 10, and 24 h after treatment and subjected to miRNA analysis (Fig. 4B). Treatment with CHX did not inhibit miRNA production in general, as miR-449b remains virus inducible regardless of CHX. After 4 h of IFN treatment, the level of miR-203 increased 2-fold, and this increase was unaffected by CHX treatment. A similar increase in miR-203 was observed during the first 4 h of Sendai virus infection, but this increase was not observed in the presence of CHX. These data indicate that protein synthesis during Sendai virus infection is required for miR-203 induction, whereas a preexisting signaling complex downstream of IFN, possibly the IFN-activated JAK-STAT pathway, mediates the initial increase in miR-203.
During prolonged stimulation with IFN-α or infection with Sendai virus (i.e., 10 h and 24 h), miR-203 continues to accumulate, but in all cases this accumulation is dramatically reduced in the presence of CHX. These data indicate that the sustained expression of miR-203 is mediated by factors that are generated during the antiviral response as a result of new protein synthesis.
IFIT1/ISG56 is a target of miR-203.
To identify potential mRNA targets of miR-203, the TargetScan algorithm was used to locate miR-203 seed matches within 3′ UTRs of cellular mRNAs (49). One potential target of miR-203 was identified as the IFN-inducible gene IFIT1/ISG56, which contains two slightly overlapping seed matches for miR-203 (Fig. 5A). To determine if miR-203 can target IFIT1/ISG56, A549 cells were transfected with miRNA mimics or antagonists in combination with IFN treatment, and the effect of these miR-203 agonists and antagonists on IFIT1/ISG56 mRNA levels were determined by RT-qPCR (Fig. 5B). The IFIT1/ISG56 mRNA is strongly induced by IFN stimulation and was unaffected by the nontargeting control miRNA mimic or antagonist. In contrast, the miR-203 mimic suppressed IFIT1/ISG56 mRNA accumulation by 43% compared to the nontargeting control. In the complementary experiment, the miR-203 antagonist increased the level of IFIT1/ISG56 mRNA by 47% compared to the control. These results demonstrate that miR-203 can specifically regulate the level of IFIT1/ISG56 mRNA even as it is undergoing potent induction during IFN stimulation.
Fig 5.

Identification and characterization of IFIT1/ISG56 as an miR-203 target. (A) Illustration of the 3′ UTR of IFIT1/ISG56, with boxes indicating the positions of two partially overlapping miR-203 seed matches. (B) A549 cells were transfected with 25 nM nontargeting miRNA mimic (Control) or nontargeting control miRNA inhibitor (Inhib) or miR-203-specific mimic or miR-203 inhibitor, as indicated. Cells were either untreated (−) or treated (+) with 1,000 units/ml IFN-α. RNA was purified from cells 10 h later and analyzed by RT-qPCR for the induction of IFIT1/ISG56 mRNAs. In parallel, levels of miR-203 were measured by TaqMan assay. Statistical significance was determined by a two-tailed t test (*, P < 0.05; **, P < 0.01). (C) A549 cells were transfected with miRNA mimics and inhibitors and then treated with IFN-α as described for panel B Whole-cell lysates were prepared 10 h later and analyzed by immunoblotting with antiserum specific for IFIT1/ISG56, MDA5, or GAPDH protein.
To verify the mRNA analysis, the effect of the miRNA agonists and antagonists on the level of IFIT1/ISG56 protein was determined by immunoblotting. A549 cells transfected with an miR-203 mimic decreased the IFIT1/ISG56 level by 34%, while miR-203 antagonist increased the IFIT1/ISG56 protein level by 51% compared to the nontargeting control mimic and inhibitor (Fig. 5C). The levels of control proteins MDA5 and GAPDH did not change due to miR-203 perturbation. These findings demonstrate that miR-203 regulates the expression of IFIT1/ISG56 at both RNA and protein levels.
Identification of additional miR-203 targets during IFN stimulation and Sendai virus infection.
