

Abstract
The coronavirus nucleocapsid protein (N) plays an essential structural role in virions through a network of interactions with positive-strand viral genomic RNA, the envelope membrane protein (M), and other N molecules. Additionally, N protein participates in at least one stage of the complex mechanism of coronavirus RNA synthesis. We previously uncovered an unanticipated interaction between N and the largest subunit of the viral replicase-transcriptase complex, nonstructural protein 3 (nsp3). This was found through analysis of revertants of a severely defective mutant of murine hepatitis virus (MHV) in which the N gene was replaced with that of its close relative, bovine coronavirus (BCoV). In the work reported here, we constructed BCoV chimeras and other mutants of MHV nsp3 and obtained complementary genetic evidence for its association with N protein. We found that the N-nsp3 interaction maps to the amino-terminal ubiquitin-like domain of nsp3, which is essential for the virus. The interaction does not require the adjacent acidic domain of nsp3, which is dispensable. In addition, we demonstrated a complete correspondence between N-nsp3 genetic interactions and the ability of N protein to enhance the infectivity of transfected coronavirus genomic RNA. The latter function of N was shown to depend on both of the RNA-binding domains of N, as well as on the serine- and arginine-rich central region of N, which binds nsp3. Our results support a model in which the N-nsp3 interaction serves to tether the genome to the newly translated replicase-transcriptase complex at a very early stage of infection.
INTRODUCTION
Coronaviruses are enveloped, positive-strand RNA viruses that infect a wide range of mammalian and avian species and mainly cause respiratory or enteric diseases (1, 2). The collection of known human coronaviruses (HCoVs), all of which are responsible for respiratory infections, has grown steadily since the emergence of the severe acute respiratory syndrome coronavirus (SARS-CoV) in 2002. The sixth pathogen to be added to this list, Middle East respiratory syndrome coronavirus (MERS-CoV), very recently emerged from an as-yet-unknown animal reservoir and is associated with a disturbingly high fatality rate (3, 4).
The murine coronavirus, mouse hepatitis virus (MHV), has long served as a model for study of both the molecular biology and pathogenesis of members of this viral family. Virions of MHV, like those of all coronaviruses, contain a canonical set of four structural proteins. Only one of these, the nucleocapsid protein (N) is found in the interior of the viral envelope, where it wraps the genomic RNA (gRNA) into a helical structure. Such a symmetry is highly unusual for positive-strand RNA animal viruses, which almost always have icosahedral capsids. The MHV N protein has been divided into multiple domains (Fig. 1), based on both genetic analyses (5–8) and extensive structural studies (references 9 and 10 and references cited in reference 8). The two largest domains, N1b and N2b, fold independently and have RNA-binding activity. N1b and N2b are also widely called the N-terminal domain (NTD) and the C-terminal domain (CTD), respectively; however, the latter nomenclature becomes confusing in descriptions that include the actual terminal domains of the molecule, N1a and N3. Flanking the RNA-binding domains are segments that are intrinsically disordered (11). Among these are the largely dispensable spacer B (5, 8) and the carboxy-terminal domain N3, which mediates binding of the nucleocapsid to the membrane protein (M) of the virion envelope (6, 7, 12, 13). Connecting the two RNA-binding domains is domain N2a, the first half of which is taken up by a serine- and arginine-rich tract designated the SR region.
Fig 1.

Interaction between the coronavirus nucleocapsid (N) protein and the nsp3 subunit of the replicase. A schematic of the genome of MHV is shown near the top; numbering in the replicase polyprotein gene (rep 1a and rep 1b) denotes the mature products nsp1 to nsp16. Beneath the genome are enlargements of the encoded N protein and nsp3 (not drawn to scale with respect to each other). Numbering indicates amino acid residues; the first amino acid of mature nsp3 is residue 833 of the unprocessed replicase polyprotein. The domains of N protein, represented as defined previously (8, 23), include the RNA-binding domains N1b (or NTD) and N2b (or CTD), the M-binding domain N3, and the SR region, which interacts with nsp3. Nomenclature for the domains of nsp3 is mostly that of Neuman and coworkers (45): Ubl, ubiquitin-like domain (27); Ac, acidic region (40); PLP, papain-like protease; ADRP, ADP-ribose-1″-phosphatase; NAB, nucleic acid-binding domain; G2M, coronavirus group 2 marker domain; TM, transmembrane segment (67, 68); Y, coronavirus highly conserved domain (40). The region between ADRP and Ubl2-PLP2 corresponds to the SARS unique domain (SUD) in SARS-CoV nsp3 (71); although the SUD comprises three structural subdomains (72), nothing is known about its highly divergent MHV counterpart. Above the genome are shown the boundaries of the cDNA clones A to G of the reverse genetics system of Yount and coworkers (42) that was used throughout this study.
In addition to its structural role in virion assembly, N protein also appears to be involved in the complex pathway of coronavirus RNA synthesis. However, studies based on replicon systems are not in agreement about the details of this participation. Some work suggests that N protein is required as an RNA chaperone to assist the template-switching steps central to coronavirus subgenomic mRNA transcription (14, 15). Other work indicates that gRNA replication, not transcription, is the process that is principally dependent upon N (16, 17). Either of these alternatives would necessitate that N protein colocalize with constituents of the viral RNA-synthetic compartment, which has been observed in multiple investigations of the early stages of MHV or SARS-CoV infection (18–22).
We recently discovered an unexpected interaction between the N protein and a specific component of the coronavirus replicative machinery (23). This finding initiated with our construction of an MHV mutant containing a substitution of the entire N gene of the closely related bovine coronavirus (BCoV). The severely defective phenotype of this chimeric virus was paradoxical, since domains of N that carry out N-RNA, N-M, or N-N interactions had been shown to be completely interchangeable between BCoV and MHV. Two classes of reverting mutations were found to rescue the BCoV N substitution mutant. The first of these mapped to the SR region of the N protein. The second type of reverting mutation mapped to nonstructural protein 3 (nsp3), a subunit of the MHV replicase-transcriptase complex. A substitution of the SARS-CoV N protein SR region in MHV N was also rescued by compensatory mutations in the SR region and in nsp3.
The coronavirus replicase-transcriptase complex is assembled from 16 nsps that become autoproteolytically processed from two large polyproteins, which are translated from gRNA via a ribosomal frameshift mechanism (1, 2). The largest of the processed replicase products, nsp3, comprises numerous separate modules (Fig. 1). For many of these modules, structures have been solved and enzymatic or RNA-binding activities have been assigned (24–32). However, the only nsp3 domains previously known to have clear functional roles in viral replication are the papain-like proteases (PLPs). Coronavirus PLPs are responsible for the three earliest of the replicase cleavage events, but different schemes have evolved to carry out this processing (33, 34). In some cases, a single PLP suffices for this role; in other cases, there are two active PLPs, which may have either specialized or overlapping assignments. For MHV, PLP1 processes the nsp1-nsp2 and nsp2-nsp3 cleavage sites, whereas PLP2 is specific for the nsp3-nsp4 site (35–39). Our genetic evidence localized the interaction between the N protein SR region and nsp3 to the first of two ubiquitin-like domains (Ubl1) (27), at the amino terminus of nsp3, and the adjacent unstructured acidic region (Ac) (40), just upstream of PLP1. In further support of these results, we showed that expressed Ubl1-Ac protein specifically bound to N protein from infected cells in a glutathione S-transferase (GST) pulldown assay. Very recently, Keane and Giedroc solved the solution structure of MHV Ubl1 and demonstrated, by isothermal titration calorimetry, the high-affinity binding of Ubl1 to an expressed N construct encompassing domain N1b and the SR region (41). In addition, they mapped the principal determinants of this binding to the SR region of N and the acidic α2 helix of Ubl1. As a step toward defining the purpose of the N-nsp3 interaction, we previously established that there was a direct correlation between the in vivo functionality of MHV or BCoV N proteins and their ability or inability, respectively, to stimulate the infectivity of transfected MHV gRNA (23). From our results, we hypothesized that the role of the association of N with nsp3 is to colocalize genomic RNA with the nascent replicase-transcriptase complex at the earliest stages of infection.
In the study presented here, we genetically probed the N-nsp3 interaction from the standpoint of nsp3. Through the construction and analysis of Ubl1 and Ac chimeras and mutants, we found that this interaction maps to the Ubl1 domain and that the Ac domain is dispensable for MHV replication. Moreover, we were able to demonstrate a complete correspondence between all genetic evidence and the requirement for N protein to stimulate the infectivity of transfected gRNA. Our results confirm the critical nature of the N-nsp3 interaction and provide further support for its proposed role in the initiation of coronavirus infection.
MATERIALS AND METHODS
Virus and cells.
Wild-type, mutant, and revertant virus stocks of MHV strain A59 were propagated in mouse 17 clone 1 (17Cl1) cells; plaque titrations and plaque purifications were performed in mouse L2 cells. Growth of both 17Cl1 and L2 monolayers was routinely carried out in Dulbecco's minimal essential medium (MEM) containing 10% fetal bovine serum. Spinner cultures of L2 cells, used for transfection of genomic RNA, were maintained at densities of between 1 × 105 and 2 × 106 cells/ml in Joklik's MEM containing 10% fetal bovine serum.
MHV mutant construction and analysis.
