

Abstract
Replication-deficient rabies viruses (RABV) are promising rabies postexposure vaccines due to their prompt and potent stimulation of protective virus neutralizing antibody titers, which are produced in mice by both T-dependent and T-independent mechanisms. To promote such early and robust B cell stimulation, we hypothesized that live RABV-based vaccines directly infect B cells, thereby activating a large pool of antigen-presenting cells (APCs) capable of providing early priming and costimulation to CD4+ T cells. In this report, we show that live RABV-based vaccine vectors efficiently infect naive primary murine and human B cells ex vivo. Infection of B cells resulted in the significant upregulation of early markers of B cell activation and antigen presentation, including CD69, major histocompatibility complex class II (MHC-II), and CD40 in murine B cells or HLA-DR and CD40 in human B cells compared to mock-infected cells or cells treated with an inactivated RABV-based vaccine. Furthermore, primary B cells infected with a live RABV expressing ovalbumin were able to prime and stimulate naive CD4+ OT-II T cells to proliferate and to secrete interleukin-2 (IL-2), demonstrating a functional consequence of B cell infection and activation by live RABV-based vaccine vectors. We propose that this direct B cell stimulation by live RABV-based vaccines is a potential mechanism underlying their induction of early protective T cell-dependent B cell responses, and that designing live RABV-based vaccines to infect and activate B cells represents a promising strategy to develop a single-dose postexposure rabies vaccine where the generation of early protective antibody titers is critical.
INTRODUCTION
Rabies virus (RABV) is a negative-sense, single-stranded RNA virus within the Rhabdoviridae family with worldwide distribution (reviewed in reference 1). Rabies is a zoonotic infectious disease that afflicts a wide range of mammalian hosts, causing a viral encephalitis that is almost invariably lethal once symptoms manifest (2). Over 3 billion people live in areas where rabies is endemic in domesticated, feral, or wild animals, with dogs being the source for the overwhelming majority of rabies exposures and fatalities globally (2). Considered a neglected disease, RABV infections are responsible for over 55,000 annual human deaths worldwide (2). Protection against lethal rabies encephalitis is conferred by virus neutralizing antibodies (VNA) to the envelope surface RABV glycoprotein (RABV-G), with sufficient titers of VNA serving to block further viral spread (3–6). The postexposure prophylaxis (PEP) regimen following suspected rabies exposure, designed to neutralize pathogenic virus before it reaches the central nervous system (CNS), consists of multiple doses of inactivated RABV-based vaccine over the course of 3 to 4 weeks, along with the injection of pooled human rabies immune globulin (RIG) immediately following exposure (5, 7, 8). While safe and highly effective if properly administered, this regimen is expensive and cumbersome in areas of the developing world where rabies is endemic; thus, there exists a need for a rabies vaccine that confers protection after a single immunization and does not require costly RIG for ensured efficacy (9). With over 15 million people treated with a course of PEP per year, and 40% of those treatments administered to children ages 5 to 14 (2), the improvement of rabies vaccine regimens has the potential for significant savings of both health care spending and years of life lost to disease.