In order to determine additional targets of miR-203, a gene expression profiling experiment was performed. A549 cells were either left untreated, treated with IFN, or infected with Sendai virus, both in the presence or absence of exogenously expressed miR-203. RNA was purified at 10 h posttreatment and used to probe a microarray to determine changes in gene expression due to miR-203. The analysis indicated that 21 genes are differentially regulated during IFN treatment and that 12 genes are regulated during Sendai virus infection by greater than 1.5-fold as a consequence of elevated miR-203 levels (Table 2). The most differentially regulated gene during IFN treatment and the second most differentially regulated gene during Sendai virus infection is LAMP1, which contains two seed matches for miR-203 and has been previously identified as an miR-203 target gene (50, 51). The identification of a known miR-203 target validates the gene expression profiling method for identifying miRNA target genes. Additional gene targets that were identified by both IFN stimulation and Sendai virus infection include RAPGEF1, SEPN1, and DNAJB6. The mRNA levels for these genes were reduced by 1.93-, 1.54-, and 1.5-fold due to miR-203 expression during IFN treatment and by 1.54-, 1.63-, 1.7-fold, respectively, due to miR-203 expression during Sendai virus infection. These three genes contain miR-203 target sites in their 3′ UTRs, suggesting that they are directly regulated by miR-203. Consistent with our direct analysis above, the level of IFIT1/ISG56 mRNA expression was affected by miR-203 during Sendai virus infection and IFN treatment; IFIT1/ISG56 expression was reduced by 22.3% and 9.6%, respectively. However, as these differences represent the net change achieved by slow miRNA destabilization compared to the rapid transcriptional activation of IFIT1/ISG56, these levels did not meet our strict cutoff requirements for the differential screen due to the limited dynamic range of hybridization-based microarrays for observation of small changes in highly abundant RNAs. Underestimation of relative mRNA expression changes during microarray experiments has been reported (52) and suggests that small changes observed during the profiling experiment are indicative of larger changes within the cell, as we observed by direct analysis.
Table 2.
MicroRNA-203-regulated genes during IFN-α treatment and Sendai virus infection
| Gene IDa | Treatment(s)b | Effect of IFN treatment |
Effect of Sendai virus infection |
No. of miR-203 seed matchese | ||
|---|---|---|---|---|---|---|
| P valuec | Fold changed | P valuec | Fold changed | |||
| LASP1 | IFN, Sendai | 9.70 × 10−13 | −1.94 | 3.89 × 10−10 | −1.83 | 2 |
| RAPGEF1 | IFN, Sendai | 1.22 × 10−11 | −1.93 | 5.09 × 10−8 | −1.54 | 2 |
| PRDX3 | IFN, Sendai | 5.89 × 10−12 | −1.82 | 2.01 × 10−10 | −2.53 | NA |
| COL4A1 | IFN | 7.62 × 10−10 | −1.71 | NA | N/A | 1 |
| TUBGCP2 | IFN, Sendai | 6.83 × 10−11 | −1.70 | 2.07 × 10−7 | −1.71 | NA |
| C12orf75 | IFN, Sendai | 5.80 × 10−10 | −1.69 | 5.78 × 10−7 | −1.62 | NA |
| SRC | IFN | 8.92 × 10−10 | −1.61 | NA | N/A | 2 |
| SELT | IFN | 3.93 × 10−8 | −1.58 | NA | N/A | 1 |
| BANF1 | IFN, Sendai | 6.23 × 10−9 | −1.55 | 2.37 × 10−9 | −1.67 | NA |
| SEPN1 | IFN, Sendai | 6.29 × 10−8 | −1.54 | 4.96 × 10−7 | −1.63 | 2 |
| POLR1E | IFN | 3.37 × 10−9 | −1.54 | NA | NA | NA |
| LAMP2 | IFN | 1.15 × 10−8 | −1.54 | NA | NA | 3 |
| TCP1 | IFN | 1.92 × 10−8 | −1.53 | NA | NA | 2 |
| CHMP3 | IFN | 7.71 × 10−7 | −1.52 | NA | NA | 2 |
| SLC12A2 | IFN | 1.15 × 10−7 | −1.51 | NA | NA | 1 |
| CHMP3 | IFN | 4.74 × 10−8 | −1.50 | NA | NA | NA |
| PHLDA3 | IFN, Sendai | 1.03 × 10−8 | −1.50 | 2.66 × 10−8 | −1.54 | NA |
| DNAJB6 | IFN, Sendai | 2.23 × 10−7 | −1.50 | 1.40 × 10−7 | −1.70 | 1 |
| PPAP2B | Sendai | NAf | NA | 1.38 × 10−8 | −1.72 | NA |
| PHLDA3 | Sendai | NA | NA | 2.66 × 10−8 | −1.54 | 1 |
| NCL | Sendai | NA | NA | 6.96 × 10−7 | −1.54 | NA |
| SLC2A3 | IFN | 2.50 × 10−7 | 1.51 | NA | NA | 1 |
| IGFBP1 | IFN | 3.63 × 10−6 | 1.57 | NA | NA | NA |
| ALDOC | IFN | 5.08 × 10−6 | 1.62 | NA | NA | NA |
Gene identifiers (IDs) that were differentially regulated by ≥1.5-fold during IFN treatment of Sendai virus infection during overexpression of miR-203.
A549 cells were treated with 1,000 units/ml IFN-α (IFN) or infected with Sendai virus (Sendai) at 5 PFU/cell in separate experiments.
Statistical significance of gene expression due to the presence of exogenous miR-203.