Mutants in this study were engineered with the infectious cDNA system for MHV A59, which was generously provided by Ralph Baric. Assembly of full-length MHV cDNA and in vitro transcription were carried out essentially as described previously (42). Virus was recovered by electroporation transfection of L2 cells grown in suspension with synthetic gRNA augmented with MHV N mRNA from vector pCK70 (see below). Virus isolations were initially performed at 33°C, based on our previous experience that many constructed mutants with impaired viability were also temperature sensitive. For verification of constructed mutants, RNA was isolated from infected cell monolayers with Ultraspec reagent (Biotecx) and reverse transcribed with a random hexanucleotide primer and avian myeloblastosis virus reverse transcriptase (Life Sciences). PCR amplification of cDNA was performed with the Expand high-fidelity PCR system (Roche). Reverse transcription-PCR (RT-PCR) products were isolated from agarose gel electrophoresis with QIAquick spin columns (Qiagen) prior to DNA sequencing.
All reverse genetics procedures reported here involved the manipulation of MHV cDNA clone A or clone G (Fig. 1). At first, to increase the number of available unique restriction sites, the cDNA insert of clone A was subcloned into a pUC8-related vector to produce clone pBar-A3. For the Bcl-Spe(BCoV) mutant, a BclI-SpeI fragment of BCoV cDNA (running from the end of nsp2 through part of nsp3 PLP1) was used to replace the corresponding fragment of pBar-A3; this yielded plasmid pA3B1. BCoV cDNA was generated by PCR amplification of random-primed cDNA produced from a stock of wild-type BCoV that was obtained from David Brian.
For Ubl1(BCoV) substitutions, the BclI-SpeI fragment of pBar-A3 was replaced with BCoV cDNA for Ubl1, which was synthesized by PCR from overlapping oligonucleotides, and a PCR-generated MHV cDNA fragment for the remainder of the interval. The resulting plasmid, pA3B2, contained (i) an exact substitution of the BCoV Ubl1 domain, as defined in Results, (ii) a unique coding-silent XbaI site created at MHV nsp2 codons 575 to 577, near the nsp2-nsp3 boundary, and (iii) a unique coding-silent SalI site created at MHV nsp3 codons 123 to 124, near the Ubl1-Ac boundary. For Ubl1-Ac(BCoV) substitutions, the XbaI-SpeI fragment of pA3B2 was replaced with BCoV cDNA for Ubl1 and Ac, which was synthesized by PCR from random-primed viral cDNA, and a PCR-generated MHV cDNA fragment for the remainder of the interval. The resulting plasmid, pA3B3, contained (i) an exact substitution of the BCoV Ubl1 and Ac domains, as defined in Results, and (ii) a unique coding-silent SacI site created at MHV nsp3 codons 234 to 235, near the downstream Ac boundary.
MHV clone A constructs for the generation of Ubl1 mutants 1 to 3 and the ΔUbl1 mutant were made by replacement of the XbaI-SalI fragment of pA3B2 with fragments synthesized by PCR from overlapping oligonucleotides. Similarly, clone A constructs for the generation of the ΔAc1 and ΔAc2 mutants were made by replacement of the XbaI-SacI fragment of pA3B3 with fragments synthesized by PCR from overlapping oligonucleotides. Finally, plasmid pG1B1, containing an exact substitution of the BCoV N gene for the MHV N gene, was created by the transfer of the EagI-PacI fragment [running from the M gene through the poly(A) region] from pMH54-BCVN (23) to replace the corresponding fragment of MHV clone G. Oligonucleotides for PCR and DNA sequencing were obtained from Integrated DNA Technologies. The overall composition of constructed plasmids was confirmed by restriction analysis, and the DNA sequences of all ligation junctions and all regions resulting from PCR amplification were verified by sequencing.
Viral growth kinetics.
To measure growth kinetics, confluent monolayers of 17Cl1 cells (75 cm2) were inoculated at a multiplicity of either 0.01 or 5.0 PFU per cell for 2 h at 37°C, with rocking every 15 min. Following removal of inocula, monolayers were washed three times with phosphate-buffered saline, and incubation was continued in fresh medium at 37°C. Sample aliquots of medium were withdrawn at various times from 2 to 48 h postinfection, and infectious titers were subsequently determined.
Genomic RNA infectivity assay.
The enhancement of the infectivity of MHV gRNA by N mRNA was measured by cotransfection of both RNA species via electroporation. Wild-type and mutant viruses were purified by ultracentrifugation in glycerol-tartrate gradients, as described in detail previously (43). Virions were disrupted with 10 mM Tris-HCl (pH 7.5), 140 mM NaCl, 1 mM MgCl2, 5 mM EDTA, 0.44% NP-40, and 1% SDS and then extracted twice with phenol-chloroform and precipitated twice with ethanol; viral gRNA was resuspended in water, quantitated by A260, aliquoted, and stored on liquid nitrogen. Capped N mRNAs were synthesized in vitro with an mMessage machine kit (Ambion). Following template removal by treatment with RNase-free DNase (Turbo DNase I; Ambion), mRNA was purified using a spin column (RNeasy MinElute kit; Qiagen). N mRNA integrity was monitored by agarose gel electrophoresis and quantitated by A260. Mixtures of gRNA and mRNA (or water control) were electroporated into 5 × 106 L2 cells, which had been grown in suspension culture (44) and washed and resuspended in calcium- and magnesium-free phosphate-buffered saline. Electroporations were carried out in 0.4-cm cuvettes at room temperature using two consecutive pulses from a Bio-Rad Gene Pulser II apparatus set at 0.3 kV and 975 μF. Transfected cells were then diluted into 1.5 × 107 fresh L2 cells that were suspended in Eagle's MEM containing 10% fetal bovine serum, and the entire mixture of cells was seeded into three 20-cm2 dishes. After 3 h at 37°C to allow for attachment and recovery, cells were overlaid with 0.9% agar in Eagle's MEM with 10% fetal bovine serum, and plaques were counted after 48 h of incubation.
Transcription vectors for MHV N-related mRNAs were generated from pCK70XB, which was derived from the previously described pCK70, a template for an exact copy of wild-type MHV subgenomic RNA7 (44). In pCK70XB, unique coding-silent XbaI and BspEI sites were made at N codons 15 to 16 and 444 to 446, respectively, by PCR mutagenesis. The vectors for the HK-SR and S-SR mutants, pCK-HKSR and pCK-SSR, were constructed by replacement of the ApaI-NgoMIV fragment of pCK70XB with the corresponding fragment of the previously described pMH54-HKSR5 or pPJ3A, respectively (23). Deletion mutant vectors pCKΔN2b, pCKΔNBd3, and pCKΔ15 were created from pCK70XB through religation of blunted ends that were produced subsequent to digestion with the pairs of restriction enzymes NgoMIV and Bpu10I, BstXI and BspEI, or BtgI and NgoMIV, respectively. Vector pCKΔ13 was made by replacement of the ApaI-SpeI fragment of pCK70XB with the corresponding fragment of the previously described pA104Δ13 (8). Deletion mutant vectors pCKΔN1a and pCKΔN1b were constructed by replacement of the XhoI-ApaI or BamHI-Bsu36I fragment of pCK70XB, respectively, with PCR-generated fragments. The transcription vector for wild-type BCoV mRNA, pB60, and its derivative pB60SL, which contains the point mutation S202L, have been described previously (23). The vector for the BCoV M-SR mutant, pB60-MSR, was obtained by replacement of the Bsu36I-SpeI fragment of pB60 with the corresponding fragment of pCK70XB.
RESULTS
Construction of an imprecise nsp3 chimera.
In previous work, we uncovered and began to characterize an interaction between nsp3 and the N protein through the construction and analysis of chimeric N protein mutants and their revertants (23). To further extend our genetic investigation of this interaction, we examined the effects of reciprocal substitutions of the BCoV Ubl1 and Ac domains in MHV. For this purpose we used the reverse genetics system of Yount and coworkers (42), in which MHV gRNA is synthesized from a full-length genomic cDNA that is assembled through the directed in vitro ligation of seven cloned cDNA fragments. Initially, we generated a mutant, designated Bcl-Spe(BCoV), in which was exchanged (at the cDNA level) a BclI-SpeI fragment running from nsp2 through the PLP1 domain of nsp3 (Fig. 2A). In this interval, the MHV and BCoV replicase proteins share 37% amino acid sequence identity, with the highest divergence falling in the Ac domain of nsp3 (see below). In contrast, the BclI and SpeI sites occur in regions of relatively high homology between the two sequences.
Fig 2.

Inexact substitution of the BCoV Ubl1 and Ac domains of nsp3. (A) Schematic of the MHV-BCoV chimeric portion of the replicase polyprotein and the MHV wild-type N protein encoded by the Bcl-Spe(BCoV) mutant. Numbering indicates amino acid residues in each protein; BCoV residue numbers are in bold. Arrows denote the positions of the most common reverting mutations in nsp2 and nsp3. The three active-site catalytic residues are indicated above the PLP1 module (35, 46). Also shown (boxed) is an alignment of the P6 through P4′ positions of the nsp2-nsp3 PLP1 cleavage sites of MHV and BCoV; shading indicates residues that differ between the two sequences. (B) Plaques of the Bcl-Spe(BCoV) mutant at 33, 37, and 39°C compared with those of the wild type. Plaque titrations were carried out on L2 cells; monolayers were stained with neutral red at 72 h postinfection and were photographed 18 h later. In the panels marked by asterisks, monolayers were inoculated with a 100-fold-higher concentration of virus than that in the other plaque titrations shown.