We previously compared RABV-specific antibody kinetics in rhesus macaques and mice immunized with recombinant replication-deficient RABV-based vaccines to kinetics in animals immunized with the commercially available inactivated human diploid cell vaccine (HDCV) (9–11). Our most promising candidate is a matrix (M) gene-deleted recombinant RABV (rRABV-ΔM) (10). RABV-M protein is crucial for viral assembly and budding, and M gene-deleted RABVs generate a 10,000-fold reduced titer of infectious virions in vitro compared to the parental rRABV grown on wild-type baby hamster kidney cells (12). rRABV-ΔM is grown to high titers (108 focus-forming units [FFU]/ml) on a cell line that supplies RABV-M in trans (10, 12). rRABV-ΔM is safe in T and B cell-deficient Rag2−/− mice and highly immunogenic in relevant animal models (10). A single inoculation of rRABV-ΔM into mice or rhesus macaques induced significantly higher titers of RABV VNAs than those induced by a commercially available HDCV (10). A particular feature of the antibody response to rRABV-ΔM is the presence of VNAs before B cells displaying a germinal center (GC) phenotype are detected, suggesting the induction of early extrafollicular antibody responses by rRABV-ΔM (13). Indeed, contrary to earlier reports citing the necessity of CD4+ T cells for protective RABV-specific B cell responses (14–18), we detected the presence of significant VNA titers within 3 days postimmunization with rRABV-ΔM and protection against lethal challenge in mice completely devoid of T cells (B6.129P2-Tcrβtm1Mom Tcrδtm1Mom/J) (13). Based on these data, we concluded that both early T cell-dependent and independent antibody responses contribute to the early protection observed with rRABV-ΔM. Furthermore, we have shown that interleukin-21 (IL-21) is critical for the induction of rapid primary antibody responses to live RABV-based vaccination (19). Beyond this, little is known about the interactions between live RABV-based vaccines and the host immune system that result in such diverse and potent RABV-specific B cell responses. A better understanding of these interactions would be of great value in the rational design of more effective RABV-based vaccines.
Attenuated RABV strains have been observed to directly interact in vitro with cells of the immune system. In vitro infections by attenuated RABV strains of mouse splenocytes and human T cell lines have been reported to result in apoptosis of infected T cells (20). In addition, murine dendritic cells (BMDCs) and monocytes are stimulated by infection with live RABV in vitro, inducing an antiviral state in the cells (21–23). Both attenuated and pathogenic RABV can establish a productive infection in primary murine bone marrow macrophages (24). Thus, attenuated RABV strains have been appreciated for their ability to infect nonneuronal immune cell types, suggesting that direct infection of host immune cells plays a role in triggering a potent immune response that neutralizes pathogenic RABV strains.
B cells are proficient in major histocompatibility complex class II (MHC-II)-restricted antigen presentation to T cells and express a suite of costimulatory surface receptors that bind to activation molecules on CD4+ T cells, which mutually enhance the interactions of the B cell-T cell pair (25–27). B cells may be activated by a range of stimuli, the most central of which is binding of cognate antigen to the B cell receptor (BCR), leading to receptor cross-linking and development of antibody-secreting or GC phenotypes (28). Activation by pathogen-associated danger signals through Toll-like receptors (TLRs), tumor necrosis factor alpha (TNF-α) family cytokines, such as B cell-activating factor (BAFF) and a proliferation-inducing ligand (APRIL), and cell-associated signals, such as CD40L on activated T cells, also provides vital survival, differentiation, and proliferation signals (25, 26, 28–30). Upon activation in the developing lymph node follicle, B cells migrate to the B cell-T cell zone interface in the paracortex and interact through MHC-II-restricted presentation with CD4+ T cells (31). The importance of B cells for priming of naive CD4+ T cells is controversial, as although this interaction has been observed (25), mice lacking B cells may show proficient activation of CD4+ T cells by other antigen-presenting cells (APCs) (32). However, recent evidence suggests that the B cell-T cell costimulatory interaction is important for fine-tuning activation of CD4+ T cells toward phenotypes most capable of supporting antibody responses (33, 34). The binding of T cell-expressed CD40L to CD40 on the B cell surface is a critical interaction in the development of a high-affinity antibody response, promoting robust B cell proliferation, development, and class switching (35, 36).
In this report, we show that an rRABV vaccine strain infects primary naive murine and human B cells in vitro, causing prompt upregulation of costimulatory molecules important for antigen presentation and effective B cell-T cell costimulatory interactions. With the confirmation that rRABV-infected B cells can prime and activate naive T cells in vitro, we propose that exploiting and enhancing direct B cell-RABV interactions in vivo represents an innovative approach to further enhance RABV-specific antibody responses to immunization. Dissecting B cell responses to live RABV provides novel insight into the highly immunogenic mechanisms underlying live RABV-based vaccine efficacy and aids in the development of more effective RABV-based vaccines.