Fold change of gene due to exogenous miR-203.
Number of miR-203 seed matches in the 3′ UTR of indicated gene as determined by at least two predictive algorithms using miRWalk (39).
NA, not applicable.
The observation that miR-203 partially reduced the level of an IFN-stimulated gene product raised the question of whether miR-203 by itself has the ability to demonstrably alter the course of Sendai virus infection under these conditions. In order to test the effect of miR-203 on Sendai virus infectivity and replication, experiments were performed to evaluate virus replication in A549 cells in the presence of miRNA mimics and antagonists. However, neither expression nor inhibition of miR-203 was found to significantly alter Sendai virus replication compared to infection alone or infection in the presence of control miRNAs (data not shown). In the absence of detectable direct effects on virus replication, it is hypothesized that miR-203 may play a role related to more subtle regulation of cellular response pathways. In order to evaluate potential effects of miR-203 on cellular gene expression, gene ontology analysis was performed on the identified miR-203-regulated genes. The miR-203-regulated genes were found to fall into three significant functional annotation clusters that include diverse cellular processes such as regulation of cytoskeleton, response to organic stimuli, regulation of cell death, and protein complex biogenesis (Table 3). This analysis indicates that miR-203 is capable of regulating diverse cellular processes during IFN stimulation and the cellular response to Sendai virus infection.
Table 3.
Functional annotation clustering of miR-203-regulated genesa
| Functional cluster no. | Enriched biological process | miR-203-regulated genes associated with the cluster |
|---|---|---|
| 1 | Response to organic substance | DANJB6, ALDOC, IGFBP1, PRDX3, SRC |
| Regulation of apoptosis | ||
| Regulation of programmed cell death | ||
| Regulation of cell death | ||
| 2 | Protein complex assembly | ALDOC, TCP1, TUBGCP2, SRC |
| Protein complex biogenesis | ||
| Macromolecular complex subunit | ||
| Macromolecular complex subunit organization | ||
| Cytoskeleton | ||
| 3 | Transmembrane receptor protein tyrosine kinase signaling pathway | RAPGEF1, IGFBP1, SLC12A2, SRC |
DISCUSSION
The mammalian antiviral response is initiated by dramatic changes in gene expression that produce effective barriers to virus replication. Many transcriptional and posttranscriptional mechanisms underlying cellular antiviral effects have been characterized in detail, and endogenous miRNA regulation of cellular responses to virus infections are an area of intense investigation. Next-generation sequencing has enabled analysis of the small-RNA profile of human A549 cells in the steady state or following Sendai virus infection. However, while the majority of cellular miRNAs maintain constant levels irrespective of infection, direct comparison has revealed specific examples of miRNAs that significantly accumulate in response to Sendai virus infection (see Table S1 in the supplemental material).
One of the Sendai virus-induced miRNAs, miR-203, was studied in detail and found to be potently induced by Sendai virus through the virus-induced IFN antiviral response. Biphasic expression of miR-203 through both direct and indirect IFN-mediated antiviral signaling pathways was found to contribute to the production of miR-203 continuously throughout Sendai virus infection. The absolute level of miRNA induction was found to vary slightly among experiments, possibly related to cell passage number, cell density, or slight differences in infection efficiencies, but miR-203 was consistently induced by Sendai virus, generally in the range of 8- to 14-fold compared to levels in mock-infected cells. Preexisting factors were found to drive the immediate expression of miR-203 following IFN stimulation, and newly synthesized downstream factors are required to mediate the continued expression of miR-203. It is noteworthy that multiple ISRE/IRF binding sites were identified within 2 kbp of the putative miR-203 precursor transcriptional start site, possibly implicating the IFN-activated JAK-STAT-ISGF3 pathway in the immediate regulation of miR-203 activation and interferon regulatory factors or other ISG products as the newly synthesized factors driving late expression. This type of regulation is reminiscent of IFN-stimulated regulation of mRNA coding genes, such as guanylate-binding protein (GBP), which is regulated by both immediate-acting STAT transcription complexes and later by IFN regulatory factors such as IRF1 (53). Activation of miR-203 by Sendai virus infection is blocked by CHX treatment at all time points, consistent with the requirement for virus-induced IFN biosynthesis to drive miRNA expression.
