

Abstract
Herpesvirus genes are temporally expressed during permissive infections, but how their expression is regulated at late times is poorly understood. Previous studies indicate that the human cytomegalovirus (CMV) gene, UL79, is required for late gene expression. However, the mechanism remains to be fully elucidated, and UL79 homologues in other CMVs have not been studied. Here, we characterized the role of the conserved murine CMV (MCMV) gene M79. We showed that M79 encoded a protein (pM79) which was expressed with early-late kinetics and localized to nuclear viral replication compartments. M79 transcription was significantly decreased in the absence of viral DNA synthesis but markedly stimulated by pM79. To investigate its role, we created the recombinant virus SMin79, in which pM79 expression was disrupted. While marker-rescued virus grew efficiently in fibroblasts, SMin79 failed to produce infectious progeny but was rescued by pM79 expression in trans. During SMin79 infection, representative viral immediate-early and early gene products as well as viral DNA accumulated sufficiently. Formation of viral replication compartments also appeared normal. Pulsed-field gel electrophoresis analysis indicated that the overall structure of replicating viral DNA was indistinguishable between wild-type and SMin79 infection. Viral tiled array and quantitative PCR analysis revealed that many late transcripts sensitive to a viral DNA synthesis inhibitor (phosphonoacetic acid) were markedly reduced by pM79 mutation. This study indicates that cytomegaloviruses use a conserved mechanism to promote transcription at late stages of infection and that pM79 is a critical regulator for at least a subset of viral DNA synthesis-dependent transcripts.
INTRODUCTION
Cytomegalovirus (CMV) is the prototypical member of the betaherpesvirus subfamily. Human CMV (HCMV) is a ubiquitous human pathogen which causes asymptomatic infection in healthy adults. However, in immunocompromised hosts, such as neonates, transplant recipients, persons with advanced AIDS, and cancer patients, HCMV is a common cause of severe and even life-threatening disease (1–4). The severity of medical problems associated with HCMV in these vulnerable populations underlies the necessity for developing safer and more effective antiviral treatments and vaccine strategies to control its infection. HCMV infection is limited to human hosts as the members of the CMV family are species specific. Murine CMV (MCMV) infection provides a tractable small-animal model to study CMV biology. MCMV shares conservation with HCMV in regard to its colinear genome, gene expression program, tissue tropism, and pathology (5, 6). Over 40% of MCMV genes have sequence or functional homologues in HCMV (7). This conservation provides us with excellent opportunities to explore MCMV as a tool to dissect the mechanistic basis of shared features of viral replication and pathogenesis. Furthermore, revealing the function of homologous viral genes in their respective hosts will allow antiviral therapeutics or vaccine candidates targeting these conserved genes to be tested in the mouse model.
Gene expression during herpesvirus lytic infection is highly coordinated and sequentially ordered such that viral genes are traditionally divided into three kinetic classes: immediate early (IE), early, and late. IE genes are transcribed following viral DNA translocation to the nucleus and require only incoming virion-associated proteins and cellular factors for their expression. Products of IE genes transactivate early genes and remodel the host cell to be permissive for virus replication. Early gene transcriptions are initiated prior to viral DNA synthesis, but some persist at late times of infection, even after the onset of DNA synthesis. Early gene products are required for both viral DNA synthesis and formation of replication compartments, which are virus-induced subnuclear structures where viral DNA synthesis occurs. Transcription of late viral genes occurs after the onset of viral DNA synthesis and peaks at late times of infection. Many late genes encode structural proteins required for virion assembly, maturation, and release. Largely consistent with this temporal regulation, transcription of many IE and early genes is resistant to viral DNA synthesis inhibitors, such as phosphonoacetic acid (PAA). Late transcripts, defined by their abundant accumulation at late times of infection and dependency on viral DNA synthesis, are largely derived from late genes. In addition, some genes have both early and late properties, as their expression initiates prior to DNA synthesis, but the transcripts continue to accumulate to high levels at late times in a DNA synthesis-dependent manner.
While IE and early transcription has been extensively studied in herpesviruses, little is known about how viral late transcription is regulated. Viral DNA synthesis is required in cis for viral late promoter activity, but the precise mechanism remains elusive (8–10). Herpes simplex virus (HSV), the prototypical alphaherpesvirus, is perhaps the best studied example. Multiple HSV proteins (ICP0, ICP4, ICP22, and ICP27) have been shown to regulate late expression (11–16). For some late genes, the TATA box as well as DNA sequences downstream of the transcription start site are also main determinants of transcription (17–19). However, a majority of these HSV genes lack homologues in betaherpesviruses (20), and evidence suggests that the requirement for these sequence elements is not universal (17, 19, 21–23).
Understanding how CMV regulates late gene expression is important to understanding its biology and identifying novel targets for antiviral therapeutics. The HCMV UL79 family is a viral gene family conserved between beta- and gammaherpesviruses (24). We along with others have recently shown that UL79 is required for HCMV late gene expression (25, 26). However, the MCMV homologue of UL79, M79, remains uncharacterized. Defining the role of M79 will set the stage for using the MCMV model to elucidate the mechanism of action for this CMV gene family and to explore novel antiviral strategies targeting this viral factor.
In this study, we characterized M79 during MCMV infection. We show that pM79, the protein product of M79, acts downstream of viral DNA synthesis to facilitate viral late transcription. Importantly, viral oligonucleotide tiled array analysis reveals at least two subsets of late transcripts. Both require viral DNA synthesis for their expression, but they have different degrees of dependence on pM79 for expression. As a result, abrogation of pM79 results in a complete failure in virus growth. These results, along with studies of HCMV UL79 and murine gammaherpesvirus 68 (MHV-68) open reading frame 18 (ORF18) (24, 26), suggest that divergent herpesviruses use similar mechanisms to promote late gene expression. Furthermore, our study provides evidence to support the model that CMV late transcription is tightly regulated beyond its dependency on viral DNA synthesis and that pM79 is a key regulator for at least a subset of MCMV late transcription, highlighting the complex regulatory mechanisms governing CMV late transcription.
MATERIALS AND METHODS
Plasmids, antibodies, and chemicals.
pYD-C245 and pYD-C571 were retroviral expression vectors derived from pRetro-EBNA (27). pYD-C245 expressed the red fluorescent protein (DsRed) (28) from an internal ribosome entry site (IRES). pYD-C571 was derived from pYD-C245. It carried the coding sequence of M79 with one copy of a FLAG tag (1×FLAG) at the C terminus expressed together with DsRed as a bicistronic transcript. pYD-C191 carried a kanamycin selection cassette bracketed by two Flp recognition target (FRT) sites. pYD-C630 was derived from pGalK (29) and carried an FRT-bracketed GalK/kanamycin dual selection cassette (30). pYD-C746 was derived from pYD-C630, where a 3×FLAG sequence preceded the FRT-bracketed selection cassette.
The primary antibodies used in this study included the following: anti-actin (clone AC15; Abcam); anti-FLAG polyclonal rabbit antibody (F7425) and monoclonal mouse antibody (F1804) (Sigma); anti-MCMV IE1 (CROMA101) and E1 (CROMA103) (generous gifts from Stipan Jonjic, University of Rijeka, Croatia); and anti-MCMV M44 (3B9.22A) and gB (2E8.21A) (generous gifts from Anthony Scalzo, University of Western Australia). The secondary antibody used for immunoblotting was horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG (Jackson Laboratory). The secondary antibodies used for immunofluorescence were Alexa Fluor 594-conjugated goat anti-mouse IgG and Alexa Fluor 488-conjugated goat anti-rabbit IgG (Invitrogen-Molecular Probes).
Other chemicals used in this study include phosphonoacetic acid (PAA) (284270-10G; Sigma-Aldrich), l-(+)-arabinose (A3256-25G; Sigma-Aldrich), and TO-PRO3 iodide (T3605; Invitrogen).
Cells and viruses.
Mouse embryonic fibroblast 10.1 cells (MEF10.1) (31) were propagated in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum (FBS), nonessential amino acids, and 1 mM sodium pyruvate. Cells were maintained at 37°C and 5% CO2 in a humidified atmosphere. To create cell lines stably expressing FLAG-tagged M79 (10.1-M79flag), MEF10.1 cells were transduced three times with retrovirus reconstituted from pYD-C571 and allowed to recover for 48 h. Clonal cells expressing DsRed were isolated by limiting dilution and expanded to produce stocks of cell lines. Individual clonal cell lines were tested by transfecting them with the recombinant MCMV bacterial artificial chromosome (BAC) clone pSMin79 (see below) and determining the titer of reconstituted virus at 5 days posttransfection. The cell line that yielded the highest titer was used in this study. Recombinant MCMV viruses SMgfp, SMrev79, and SM79flag (see below) were reconstituted from electroporation of corresponding BAC clones into MEF10.1 cells. Recombinant virus SMin79 (see below) was reconstituted from electroporation of the BAC clone pSMin79 into 10.1-M79flag cells.
BAC recombineering.