MATERIALS AND METHODS
Viral vaccines and mice.
The construction of rRABV and rRABV-ΔM used in this study was described elsewhere, and the vaccines were previously named SPBN and SPBN-ΔM, respectively (10). Each vaccine is a molecular clone derived from the attenuated SAD-B19 vaccine strain of RABV (37). Virus stocks of rRABV were propagated in serum-free medium on baby hamster kidney cells and then concentrated and purified over a 20% sucrose cushion. rRABV-ΔM was propagated on baby hamster kidney cells stably expressing RABV-M (12) as described previously (10). rRABV-UV is rRABV that was inactivated by UV irradiation, and inactivation was verified by inoculating baby hamster kidney cells with an aliquot of rRABV-UV followed by immunostaining for RABV nucleoprotein 48 h postinoculation. The detection limit for inactivation is <10 FFU/ml (10, 11). In addition, neuroblastoma (NA) cells were treated with a volume of rRABV-UV equivalent to a multiplicity of infection (MOI) of 0.1, and the culture supernatant was passaged three times at a 1:10 ratio onto fresh NA cells at 3-day intervals. Immunostaining cells of the final passage for RABV-N confirmed inactivation of the rRABV-UV stock used in these studies.
rRABV expressing the gene for chicken ovalbumin (rRABV-OVA) was constructed using mRNA collected from the constitutively OVA-expressing E.G7-OVA cell line (ATCC) using the RNeasy Mini kit according to the manufacturer's instructions (Qiagen). OVA cDNA was generated with the OneStep reverse transcription-PCR (RT-PCR) kit (Qiagen) and amplified using primers OVA-Forward (5′-TTTCGTACGATAATGATATCTCAAGCTGTCCATG-3′) and OVA-Reverse (5′-AAAGCTAGCTTAAGGGGAAACACATCTGCC-3′). OVA cDNA was cloned into rRABV plasmid using the inserted flanking restriction enzyme sites BsiWI and NheI (New England Biosciences) in the viral genome, resulting in rRABV-OVA. The virus was then recovered using techniques previously described (38), and OVA expression was confirmed by immunohistochemistry of infected baby hamster kidney cells and Western blotting of infected NA cell culture supernatants. Supernatants were transferred onto a polyvinylidene difluoride (PVDF) membrane and probed using a polyclonal rabbit anti-chicken ovalbumin primary antibody (Acris) at a dilution of 1:2,500 in phosphate-buffered saline (PBS)–0.05% Tween-20 and a donkey anti-rabbit IgG horseradish peroxidase-conjugated secondary antibody at a dilution of 1:100,000 in PBS–0.05% Tween 20. The membrane was developed using ECL Western blotting substrate (Pierce). Infection of primary murine B cells by rRABV-OVA was confirmed as described below (mouse B cell collection, in vitro culture and staining).
Female C57BL/6 mice aged 6 to 12 weeks were obtained from the National Cancer Institute. Female OT-II [B6.Cg-Tg(TcrαTcrβ)425Cbn/J] mice, aged 10 to 12 weeks, used in the B cell-T cell coculture assay, were purchased from the Jackson Laboratory.
Mouse B cell collection, in vitro culture, and staining.