Computer-aided target analysis identified the IFIT1/ISG56 mRNA as a potential miR-203 target, and the ability of miR-203 to destabilize the mRNA was demonstrated using miRNA agonists and antagonists. Together, these results indicate that an IFN-inducible miRNA can regulate the abundance of an IFN-inducible target gene. Although this arrangement of coregulated antiviral activators and their inhibitors may seem counterintuitive, this pattern of regulation resembles a classic example of an incoherent feed-forward loop, in which a single signal (i.e., infection or IFN) activates two independent responders (IFIT1/ISG56 and miR-203), and one responder (miR-203) serves to negatively regulate the other (IFIT1/ISG56) (54). The results described here fit well with such a model for an antiviral regulatory pathway (Fig. 6). After infection with Sendai virus, cells mount an antiviral response by producing type I IFN, which signals to activate the transcription of a variety of ISGs, including IFIT1/ISG56, miR-203, and other mediators such as IRF family proteins. miR-203 is potentially positively regulated by IRFs for sustained expression in a positive feed-forward loop during the antiviral response. The IFIT1/ISG56 mRNA is regulated by miR-203 in an incoherent feed-forward loop, thereby modulating the production of IFIT1/ISG56 protein (Fig. 6). miR-203 expression was found to be sensitive to CHX treatment at two points during Sendai virus infection. The production of type I IFN requires translation of nascent mRNAs during the early phase of infection and the production of additional signaling molecules that participate in miR-203 regulation during late stages of infection; these are represented in the model as IFN regulatory factors.
Fig 6.
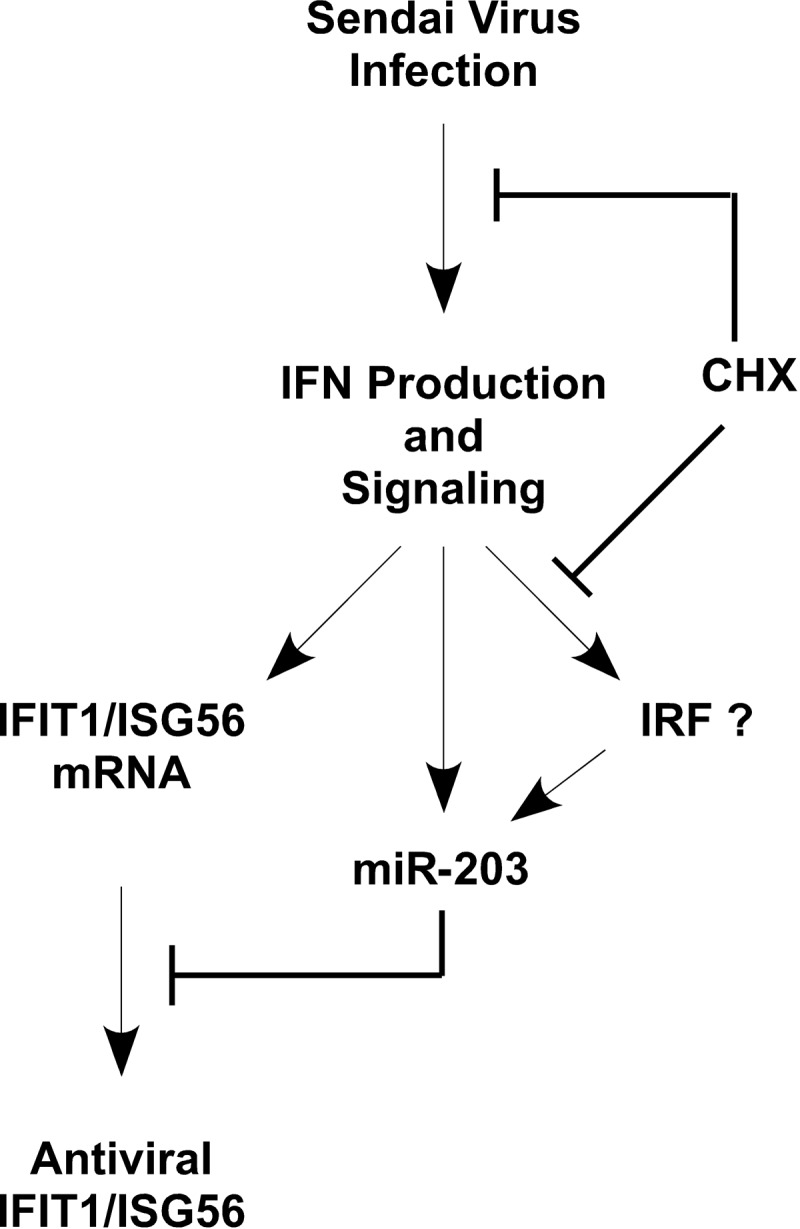
Model of the regulation of miR-203 and IFIT1/ISG56 during Sendai virus infection. Cells infected with Sendai virus generate an antiviral response by producing type I IFN, which signals through the JAK-STAT-ISGF3 pathway to induce IFN-stimulated gene products including IFIT1/ISG56, miR-203, and IFN regulatory factors (IRFs). Both IFN production and IRF induction are sensitive to treatment with CHX. miR-203 can be induced by IFN initially by preexisting proteins, but sustained miR-203 expression requires newly synthesized signaling proteins, potentially, but not limited to, IRF family members (IRF?). miR-203 accumulation functions to destabilize IFIT1/ISG56 mRNA and regulate its translation. See the text for more details of the model.