Recombinant MCMV BAC clones used in this study were derived from the self-excisable parental MCMV BAC clone, pSM3fr, which carried a full-length genome of the MCMV Smith strain (32). All recombinant MCMV BAC clones in this study were created using the linear recombination-based BAC recombineering protocol that we have previously established (30). Recombination was carried out in Escherichia coli strain SW105 that harbored an MCMV BAC clone and expressed an arabinose-inducible flippase gene for transient expression of Flp recombinase (29). We inserted the green fluorescent protein (GFP) expression cassette at the C terminus of the IE2 loci within pSM3fr to produce the BAC clone pSMgfp. This clone was used to produce wild-type virus SMgfp as IE2 has been shown to be dispensable for MCMV infection in vivo and in vitro (33–35). We independently confirmed that the insertion of the GFP cassette at this locus had no deleterious consequences on virus growth in our infection system (data not shown). The BAC clone pSMin79 carried a frameshift mutation in the viral gene M79 (Fig. 1A). To construct pSMin79, the FRT-bracketed GalK/kanamycin cassette was PCR amplified from pYD-C630 and recombined into pSMgfp 403 nucleotides (nt) downstream of the start codon of the M79 coding sequence. Transformants were selected by kanamycin resistance. The selection cassette was removed by arabinose induction of Flp recombinase and subsequent Flp-FRT recombination (30), leaving an 88-nt insert within the M79 coding sequence and creating a frameshift mutation. The BAC clone pSM79flag contained a C-terminally 3×FLAG-tagged M79 (Fig. 1A). To construct pSM79flag, a DNA fragment that contained the FRT-bracketed GalK/kanamycin selection cassette preceded by a 3×FLAG sequence was PCR amplified from pYD-C746 and recombined into the C terminus of the M79 coding sequence. The selection cassette was subsequently removed by Flp-FRT recombination, resulting in the 3×FLAG fused in frame with the M79 coding sequence. The BAC clone pSMrev79 was derived from pSMin79 and contained the repaired M79 coding sequence. To create pSMrev79, a patched PCR fragment containing the wild-type M79 coding sequence followed by a FRT-bracketed kanamycin selection cassette was recombined into pSMin79 to replace the M79 frameshift mutation. The selection cassette was subsequently removed by Flp-FRT recombination, leaving an 81-bp sequence insert after the stop codon of the M79 coding sequence, which had no deleterious effect on virus replication (Fig. 1D). All the final BAC clones were validated by restriction digestion, PCR analysis, and direct sequencing as previously described (36).
Fig 1.

Gene M79 is essential for MCMV growth in fibroblasts. (A) Diagram depicting recombinant MCMV BAC clones used in this study. The BAC clone pSM79flag carried a 3×FLAG tag that was fused in frame at the C terminus of the M79 coding sequence (indicated by the shaded region). The BAC clone pSMin79 carried an 88-bp insert (indicated by the black region) at 403 nt downstream of the start codon of the M79 coding sequence, resulting in a frameshift mutation. See Materials and Methods for details. (B) Growth of SMin79 virus on MEF10.1 cells expressing FLAG-tagged M79 (10.1-M79flag). The left panel shows virus-driven GFP expression in normal MEF10.1 cells or 10.1-M79flag cells at 7 days posttransfection with pSMin79. The right panel shows titers of wild-type virus (SMgfp) and M79 mutant virus (SMin79) produced at 72 hpi in 10.1-M79flag cells that were infected at an MOI of 2. Shown is a representative result from at least two reproducible, independent experiments. (C and D) Growth kinetic analysis of M79 recombinant viruses used in this study. Normal MEF10.1 cells were infected at an MOI of 2, cell-free and cell-associated viruses were collected at indicated times, and viral titers were determined by 50% tissue culture infectious dose (TCID50) assay in 10.1-M79flag cells. The detection limit of the TCID50 assay is indicated by a dashed line. Shown is a representative result from two reproducible, independent experiments.
To reconstitute recombinant viruses that did not require complementation to grow, confluent MEF10.1 cells were electroporated with 5 μg of MCMV BAC DNA and plated on a 10-cm plate. Culture medium was changed at 24 h posttransfection, and virus was harvested by collecting cell-free culture medium after the entire monolayer of cells was lysed. Alternatively, virus stocks were produced by collecting cell-free supernatant from infected culture at a multiplicity of infection (MOI) of 0.001. Virus titers were determined in duplicate by a 50% tissue culture infectious dose (TCID50) assay in MEF10.1 cells. To reconstitute, propagate, and determine titers of SMin79 virus, 10.1-M79flag cells were used as described above. In experiments where comparative analysis was performed between SMin79 and other recombinant viruses, titers of all viruses were determined in 10.1-M79flag cells.
Viral growth analysis.
MEF10.1 cells were seeded in 12-well plates overnight to produce a confluent monolayer. Cells were inoculated with recombinant MCMV viruses for 1 h at an MOI of 2 for single-step or of 0.01 for multistep growth analysis. The inoculum was removed, the infected monolayer was rinsed with phosphate-buffered saline (PBS), and fresh medium was replenished. At various times postinfection, cell-free virus was collected in duplicate by harvesting medium from infected cultures. Cell-associated virus was collected by rinsing infected cells once with PBS and scraping cells into fresh medium. Cells were lysed by one freeze-thaw cycle followed by sonication. Lysates were cleared of cell debris by low-speed centrifugation, and supernatants were saved as cell-associated virus. Virus titers were determined by TCID50 assay.
DNA and RNA analysis.
Intracellular DNA was measured by quantitative PCR (qPCR) as previously described (36). Briefly, MCMV-infected cells were collected in a lysis buffer (200 mM NaCl, 20 mM Tris [pH 8.0], 20 mM EDTA, 0.2 mg/ml proteinase K, 0.4% sodium dodecyl sulfate [SDS]) and lysed by incubation at 55°C overnight. DNA was extracted with phenol-chloroform and treated with RNase A (100 μg/ml) at 37°C for 1 h. Samples were extracted again with phenol-chloroform, precipitated with ethanol, and resuspended in nuclease-free water (Ambion). Viral DNA was quantified by qPCR using SYBR Advantage qPCR Premix (Clontech) and a primer pair specific for the MCMV IE1 or M55 gene (Table 1). Cellular DNA was quantified using a primer pair specific for the mouse actin gene (Table 1) (37). A standard curve was generated using serially diluted pSMgfp BAC DNA or DNA from infected cells and used to calculate relative amounts of viral or cellular DNA in a sample. The amount of viral DNA was normalized by dividing the number of IE1 or M55 equivalents by the number of actin equivalents. The normalized amount of viral DNA in SMgfp-infected cells at 2 h postinfection (hpi) was set at 1.
Table 1.
Primers used in quantitative PCR analysis
| Primer name | Primer sequence |
|---|---|
| MCMV IE1 forward | 5′-CAGGGTGGATCATGAAGCCT-3′ |
| MCMV IE1 reverse | 5′-AGCGCATCGAAAGACAACG-3′ |
| MCMV M25 forward | 5′-AAGACATGTCACGCGACGGA-3′ |
| MCMV M25 reverse | 5′-CTATTGCCCATCATCGCCCG-3′ |
| MCMV gB (M55) forward | 5′-GCGATGTCCGAGTGTGTCAAG-3′ |
| MCMV gB (M55) reverse | 5′-CGACCAGCGGTCTCGAATAAC-3′ |
| MCMV M74 forward | 5′-AGGAGGCTGTGACTTTGAAA-3′ |
| MCMV M74 reverse | 5′-CTCATCAGCCGTTACTCGAG-3′ |
| MCMV M79 forward | 5′-CTACCTGATCGCCTGGAAAAAG-3′ |
| MCMV M79 reverse | 5′-TAGTCCTGGATCAGGAAGGAAAAG-3′ |
| MCMV M112/113 (E1) forward | 5′-GAATCCGAGGAGGAAGACGAT-3′ |
| MCMV M112/113 (E1) reverse | 5′-GGTGAACGTTTGCTCGATCTC-3′ |
| MCMV M116 forward | 5′-TCCTTGGTGGTGATGGCGGT-3′ |
| MCMV M116 reverse | 5′-GCATCCCGTACCTGACCACA-3′ |
| MCMV M121 forward | 5′-CCCGTTCGCTTCTGAAACTG-3′ |
| MCMV M121 reverse | 5′-GCTTCTCGAGGCAGCAGCAA-3′ |
| Mouse actin forward | 5′-GCTGTATTCCCCTCCATCGTG-3′ |
| Mouse actin reverse | 5′-CACGGTTGGCCTTAGGGTTCA-3′ |
Intracellular RNA was determined by reverse transcription-coupled qPCR (RT-qPCR) as previously described (36). Total RNA was extracted by TRIzol reagent (Invitrogen) and treated with Turbo DNA-free reagents (Ambion) to remove contaminating DNA. First-strand cDNA synthesis was performed with a High Capacity cDNA Reverse Transcription Kit using random hexamer primers with total RNA (Applied Biosystems). Each sample also included a control without the addition of reverse transcriptase to determine the level of residual contaminating DNA. cDNA was quantified using SYBR Advantage qPCR Premix (Clontech) and primer pairs specific for viral genes or the mouse actin gene (Table 1). A standard curve was generated for each gene using serially diluted cDNA from infected cells and used to calculate the relative amount of a transcript in each sample. The amounts of viral transcript were normalized by dividing the number of viral transcript equivalents by the number of actin equivalents. The normalized amount of transcript during SMgfp infection at 10 hpi in the absence of PAA was set to 1.
Tiled array design, experimental procedure, and analysis.
The array was designed using Agilent's eArray package. The array consisted of 103,347 60-mer oligonucleotide probes for each strand of the MCMV genome, and each probe advanced 4 to 5 nt. In addition, the array also contained probes complementary to Agilent's RNA Spike-Ins and 400 negative-control probes against Arabidopsis with no homology to mouse or MCMV genomes.