Single-cell suspensions (106 cells/ml) from spleens or inguinal lymph nodes of naive female C57BL/6 mice were cultured in splenocyte medium (RPMI 1640 containing 10% fetal bovine serum [FBS], 50 μM beta-mercaptoethanol, 100 IU/ml penicillin-streptomycin, and 100 mM HEPES). In experiments using purified B cell cultures, B cells were magnetically isolated from homogenized naive primary mouse splenocytes using the negative-selection-principle MagCellect mouse B cell isolation kit according to the manufacturer's protocol (R&D Systems). Isolated B cell purity was determined by flow cytometry analysis as >95% B220+ cells. Cell suspensions were infected at an MOI of 5 with rRABV, an equivalent volume of the same stock of rRABV-UV, or splenocyte medium and incubated at 37°C with 5% CO2 for 1 to 4 days as indicated in the figures. In separate experiments, total splenocytes were mock infected or infected at an MOI of 5 with rRABV-ΔM for 2 days under the culture conditions described above. Cells were harvested, and 106 cells/sample well were plated on a 96-well plate, pelleted at 300 × g, washed in PBS–2% FBS, and incubated with CD16/32 Fc block (BD Biosciences). Cells were washed as described above and then stained with a surface antibody cocktail, including anti-B220-peridinin chlorophyll protein (PerCP), anti-CD69-Pacific Blue (both from BD Biosciences), anti-MHC-II-Alexa Fluor 700, and anti-CD40-allophycocyanin (both from eBioscience). Cells were fixed in 3% paraformaldehyde in PBS, resuspended in PBS–2% FBS, and stored overnight at 4°C. Samples were then permeabilized using BD Perm/Wash (BD Biosciences) for intracellular staining using monoclonal anti-RABV-N-fluorescein isothiocyanate (FITC) antibody (FujiRebio Diagnostics). Cells were resuspended in PBS–2% FBS for immediate acquisition on a BD LSRII flow cytometer. Data were analyzed using FlowJo (Tree Star) software, and statistical significance was calculated using an unpaired, two-tailed Student's t test in Prism 5 (GraphPad) software.
Human PBMC in vitro culture and staining.
Peripheral blood mononuclear cells (PBMCs) from anonymous healthy adult male donors (Bioreclamation, Inc.) were washed twice in splenocyte medium and cultured at 106 cells/ml of splenocyte medium. Cell suspensions were mock infected with splenocyte medium or infected at an MOI of 5 with rRABV, or an equivalent volume of the same stock of rRABV-UV, and cultured at 37°C and 5% CO2 for 2 or 4 days. Cells were stained in PBS containing aqua blue fixable LIVE/DEAD cell stain (Invitrogen), followed by staining in PBS–2% FBS containing anti-human CD19-phycoerythrin (PE)-Cy7, anti-human HLA-DR-PE, anti-human CD40-allophycocyanin, anti-human CD4-Alexa Fluor 700, and anti-human CD28-eFluor 450 (all eBioscience). Cells were fixed in 3% paraformaldehyde in PBS before permeabilization in BD Perm/Wash (BD Biosciences) for intracellular staining using anti-RABV-N-FITC antibody (FujiRebio Diagnostics). Cells were resuspended in PBS–2% FBS for immediate acquisition on a BD LSRII flow cytometer. Data were analyzed using FlowJo (Tree Star) software, and statistical significance was calculated using an unpaired, two-tailed Student's t test in Prism 5 (GraphPad) software.
Real time-PCR for rabies viral RNA.
B cells were magnetically isolated from homogenized naive primary mouse splenocytes using the MagCellect mouse B cell isolation kit (R&D Systems) and cultured and infected in vitro as described above for 2 or 4 days. Total RNA was purified from the cultured, isolated cells using an RNeasy kit as described by the manufacturer (Qiagen). cDNA was prepared from 2 μg of purified RNA per sample with the Omniscript RT kit (Qiagen) using primers RABV-Genome (5′-CATGGAACTGACAAGAGA-3′) and RABV-Message (5′-TTTTTTTTTTTTTTTTTTTTV-3′; V = G, C, or A) (both from Invitrogen) as described in reference 39. Quantitative real-time PCR was completed on the resulting cDNA using custom forward (5′-CATGGAACTGACAAGAGA-3′) and reverse (5′-TGCTCAACCTATACAGAC-3′) primers for RABV-N antisense genome and RABV-N sense message, respectively, and the custom TaqMan probe (5′-6-carboxyfluorescein [FAM]ATGCGTCCTTAGTCGGTCTTCTC-6-carboxytetramethylrhodamine [TAMRA]-3′) (Applied Biosystems), also as described in reference 39. rRABV plasmid DNA was used as a standard for absolute quantification of gene copy number, and all samples were analyzed at normal speed for 45 cycles on a 0.1-ml 96-well plate in a Life Technologies StepOnePlus thermal cycler. Data were analyzed by the CT method to determine copy number and an unpaired, two-tailed Student's t test to determine statistical significance using StepOne (Life Technologies) and Prism 5 (GraphPad) software.