Two independent isolates of the Bcl-Spe(BCoV) mutant were obtained, one of which is shown in Fig. 2B. Both isolates exhibited the same severely impaired growth phenotype compared to an otherwise isogenic wild-type MHV that was constructed by the same technique. The Bcl-Spe(BCoV) mutant formed plaques at 33°C that were noticeably smaller than those of the wild type. At 37°C, plaques of the mutant were tiny, and mutant titers were some 100-fold lower than at 33°C; at 39°C, the mutant was incapable of growth. To explore the basis for this phenotype, we isolated nine revertants that formed much larger and more numerous plaques at 37°C, following serial passaging of the mutant at 37°C and then 39°C. Revertant genomes were sequenced from upstream of the substitution boundary in nsp2 through part of the ADP-ribose-1″-phosphatase (ADRP) domain in nsp3, as well as in the part of the N protein spanning the SR region. No second-site mutations were found in N. However, all revertants had mutations within or near the BCoV substitution, and, with one exception, these fell into two classes (Fig. 2A). First, four revertants contained either F521C or F545C in the MHV portion of nsp2. This suggested to us that the chimeric nsp2 in the Bcl-Spe(BCoV) mutant had an unpaired cysteine that may have created a protein conformation that impeded replicase folding or processing. More significantly, in the BCoV portion of PLP1, the mutation K234T arose in eight of the nine revertants, which collectively represented three independent sets. This strongly implied that in the Bcl-Spe(BCoV) mutant there was an incompatibility between the mostly MHV PLP1 and one of its substrates, the BCoV nsp2-nsp3 cleavage site, which varies from its MHV counterpart at three of 10 positions (Fig. 2A). Although the sequence requirements of MHV PLP1 have been extensively examined, both in vitro (36–38) and in vivo (34, 39), the available data did not allow us to predict the effects of the specific sequence changes introduced by our substitution of the BCoV nsp2-nsp3 cleavage site in place of that of MHV. From these results we concluded that, for the most part, the phenotype of the Bcl-Spe(BCoV) mutant was unrelated to the N-nsp3 interaction. Importantly, this outcome showed us that in order to acquire informative results from the creation of nsp3 chimeras, it would be necessary to carefully regard domain boundaries and proteinase cleavage site preferences.
Construction of exact nsp3 domain chimeras.
We next generated two nsp3 chimeras, designated Ubl1(BCoV) and Ubl1-Ac(BCoV), in which the BCoV Ubl1 domain alone or else both the BCoV Ubl1 and Ac domains were precisely substituted for their MHV counterparts (Fig. 3A). An alignment of the amino-terminal regions of MHV and BCoV nsp3 is shown in Fig. 3B. Our assignment of the boundaries of the Ubl1 and Ac domains was based on alignment of the nsp3 sequences of numerous betacoronaviruses (45, 46), as well as the available structure of SARS-CoV Ubl1 (27) and in vitro mapping of the upstream limit of MHV PLP1 (35, 38). Additionally, in both mutants, the Ubl1 domain substitution was designed to begin at the fourth residue of BCoV nsp3, thereby maintaining all MHV P6-P4′ residues at the nsp2-nsp3 junction. In this manner, the MHV PLP1 of nsp3 continued to be paired with its cognate cleavage site.
Fig 3.

Exact substitution of the BCoV Ubl1 and Ac domains of nsp3. (A) Schematics of the MHV-BCoV chimeric portion of nsp3 and the MHV wild-type N protein encoded by the Ubl1(BCoV) and Ubl1-Ac(BCoV) mutants. Numbering indicates amino acid residues in each protein; BCoV residue numbers are in bold. (B) Alignment of the amino-terminal regions of MHV and BCoV nsp3; shading indicates identical residues. (C) Plaques of the Ubl1(BCoV) and Ubl1-Ac(BCoV) mutants at 37°C compared with those of the wild type. Plaque titrations were carried out on L2 cells; monolayers were stained with neutral red at 72 h postinfection and were photographed 18 h later.
Two independent isolates of the Ubl1(BCoV) mutant were obtained, and both were initially verified to have the intended substitution in nsp3 and wild-type MHV sequence in the portion of the N protein that contains the SR region. One of the isolates formed plaques that were indistinguishable from those of the wild type at 33, 37, and 39°C. The other had smaller plaques than the wild type at all three temperatures. However, upon sequencing the entire genomes of the latter virus and of a large-plaque variant derived therefrom, we found that the partially defective phenotype of the second isolate was due to an extraneous mutation near the cleavage site of the S protein (A703D), apparently an artifact of the reverse genetics process. Upon reversion of the S gene mutation, the second Ubl1(BCoV) isolate had the same wild-type-like phenotype as did the first; plaques of the (restored A703) second isolate at 37°C are shown in Fig. 3C. This result, which ruled out the possibility of compensatory second-site mutations anywhere else in the genome, showed that exact substitution of the BCoV Ubl1 domain was not harmful to MHV replication.
Similarly, we obtained two independent isolates of the Ubl1-Ac(BCoV) mutant. Both of these were determined to have the constructed substitution in nsp3 (with no additional mutations there or in flanking regions of 90 residues) as well as no mutations within a 370-residue span encompassing the SR region of the MHV N protein. The two Ubl1-Ac(BCoV) mutants formed plaques that were identical to those of the wild type at 33, 37, and 39°C; plaques of one isolate at 37°C are shown in Fig. 3C. Moreover, both the Ubl1(BCoV) mutant and the Ubl1-Ac(BCoV) mutant grew to high titers (9.4 × 107 and 1.0 × 108 PFU/ml, respectively, at passage 3) that were comparable to those of wild-type MHV. Thus, contrary to our initial expectations, exact substitutions of the heterologous BCoV Ubl1 or Ac domain were fully tolerated by MHV. In contrast to the previously observed incompatibility of the BCoV N protein with MHV nsp3 (23), the MHV N protein was entirely competent to interact with nsp3 containing either BCoV Ubl1 alone or BCoV Ubl1 plus Ac.
Surprisingly, this nonreciprocal relationship extended across an even greater phylogenetic distance. We were also able to construct MHV mutants with analogous substitutions of the SARS-CoV nsp3 Ubl1 and Ac domains. These viruses formed plaques that were identical to those of wild-type MHV [the Ubl1(SARS-CoV) mutant] or only slightly smaller than those of wild-type MHV [the Ubl1-Ac(SARS-CoV) mutant] at 33, 37, and 39°C (data not shown). This result was somewhat striking in light of our previous finding that an MHV mutant N protein harboring the SR region of the SARS-CoV N protein was nonfunctional unless it additionally contained compensating mutations in the SR region and in nsp3 (23).
Complementation of N(BCoV) by Ubl1(BCoV).
We next sought to determine if the BCoV Ubl1 domain, either alone or in conjunction with the BCoV Ac domain, could rescue the severe defect caused by substitution of the BCoV N protein in MHV. To accomplish this, we first reconstructed the BCoV N substitution mutant using the full-length cDNA reverse genetics system (42) to engineer an exact gene-for-gene replacement of MHV N by its BCoV counterpart. We obtained two independent isolates of this mutant, which was designated N(BCoV) (Fig. 4A). One isolate had a low-to-undetectable frequency of revertants in early-passage stocks. As expected, this virus formed tiny plaques at 33, 37, and 39°C and had a pronounced temperature sensitivity (Fig. 4B). Sequence analysis confirmed that this isolate contained the entire wild-type BCoV N gene and had no mutations in the first 650 amino acids of MHV nsp3. Two large-plaque revertants originating from this isolate were found to have mutations in the SR region of the BCoV N protein. One of them, an insertion of KKP following BCoV N residue S210, we had not observed before; the second, S206I, had arisen twice independently in our previous study (23). The other N(BCoV) isolate was completely overrun by a revertant at the earliest passage. This virus was found to have a novel reverting mutation, S200Y, in the SR region of the BCoV N protein. None of the three revertants had mutations in nsp3. Thus, in both its phenotype and its propensity to rapidly gain suppressor mutations in the SR region of the N protein, the reconstructed N(BCoV) mutant exactly recapitulated the phenotype of the previously created BCoV N mutant (Alb613) (23), which had been constructed by targeted RNA recombination.
Fig 4.

Complementation of the N(BCoV) substitution by the Ubl1(BCoV) substitution. (A and C) Schematics of the MHV or MHV-BCoV chimeric portion of nsp3 and the BCoV N protein encoded by the N(BCoV), Ubl1/N(BCoV), and Ubl1-Ac/N(BCoV) mutants. Numbering indicates amino acid residues in each protein; BCoV residue numbers are in bold. In panel A, arrows denote the positions of three reverting mutations that arose in isolates of the N(BCoV) mutant. (B and D) Plaques of the N(BCoV) and Ubl1/N(BCoV) mutants at 33, 37, and 39°C compared with those of the wild type. Plaque titrations were carried out on L2 cells; monolayers were stained with neutral red at 72 h postinfection and were photographed 18 h later.