To prepare viral RNA, total RNA was harvested from infected MEF10.1 cells at 20 hpi using TRIzol (Invitrogen), and mRNA was purified using RNeasy columns (Qiagen) according to the manufacturer's instructions. cDNA probes were synthesized, fluorescently labeled by random hexamer-primed polymerization using a SuperScript Plus Indirect cDNA Labeling module (Invitrogen), and hybridized to the array chip at Genome Technology Access Center of Washington University School of Medicine (GTAC). Agilent's RNA Spike-In Kit was used to monitor the linearity, sensitivity, and accuracy of the array.
The total signal intensity for each probe was log2 transformed, and the mock signal was subtracted from the experimental signal after values were normalized to each other using the spike-in RNA signals. To enable comparisons among samples, raw data from each MCMV-infected sample were normalized using spike-in controls. This normalization was further refined and validated using qRT-PCR data for several viral probes. Normalized intensities of experimental probes were mapped back to the MCMV genome, and mean fluorescence of each nucleotide was calculated from all overlapping probes. Changes in fluorescence intensity greater than 3-fold between compared samples were used for data interpretation. MCMV open reading frames (ORFs) were annotated based on the studies by Rawlinson and coworkers (7) and by Cheng and coworkers (38) and updated with details from additional publications whenever possible. Data were converted to gff3 file format and visualized using gBrowse (39).
PFGE and Southern blot analysis.
Pulsed-field gel electrophoresis (PFGE) was performed on a Bio-Rad CHEF Mapper XA pulsed-field electrophoresis system. To prepare DNA from infected cells, MEF10.1 cells were seeded onto a 60-mm dish at a density of 1.4 × 106 cells per dish and infected at an MOI of 2. At 36 hpi, cells were scraped off the dish, collected by centrifugation at 200 × g for 5 min, and resuspended in 180 μl of 55°C 1% low-melting-point agarose (NuSieve GTG Agarose, Lonza) in PBS. Ninety microliters of cell suspension was cast into a disposable casting mold (Bio-Rad) and solidified at 4°C for 15 min. To prepare DNA from cell-free virions, liquid viral stock equivalent to 106 PFU was cast in one low-melting-point agarose block. Agarose blocks were transferred into lysis buffer (20 mM Tris-Cl, pH 8.0, 200 mM NaCl, 400 mM EDTA, 1% SDS, 1 mg/ml proteinase K) to lyse imbedded samples by incubation at 37°C overnight. Blocks were then rinsed five times with TE buffer (10 mM Tris-HCl, pH 8.0, 0.1 mM EDTA) at 50°C for 15 min each and stored in TE buffer at 4°C.
Restriction enzyme digestions were carried out by incubating one-half of a block (∼45 μl) in 200 μl of digestion buffer containing 50 units of PacI at 4°C overnight and then at 37°C for 6 h. Digested blocks were loaded into wells of a 1% megabase agarose gel (Bio-Rad Pulse Field Certified Agarose) in 0.5× TBE buffer (0.045 M Tris, pH 8.0, 0.045 M boric acid, 0.001 M EDTA, pH 8.0) and sealed with 1% low-melting-point agarose in 0.5× TBE buffer. PFGE was performed at 6 V/cm at 14°C for 16 h, with a linear pulse increase from 0.26 to 20.01 s over the course of the run.
Resolved viral fragments were analyzed by Southern blotting as previously described (40). Briefly, the gel was transferred to a Nytran SuPerCharge membrane as described in the Turboblotter transfer system protocol (VWR), and the membrane was air dried and cross-linked by UV at 125 mJ. The 32P-labeled probe was generated by random hexamer priming of pSMgfp BAC DNA using a Prime-It II Labeling Kit (Stratagene), purified with ProbeQuant G-50 microcolumns (Amersham), and denatured in 10 mM EDTA at 90°C for 10 min before use. The membrane was prehybridized with 10 ml of ULTRAhyb solution (Ambion) at 42°C for 1 h and hybridized with 107 cpm of denatured probes at 42°C for 3 h. The membrane was washed once with buffer I (2× SSC and 0.1% SDS [1× SSC is 0.15 M NaCl plus 0.015 M sodium citrate]) for 10 min, followed by two washes with buffer II (0.1× SSC and 0.1% SDS) for 15 min before exposure on a Kodak film.
Protein analysis.
Protein accumulation was analyzed by immunoblotting. Cells were washed, and lysates were collected in sodium dodecyl sulfate (SDS)-containing sample buffer. Proteins were resolved by SDS-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene difluoride (PVDF) membrane. Proteins of interest were detected by hybridizing the membrane with specific primary antibodies followed by horseradish peroxidase (HRP)-coupled secondary antibodies and visualized by using SuperSignal West Pico enhanced chemiluminescent (ECL) substrate (Thermo Scientific).
Intracellular localization of proteins of interest was analyzed by immunofluorescence assay. Cells were seeded onto coverslips at 24 h prior to infection. At various times, cells were washed with PBS, fixed and permeabilized with methanol (−20°C) for 10 min, and blocked with 5% FBS in PBS at room temperature for 1 h. Cells were incubated with primary antibodies for 30 min at room temperature and subsequently labeled with secondary antibodies coupled to Alexa Fluor 488 or Alexa Fluor 594 (Invitrogen-Molecular Probes). Cells were counterstained with TO-PRO3 and mounted on slides with Prolong Gold antifade reagent (Invitrogen-Molecular Probes). Confocal microscopic images were captured by a Zeiss LSM510 Meta confocal laser scanning microscope.
Microarray data accession number.
The data have been deposited in the Gene expression Omnibus (GEO) database under series entry GSE47586.
RESULTS
M79 is essential for MCMV replication.
The homolog of M79 in HCMV, UL79, is essential for virus replication (25, 26), but the role of M79 has not been characterized. To investigate this, we created a mutant BAC clone of the MCMV Smith strain, pSMin79, by BAC recombineering. In this mutant clone, an 88-nt insertion was introduced at 403 nt downstream of the start codon of the M79 coding sequence, resulting in a frameshift mutation (Fig. 1A). We anticipated that this site of insertion made it unlikely to interfere with expression of neighboring genes, particularly M80, an essential gene that overlaps with M79 at its N terminus (41–43).
To reconstitute recombinant virus, both pSMin79 and its wild-type parental clone, pSMgfp, were electroporated into 10.1 mouse embryonic fibroblasts (MEF10.1). Cells transfected with pSMgfp readily initiated virus production and spread. By 5 days posttransfection, the monolayer of transfected cells demonstrated complete cytopathic effect (CPE) and full virus spread, indicated by virus-driven GFP. However, even though pSMin79 transfection was efficient, evidenced by the presence of individual GFP-positive cells upon initial inspection at day 2, it repeatedly failed to produce any CPE or GFP spread even at 2 weeks posttransfection (Fig. 1B). Transfection of multiple independently isolated pSMin79 clones yielded the same result, suggesting that M79 is essential for MCMV replication.
To provide a means to propagate BAC-derived M79 mutant virus, we created multiple clonal MEF10.1 cell lines (10.1-M79flag) that stably expressed C-terminally FLAG-tagged M79 by retroviral transduction. Transfection of pSMin79 into 10.1-M79flag cells supported efficient virus reconstitution, producing complete CPE and full spread of virus-driven GFP expression on the monolayer (Fig. 1B). Importantly, transfection of pSMin79 in 10.1-M79flag cells produced virus with titers similar to the titer of reconstituted wild-type virus, SMgfp (Fig. 1B).
Finally, to provide definitive evidence for the essential role of M79 in MCMV infection, we performed growth curve analysis of SMin79 in MEF10.1 cells. For this experiment, we also created SMrev79, a marker-rescued virus of SMin79. The SMin79 virus failed to produce cell-free or cell-associated progeny virus in both multistep (data not shown) and single-step growth analyses (Fig. 1C). On the other hand, SMrev79 replicated indistinguishably from wild-type virus (Fig. 1D). Together, our results indicate that the defect of SMin79 is the direct result of M79 ablation and that M79 is essential for MCMV replication at steps prior to virus release.
Expression of M79 gene products is markedly enhanced by viral DNA synthesis and protein pM79.
To better understand the function of the M79 gene, we first characterized its potential protein product (pM79). M79 is predicted to encode a protein of 258 amino acids (aa) with a molecular mass of 29 kDa. As no specific antibody to the M79 protein was available, we created a recombinant virus, SM79flag, in which the M79 coding sequence was tagged with 3×FLAG at the C terminus (Fig. 1A). SM79flag was reconstituted efficiently from BAC transfection and grew indistinguishably from wild-type virus (Fig. 1D), indicating that the 3×FLAG tag did not interfere with M79 function. To determine expression of pM79 during infection, we infected MEF10.1 cells with SM79flag and analyzed the accumulation of FLAG-tagged pM79 over the course of a single viral replication cycle by immunoblotting (Fig. 2A). FLAG-tagged pM79 migrated at an apparent molecular mass of 31 kDa, consistent with the predicted size, and was detected at 24 to 36 h postinfection (hpi) (Fig. 2A). We then profiled M79 transcription by reverse transcription-coupled quantitative PCR analysis (RT-qPCR) to more precisely characterize M79 expression (Fig. 2B). When viral DNA synthesis was inhibited by PAA, low levels of M79 transcription persisted and increased modestly from 10 hpi to 30 hpi, suggesting that a small amount of M79 transcript was produced independent of viral DNA synthesis. Importantly, however, the majority of M79 transcription was inhibited by PAA, and in the absence of PAA, M79 transcript levels accumulated to high abundance at late times postinfection (20 to 30 hpi). Together, our results indicate that low levels of M79 expression occur independent of viral DNA synthesis but that the majority of its expression requires viral DNA synthesis, similar to the previously described viral gene expression pattern of early-late kinetics (10).