B cell-T cell in vitro costimulation studies.
B cell-T cell costimulation studies were completed as described by Quah et al. (40), with modifications. Briefly, spleens were harvested from naive female C57BL/6 mice, homogenized, and resuspended in splenocyte medium. B cells were isolated using the negative-selection MagCellect mouse B cell isolation kit as described by the manufacturer (R&D Systems). Purified B cells were cultured in splenocyte medium at 2.5 × 106 cells/ml at 37°C and 5% CO2 with either rRABV-OVA, rRABV-OVA plus 5 μg/ml MHC-II I-Ab blocking antibody (BD Biosciences), rRABV-UV-OVA (all at an MOI of 5), or an equivalent volume of medium. On the same day, spleens were harvested from 10- to 12-week-old naive female OT-II mice, homogenized, and resuspended in splenocyte medium. CD4+ T cells from the OT-II splenocyte suspensions were isolated using the negative-selection MagCellect mouse CD4+ T cell isolation kit (R&D Systems) as instructed by the manufacturer. The purity of T cell isolates was confirmed by flow cytometry analysis (>92% CD3e+ CD4+). Purified CD4+ T cells were washed once in 20 ml of PBS and resuspended at 106 cells/ml in PBS for staining using 5 mM CellTrace violet (Invitrogen) for 20 min at 37°C. Dye was quenched using a 5× staining volume of splenocyte medium before cells were pelleted at 300 × g and resuspended in splenocyte medium. Purified B cells were mixed in a 9:1 ratio with OT-II CD4+ T cells at a combined cell density of 2.5 × 106 cells/ml in 800 μl total culture medium/well of a 24-well plate. B cell-T cell cocultures were cultured at 37°C and 5% CO2 for 120 h. On the day of harvest, BD GolgiStop (BD Biosciences) was added to all wells at a concentration of 0.5 μl GolgiStop per 800 μl medium, and cells were incubated for a further 5 h at 37°C and 5% CO2. Cells were then harvested, washed, and resuspended at 106 cells/ml for staining with LIVE/DEAD fixable aqua blue dead cell stain (Invitrogen). Cells were pelleted at 300 × g, resuspended in PBS–2% FBS, blocked with CD16/32 Fc block (BD Biosciences), and then stained with a surface antibody cocktail including anti-B220-PerCP (BD Biosciences), anti-CD3e-PE, and anti-CD4-allophycocyanin (both eBioscience). Cells were fixed in 3% paraformaldehyde and stored overnight at 4°C in PBS–2% FBS. Samples then were permeabilized using BD Perm/Wash (BD Biosciences) for intracellular staining using anti-RABV-N-FITC antibody (FujiRebio Diagnostics) and anti-IL-2-Alexa Fluor 700 (BD Biosciences). Cells were resuspended in PBS–2% FBS for immediate acquisition on a BD LSRII flow cytometer. Data were analyzed using FlowJo (Tree Star) software, and statistical significance was calculated using an unpaired, two-tailed Student's t test in Prism 5 (GraphPad) software.
RESULTS
Live RABV-based vaccines infect primary murine B cells.