In two further constructs, designated Ubl1/N(BCoV) and Ubl1-Ac/N(BCoV), we combined the BCoV nsp3 substitutions with the BCoV N substitution (Fig. 4C). Both independent isolates that we obtained for the Ubl1/N(BCoV) mutant formed plaques that approached wild-type size at 33 and 37°C, but at 39°C their plaques were just marginally larger than those of the N(BCoV) mutant. To determine the basis for this phenotype, we selected from one of the Ubl1/N(BCoV) isolates a variant that formed large plaques at 39°C. Sequence analysis of the entire genome of this variant revealed that it had the expected sequence for the Ubl1/N(BCoV) mutant. We therefore concluded that this virus represented the correct Ubl1/N(BCoV) mutant, and we found that at 33, 37, and 39°C it formed plaques that were only slightly smaller than those of the wild type (Fig. 4D). Apparently, as had occurred with earlier mutants (see above), the original Ubl1/N(BCoV) isolate contained a spurious mutation arising from the reverse genetics manipulations that was responsible for its reduced plaque size at 39°C. Similarly, two independent isolates of the Ubl1-Ac/N(BCoV) mutant also appeared wild-type-like at 33 and 37°C but not at 39°C. For both of these we selected variants that formed large plaques at 39°C. However, because of the results obtained with the Ubl1/N(BCoV) mutant, we did not exhaustively pursue the basis for the reduced plaque size of the original Ubl1-Ac/N(BCoV) isolates at 39°C.
To more closely examine the extent of complementation of the substituted BCoV N protein by the substituted BCoV Ubl1 domain, we compared the growth kinetics of the Ubl1/N(BCoV) mutant and wild-type MHV at 37°C. At a high multiplicity of infection (5.0 PFU per cell), growth of the Ubl1/N(BCoV) mutant was only slightly delayed with respect to that of the wild type, and both viruses reached comparable peak titers at 12 to 16 h postinfection (Fig. 5A). [It was not possible to obtain a sufficiently high-titer stock of the N(BCoV) mutant to include it in this comparison.] In multiple-cycle growth begun at a low multiplicity of infection (0.01 PFU per cell), the Ubl1/N(BCoV) mutant lagged about 2 h behind the wild type, and the viruses reached nearly identical maximal titers at 28 to 36 h postinfection (Fig. 5B). In contrast, growth of the N(BCoV) mutant lagged at least 8 h behind that of the other two viruses, and its optimal titers were at least 100-fold lower. The growth curve for the N(BCoV) mutant in Fig. 5B represents an upper limit for the actual growth of that virus, because the infecting inoculum already consisted of 10% revertants. It is likely that even the curve corrected for revertant plaques (dashed line in Fig. 5B) is an overestimate at times after 12 h, due to the rescue of mutant genomes in infected cells by revertant N protein. Data from a second, independent growth experiment were identical to those shown in Fig. 5. From these results, we concluded that substitution of the BCoV Ubl1 domain is sufficient for nearly complete complementation of the severe defect caused by substitution of the BCoV N protein in MHV.
Fig 5.

Growth kinetics of the Ubl1/N(BCoV) and N(BCoV) mutants. Confluent monolayers of 17Cl1 cells were infected at 37°C with Ubl1/N(BCoV) or wild-type virus at a multiplicity of infection (moi) of 5.0 PFU per cell (A) or with Ubl1/N(BCoV), N(BCoV), or wild-type virus at a multiplicity of 0.01 PFU per cell (B). At the indicated times postinfection, aliquots of medium were removed, and infectious titers were determined by plaque assay on L2 cells. In panel B the broken line represents the growth curve for the N(BCoV) mutant corrected for revertants, obtained by subtracting the number of large plaques from the total number of plaques.
MHV Ubl1 and Ac mutants.
To further dissect the N-nsp3 interaction in a purely MHV background, we generated a series of mutants of the Ubl1 and Ac domains. We designed Ubl1 point mutations that would change surface clusters of charged residues to alanines (Fig. 6A), based on the positions of aligned MHV residues on the SARS-CoV Ubl1 structure (27). (At the time that these mutants were constructed, the MHV Ubl1 structure [41] had not yet been reported.) Ubl1-mut1 (K22A R104A) altered two conserved basic residues that are juxtaposed where β-strands 1 and 4 cross each other. In Ubl1-mut2 (D94A E95A D98A E99A), four conserved and adjacent acidic residues, located in the loop between β-strands 3 and 4, were changed to alanines. In Ubl1-mut3 (K9A K11A R13A K14A), four basic residues that fall in the flexibly disordered amino terminus and are unique to MHV were changed to alanines. Ubl1-mut1 and Ubl1-mut3 were found to be viable and had wild-type or nearly wild-type phenotypes. Plaques formed by these mutants at 37°C are shown in Fig. 6B; plaques of Ubl1-mut1 were slightly smaller than those of the wild type at 39°C (data not shown). Isolates of both Ubl1-mut1 and Ubl1-mut3 were confirmed to have the expected mutations, and there were no additional mutations in the first 600 residues of nsp3 or in the entire N protein.
Fig 6.

MHV Ubl1 and Ac mutants. (A) Sequence of the amino-terminal region of MHV nsp3. Constructed Ubl1 and Ac point mutations and deletions are indicated; lethal mutations are boxed. (B) Plaques of viable Ubl1 and Ac mutants at 37°C compared with those of the wild type. (C) Failure of the ΔAc2 mutation to complement the BCoV N substitution. Plaques of the ΔAc2(MHV)/N(BCoV) mutant at 37°C are compared with those of the wild type. Plaque titrations for panels B and C were carried out on L2 cells; monolayers were stained with neutral red at 72 h postinfection and were photographed 18 h later.
In contrast, the mutations in Ubl1-mut2 were lethal. Likewise, deletion of all structured segments of Ubl1 (residues R19 to A111) in mutant ΔUbl1 was lethal. It should be noted that the ΔUbl1 deletion left the nsp2-nsp3 cleavage site fully intact (Fig. 6A). For both Ubl1-mut2 and ΔUbl1, we could not obtain viable virus in five independent trials in which other mutants or the wild type were successfully recovered. The lethality of these two mutants demonstrated that the Ubl1 domain is an essential part of the coronavirus replicase-transcriptase complex.
Unlike Ubl1, the Ac domain was found to be completely dispensable for viral replication. Ac is one of the least conserved regions of nsp3, not only among closely related coronavirus species but even among strains of the same species. The Ac domain of BCoV diverges considerably from, and is much smaller than, that of MHV (Fig. 3B). Similarly, the Ac domain of HCoV-HKU1 is poorly conserved and harbors from 2 to 17 copies of an acidic decapeptide repeat (47, 48). Thus, to test the importance of this segment of nsp3, we constructed two deletion mutants (Fig. 6A). The first, ΔAc1, was patterned on the aligned sequence of MHV strain 2 (49) and contained two closely spaced deletions (residues E155 to E198 and K204 to V212). The deletion in the second, ΔAc2, was more extensive, removing almost the entirety of the Ac region (residues A125 to D233). Two or three independent isolates of each mutant were obtained, and all had phenotypes that were similar or identical to that of the wild type at 33, 37, and 39°C; plaques of one isolate of each mutant at 37°C are shown in Fig. 6B. None of isolates had additional mutations in the first 600 residues of nsp3 or in the entire N protein. The viability of the ΔAc1 and ΔAc2 constructs revealed that, unlike Ubl1, the Ac domain is nonessential. Taken together with the BCoV chimera complementation results (Fig. 4 and 5), this showed that the interaction between nsp3 and the N protein is principally or exclusively confined to the Ubl1 domain of nsp3.
The pronounced size difference between the Ac domains of MHV and BCoV (Fig. 3B) raised the possibility that the larger MHV Ac domain was somehow responsible for the inability of the BCoV N protein to interact with the Ubl1 domain of MHV nsp3. Consistent with this idea was our finding that one of the revertants of the original BCoV N mutant contained a 21-amino-acid deletion in Ac (23). To test this notion, we constructed a mutant, designated ΔAc2(MHV)/N(BCoV), in which the ΔAc2 deletion was paired with the BCoV N substitution (Fig. 6C). The phenotype of this mutant was identical to that of the N(BCoV) mutant, which demonstrated that the MHV Ac domain does not exert a dominant negative effect over the BCoV N protein. This result ruled out the possibility that the failure of the BCoV N protein to interact with MHV Ubl1 is due to obstruction by the MHV Ac domain, and it reinforced the conclusion that the N-nsp3 interaction maps solely to the Ubl1 domain of nsp3.
Complete correlation between N-nsp3 genetic interactions and the ability of N protein to enhance the infectivity of coronavirus genomic RNA.
In contrast to almost all other positive-strand RNA viral genomes, coronavirus gRNA is only minimally infectious upon transfection into susceptible host cells. It has been a long-standing observation that the infectivity of coronavirus gRNA is markedly increased by the cotransfection of mRNA encoding the N protein (9, 42, 44, 50–53). We have previously shown that cotransfected N mRNA needs to be translated into N protein in order to be active in this regard. Moreover, BCoV N mRNA was not able to enhance the infectivity of MHV gRNA, but BCoV N mRNA acquired activity if it contained an SR region-reverting mutation (23). We thus concluded that there was a critical relationship between the N-nsp3 interaction and the initiation of coronavirus infection. To extend our prior results, we investigated the correspondence between the genetic interactions of chimeric N and nsp3 proteins and the stimulatory activity of cotransfected N mRNA. For this purpose, we first improved our infectivity assay, carrying out gRNA-mRNA cotransfections by electroporation, as described in Materials and Methods. This method typically produced infectious titers of greater than 4 × 103 PFU per μg gRNA at the optimal N mRNA concentration (Fig. 7A), which represented a 100- to 300-fold-higher efficiency than was obtained previously with chemical cotransfections (23). Note that in these experiments wild-type MHV gRNA was isolated from highly purified virions of the previously described Alb240 (54), which is nearly identical in sequence to wild-type MHV reconstructed by the full-length cDNA reverse genetics system (42).
Fig 7.