Fig 2.
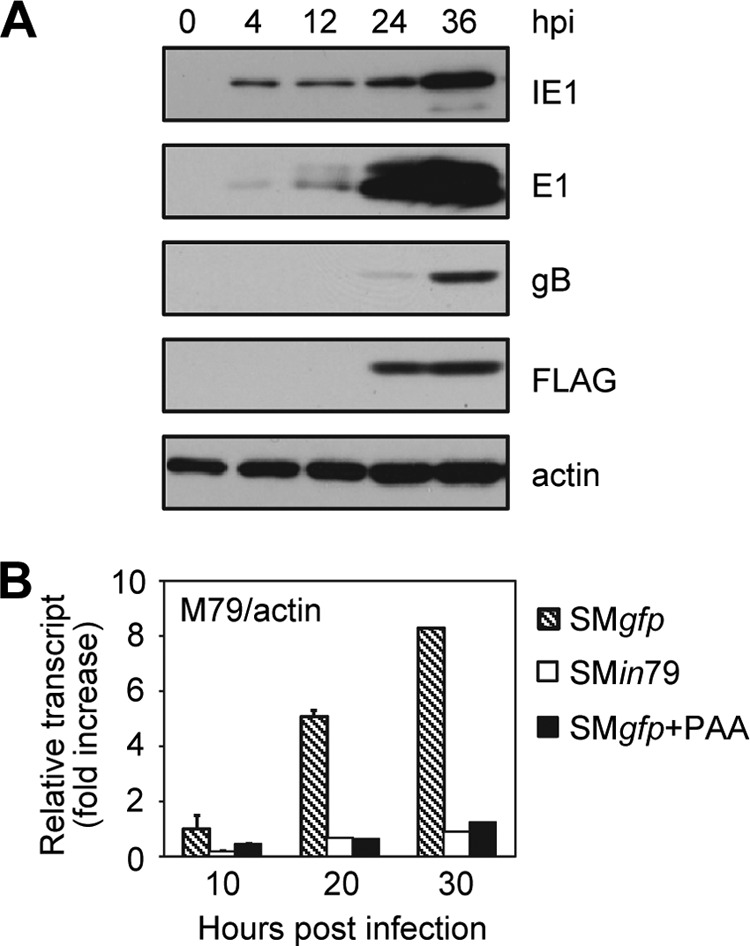
Expression of M79 gene products is markedly enhanced by viral DNA synthesis and protein pM79. (A) Accumulation of the M79 protein product during MCMV infection. MEF10.1 cells were infected with SM79flag virus at an MOI of 2, and total cell lysates were collected at indicated times and analyzed by immunoblotting. The M79 protein was detected with anti-FLAG antibody. Actin was used as a loading control, and viral proteins IE1, E1, and gB were used as representative immediate-early, early, and late proteins, respectively. Shown is a representative result from three reproducible, independent experiments. (B) Accumulation of the M79 transcript during MCMV infection. MEF10.1 cells were infected with SMgfp in the presence or absence of viral DNA synthesis inhibitor phosphonoacetic acid (PAA) (200 μg/ml) or with SMin79 at an MOI of 2. Total RNA was isolated at the indicated times, and the amount of M79 transcript was measured by reverse transcription-coupled quantitative PCR (RT-qPCR) analysis with the primers listed in Table 1. The values were normalized to those of actin, and the normalized amount of M79 transcript during SMgfp infection at 10 hpi in the absence of PAA was set to 1. Shown is a representative result from three reproducible, independent experiments.
It was interesting that during SMin79 infection, M79 transcript levels were drastically reduced relative to the levels of SMgfp virus infection (Fig. 2B). As the small insertion mutation in SMin79 was designed to abolish only the M79 protein but not its transcript, we interpreted this to suggest that pM79 enhances its own transcription, particularly at late times of infection, correlating with the previous report of its homolog UL79 during HCMV infection (26).
M79 protein localizes to viral nuclear replication compartments.
We next examined the intracellular localization of pM79 during infection of SM79flag using a rabbit anti-FLAG antibody. At 24 hpi, FLAG staining was found exclusively in the nucleus, closely colocalized with the viral polymerase processivity factor pM44 in the nuclei of infected cells (Fig. 3A). As pM44 is a widely used marker of viral replication compartments, this result indicates that the nuclear pM79 localizes within replication compartments. The rabbit anti-FLAG antibody also produced a diffuse, weak cytoplasmic staining, which was likely nonspecific as it was also present in SMgfp-infected control cells (Fig. 3A). To test this, a separate set of infected cells was stained with a mouse anti-FLAG antibody even though this strategy precluded us from costaining cells with the mouse anti-pM44 antibody to mark replication compartments (Fig. 3B). Nonetheless, in these cells there was only specific staining in the nuclei resembling replication compartments, and there was no cytoplasmic background staining observed. Collectively, we conclude that pM79 is a nuclear protein that localizes to replication compartments during MCMV infection.
Fig 3.

pM79 localizes to replication compartments during infection. MEF10.1 cells were mock infected or infected with SMgfp or SM79flag at an MOI of 2. At 24 hpi, cells were fixed with methanol (which quenches GFP fluorescence) and stained with either rabbit polyclonal (A) or mouse monoclonal (B) anti-FLAG antibody for detection of the tagged M79 protein (green). In the experiment shown in panel A, cells were costained with antibody to pM44 to mark replication compartments (red). Images in the last row of panel A are magnified views of an infected nucleus where the M79 protein (pM79) localized. Cells were counterstained with TO-PRO3 to visualize the nuclei (blue). Scale bar, 20 μm. Shown is a representative result from four reproducible, independent experiments.
M79 is not required for viral DNA synthesis or development of nuclear replication compartments.
As pM79 localized to viral replication compartments during infection, we next determined if this protein was involved in viral DNA replication processes, particularly the ability of the virus to synthesize its genome and form nuclear replication compartments. To test if pM79 was required for viral DNA synthesis, we first examined the kinetics of viral DNA accumulation during SMin79 infection by quantitative PCR (qPCR) analysis. Viral DNA accumulation over the course of SMin79 infection was comparable to that in SMgfp infection, indicating that pM79 is not required for the virus to synthesize its DNA (Fig. 4A).
Fig 4.
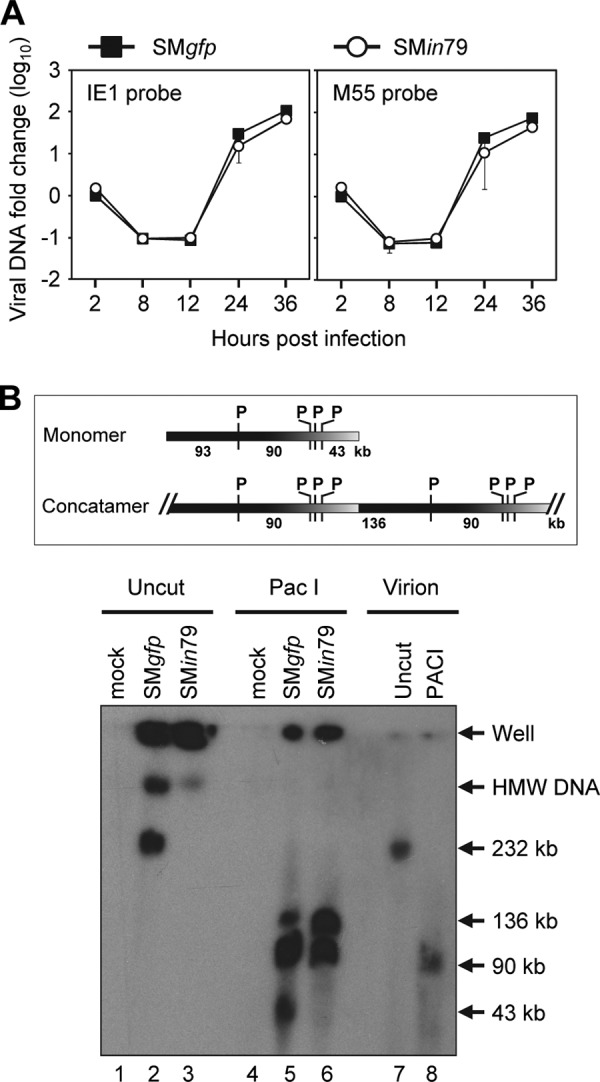
pM79 is not required for viral DNA synthesis. (A) Accumulation of viral DNA during pM79-deficient virus infection. MEF10.1 cells were infected with SMgfp or SMin79 at an MOI of 2, and total DNA was harvested from infected cells at indicated times. Viral DNA accumulation was analyzed by qPCR using primers specific for viral genes IE1 or M55 (Table 1 gives primer sequences), and the values were normalized to the value of actin. The normalized values of viral DNA during SMgfp infection at 2 hpi were set to 1. Shown is a representative result from five reproducible, independent experiments. (B) PFGE analysis of intracellular viral DNA during M79-deficient virus infection. MEF10.1 cells were infected with SMgfp or SMin79 at an MOI of 2 and collected at 36 hpi. Cell-free virions or infected cells were suspended in a low-melting-point agarose block, lysed, digested with PacI, and subjected to pulsed-field gel electrophoresis. The gel was then transferred to a membrane and hybridized with a 32P-labeled probe specific to the entire SMgfp BAC sequence. The positions of wells, high-molecular-weight viral DNA (HMW DNA), 232-kb monomer viral DNA, and prominent digested viral DNA fragments are indicated. Shown is a representative result from two reproducible, independent experiments. The top panel shows the schematic diagram of monomer and concatemer viral DNA with PacI sites (P) indicated.