To test our hypothesis that live attenuated rRABV infects B lymphocytes, naive murine primary splenocytes were infected at an MOI of 5 with rRABV or rRABV-UV or were mock infected for 1 to 4 days in vitro. rRABV-UV was included as a negative control to represent current inactivated RABV-based vaccines used in humans and to verify the detection of active infection by live virus, as opposed to the uptake of viral particles not resulting in infection. In an attempt to maintain the B cells and accessory splenocytes in a resting state similar to that in which they would exist in vivo at the time of initial immunization, no additional mitogens were added to the culture. Figure 1A shows a representative gating strategy of total live splenocytes stained for intracellular RABV nucleoprotein (RABV-N) as a marker of infection and the B cell marker B220 (CD45R) (13, 19). By 4 days postinfection, approximately 60% of the splenocyte cultures were B220+ B cells, 10% were CD4+ T cells, and 30% were non-B/non-T cells. Over 10% of B220+ B cells were RABV-N+ as early as 1 day postinfection with live rRABV, increasing to approximately 30% by day 4 postinfection (Fig. 1B). At all time points tested, the proportion of RABV-N+ B220+ cells was significantly higher in rRABV-infected cultures than in cultures treated with rRABV-UV or PBS alone (P < 0.001). A significant proportion of B cells treated with rRABV-UV also stained positive for RABV-N compared to mock-treated cultures. A small proportion of non-B220+ cells stained positive for RABV-N following treatment with rRABV and rRABV-UV, but B cells comprised the majority of the RABV-N+ cells at all time points (Fig. 1A and data not shown).
Fig 1.

rRABV-based vaccines infect naive mouse B220+ B cells from in vitro cultures of primary splenocytes, isolated splenic B cells, or primary lymph node cells. Naive mouse splenocytes or lymph node cells were cultured at 106 cells/ml and infected in vitro with rRABV or rRABV-UV (A to E) or rRABV-ΔM (F) at an MOI of 5 or were mock infected with medium. At the indicated time points postinfection, cells were stained for surface B220 and intracellular RABV-N and analyzed by flow cytometry. Data shown incorporate 5 (B and E) or 3 (D and F) independent cultures per treatment for each time point. (A) Representative gating strategy for total mouse splenocytes gated on live cells and stained for B220 and RABV-N that had been mock infected (left) or infected with rRABV (right). (B) Percentages of live B220+ cells from total splenocyte cultures that were mock infected or infected with rRABV or rRABV-UV and stained as RABV-N+ at the indicated time points. (C) Representative gating strategy for magnetically isolated splenic B cell cultures (>95% B220+ postisolation) gated on live cells and stained for B220 and RABV-N after mock infection (left) or infection with rRABV (right). (D) Percentages of live B220+ cells from isolated splenic B cell cultures that were mock infected or infected with rRABV or rRABV-UV and stained as RABV-N+ at the indicated time points. (E) Percentages of live B220+ cells from total lymph node cell cultures that were mock infected or infected with rRABV or rRABV-UV and stained as RABV-N+ at 4 days postinfection. (F) Percentages of RABV-N+ B220+ cells from total live splenocyte cultures infected with rRABV-ΔM or mock treated at 2 days postinfection. To compare two groups of data, we used an unpaired, two-tailed Student's t test (**, P < 0.01; ***, P < 0.001).
To determine whether rRABV directly infects B cells in the absence of other cell types in culture, isolated primary murine splenic B cells were infected at an MOI of 5 with rRABV or rRABV-UV or were mock treated with PBS alone. Figure 1C shows a representative gating strategy of isolated B cells stained for RABV-N and B220. A significant proportion of B220+ B cells stained positive for RABV-N following exposure to both rRABV and rRABV-UV compared to mock-treated cells (P < 0.01) (Fig. 1D), although it was a lower overall proportion of RABV-N+ cells than total splenocyte cultures (compare Fig. 1B to D). Consistent with our observations with total splenocytes, the percentage of RABV-N staining was significantly greater in isolated B220+ B cells infected with live rRABV than in B cells treated with rRABV-UV (P < 0.001).