Beyond regulation of IFIT1, several other mRNA targets of miR-203 were identified using a microarray screening approach. Despite these connections, miR-203 expression or antagonism failed to reveal dramatic effects on Sendai virus replication, kinetics, or cytopathicity. These findings are distinct from cases of virus-hijacked microRNA variants that have been linked to efficient virus and cellular regulatory phenomena (55) and may indicate that miR-203 serves to fine-tune cellular protein expression during the cellular response to virus infection without directly limiting Sendai virus replication. A role for miR-203 as a mediator of more subtle alterations to its protein target is consistent with the current understanding of miRNA action (56). By analogy, while it is well established that IFN is a potent antiviral cytokine, not all of the virus-inducible IFN target genes imitate this antiviral activity when they are expressed alone (57). Therefore, it is not surprising that the partial attenuation of IFIT1/ISG56 does not have a measurable impact on Sendai virus replication under these conditions. Both IFIT1/ISG56 and miR-203 are regulated with distinct kinetics and to various degrees by both ISGF3 and other virus-induced transcription factors (58–60), supporting a model in which miR-203 functions as a regulator rather than an on-off switch to regulate rather than eliminate IFIT1/ISG56 expression. Additional miR-203 targets identified here and in other studies have implicated miR-203 in diverse processes related to virus replication and immune regulation, including modulation of MyD88 (61), tumor necrosis factor alpha (TNF-α), interleukin-24 (IL-24) (62), suppressor of cytokine signaling 1 (SOCS1) and SOCS3 (63), regulation of papillomavirus infection in keratinocytes (64, 65), and regulation of miR-203 abundance during rabies virus (30), Epstein-Barr virus (66), and coxsackie B3 virus infection (67). It is likely that IFN and virus induction of miR-203 play both general and tissue-specific roles in the regulation of antiviral responses.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the Northwestern University Genomics Core Facility for assistance with the deep-sequencing experiment and associated bioinformatics, Robert A. Lamb for the generous gift of influenza virus strains and expertise in propagating Sendai virus, and members of the Horvath lab for critical and helpful discussions.
W.A.B. is supported by the Malkin Scholars Fund and the Cell and Molecular Basis of Disease Predoctoral training grant (T32 GM008061). This work was supported by NIH Grant U01AI082984 from the NIAID IMVC program.
Footnotes
Published ahead of print 19 June 2013
Supplemental material for this article may be found at http://dx.doi.org/10.1128/JVI.01064-13.
REFERENCES
- 1. Ishii KJ, Koyama S, Nakagawa A, Coban C, Akira S. 2008. Host innate immune receptors and beyond: making sense of microbial infections. Cell Host Microbe 3:352–363 [DOI] [PubMed] [Google Scholar]
- 2. Levy DE, Kessler DS, Pine R, Darnell JE., Jr 1989. Cytoplasmic activation of ISGF3, the positive regulator of interferon-alpha-stimulated transcription, reconstituted in vitro. Genes Dev. 3:1362–1371 [DOI] [PubMed] [Google Scholar]
- 3. Levy DE, Kessler DS, Pine R, Reich N, Darnell JE., Jr 1988. Interferon-induced nuclear factors that bind a shared promoter element correlate with positive and negative transcriptional control. Genes Dev. 2:383–393 [DOI] [PubMed] [Google Scholar]
- 4. Peng X, Gralinski L, Ferris MT, Frieman MB, Thomas MJ, Proll S, Korth MJ, Tisoncik JR, Heise M, Luo S, Schroth GP, Tumpey TM, Li C, Kawaoka Y, Baric RS, Katze MG. 2011. Integrative deep sequencing of the mouse lung transcriptome reveals differential expression of diverse classes of small RNAs in response to respiratory virus infection. mBio 2(6):e00196–11. 10.1128/mBio.00198-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Li Y, Chan EY, Li J, Ni C, Peng X, Rosenzweig E, Tumpey TM, Katze MG. 2010. MicroRNA expression and virulence in pandemic influenza virus-infected mice. J. Virol. 84:3023–3032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buggele WA, Johnson KE, Horvath CM. 2012. Influenza A virus infection of human respiratory cells induces primary microRNA expression. J. Biol. Chem. 287:31027–31040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Carthew RW, Sontheimer EJ. 2009. Origins and mechanisms of miRNAs and siRNAs. Cell 136:642–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Miyoshi K, Uejima H, Nagami-Okada T, Siomi H, Siomi MC. 2008. In vitro RNA cleavage assay for Argonaute-family proteins. Methods Mol. Biol. 442:29–43 [DOI] [PubMed] [Google Scholar]