To probe if its overall structure was altered in the absence of pM79, viral DNA from infected cells was analyzed by pulsed-field gel electrophoresis (PFGE) (Fig. 4B). SMgfp-infected cells produced both concatemeric replicating viral DNA, which was retained in the well, and cleaved 232-kb monomeric viral genome, which migrated into the gel (Fig. 4B, lane 2). In addition, a minor population of viral DNA with an apparent molecular size greater than 232 kb also migrated into the gel, likely representing polymeric DNA intermediates. In contrast, SMin79-infected cells produced only concatemeric viral DNA and polymeric DNA intermediates without any monomeric viral genomes (Fig. 4B, lane 3). This suggests either a failure of the mutant virus to cleave replicating viral DNA into mature viral genomes or altered viral DNA structures that prevent monomeric viral genomes from migrating into the gel. To differentiate these possibilities, intracellular viral DNA was digested with PacI, a restriction enzyme that cuts the MCMV genome four times. As expected, PacI cut the 232-kb monomeric virion genome (Fig. 4B, lane 7) into linear fragments of 92.8 kb, 90.3 kb (which comigrated together), 43.0 kb, 1.3 kb, and 4.2 kb (the last two run off the gel) (lane 8). Furthermore, PacI-digested intracellular DNA from SMgfp-infected cells produced an additional 135.8-kb linear fragment that resulted from joint ends within concatemeric viral DNA (Fig. 4B, lane 5). PacI digestion of SMin79-infected cells released the same fragments of 90.3 kb and 135.8 kb from concatemeric DNA but did not produce the monomer-derived fragments (lane 6). Therefore, the overall structure of replicating viral DNA produced by pM79-deficient virus was not appreciably different from that of wild-type virus. Together, our data indicate that loss of M79 does not have a deleterious effect on viral genomic amplification.
We also tested if pM79 was involved in the development of replication compartments, virus-induced nuclear structures critical to successful viral DNA replication (44). We infected cells with wild-type or M79-deficient virus and monitored the formation and maturation of replication compartments marked by pM44 staining. pM44 staining became evident at 12 hpi indicating the formation of multiple small nuclear foci (i.e., prereplicative compartments), and these foci coalesced into larger and fewer mature replication compartments at 24 hpi (Fig. 5). Importantly, no appreciable difference was observed in this temporal progression of replication compartments between infections of SMgfp and SMin79 virus (Fig. 5). Therefore, pM79 does not appear to play a role in the development of replication compartments.
Fig 5.
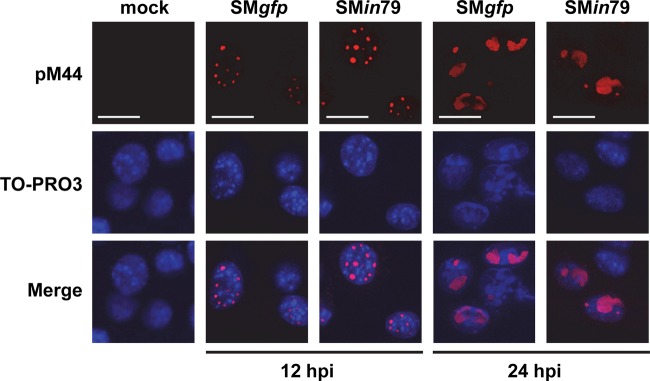
pM79 is not required for the maturation of viral replication compartments. MEF10.1 cells were infected with SMgfp or SMin79 virus at an MOI of 2. At 12 and 24 hpi, cells were fixed with methanol and stained with antibody to pM44 to mark replication compartments (red). Cells were also counterstained with TO-PRO3 to visualize the nuclei (blue). Scale bar, 20 μm. Shown is a representative result from three reproducible, independent experiments.
These data taken together suggest that the defect seen in SMin79 infection occurs downstream of viral DNA synthesis. Moreover, the failure of mutant virus to process replicating viral DNA into monomeric genomes suggests that the defect occurs at or prior to genome cleavage and packaging, such as the step of late gene expression or capsid assembly.
M79-deficient virus is defective in the efficient accumulation of representative late viral gene products.
To continue to define the stage of the viral replication cycle where pM79 acts, we analyzed the accumulation of representative viral proteins from each kinetic class: immediate-early protein IE1, early protein E1 (M112/113), and late protein gB (M55). M79-deficient virus demonstrated two defects in viral protein accumulation (Fig. 6A). One was a modest delay in IE1 and E1 protein accumulation, whereas the other was the complete loss of gB. As this modest delay in IE1 and E1 had no deleterious consequence on viral DNA synthesis (Fig. 4), it was unlikely responsible for defects in events beyond viral DNA synthesis, such as late protein accumulation and ultimately virus growth. The delay in IE1 and E1 expression might be due to the characteristics of the mutant viral stocks, such as tegumentation or virion composition, which could be slightly different from wild-type stocks because of the efficiency of complementation. To rule out any effect of IE1 and E1 expression, we infected cells with SMin79 at an MOI of 5 and SMgfp at an MOI of 1 to compensate for IE1 and E1 expression levels. Despite elevated IE1 and E1 accumulation in SMin79 infection under this condition, gB remained absent even after 48 hpi (Fig. 6B). We interpreted this result to indicate that the major defect of SMin79 was the inability to express viral gene products at late times of infection.
Fig 6.

pM79 is required for efficient expression of a representative viral late gene. (A) Accumulation of representative viral proteins during pM79-deficient virus infection. MEF10.1 cells were infected with SMgfp or SMin79 at an MOI of 2, and the accumulation of viral proteins IE1, E1, and gB at indicated times was analyzed by immunoblotting. Shown is a representative result from five reproducible, independent experiments. (B) Failure of gB accumulation during pM79-deficient virus infection cannot be rescued by elevating expression of viral early genes. Cells were infected with SMgfp or SMin79 at an MOI of 1 or 5, respectively, so that SMin79-infected cells expressed E1 protein at levels comparable to those in SMgfp-infected cells. Cell lysates were collected at indicated times and analyzed by immunoblotting. Shown is a representative result from three reproducible, independent experiments. (C) Accumulation of viral transcripts during pM79-deficient virus infection. Cells were infected with SMgfp in the presence or absence of 200 μg/ml PAA or with SMin79 as described for panel A. Total RNA was isolated at indicated times, and amounts of viral E1 and M55 transcript were measured by RT-qPCR with the primers listed in Table 1. Shown is a representative result from at least three reproducible, independent experiments. The values were normalized to that of actin, and normalized values of viral transcript during SMgfp infection at 10 hpi in the absence of PAA were set to 1.
To test if this defect was at the transcriptional level, we analyzed the accumulation of E1 and M55 transcripts during wild-type and M79 mutant virus infection. E1 transcript accumulated at comparable levels during SMin79 and SMgfp infection, particularly at early times (10 hpi) (Fig. 6C). In contrast, accumulation of M55 transcript in SMin79-infected cells was effectively reduced to levels comparable to those under PAA treatment. All transcripts detected were specific and were not the result of genomic DNA contamination as mock cells and reactions done in the absence of reverse transcriptase failed to produce any products (data not shown). This result supports the hypothesis that both pM79 and viral DNA synthesis are required for efficient viral late transcription.
pM79 regulates the accumulation of a subset of viral late transcripts.
The failure of SMin79 to express a representative late transcript could be due to a global downregulation of MCMV late transcription, or it could be due to a downregulation of only a subset of late transcripts. To differentiate these two possibilities, we profiled the entire MCMV transcriptome with or without pM79. We designed a high-density oligonucleotide tiled array with probes to both the forward and reverse strands of the MCMV genome, allowing us to measure transcription across the entire viral genome. We first determined viral regions where transcription was dependent on viral DNA synthesis. To test this, MEF10.1 cells were infected with MCMV in the presence or absence of PAA. At 20 hpi, RNA was harvested, converted to fluorescently labeled cDNA, and hybridized to the MCMV DNA array. The normalized mean intensity of probes across the genome was plotted as shown in Fig. 7. The sensitivity of viral transcription to PAA was variable across the genome, ranging from no change to a greater than 100-fold reduction. Although most viral regions exhibited some sensitivity to PAA, many regions that were highly regulated did not correspond to annotated ORFs. To minimize false positives, we considered a region of transcription to be dependent on viral DNA synthesis only if the intensity of the PAA-treated sample was at least 3-fold lower than that of the untreated sample in this analysis. By this criterion, among viral regions corresponding to the 172 annotated ORFs examined, efficient transcription of 115 ORFs was dependent on viral DNA synthesis (Table 2). Among them, 80 ORFs have been previously analyzed for their temporal expression, and 61 have been found to be expressed at elevated levels at late stages of MCMV infection (i.e., late genes) (45, 46). Therefore, transcripts of a large set (76%) of annotated ORFs sensitive to PAA are also known to be highly expressed at late times, consistent with the notion that most viral DNA synthesis-dependent transcripts are derived from late genes.
Fig 7.