Abilities of interspecies chimeric N proteins to enhance the infectivity of genomic RNA. (A, C, and D) L2 cells in suspension were transfected by electroporation with wild-type (wt) MHV gRNA plus added synthetic N mRNA or a water control. Transfected cells were diluted into fresh L2 cells in suspension and then seeded into dishes. Following attachment of cells, monolayers were overlaid with agar, and plaques were counted after 48 h of incubation at 37°C. Each histogram represents the mean infectious titer (± standard deviation) from four separate transfections. (A) Optimization of cotransfected N mRNA concentration. (B) Schematic of chimeric N proteins encoded by cotransfected mRNAs. (C) Cotransfections of wild-type MHV gRNA with mRNAs for wild-type MHV N, wild-type BCoV N, BCoV N containing the SR region of MHV N (BCoV M-SR), and BCoV N containing the reverting mutation S202L. (D) Cotransfections of wild-type MHV gRNA with mRNAs for wild-type MHV N, MHV N containing the SR region of HCoV-HKU1 N (MHV HK-SR), MHV N containing the SR region of SARS-CoV N (MHV S-SR), and MHV N containing a deletion of spacer B and domain N3 (MHV NΔB3).
Initially, we replicated previous results using the new assay (Fig. 7B and C). As observed before (9, 23), cotransfected wild-type MHV N mRNA stimulated the infectivity of wild-type MHV gRNA at least 25-fold. In contrast, wild-type BCoV mRNA was nearly inert, although we were now able to detect some small residual activity relative to that of the negative control that had no cotransfected mRNA. Infectivity was partially stimulated by BCoV mRNA containing S202L, a reverting mutation of the original BCoV N substitution mutant. Still-higher levels of gRNA infectivity were restored with mRNA encoding BCoV N in which the entire SR region was replaced with that of MHV N (M-SR mutant mRNA). These results were expanded by examination of two other SR region substitutions for which we had previously constructed viral mutants (Fig. 7B and D). One of them, a mutant designated HK-SR, in which the MHV N protein SR region was replaced by that from HCoV-HKU1, had a phenotype indistinguishable from that of wild-type MHV (23). In accord with this prior finding, HK-SR N mRNA was as capable as wild-type MHV N mRNA in enhancing the infectivity of MHV gRNA. In a mutant designated S-SR, the replacement of the MHV N SR region with that from SARS-CoV N was previously found to be lethal (23). In an analogous manner, S-SR N mRNA showed only a minimal ability to augment the infectivity of MHV gRNA. Thus, the efficacies of SR-substituted N mRNAs in promoting the infectivity of MHV gRNA entirely reflected the status of MHV mutants harboring the same N protein substitutions.
The availability of genomes encoding chimeric nsp3 molecules allowed us to directly test whether nsp3 was relevant to the requirement for N protein for optimal gRNA infectivity. Accordingly, we carried out cotransfection experiments with gRNA purified from virions of the Ubl1(BCoV) mutant or the Ubl1-Ac (BCoV) mutant (Fig. 8). For both mutant gRNAs, we observed a high degree of stimulation by wild-type BCoV N mRNA, almost to the same levels obtained with wild-type MHV N mRNA. This finding sharply contrasted with the inability of BCoV N mRNA to enhance the infectivity of wild-type MHV gRNA (Fig. 7C) (23). More to the point, it exactly paralleled the observed complementation between the Ubl1(BCoV) and N(BCoV) substitutions (Fig. 4 and 5). These results demonstrated that nsp3, specifically the Ubl1 domain of nsp3, is required in conjunction with N protein for the initiation of infection by coronavirus gRNA.
Fig 8.

Complementation between Ubl1(BCoV) and N(BCoV) correlates with the ability of N protein to enhance the infectivity of genomic RNA. Cotransfections of gRNA from the Ubl1(BCoV) mutant (A) or the Ubl1-Ac (BCoV) mutant (B) with either wild-type MHV N mRNA or wild-type BCoV N mRNA were carried out as described in the legend to Fig. 7. Above each panel is a schematic of the chimeric nsp3 of the respective mutant. Each histogram represents the mean infectious titer (± standard deviation) from four separate transfections.
Finally, to dissect the basis for the stimulation of gRNA infectivity by N protein, we constructed a set of mRNA deletion mutants that scanned all domains of N (Fig. 9). We found that truncation of either the amino terminus (mutant NΔ1a) or the carboxy terminus (mutant NΔB3) had little or no effect on the enhancement of infectivity by N (also see Fig. 7D). The function of domain N1a, if any, is unknown. Domain N3 is required for interaction with the viral membrane protein (M) (6, 7, 12, 13), and it is also involved in N-N interactions (8), so it might not be expected to also participate in the N-nsp3 interaction. On the other hand, removal of either of the RNA-binding domains of N (mutants NΔ1b and NΔ2b) abolished mRNA stimulatory activity. This outcome was consistent with previous demonstrations by others that any one of three lethal point mutations in domain N1b—Y127A, R125A, or Y190A—eliminated both the RNA-binding activity of N and its ability to stimulate gRNA infectivity (9, 55). For knockout of the SR region of N protein, we chose two deletion mutations, NΔ13 and NΔ15, that we had previously shown were fully competent to bind RNA (8). Both of these mutations abolished mRNA stimulatory activity. Notably, we also determined through targeted RNA recombination (56) that the NΔ13 and NΔ15 mutations are lethal in MHV. Taken together, these results show that the SR region as well as both RNA-binding domains of N are required for its enhancement of gRNA infectivity.
Fig 9.
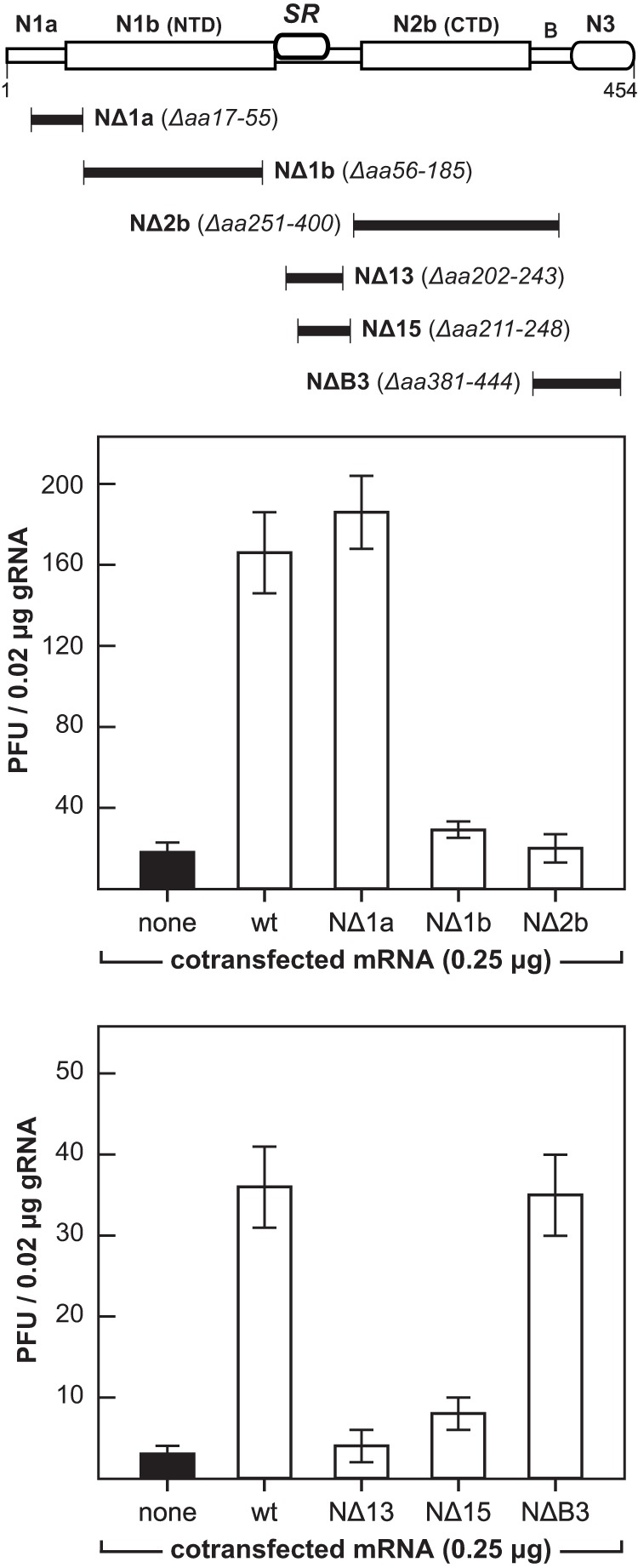
Requirements on N protein for enhancement of the infectivity of genomic RNA. Top, schematic indicating the boundaries of deletion mutants scanning the N protein. Cotransfections of wild-type MHV gRNA with wild-type MHV N mRNA or mRNA encoding each of the N deletion mutants were carried out as described in the legend to Fig. 7. Each histogram represents the mean infectious titer (± standard deviation) from four separate transfections.
DISCUSSION
Multiple lines of genetic support for the N-nsp3 interaction.