- 9. Davis-Dusenbery BN, Hata A. 2010. Mechanisms of control of microRNA biogenesis. J. Biochem. 148:381–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Huntzinger E, Izaurralde E. 2011. Gene silencing by microRNAs: contributions of translational repression and mRNA decay. Nat. Rev. Genet. 12:99–110 [DOI] [PubMed] [Google Scholar]
- 11. Bartel DP. 2004. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell 116:281–297 [DOI] [PubMed] [Google Scholar]
- 12. Baek D, Villen J, Shin C, Camargo FD, Gygi SP, Bartel DP. 2008. The impact of microRNAs on protein output. Nature 455:64–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wu L, Fan J, Belasco JG. 2006. MicroRNAs direct rapid deadenylation of mRNA. Proc. Natl. Acad. Sci. U. S. A. 103:4034–4039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Giraldez AJ, Mishima Y, Rihel J, Grocock RJ, Van Dongen S, Inoue K, Enright AJ, Schier AF. 2006. Zebrafish miR-430 promotes deadenylation and clearance of maternal mRNAs. Science 312:75–79 [DOI] [PubMed] [Google Scholar]
- 15. Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, Bartel DP, Linsley PS, Johnson JM. 2005. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature 433:769–773 [DOI] [PubMed] [Google Scholar]
- 16. Guo H, Ingolia NT, Weissman JS, Bartel DP. 2010. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466:835–840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Rehwinkel J, Behm-Ansmant I, Gatfield D, Izaurralde E. 2005. A crucial role for GW182 and the DCP1:DCP2 decapping complex in miRNA-mediated gene silencing. RNA 11:1640–1647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ding SW. 2010. RNA-based antiviral immunity. Nat. Rev. Immunol. 10:632–644 [DOI] [PubMed] [Google Scholar]
- 19. Ding SW, Voinnet O. 2007. Antiviral immunity directed by small RNAs. Cell 130:413–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Umbach JL, Cullen BR. 2009. The role of RNAi and microRNAs in animal virus replication and antiviral immunity. Genes Dev. 23:1151–1164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Otsuka M, Jing Q, Georgel P, New L, Chen J, Mols J, Kang YJ, Jiang Z, Du X, Cook R, Das SC, Pattnaik AK, Beutler B, Han J. 2007. Hypersusceptibility to vesicular stomatitis virus infection in Dicer1-deficient mice is due to impaired miR24 and miR93 expression. Immunity 27:123–134 [DOI] [PubMed] [Google Scholar]
- 22. Hou J, Wang P, Lin L, Liu X, Ma F, An H, Wang Z, Cao X. 2009. MicroRNA-146a feedback inhibits RIG-I-dependent Type I IFN production in macrophages by targeting TRAF6, IRAK1, and IRAK2. J. Immunol. 183:2150–2158 [DOI] [PubMed] [Google Scholar]
- 23. Taganov KD, Boldin MP, Chang KJ, Baltimore D. 2006. NF-κB-dependent induction of microRNA miR-146, an inhibitor targeted to signaling proteins of innate immune responses. Proc. Natl. Acad. Sci. U. S. A. 103:12481–12486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Lagos D, Pollara G, Henderson S, Gratrix F, Fabani M, Milne RS, Gotch F, Boshoff C. 2010. miR-132 regulates antiviral innate immunity through suppression of the p300 transcriptional co-activator. Nat. Cell Biol. 12:513–519 [DOI] [PubMed] [Google Scholar]
- 25. Jopling CL, Yi M, Lancaster AM, Lemon SM, Sarnow P. 2005. Modulation of hepatitis C virus RNA abundance by a liver-specific microRNA. Science 309:1577–1581 [DOI] [PubMed] [Google Scholar]
- 26. Filipowicz M. 2011. Prognostic potential of hepatic miR-122 measurements and antisense strategies targeting miR-122 as a therapeutic approach in viral hepatitis. Liver Int. 31:437–439 [DOI] [PubMed] [Google Scholar]