Tiled array analysis of genome-wide transcription during MCMV infection. MEF10.1 cells were infected with SMgfp in the presence or absence of PAA or with SMin79 at an MOI of 2. Total RNA was isolated at 20 hpi and reverse transcribed, and labeled cDNAs were hybridized to an oligonucleotide tiled array of the MCMV genome. The mean fluorescence of probes overlapping each nucleotide position is plotted on a log2 scale underneath the annotated viral genomic sequence. Blue or red arrows represent annotated open reading frames on the positive or negative strand of the viral genome, respectively. The colored lines represent the transcriptional signals from SMgfp infection (black), SMgfp infection with PAA (red), and SMin79 infection (blue). These probe intensities were compared on a nucleotide-by-nucleotide basis between SMgfp infections with and without PAA treatment or between infections of SMgfp and SMin79. Regions in which the fluorescence intensity was reduced by greater than 3-fold by PAA treatment (PAA) or mutation of M79 (in79) are plotted on a linear scale as fold reduction below the transcriptional intensity plots. Genomic sequences are as follows: 1 to 80 kb (A), 80 to 160 kb (B), and 160 to 230 kb (C). Shown is a representative result from two reproducible, independent experiments.
Table 2.
M79-dependent expression of MCMV ORFs
| ORF | Stranda | Downregulation by:b |
Kineticsc | |
|---|---|---|---|---|
| PAA treatment | M79 mutation | |||
| m011 | Y | Y | L | |
| m012 | Y | Y | L | |
| m014 | Y | Y | L | |
| M032 | C | Y | Y | L |
| M035 | Y | Y | L | |
| m039 | C | Y | Y | L |
| m040 | C | Y | Y | ND |
| M046 | C | Y | Y | |
| M053 | Y | Y | L | |
| M055 | C | Y | Y | L |
| M056 | C | Y | Y | L |
| M072 | C | Y | Y | L |
| M073 | Y | Y | L | |
| M074 | C | Y | Y | L |
| M075 | C | Y | Y | L |
| M076 | Y | Y | ND | |
| M077 | Y | Y | L | |
| M080 | Y | Y | L | |
| M084 | C | Y | Y | ND |
| M085 | C | Y | Y | L |
| M086 | C | Y | Y | L |
| M096 | Y | Y | L | |
| M100 | C | Y | Y | L |
| M104 | C | Y | Y | L |
| M114 | C | Y | Y | L |
| M115 | C | Y | Y | L |
| M116 | C | Y | Y | L |
| M118 | C | Y | Y | L |
| m119.1 | C | Y | Y | L |
| m131 | C | Y | Y | L |
| m155 | C | Y | Y | L |
| m156 | C | Y | Y | L |
| m157 | C | Y | Y | ND |
| m158 | C | Y | Y | ND |
| m159 | C | Y | Y | L |
| m160 | C | Y | Y | L |
| m161 | C | Y | Y | L |
| m162 | C | Y | Y | L |
| m163 | C | Y | Y | |
| m165 | C | Y | Y | |
| m168 | Y | Y | L | |
| m164 | C | N | Y | |
| m166 | C | N | Y | |
| m001 | C | Y | N | ND |
| m002 | Y | N | ND | |
| m003 | Y | N | ||
| m007 | Y | N | L | |
| m015 | Y | N | L | |
| m016 | Y | N | L | |
| m018 | C | Y | N | |
| m019 | Y | N | ND | |
| m021 | Y | N | ND | |
| m022 | Y | N | ND | |
| M023 | C | Y | N | |
| m023.1 | Y | N | ND | |
| M024 | C | Y | N | ND |
| M025 | Y | N | L | |
| m025.1 | C | Y | N | L |
| m025.2 | C | Y | N | L |
| m025.3 | C | Y | N | ND |
| m025.4 | C | Y | N | ND |
| M026 | C | Y | N | |
| M027 | C | Y | N | |
| m029 | Y | N | ||
| m030 | Y | N | ||
| M031 | Y | N | ||
| M031b | Y | N | ND | |
| M033 | Y | N | ND | |
| M044.1 | Y | N | ND | |
| M044.3 | Y | N | ND | |
| m045.2 | C | Y | N | ND |
| M050 | C | Y | N | L |
| M052 | Y | N | L | |
| m069.1 | Y | N | ND | |
| M071 | Y | N | L | |
| M073x2d | Y | N | ND | |
| M082 | C | Y | N | L |
| M083 | C | Y | N | L |
| M087 | Y | N | ||
| M088 | Y | N | ||
| M089x1 | C | Y | N | L |
| m090 | C | Y | N | L |
| M091 | Y | N | ||
| M092 | Y | N | ||
| M093 | Y | N | L | |
| M094 | Y | N | L | |
| M095 | Y | N | L | |
| M097 | Y | N | ||
| M098 | Y | N | ||
| M099 | Y | N | L | |
| M103 | C | Y | N | L |
| m106 | C | Y | N | L |
| m106.1 | C | Y | N | ND |
| m106.3 | C | Y | N | ND |
| m107 | Y | N | L | |
| m108 | C | Y | N | L |
| m117 | C | Y | N | ND |
| m117.1 | Y | N | ND | |
| m119.5 | Y | N | L | |
| M121 | C | Y | N | L |
| m123x2 | C | Y | N | ND |
| m124 | Y | N | L | |
| m124.1 | C | Y | N | ND |
| m125 | Y | N | ND | |
| m126 | Y | N | ND | |
| m127 | C | Y | N | ND |
| m128x3 | Y | N | ||
| m129 | C | Y | N | L |
| m130 | Y | N | ND | |
| m134 | Y | N | ND | |
| m136 | C | Y | N | ND |
| m137 | C | Y | N | |
| m144 | C | Y | N | ND |
| m148 | Y | N | L | |
| m149 | Y | N | ND | |
| m167 | C | Y | N | L |
| m170 | C | Y | N | L |
C, complementary strand; blank entries, forward strand.
The mean intensity of a transcriptional region was (Y) or was not (N) reduced by at least 3-fold in array analysis.
Transcription of an ORF is considered to be late kinetics (L) when it is elevated by at least 50% from 12 to 24 hpi, as described by Marcinowski and coworkers (46), or absent at 6.5 hpi but detectable at 24 hpi, as described by Lacaze and coworkers (45). Blank entries, early kinetics. ND, kinetics class has not been previously described.
x, exon.
We then examined the transcriptome profile in SMin79 infection. Overall, the effect of pM79 mutation on viral transcription was less pronounced than that of PAA (Fig. 7). Compared to PAA treatment, fewer regions of transcription were reduced by greater than 3-fold in the absence of pM79 in array analysis. Interestingly, many unannotated regions of RNA expression were affected by PAA but not by pM79 mutation. Transcription from regions corresponding to 43 ORFs was reduced by greater than 3-fold in the absence of pM79 by array analysis, and 41 of them (95%) were also reduced by PAA treatment (Table 2). Transcription from the regions corresponding to the remaining 74 PAA-sensitive ORFs was less affected by pM79 mutation, based on the criterion used in our analysis. These results suggest that at least a subset of DNA synthesis-dependent viral transcripts also have a high dependence on pM79 for their expression.
This differentiated dependency of late transcripts on pM79 was validated by RT-qPCR analysis. Transcription of M74 and M116, which showed a greater dependency on pM79 in array analysis (Table 2), was reduced by 9.8- and 37.7-fold by pM79 mutation in qPCR analysis (Fig. 8A). Conversely, transcription of M25 and M121 was reduced by only 5.5 and 4.5-fold, respectively, in pM79 mutant virus infection (Fig. 8B), consistent with the result of array analysis (Table 2) and thus showing relatively less dependency on pM79.
Fig 8.
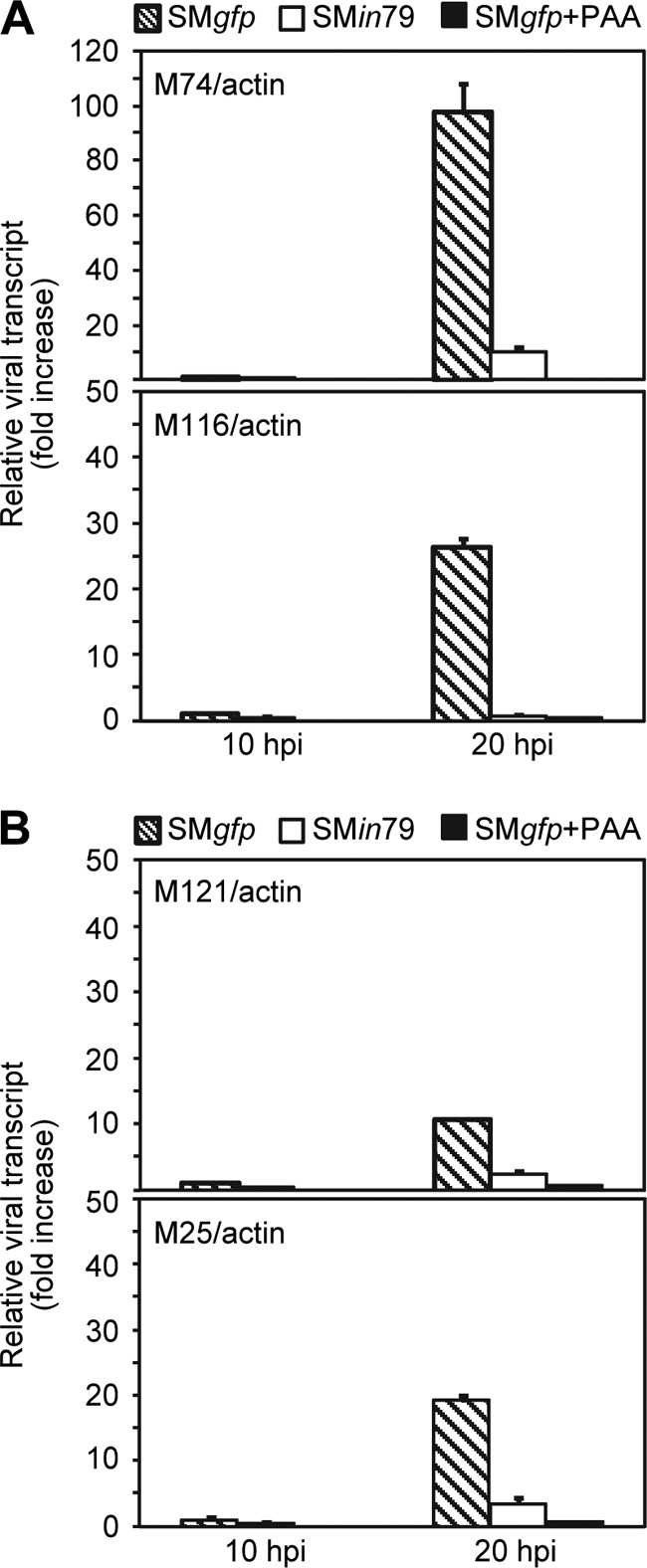
RT-qPCR analysis of representative PAA-sensitive transcripts in MCMV infection. MEF10.1 cells were infected with SMgfp in the presence or absence of 200 μg/ml PAA or with SMin79 at an MOI of 2. Total RNA was isolated at the indicated times, and amounts of indicated late transcripts were measured by RT-qPCR with the primers listed in Table 1. The values were normalized to the value for actin, and normalized values of viral transcript during SMgfp infection at 10 hpi in the absence of PAA were set to 1.