In this study, we have obtained a broad range of genetic evidence confirming the previously discovered interaction between the coronavirus structural protein N and the largest subunit of the replicase-transcriptase complex, nsp3. Whereas our prior investigation originated with N protein substitutions (23), in the work reported here we constructed domain substitutions and mutations in nsp3. Our initial attempt to create an interspecies chimeric nsp3, the Bcl-Spe(BCoV) mutant, produced a virus with a significantly defective phenotype (Fig. 2). However, the growth properties of that mutant turned out to be unrelated to the characteristics of the N-nsp3 interaction. This outcome alerted us to the possibility that imprecise substitutions could give rise to spurious results stemming from incompatibilities between MHV PLP1 and a heterologous nsp2-nsp3 cleavage site. Presumably, a similar caution would apply to other chimeric substitutions among the 16 subunits of the coronavirus replicase-transcriptase complex, all of which are proteolytically processed from polyprotein precursors.
Precise substitutions of the BCoV Ubl1 or Ubl1-Ac domain, surprisingly, were completely tolerated by MHV (Fig. 3). We had expected that these replacements, like the original BCoV N substitution at the other end of the genome (23), would generate mutants with extremely impaired phenotypes. The wild-type-like phenotypes of the Ubl1(BCoV) and Ubl1-Ac(BCoV) mutants suggest that BCoV nsp3 is less discriminating than MHV nsp3 in binding to its own N protein or to related N proteins. When more is understood about the configuration of the N protein SR region, then perhaps the molecular basis for this nonreciprocal relationship will become clear. Most significantly, the Ubl1(BCoV) substitution [and the Ubl1-Ac(BCoV) substitution] almost completely suppressed the severe defect caused by placement of the BCoV N protein in MHV (Fig. 4 and 5). This complementation clearly established that the BCoV N protein is fully functional in MHV, provided that it is able to interact with its cognate Ubl1.
To further probe the importance of the N-nsp3 interaction, we constructed mutants of the MHV Ubl1 and Ac domains (Fig. 6). Although some point mutations of Ubl1 had no effect on the virus, a deletion of most of this domain (in ΔUbl1) and one set of clustered charged-to-alanine mutations (in Ubl1-mut2) were found to be lethal. These results suggest that the N-nsp3 interaction is essential and that the N(BCoV) mutant, despite its dramatic impairment, must retain some vestige of activity. Alternatively, we cannot yet rule out the possibility that there exists some other critical role for Ubl1 that contributes to the lethality of the ΔUbl1 and Ubl1-mut2 mutants. Among three protein-protein interaction studies of the coronavirus replicase-transcriptase complex (57–59), one found potential interacting partners for the Ubl1-Ac region of nsp3. Binding to nsp6 was detected by a yeast two-hybrid assay, and binding to nsp8 and nsp9 was observed by a pulldown assay (57). Our construction of MHV Ubl1 point mutants in the present study was based on alignment to the reported structure of SARS-CoV Ubl1 (27). In the future we hope to design mutants based on the recently reported structure of MHV Ubl1 (41) that will allow us to uncover genetic cross talk with N and, possibly, other viral components.
In contrast to the Ubl1 domain, the Ac domain was found to be nonessential for viral replication, as it could be entirely deleted (in the ΔAc2 mutant) with no effect on viral phenotype (Fig. 6). This finding, coupled with the complementation between BCoV Ubl1 and BCoV N, as well as with the in vitro binding of Ubl1 to N demonstrated by Keane and Giedroc (41), solidifies the conclusion that the N-nsp3 interaction maps to Ubl1 alone in nsp3. An incidental property of the ΔAc2 mutant is that it completely abolishes a 58-nucleotide (nt) cis-acting RNA element that appears to be required for the replication of some MHV defective interfering RNAs (60) but not for others (44). Therefore, while this RNA element may enhance defective interfering RNA synthesis under particular circumstances (61), it cannot be playing a consequential role in native viral RNA synthesis.
Relatively little is known about whether other domains of nsp3 are essential components of the RNA synthetic machinery or are dispensable as such, even though they may act in separate capacities, such as evasion of innate immunity (62–64). An active-site catalytic residue mutant of MHV PLP1 was shown to be only minimally viable, but it was rescued to a great extent by deletion of its substrates, the nsp1-nsp2 and nsp2-nsp3 cleavage sites (39). This implied that the inactive PLP1 in the original mutant was obstructing other crucial functions of nsp3 by remaining bound to one or both of its cleavage sites. Analysis of an active-site mutant of the ADRP domain of HCoV-229E demonstrated that loss of the ADP-ribose-1″-phosphatase activity of ADRP does not affect viral replication in tissue culture (65). Intriguingly, an MHV conditional-lethal mutation that maps at the boundary of the PLP1 and ADRP domains has been described (66); however, it is not yet clear whether this mutation affects a known or a novel activity of nsp3. Future reverse genetic analysis of nsp3 might allow us to determine which modules of this large protein are directly involved in viral RNA synthesis and which provide accessory functions.
Function of the N-nsp3 interaction.
In addition to further establishing the interaction between N and nsp3, the work presented here shows that this interaction is intrinsic to a function of N protein that is required early in infection. Investigations using replicon systems have documented a marked requirement for N protein in coronavirus RNA synthesis, although it is not agreed upon whether the basis for this effect resides in genome replication or transcription (14–17). One manifestation of the requirement for N is that transfected coronavirus gRNA is only minimally infectious unless supplemented with a source of N protein, usually as cotransfected N mRNA (9, 42, 44, 50–53). This need for N protein must occur at a very early step in the initiation of a productive infection, since transfected gRNA eventually generates its own subgenomic N mRNA. We previously showed that there was a fundamental relationship between the N-nsp3 interaction and the ability of N protein to enhance the infectivity of gRNA (23). The severe defectiveness of the BCoV N substitution mutant was mirrored by the inability of BCoV N mRNA to support MHV gRNA infectivity. However, reverting mutations in the SR region of the BCoV N protein compensated for the defect of the BCoV N mutant, and they were also able to partially rescue the activity of BCoV N mRNA with MHV gRNA.
We have now shown that this correlation extends, without exception, to other genetic constructs (Fig. 7). BCoV N mRNA in which the entire SR region was replaced with the MHV N SR region regained the ability to stimulate MHV gRNA infectivity to a relatively high level. Similarly, MHV N mRNA substituted with the HCoV-HKU1 SR region was highly active, reflecting the wild-type-like phenotype of the previously obtained HK-SR viral mutant (23). Likewise, MHV N mRNA substituted with the SARS-CoV SR region lost almost all activity, consistent with the lethality of the corresponding S-SR viral mutant. Moreover, experiments with viral genomes of nsp3 chimeric mutants provided the direct counterpart of our genetic complementation results (Fig. 8). Wild-type BCoV N mRNA was found to be fully active in stimulating the infectivity of gRNA from either the Ubl1(BCoV) mutant or the Ubl1-Ac(BCoV) mutant. This demonstrated that BCoV N mRNA was indeed functional when paired with complementary gRNA, i.e., gRNA that encodes a compatible version of nsp3. This result additionally ruled out the possibility that the consistently observed inactivity of BCoV N mRNA with wild-type MHV gRNA was trivially due to some undetected flaw in the mRNA or its vector. Wild-type MHV N mRNA was also highly active with Ubl1(BCoV) and Ubl1-Ac(BCoV) gRNA, in accord with the wild-type-like phenotypes of the corresponding viral mutants.
To shed more light on the requirements on N protein for the enhancement of gRNA infectivity, we produced a series of N mRNAs deleting each of the previously defined domains of the N molecule (Fig. 9). This revealed that the two termini of N protein, domains N1a and N3, were dispensable for its activity in the cotransfection assay, although domain N3 is clearly essential for other protein-protein interactions in viral assembly (6–8, 12, 13). Conversely, we found that N needed to possess both of its RNA-binding domains, N1b (NTD) and N2b (CTD), in order to be able to stimulate gRNA infectivity. Not surprisingly, deletions removing most of the SR region also rendered N mRNA inactive. Work is currently in progress to try to identify key residues in the SR region that are determinants of the N-nsp3 interaction.
The fact that N protein must retain its RNA-binding ability, in addition to its SR region, is consistent with our previously posited model that (in an infection, rather than a transfection) the role of the N-nsp3 interaction is to tether the infecting nucleocapsid to the newly translated replicase-transcriptase complex. Although N protein must be displaced from the 5′ two-thirds of the incoming viral gRNA in order to allow translation, residual nucleocapsid structure at the 3′ end of the genome would allow its immediate association with nsp3, the first translation product to become anchored to the membrane (67, 68). This continued association between the genome and nsp3 could serve a number of purposes. First, it could confine repeated rounds of genome translation to a restricted locus, thereby enabling the concentration of replicase components in a single region of the endoplasmic reticulum (ER) membrane. Second, it could recruit the genome to the replication compartment while that compartment is beginning to be shaped through remodeling of intracellular membranes (69). Finally, it could assist in the formation of an initiation complex at the 3′ end of the genome, which is the first step in both RNA replication and transcription (70). Further dissection of the N-nsp3 interaction may provide a starting point for unraveling a large number of protein-protein and protein-RNA interactions essential to coronavirus RNA synthesis.
ACKNOWLEDGMENTS
We are grateful to Ralph Baric and Amy Sims (University of North Carolina, Chapel Hill, NC) for generously providing cloned cDNAs and protocols for the MHV full-length cDNA reverse genetics system. We thank David Brian (University of Tennessee, Knoxville, TN) for providing bovine coronavirus. We thank the Applied Genomics Technology Core Facility of the Wadsworth Center for DNA sequencing.
This work was supported by National Institutes of Health (National Institute of Allergy and Infectious Diseases) grants R01 AI064603 and R56 AI064603.