- 27. Pedersen IM, Cheng G, Wieland S, Volinia S, Croce CM, Chisari FV, David M. 2007. Interferon modulation of cellular microRNAs as an antiviral mechanism. Nature 449:919–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Zhang X, Daucher M, Armistead D, Russell R, Kottilil S. 2013. MicroRNA expression profiling in HCV-infected human hepatoma cells identifies potential anti-viral targets induced by interferon-alpha. PLoS One 8:e55733. 10.1371/journal.pone.0055733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Thornburg NJ, Hayward SL, Crowe JE., Jr 2012. Respiratory syncytial virus regulates human microRNAs by using mechanisms involving beta interferon and NF-κB. mBio 3(6):e00220-12. 10.1128/mBio.00220-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhao P, Zhao L, Zhang K, Feng H, Wang H, Wang T, Xu T, Feng N, Wang C, Gao Y, Huang G, Qin C, Yang S, Xia X. 2012. Infection with street strain rabies virus induces modulation of the microRNA profile of the mouse brain. Virol. J. 9:159. 10.1186/1743-422X-9-159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cameron JE, Yin Q, Fewell C, Lacey M, McBride J, Wang X, Lin Z, Schaefer BC, Flemington EK. 2008. Epstein-Barr virus latent membrane protein 1 induces cellular MicroRNA miR-146a, a modulator of lymphocyte signaling pathways. J. Virol. 82:1946–1958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Song L, Liu H, Gao S, Jiang W, Huang W. 2010. Cellular microRNAs inhibit replication of the H1N1 influenza A virus in infected cells. J. Virol. 84:8849–8860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Patel D, Nan Y, Shen M, Ritthipichai K, Zhu X, Zhang YJ. 2010. Porcine reproductive and respiratory syndrome virus inhibits type I interferon signaling by blocking STAT1/STAT2 nuclear translocation. J. Virol. 84:11045–11055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sung TL, Rice AP. 2009. miR-198 inhibits HIV-1 gene expression and replication in monocytes and its mechanism of action appears to involve repression of cyclin T1. PLoS Pathog. 5:e1000263. 10.1371/journal.ppat.1000263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lecellier CH, Dunoyer P, Arar K, Lehmann-Che J, Eyquem S, Himber C, Saib A, Voinnet O. 2005. A cellular microRNA mediates antiviral defense in human cells. Science 308:557–560 [DOI] [PubMed] [Google Scholar]
- 36. Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25:402–408 [DOI] [PubMed] [Google Scholar]
- 37. Du P, Kibbe WA, Lin SM. 2008. lumi: a pipeline for processing Illumina microarray. Bioinformatics 24:1547–1548 [DOI] [PubMed] [Google Scholar]
- 38. Bolstad BM, Irizarry RA, Astrand M, Speed TP. 2003. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics 19:185–193 [DOI] [PubMed] [Google Scholar]
- 39. Dweep H, Sticht C, Pandey P, Gretz N. 2011. miRWalk-database: prediction of possible miRNA binding sites by “walking” the genes of three genomes. J. Biomed. Inform. 44:839–847 [DOI] [PubMed] [Google Scholar]
- 40. Breuer K, Foroushani AK, Laird MR, Chen C, Sribnaia A, Lo R, Winsor GL, Hancock RE, Brinkman FS, Lynn DJ. 2013. InnateDB: systems biology of innate immunity and beyond—recent updates and continuing curation. Nucleic Acids Res. 41:D1228–D1233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Lynn DJ, Winsor GL, Chan C, Richard N, Laird MR, Barsky A, Gardy JL, Roche FM, Chan TH, Shah N, Lo R, Naseer M, Que J, Yau M, Acab M, Tulpan D, Whiteside MD, Chikatamarla A, Mah B, Munzner T, Hokamp K, Hancock RE, Brinkman FS. 2008. InnateDB: facilitating systems-level analyses of the mammalian innate immune response. Mol. Syst. Biol. 4:218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Huang da W, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 4:44–57 [DOI] [PubMed] [Google Scholar]
- 43. Huang da W, Sherman BT, Lempicki RA. 2009. Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 45. Baum A, Sachidanandam R, Garcia-Sastre A. 2010. Preference of RIG-I for short viral RNA molecules in infected cells revealed by next-generation sequencing. Proc. Natl. Acad. Sci. U. S. A. 107:16303–16308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kolakofsky D. 1976. Isolation and characterization of Sendai virus DI-RNAs. Cell 8:547–555 [DOI] [PubMed] [Google Scholar]
- 47. Desmyter J, Melnick JL, Rawls WE. 1968. Defectiveness of interferon production and of rubella virus interference in a line of African green monkey kidney cells (Vero). J. Virol. 2:955–961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Emeny JM, Morgan MJ. 1979. Regulation of the interferon system: evidence that Vero cells have a genetic defect in interferon production. J. Gen. Virol. 43:247–252 [DOI] [PubMed] [Google Scholar]
- 49. Lewis BP, Burge CB, Bartel DP. 2005. Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 120:15–20 [DOI] [PubMed] [Google Scholar]