Together, our results show that M79 is critical for the accumulation of at least a subset of DNA synthesis-dependent viral late products (Fig. 8). These results, along with the observations of UL79 in HCMV and ORF18 in MHV-68 (24–26), underscore a conserved function of the UL79 family of genes in beta- and gammaherpesvirus replication cycles.
DISCUSSION
MCMV is the commonly used model virus for HCMV, so revealing HCMV genes that are functionally conserved in MCMV will allow the use of the robust mouse genetic system to elucidate their role and test novel antivirals targeting these products. Previously, we have found that the HCMV gene UL79 regulates viral late gene expression (26). Here, we characterized the function of its MCMV sequence homologue, M79, by analyzing mutant MCMV virus in which pM79 expression was disrupted. pM79 accumulated with early-late kinetics in nuclear viral replication compartments and, if disrupted, abrogated the ability of MCMV to replicate. In particular, we showed that pM79 was critical for MCMV to promote expression of a set of late transcripts. Moreover, not only was MCMV DNA synthesized efficiently in the absence of pM79, but PFGE analysis also showed that the overall structure of replicating viral DNA was unimpaired. The development of viral replication compartments during mutant virus infection was indistinguishable from that during wild-type virus infection. This body of evidence further excludes the involvement of pM79 in viral DNA synthesis or other events preceding viral late transcription. Furthermore, the failure of pM79-deficient virus to cleave viral concatameric replicating DNA is consistent with its defect in late viral transcription. For example, genes required for genomic cleavage (e.g., M56 and M104) or capsid assembly (e.g., M80, M85, and M86) are downregulated during SMin79 infection (Table 2). Our work, together with reports of the role of the pM79 homologues, including pUL79 in HCMV and ORF18 in MHV-68 (24–26), suggests a common mechanism governing late transcription among beta- and gammaherpesviruses. How viral late transcription is regulated remains largely unknown, and viral/host factors involved are poorly defined. The pM79/pUL79 protein family represents an invaluable tool to gain insight into this key viral process.
Our oligonucleotide tiled array analysis has identified a set of annotated genes whose transcription is substantially reduced at late stages of virus infection when pM79 is abrogated (Fig. 7). Comparative analysis of viral transcriptomes among MCMV infections with PAA, without PAA, or in the absence of pM79 leads to two interesting observations. First, while viral DNA synthesis is required for transcription from genomic regions containing many previously reported late genes, it also facilitates the continued transcription from genomic regions containing several previously reported early genes at late times of infection. Second, viral transcripts that are dependent on viral DNA synthesis also have a dependency on pM79 for their accumulation. There seems to be a striation in dependence, such that without pM79 some transcripts are markedly reduced, whereas others are reduced to a much less extent. Thus, pM79 is a key viral regulator of late transcription in MCMV infection. In future studies, similar transcriptome analysis should be applied to other viral genes known or predicted to be regulators of late gene expression. Examples are HCMV UL79 and MHV-68 ORF18, as well as MCMV M87 and M95 (homologues of HCMV UL87 and UL95). Such analysis will reveal whether these viral regulators control an overlapping or distinct set of viral late gene expression.
How does pM79 regulate expression of late transcripts? A late transcript regulator could be involved in epigenetic regulation of replicating viral DNA. For instance, viral DNA-associated histones are modified when herpesviruses replicate their genomes, and this may render late promoters accessible for transcription (47–49). However, the pM79 coding sequence does not resemble those of histone-modifying enzymes, such as histone acetyltransferase or histone deacetylase (data not shown). If pM79 had a role in epigenetic regulation of gene expression, it would likely act indirectly, for instance, by recruiting modification enzymes to histones associated with late viral promoters. Late regulators may also act as transcription factors. However, pM79 does not contain any identifiable putative DNA binding domains (data not shown). Therefore, in this capacity pM79 would have to act as a modulator of cellular and viral transcriptional regulators or RNA polymerase to facilitate viral late gene transcription. Finally, it is tempting to speculate that regulation of late gene expression may function as a switch to decide a lytic or latent viral infection. Cellular or viral repressors may associate with viral late promoters by default to keep them silent, but viral regulators such as pM79 could displace these repressors to favor a productive, lytic infection. Work is under way to identify viral and cellular binding partners of pM79 in order to reveal the mechanism of its activity.
The identification of pM79 as a protein essential for MCMV late transcription indicates that it is a functional homologue of HCMV pUL79. MCMV infection in mice is an important model for preclinical evaluation of antiviral compounds, most of which have been directed at highly conserved viral DNA replication proteins. The pM79/pUL79 protein family presents an attractive, alternative target for therapeutic intervention in CMV disease. The functional conservation between pM79 and pUL79 justifies MCMV as a credible model to test this novel antiviral strategy in vivo.
ACKNOWLEDGMENTS
We thank Herbert Virgin and the members of his laboratory for helpful discussions and invaluable advice, Ulrich Koszinowski (Max von Pettenkofer Institute, Ludwig Maximilian University, Germany), Martin Messerle (Hannover Medical School, Hannover, Germany), and Wolfram Brune (Heinrich Pette Institute, Leibniz Institute for Experimental Virology, Germany) for the MCMV BAC clone pSM3fr, Anthony Scalzo (University of Western Australia) for M44 and gB antibodies, Stipan Jonjic (University of Rijeka, Croatia) for IE1 and E1 antibodies, Jian Gao (Washington University) for assistance in array analysis, and members of the Yu lab for critical readings of the manuscript.
We thank the Genome Technology Access Center in the Department of Genetics at Washington University School of Medicine for help with genomic analysis. The Center is partially supported by NCI Cancer Center Support grant P30 CA91842 to the Siteman Cancer Center and by ICTS/CTSA grant UL1RR024992 from the National Center for Research Resources (NCRR), a component of the National Institutes of Health (NIH), and NIH Roadmap for Medical Research.
This study was supported by Public Health Service grants (RO1CA120768 and RO1AI51345). D.Y. holds an Investigators in the Pathogenesis of Infectious Disease award from the Burroughs Wellcome Fund, and W.M.Y. is an investigator of the Howard Hughes Medical Institute.
This publication is solely the responsibility of the authors and does not necessarily represent the official view of NCRR or NIH.