Footnotes
Published ahead of print 12 June 2013
REFERENCES
- 1. Masters PS. 2006. The molecular biology of coronaviruses. Adv. Virus Res. 66:193–292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Perlman S, Netland J. 2009. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol. 7:439–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. van Boheemen S, de Graaf M, Lauber C, Bestebroer TM, Raj VS, Zaki AM, Osterhaus AD, Haagmans BL, Gorbalenya AE, Snijder EJ, Fouchier RA. 2012. Genomic characterization of a newly discovered coronavirus associated with acute respiratory distress syndrome in humans. mBio 3(6):e00473–12. 10.1128/mBio.00473-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Groot RJ, Baker SC, Baric RS, Brown CS, Drosten C, Enjuanes L, Fouchier RA, Galiano M, Gorbalenya AE, Memish Z, Perlman S, Poon LL, Snijder EJ, Stephens GM, Woo PC, Zaki AM, Zambon M, Ziebuhr J. 15 May 2013. Middle East respiratory syndrome coronavirus (MERS-CoV); announcement of the Coronavirus Study Group. J. Virol. 10.1128/JVI.01244-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Koetzner CA, Parker MM, Ricard CS, Sturman LS, Masters PS. 1992. Repair and mutagenesis of the genome of a deletion mutant of the coronavirus mouse hepatitis virus by targeted RNA recombination. J. Virol. 66:1841–1848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kuo L, Masters PS. 2002. Genetic evidence for a structural interaction between the carboxy termini of the membrane and nucleocapsid proteins of mouse hepatitis virus. J. Virol. 76:4987–4999 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hurst KR, Kuo L, Koetzner CA, Ye R, Hsue B, Masters PS. 2005. A major determinant for membrane protein interaction localizes to the carboxy-terminal domain of the mouse coronavirus nucleocapsid protein. J. Virol. 79:13285–13297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hurst KR, Koetzner CA, Masters PS. 2009. Identification of in vivo-interacting domains of the murine coronavirus nucleocapsid protein. J. Virol. 83:7221–7234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Grossoehme NE, Li L, Keane SC, Liu P, Dann CE, III, Leibowitz JL, Giedroc DP. 2009. Coronavirus N protein N-terminal domain (NTD) specifically binds the transcriptional regulatory sequence (TRS) and melts TRS-cTRS RNA duplexes. J. Mol. Biol. 394:544–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ma Y, Tong X, Xu X, Li X, Lou Z, Rao Z. 2010. Structures of the N- and C-terminal domains of MHV-A59 nucleocapsid protein corroborate a conserved RNA-protein binding mechanism in coronavirus. Protein Cell 1:688–697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang CK, Hsu YL, Chang YH, Chao FA, Wu MC, Huang YS, Hu CK, Huang TH. 2009. Multiple nucleic acid binding sites and intrinsic disorder of severe acute respiratory syndrome coronavirus nucleocapsid protein: implications for ribonucleocapsid protein packaging. J. Virol. 83:2255–2264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Verma S, Bednar V, Blount A, Hogue BG. 2006. Identification of functionally important negatively charged residues in the carboxy end of mouse hepatitis coronavirus A59 nucleocapsid protein. J. Virol. 80:4344–4355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Verma S, Lopez LA, Bednar V, Hogue BG. 2007. Importance of the penultimate positive charge in mouse hepatitis coronavirus A59 membrane protein. J. Virol. 81:5339–5348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Almazán F, Galán C, Enjuanes L. 2004. The nucleoprotein is required for efficient coronavirus genome replication. J. Virol. 78:12683–12688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Zúñiga S, Cruz JL, Sola I, Mateos-Gómez PA, Palacio L, Enjuanes L. 2010. Coronavirus nucleocapsid protein facilitates template switching and is required for efficient transcription. J. Virol. 84:2169–2175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Thiel V, Herold J, Schelle B, Siddell SG. 2001. Viral replicase gene products suffice for coronavirus discontinuous transcription. J. Virol. 75:6676–6681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schelle B, Karl N, Ludewig B, Siddell SG, Thiel V. 2005. Selective replication of coronavirus genomes that express nucleocapsid protein. J. Virol. 79:6620–6630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Denison MR, Spaan WJ, van der Meer Y, Gibson CA, Sims AC, Prentice E, Lu XT. 1999. The putative helicase of the coronavirus mouse hepatitis virus is processed from the replicase gene polyprotein and localizes in complexes that are active in viral RNA synthesis. J. Virol. 73:6862–6871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. van der Meer Y, Snijder EJ, Dobbe JC, Schleich S, Denison MR, Spaan WJ, Locker JK. 1999. Localization of mouse hepatitis virus nonstructural proteins and RNA synthesis indicates a role for late endosomes in viral replication. J. Virol. 73:7641–7657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sims AC, Ostermann J, Denison MR. 2000. Mouse hepatitis virus replicase proteins associate with two distinct populations of intracellular membranes. J. Virol. 74:5647–5654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stertz S, Reichelt M, Spiegel M, Kuri T, Martínez-Sobrido L, García-Sastre A, Weber F, Kochs G. 2007. The intracellular sites of early replication and budding of SARS-coronavirus. Virology 361:304–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Verheije MH, Hagemeijer MC, Ulasli M, Reggiori F, Rottier PJ, Masters PS, de Haan CA. 2010. The coronavirus nucleocapsid protein is dynamically associated with the replication-transcription complexes. J. Virol. 84:11575–11579 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hurst KR, Ye R, Goebel SJ, Jayaraman P, Masters PS. 2010. An interaction between the nucleocapsid protein and a component of the replicase-transcriptase complex is crucial for the infectivity of coronavirus genomic RNA. J. Virol. 84:10276–10288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saikatendu KS, Joseph JS, Subramanian V, Clayton T, Griffith M, Moy K, Velasquez J, Neuman BW, Buchmeier MJ, Stevens RC, Kuhn P. 2005. Structural basis of severe acute respiratory syndrome coronavirus ADP-ribose-1″-phosphate dephosphorylation by a conserved domain of nsP3. Structure. 13:1665–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Egloff MP, Malet H, Putics A, Heinonen M, Dutartre H, Frangeul A, Gruez A, Campanacci V, Cambillau C, Ziebuhr J, Ahola T, Canard B. 2006. Structural and functional basis for ADP-ribose and poly(ADP-ribose) binding by viral macro domains. J. Virol. 80:8493–8502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Ratia K, Saikatendu KS, Santarsiero BD, Barretto N, Baker SC, Stevens RC, Mesecar AD. 2006. Severe acute respiratory syndrome coronavirus papain-like protease: structure of a viral deubiquitinating enzyme. Proc. Natl. Acad. Sci. U. S. A. 103:5717–5722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Serrano P, Johnson MA, Almeida MS, Horst R, Herrmann T, Joseph JS, Neuman BW, Subramanian V, Saikatendu KS, Buchmeier MJ, Stevens RC, Kuhn P, Wüthrich K. 2007. Nuclear magnetic resonance structure of the N-terminal domain of nonstructural protein 3 from the severe acute respiratory syndrome coronavirus. J. Virol. 81:12049–12060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chatterjee A, Johnson MA, Serrano P, Pedrini B, Joseph JS, Neuman BW, Saikatendu K, Buchmeier MJ, Kuhn P, Wüthrich K. 2009. Nuclear magnetic resonance structure shows that the severe acute respiratory syndrome coronavirus-unique domain contains a macrodomain fold. J. Virol. 83:1823–1836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Piotrowski Y, Hansen G, Boomaars-van der Zanden AL, Snijder EJ, Gorbalenya AE, Hilgenfeld R. 2009. Crystal structures of the X-domains of a group-1 and a group-3 coronavirus reveal that ADP-ribose-binding may not be a conserved property. Protein Sci. 18:6–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tan J, Vonrhein C, Smart OS, Bricogne G, Bollati M, Kusov Y, Hansen G, Mesters JR, Schmidt CL, Hilgenfeld R. 2009. The SARS-unique domain (SUD) of SARS coronavirus contains two macrodomains that bind G-quadruplexes. PLoS Pathog. 5:e1000428. 10.1371/journal.ppat.1000428 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Xu Y, Cong L, Chen C, Wei L, Zhao Q, Xu X, Ma Y, Bartlam M, Rao Z. 2009. Crystal structures of two coronavirus ADP-ribose-1″-monophosphatases and their complexes with ADP-ribose: a systematic structural analysis of the viral ADRP domain. J. Virol. 83:1083–1092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Serrano P, Johnson MA, Chatterjee A, Neuman BW, Joseph JS, Buchmeier MJ, Kuhn P, Wüthrich K. 2009. NMR structure of the nucleic acid-binding domain of the SARS coronavirus nonstructural protein 3. J. Virol. 83:12998–13008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ziebuhr J, Schelle B, Karl N, Minskaia E, Bayer S, Siddell SG, Gorbalenya AE, Thiel V. 2007. Human coronavirus 229E papain-like proteases have overlapping specificities but distinct functions in viral replication. J. Virol. 81:3922–3932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gadlage MJ, Denison MR. 2010. Exchange of the coronavirus replicase polyprotein cleavage sites alters protease specificity and processing. J. Virol. 84:6894–6898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baker SC, Yokomori K, Dong S, Carlisle R, Gorbalenya AE, Koonin EV, Lai MM. 1993. Identification of the catalytic sites of a papain-like cysteine proteinase of murine coronavirus. J. Virol. 