- 50. Takeshita N, Mori M, Kano M, Hoshino I, Akutsu Y, Hanari N, Yoneyama Y, Ikeda N, Isozaki Y, Maruyama T, Akanuma N, Miyazawa Y, Matsubara H. 2012. miR-203 inhibits the migration and invasion of esophageal squamous cell carcinoma by regulating LASP1. Int. J. Oncol. 41:1653–1661 [DOI] [PubMed] [Google Scholar]
- 51. Wang C, Zheng X, Shen C, Shi Y. 2012. MicroRNA-203 suppresses cell proliferation and migration by targeting BIRC5 and LASP1 in human triple-negative breast cancer cells. J. Exp. Clin. Cancer Res. 31:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yuen T, Wurmbach E, Pfeffer RL, Ebersole BJ, Sealfon SC. 2002. Accuracy and calibration of commercial oligonucleotide and custom cDNA microarrays. Nucleic Acids Res. 30:e48. 10.1093/nar/30.10.e48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Kimura T, Nakayama K, Penninger J, Kitagawa M, Harada H, Matsuyama T, Tanaka N, Kamijo R, Vilcek J, Mak TW, Taniguchi T. 1994. Involvement of the IRF-1 transcription factor in antiviral responses to interferons. Science 264:1921–1924 [DOI] [PubMed] [Google Scholar]
- 54. Osella M, Bosia C, Cora D, Caselle M. 2011. The role of incoherent microRNA-mediated feedforward loops in noise buffering. PLoS Comput. Biol. 7:e1001101. 10.1371/journal.pcbi.1001101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Cullen BR. 2011. Viruses and microRNAs: RISCy interactions with serious consequences. Genes Dev. 25:1881–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Selbach M, Schwanhausser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. 2008. Widespread changes in protein synthesis induced by microRNAs. Nature 455:58–63 [DOI] [PubMed] [Google Scholar]
- 57. Schoggins JW, Wilson SJ, Panis M, Murphy MY, Jones CT, Bieniasz P, Rice CM. 2011. A diverse range of gene products are effectors of the type I interferon antiviral response. Nature 472:481–485 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yang Z, Dong D, Zhang Z, Crabbe MJ, Wang L, Zhong Y. 2012. Preferential regulation of stably expressed genes in the human genome suggests a widespread expression buffering role of microRNAs. BMC Genomics 13(Suppl 7):S14. 10.1186/1471-2164-13-S7-S14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Vaz C, Mer AS, Bhattacharya A, Ramaswamy R. 2011. MicroRNAs modulate the dynamics of the NF-κB signaling pathway. PLoS One 6:e27774. 10.1371/journal.pone.0027774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Martinez NJ, Walhout AJ. 2009. The interplay between transcription factors and microRNAs in genome-scale regulatory networks. Bioessays 31:435–445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wei J, Huang X, Zhang Z, Jia W, Zhao Z, Zhang Y, Liu X, Xu G. 2013. 21 March 2013. MyD88 as a target of microRNA-203 in regulation of lipopolysaccharide or Bacille Calmette-Guerin induced inflammatory response of macrophage RAW264.7 cells. Mol. Immunol. 55:303–309 [Epub ahead of print]. 10.1016/j.molimm.2013.03.004 [DOI] [PubMed] [Google Scholar]
- 62. Primo MN, Bak RO, Schibler B, Mikkelsen JG. 2012. Regulation of pro-inflammatory cytokines TNFα and IL24 by microRNA-203 in primary keratinocytes. Cytokine 60:741–748 [DOI] [PubMed] [Google Scholar]
- 63. Moffatt CE, Lamont RJ. 2011. Porphyromonas gingivalis induction of microRNA-203 expression controls suppressor of cytokine signaling 3 in gingival epithelial cells. Infect. Immun. 79:2632–2637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sonkoly E, Wei T, Pavez Lorie E, Suzuki H, Kato M, Torma H, Stahle M, Pivarcsi A. 2010. Protein kinase C-dependent upregulation of miR-203 induces the differentiation of human keratinocytes. J. Investig. Dermatol. 130:124–134 [DOI] [PubMed] [Google Scholar]
- 65. Melar-New M, Laimins LA. 2010. Human papillomaviruses modulate expression of microRNA 203 upon epithelial differentiation to control levels of p63 proteins. J. Virol. 84:5212–5221 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Yu H, Lu J, Zuo L, Yan Q, Yu Z, Li X, Huang J, Zhao L, Tang H, Luo Z, Liao Q, Zeng Z, Zhang J, Li G. 2012. Epstein-Barr virus downregulates microRNA 203 through the oncoprotein latent membrane protein 1: a contribution to increased tumor incidence in epithelial cells. J. Virol. 86:3088–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Hemida MG, Ye X, Zhang HM, Hanson PJ, Liu Z, McManus BM, Yang D. 2013. MicroRNA-203 enhances coxsackievirus B3 replication through targeting zinc finger protein-148. Cell. Mol. Life Sci. 70:277–291 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.