Footnotes
Published ahead of print 12 June 2013
REFERENCES
- 1. Britt WJ, Alford CA. 1996. Cytomegalovirus, p 2493–2523 In Fields BN, Knipe DM, Howley PM. (ed), Fields virology, 3rd ed Lippincott-Raven, Philadelphia, PA [Google Scholar]
- 2. Crough T, Khanna R. 2009. Immunobiology of human cytomegalovirus: from bench to bedside. Clin. Microbiol. Rev. 22:76–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fowler KB, Stagno S, Pass RF, Britt WJ, Boll TJ, Alford CA. 1992. The outcome of congenital cytomegalovirus infection in relation to maternal antibody status. N. Engl. J. Med. 326:663–667 [DOI] [PubMed] [Google Scholar]
- 4. Selik RM, Chu SY, Ward JW. 1995. Trends in infectious diseases and cancers among persons dying of HIV infection in the United States from 1987 to 1992. Ann. Intern. Med. 123:933–936 [DOI] [PubMed] [Google Scholar]
- 5. Reddehase MJ, Simon CO, Seckert CK, Lemmermann N, Grzimek NK. 2008. Murine model of cytomegalovirus latency and reactivation. Curr. Top. Microbiol. Immunol. 325:315–331 [DOI] [PubMed] [Google Scholar]
- 6. Scalzo AA, Corbett AJ, Rawlinson WD, Scott GM, Degli-Esposti MA. 2007. The interplay between host and viral factors in shaping the outcome of cytomegalovirus infection. Immunol. Cell Biol. 85:46–54 [DOI] [PubMed] [Google Scholar]
- 7. Rawlinson WD, Farrell HE, Barrell BG. 1996. Analysis of the complete DNA sequence of murine cytomegalovirus. J. Virol. 70:8833–8849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Johnson PA, Everett RD. 1986. DNA replication is required for abundant expression of a plasmid-borne late US11 gene of herpes simplex virus type 1. Nucleic Acids Res. 14:3609–3625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mavromara-Nazos P, Roizman B. 1987. Activation of herpes simplex virus 1 gamma 2 genes by viral DNA replication. Virology 161:593–598 [DOI] [PubMed] [Google Scholar]
- 10. Mocarski ES, Shenk T, Pass RF. 2007. Cytomegaloviruses, p 2701–2772 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed, vol 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 11. Chen J, Silverstein S. 1992. Herpes simplex viruses with mutations in the gene encoding ICP0 are defective in gene expression. J. Virol. 66:2916–2927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Poffenberger KL, Raichlen PE, Herman RC. 1993. In vitro characterization of a herpes simplex virus type 1 ICP22 deletion mutant. Virus Genes 7:171–186 [DOI] [PubMed] [Google Scholar]
- 13. Rice SA, Knipe DM. 1990. Genetic evidence for two distinct transactivation functions of the herpes simplex virus alpha protein ICP27. J. Virol. 64:1704–1715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Smith CA, Bates P, Rivera-Gonzalez R, Gu B, DeLuca NA. 1993. ICP4, the major transcriptional regulatory protein of herpes simplex virus type 1, forms a tripartite complex with TATA-binding protein and TFIIB. J. Virol. 67:4676–4687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zabierowski S, DeLuca NA. 2004. Differential cellular requirements for activation of herpes simplex virus type 1 early (tk) and late (gC) promoters by ICP4. J. Virol. 78:6162–6170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhu Z, Schaffer PA. 1995. Intracellular localization of the herpes simplex virus type 1 major transcriptional regulatory protein, ICP4, is affected by ICP27. J. Virol. 69:49–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Depto AS, Stenberg RM. 1992. Functional analysis of the true late human cytomegalovirus pp28 upstream promoter: cis-acting elements and viral trans-acting proteins necessary for promoter activation. J. Virol. 66:3241–3246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Johnson PA, Everett RD. 1986. The control of herpes simplex virus type-1 late gene transcription: a “TATA-box”/cap site region is sufficient for fully efficient regulated activity. Nucleic Acids Res. 14:8247–8264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. McWatters BJ, Stenberg RM, Kerry JA. 2002. Characterization of the human cytomegalovirus UL75 (glycoprotein H) late gene promoter. Virology 303:309–316 [DOI] [PubMed] [Google Scholar]
- 20. Alba MM, Das R, Orengo CA, Kellam P. 2001. Genomewide function conservation and phylogeny in the Herpesviridae. Genome Res. 11:43–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Isomura H, Stinski MF, Kudoh A, Murata T, Nakayama S, Sato Y, Iwahori S, Tsurumi T. 2008. Noncanonical TATA sequence in the UL44 late promoter of human cytomegalovirus is required for the accumulation of late viral transcripts. J. Virol. 82:1638–1646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leach FS, Mocarski ES. 1989. Regulation of cytomegalovirus late-gene expression: differential use of three start sites in the transcriptional activation of ICP36 gene expression. J. Virol. 63:1783–1791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Serio TR, Cahill N, Prout ME, Miller G. 1998. A functionally distinct TATA box required for late progression through the Epstein-Barr virus life cycle. J. Virol. 72:8338–8343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Arumugaswami V, Wu TT, Martinez-Guzman D, Jia Q, Deng H, Reyes N, Sun R. 2006. ORF18 is a transfactor that is essential for late gene transcription of a gammaherpesvirus. J. Virol. 80:9730–9740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Isomura H, Stinski MF, Murata T, Yamashita Y, Kanda T, Toyokuni S, Tsurumi T. 2011. The human cytomegalovirus gene products essential for late viral gene expression assemble into prereplication complexes before viral DNA replication. J. Virol. 85:6629–6644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Perng YC, Qian Z, Fehr AR, Xuan B, Yu D. 2011. The human cytomegalovirus gene UL79 is required for the accumulation of late viral transcripts. J. Virol. 85:4841–4852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kinsella TM, Nolan GP. 1996. Episomal vectors rapidly and stably produce high-titer recombinant retrovirus. Hum. Gene Ther. 7:1405–1413 [DOI] [PubMed] [Google Scholar]
- 28. Baird GS, Zacharias DA, Tsien RY. 2000. Biochemistry, mutagenesis, and oligomerization of DsRed, a red fluorescent protein from coral. Proc. Natl. Acad. Sci. U. S. A. 97:11984–11989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 33:e36. 10.1093/nar/gni035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Paredes AM, Yu D. 2012. Human cytomegalovirus: bacterial artificial chromosome (BAC) cloning and genetic manipulation. Curr. Protoc Microbiol. Chapter 14:Unit 14E.14. 10.1002/9780471729259.mc14e04s24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Harvey DM, Levine AJ. 1991. p53 alteration is a common event in the spontaneous immortalization of primary BALB/c murine embryo fibroblasts. Genes Dev. 5:2375–2385 [DOI] [PubMed] [Google Scholar]
- 32. Wagner M, Jonjic S, Koszinowski UH, Messerle M. 1999. Systematic excision of vector sequences from the BAC-cloned herpesvirus genome during virus reconstitution. J. Virol. 73:7056–7060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Busche A, Angulo A, Kay-Jackson P, Ghazal P, Messerle M. 2008. Phenotypes of major immediate-early gene mutants of mouse cytomegalovirus. Med. Microbiol. Immunol. 197:233–240 [DOI] [PubMed] [Google Scholar]
- 34. Cardin RD, Abenes GB, Stoddart CA, Mocarski ES. 1995. Murine cytomegalovirus IE2, an activator of gene expression, is dispensable for growth and latency in mice. Virology 209:236–241 [DOI] [PubMed] [Google Scholar]
- 35. Manning WC, Mocarski ES. 1988. Insertional mutagenesis of the murine cytomegalovirus genome: one prominent alpha gene (ie2) is dispensable for growth. Virology 167:477–484 [PubMed] [Google Scholar]
- 36. Xuan B, Qian Z, Torigoi E, Yu D. 2009. Human cytomegalovirus protein pUL38 induces ATF4 expression, inhibits persistent JNK phosphorylation, and suppresses endoplasmic reticulum stress-induced cell death. J. Virol. 83:3463–3474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wheat RL, Clark PY, Brown MG. 2003. Quantitative measurement of infectious murine cytomegalovirus genomes in real-time PCR. J. Virol. Methods 112:107–113 [DOI] [PubMed] [Google Scholar]
- 38. Cheng TP, Valentine MC, Gao J, Pingel JT, Yokoyama WM. 2010. Stability of murine cytomegalovirus genome after in vitro and in vivo passage. J. Virol. 84:2623–2628 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Stein LD, Mungall C, Shu S, Caudy M, Mangone M, Day A, Nickerson E, Stajich JE, Harris TW, Arva A, Lewis S. 2002. The generic genome browser: a building block for a model organism system database. Genome Res. 12:1599–1610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Qian Z, Xuan B, Hong TT, Yu D. 2008. The full-length protein encoded by human cytomegalovirus gene UL117 is required for the proper maturation of viral replication compartments. J. Virol. 82:3452–3465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Bai Y, Trang P, Li H, Kim K, Zhou T, Liu F. 2008. Effective inhibition in animals of viral pathogenesis by a ribozyme derived from RNase P catalytic RNA. Proc. Natl. Acad. Sci. U. S. A. 105:10919–10924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Loutsch JM, Galvin NJ, Bryant ML, Holwerda BC. 1994. Cloning and sequence analysis of murine cytomegalovirus protease and capsid assembly protein genes. Biochem. Biophys. Res. Commun. 203:472–478 [DOI] [PubMed] [Google Scholar]
- 43. Yu D, Silva MC, Shenk T. 2003. Functional map of human cytomegalovirus AD169 defined by global mutational analysis. Proc. Natl. Acad. Sci. U. S. A. 100:12396–12401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Penfold ME, Mocarski ES. 1997. Formation of cytomegalovirus DNA replication compartments defined by localization of viral proteins and DNA synthesis. Virology 239:46–61 [DOI] [PubMed] [Google Scholar]
- 45. Lacaze P, Forster T, Ross A, Kerr LE, Salvo-Chirnside E, Lisnic VJ, Lopez-Campos GH, Garcia-Ramirez JJ, Messerle M, Trgovcich J, Angulo A, Ghazal P. 2011. Temporal profiling of the coding and noncoding murine cytomegalovirus transcriptomes. J. Virol. 85:6065–6076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Marcinowski L, Lidschreiber M, Windhager L, Rieder M, Bosse JB, Radle B, Bonfert T, Gyory I, de Graaf M, Prazeres da Costa O, Rosenstiel P, Friedel CC, Zimmer R, Ruzsics Z, Dolken L. 2012. Real-time transcriptional profiling of cellular and viral gene expression during lytic cytomegalovirus infection. PLoS Pathog. 8:e1002908. 10.1371/journal.ppat.1002908 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Liang Y, Vogel JL, Narayanan A, Peng H, Kristie TM. 2009. Inhibition of the histone demethylase LSD1 blocks alpha-herpesvirus lytic replication and reactivation from latency. Nat. Med. 15:1312–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lieberman PM. 2008. Chromatin organization and virus gene expression. J. Cell. Physiol. 216:295–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sinclair J. 2010. Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection. Biochim. Biophys. Acta 1799:286–295 [DOI] [PubMed] [Google Scholar]