67:6056–6063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Dong S, Baker SC. 1994. Determinants of the p28 cleavage site recognized by the first papain-like cysteine proteinase of murine coronavirus. Virology 204:541–549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hughes SA, Bonilla PJ, Weiss SR. 1995. Identification of the murine coronavirus p28 cleavage site. J. Virol. 69:809–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bonilla PJ, Hughes SA, Weiss SR. 1997. Characterization of a second cleavage site and demonstration of activity in trans by the papain-like proteinase of the murine coronavirus mouse hepatitis virus strain A59. J. Virol. 71:900–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Graham RL, Denison MR. 2006. Replication of murine hepatitis virus is regulated by papain-like proteinase 1 processing of nonstructural proteins 1, 2, and 3. J. Virol. 80:11610–11620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ziebuhr J, Thiel V, Gorbalenya AE. 2001. The autocatalytic release of a putative RNA virus transcription factor from its polyprotein precursor involves two paralogous papain-like proteases that cleave the same peptide bond. J. Biol. Chem. 276:33220–33232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Keane SC, Giedroc DP. 2013. Solution structure of mouse hepatitis virus (MHV) nsp3a and determinants of the interaction with MHV nucleocapsid (N) protein. J. Virol. 87:3502–3515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yount B, Denison MR, Weiss SR, Baric RS. 2002. Systematic assembly of a full-length infectious cDNA of mouse hepatitis virus strain A59. J. Virol. 76:11065–11078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ye R, Montalto-Morrison C, Masters PS. 2004. Genetic analysis of determinants for spike glycoprotein assembly into murine coronavirus virions: distinct roles for charge-rich and cysteine-rich regions of the endodomain. J. Virol. 78:9904–9917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Masters PS, Koetzner CA, Kerr CA, Heo Y. 1994. Optimization of targeted RNA recombination and mapping of a novel nucleocapsid gene mutation in the coronavirus mouse hepatitis virus. J. Virol. 68:328–337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Neuman BW, Joseph JS, Saikatendu KS, Serrano P, Chatterjee A, Johnson MA, Liao L, Klaus JP, Yates JR, III, Wüthrich K, Stevens RC, Buchmeier MJ, Kuhn P. 2008. Proteomics analysis unravels the functional repertoire of coronavirus nonstructural protein 3. J. Virol. 82:5279–5294 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Barretto N, Jukneliene D, Ratia K, Chen Z, Mesecar AD, Baker SC. 2005. The papain-like protease of severe acute respiratory syndrome coronavirus has deubiquitinating activity. J. Virol. 79:15189–15198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Woo PC, Lau SK, Chu CM, Chan KH, Tsoi HW, Huang Y, Wong BH, Poon RW, Cai JJ, Luk WK, Poon LL, Wong SS, Guan Y, Peiris JS, Yuen KY. 2005. Characterization and complete genome sequence of a novel coronavirus, coronavirus HKU1, from patients with pneumonia. J. Virol. 79:884–895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Woo PC, Lau SK, Yip CC, Huang Y, Tsoi HW, Chan KH, Yuen KY. 2006. Comparative analysis of 22 coronavirus HKU1 genomes reveals a novel genotype and evidence of natural recombination in coronavirus HKU1. J. Virol. 80:7136–7145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Das Sarma J, Fu L, Hingley ST, Lavi E. 2001. Mouse hepatitis virus type-2 infection in mice: an experimental model system of acute meningitis and hepatitis. Exp. Mol. Pathol. 71:1–12 [DOI] [PubMed] [Google Scholar]
- 50. Yount B, Curtis KM, Baric RS. 2000. Strategy for systematic assembly of large RNA and DNA genomes: transmissible gastroenteritis virus model. J. Virol. 74:10600–10611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Casais R, Thiel V, Siddell SG, Cavanagh D, Britton P. 2001. Reverse genetics system for the avian coronavirus infectious bronchitis virus. J. Virol. 75:12359–12369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yount B, Curtis KM, Fritz EA, Hensley LE, Jahrling PB, Prentice E, Denison MR, Geisbert TW, Baric RS. 2003. Reverse genetics with a full-length infectious cDNA of severe acute respiratory syndrome coronavirus. Proc. Natl. Acad. Sci. U. S. A. 100:12995–13000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Coley SE, Lavi E, Sawicki SG, Fu L, Schelle B, Karl N, Siddell SG, Thiel V. 2005. Recombinant mouse hepatitis virus strain A59 from cloned, full-length cDNA replicates to high titers in vitro and is fully pathogenic in vivo. J. Virol. 79:3097–3106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kuo L, Masters PS. 2003. The small envelope protein E is not essential for murine coronavirus replication. J. Virol. 77:4597–4608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Keane SC, Liu P, Leibowitz JL, Giedroc DP. 2012. Functional transcriptional regulatory sequence (TRS) RNA binding and helix destabilizing determinants of murine hepatitis virus (MHV) nucleocapsid (N) protein. J. Biol. Chem. 287:7063–7073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Kuo L, Godeke GJ, Raamsman MJ, Masters PS, Rottier PJ. 2000. Retargeting of coronavirus by substitution of the spike glycoprotein ectodomain: crossing the host cell species barrier. J. Virol. 74:1393–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Imbert I, Snijder EJ, Dimitrova M, Guillemot JC, Lécine P, Canard B. 2008. The SARS-coronavirus PLnc domain of nsp3 as a replication/transcription scaffolding protein. Virus Res. 133:136–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pan J, Peng X, Gao Y, Li Z, Lu X, Chen Y, Ishaq M, Liu D, Dediego ML, Enjuanes L, Guo D. 2008. Genome-wide analysis of protein-protein interactions and involvement of viral proteins in SARS-CoV replication. PLoS One 3:e3299. 10.1371/journal.pone.0003299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. von Brunn A, Teepe C, Simpson JC, Pepperkok R, Friedel CC, Zimmer R, Roberts R, Baric R, Haas J. 2007. Analysis of intraviral protein-protein interactions of the SARS coronavirus ORFeome. PLoS One 2:e459. 10.1371/journal.pone.0000459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Repass JF, Makino S. 1998. Importance of the positive-strand RNA secondary structure of a murine coronavirus defective interfering RNA internal replication signal in positive-strand RNA synthesis. J. Virol. 72:7926–7933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Johnson RF, Feng M, Liu P, Millership JJ, Yount B, Baric RS, Leibowitz JL. 2005. Effect of mutations in the mouse hepatitis virus 3′(+)42 protein binding element on RNA replication. J. Virol. 79:14570–14585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Eriksson KK, Cervantes-Barragán L, Ludewig B, Thiel V. 2008. Mouse hepatitis virus liver pathology is dependent on ADP-ribose-1″-phosphatase, a viral function conserved in the alpha-like supergroup. J. Virol. 82:12325–12334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Frieman M, Ratia K, Johnston RE, Mesecar AD, Baric RS. 2009. Severe acute respiratory syndrome coronavirus papain-like protease ubiquitin-like domain and catalytic domain regulate antagonism of IRF3 and NF-kappaB signaling. J. Virol. 83:6689–6705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Clementz MA, Chen Z, Banach BS, Wang Y, Sun L, Ratia K, Baez-Santos YM, Wang J, Takayama J, Ghosh AK, Li K, Mesecar AD, Baker SC. 2010. Deubiquitinating and interferon antagonism activities of coronavirus papain-like proteases. J. Virol. 84:4619–4629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Putics A, Filipowicz W, Hall J, Gorbalenya AE, Ziebuhr J. 2005. ADP-ribose-1″monophosphatase: a conserved coronavirus enzyme that is dispensable for viral replication in tissue culture. J. Virol. 79:12721–12731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Stokes HL, Baliji S, Hui CG, Sawicki SG, Baker SC, Siddell SG. 2010. A new cistron in the murine hepatitis virus replicase gene. J. Virol. 84:10148–10158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Oostra M, Hagemeijer MC, van Gent M, Bekker CP, te Lintelo EG, Rottier PJ, de Haan CA. 2008. Topology and membrane anchoring of the coronavirus replication complex: not all hydrophobic domains of nsp3 and nsp6 are membrane spanning. J. Virol. 82:12392–12405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Kanjanahaluethai A, Chen Z, Jukneliene D, Baker SC. 2007. Membrane topology of murine coronavirus replicase nonstructural protein 3. Virology 361:391–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Knoops K, Kikkert M, Worm SH, Zevenhoven-Dobbe JC, van der Meer Y, Koster AJ, Mommaas AM, Snijder EJ. 2008. SARS-coronavirus replication is supported by a reticulovesicular network of modified endoplasmic reticulum. PLoS Biol. 6:e226. 10.1371/journal.pbio.0060226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Züst R, Miller TB, Goebel SJ, Thiel V, Masters PS. 2008. Genetic interactions between an essential 3′ cis-acting RNA pseudoknot, replicase gene products, and the extreme 3′ end of the mouse coronavirus genome. J. Virol. 82:1214–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Snijder EJ, Bredenbeek PJ, Dobbe JC, Thiel V, Ziebuhr J, Poon LL, Guan Y, Rozanov M, Spaan WJ, Gorbalenya AE. 2003. Unique and conserved features of genome and proteome of SARS-coronavirus, an early split-off from the coronavirus group 2 lineage. J. Mol. Biol. 331:991–1004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Johnson MA, Chatterjee A, Neuman BW, Wüthrich K. 2010. SARS coronavirus unique domain: three-domain molecular architecture in solution and RNA binding. J. Mol. Biol. 400:724–742 [DOI] [PMC free article] [PubMed] [Google Scholar]