To account for heterogeneous B cell populations within the spleen and lymph node (29), the experiments were repeated using homogenized total lymph node cells. By day 4 postinfection, significant proportions of RABV-N+ B220+ B cells were detected in lymph node cultures infected with rRABV compared to cultures treated with rRABV-UV or PBS alone (P < 0.001) (Fig. 1E). Lymph node B cells exhibited a small but significant population of RABV-N+ cells on exposure to rRABV-UV, similar to our results with spleen-derived B cells (compare Fig. 1E to B and D). Overall, our flow cytometry data suggest that primary murine B cell populations within the spleen and lymph node are susceptible to infection with live rRABV and that they are able to take up low levels of rRABV-UV.
As noted previously, rRABV-ΔM is emerging as a promising replication-deficient, RABV-based vaccine to replace current inactivated vaccines. To measure the susceptibility of B cells to infection with rRABV-ΔM, the above-described experiments were repeated using total murine splenocytes that were mock infected or infected at an MOI of 5 with rRABV-ΔM. As shown in Fig. 1F, rRABV-ΔM infects primary murine B cells at a rate comparable to that of rRABV, as detected by intracellular RABV-N staining. Together, these data indicate that both live replication-deficient and replication-competent rRABV-based vaccines infect primary murine B cells.
The potential exists that the RABV-N staining described above in rRABV-infected cultures represents passively engulfed viral particles rather than true infection. Therefore, we examined the expression of viral genome and mRNA in rRABV-exposed B cells using quantitative RT-PCR (qRT-PCR). As expected, no viral genomic (Fig. 2A) or mRNA (Fig. 2B) was detected in mock-treated B cell cultures. Significant levels of viral genomic RNA and mRNA were detected in purified B cells infected with rRABV and B cells treated with rRABV-UV, although higher levels of both genomic RNA and mRNA in live rRABV-infected B cells demonstrate that active replication and viral gene transcription occur within infected B cells. However, the infection of B cells is nonproductive, since passaging supernatants from infected splenocytes onto a susceptible cell line (baby hamster kidney cells) did not produce significant titers of virus (data not shown). Low levels of viral genomic RNA and mRNA were detected in B cells exposed to rRABV-UV. The detection of positive-sense RABV RNA in rRABV-UV-treated B cells is consistent with the uptake of inactive viral particles by B cells. Finke et al. demonstrated that both genomic (negative-sense) and antigenomic (positive-sense) RNA are detected at similar ratios in virus particles and infected cells (41). Of note, as the qRT-PCR primers used in our protocol do not discriminate between positive-sense RABV mRNA and positive-sense RABV antigenome, both of these molecules would be detected using sensitive qRT-PCR methods. Therefore, the low levels of positive-sense mRNA detected in rRABV-UV-exposed B cells are consistent with the detection of RABV antigenome. These studies provide genetic evidence that rRABV directly infects B cells, confirming data from our RABV-N staining-based flow cytometry protocol for detection of infection.
Fig 2.
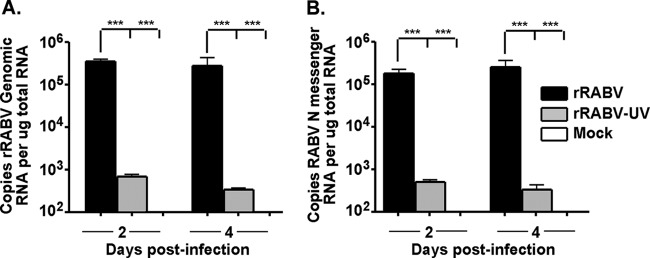
Genomic and messenger rRABV RNA are detected from isolated naive mouse splenic B cells infected with rRABV. B cells were purified from naive total mouse splenocytes using a magnetic selection protocol (>95% B220+ postisolation) and infected at an MOI of 5 with rRABV or rRABV-UV or mock infected with medium. RNA was purified from cells at the indicated time points, and absolute quantitative PCR was performed on samples in triplicate as described in Materials and Methods. Data shown are a combination of 3 independent cultures per treatment for each time point. (A) Copy number of genomic rRABV RNA in isolated B cells infected with rRABV or rRABV-UV or mock infected at the given time points, normalized to 1 μg of total input RNA. (B) Copy number of messenger rRABV RNA in isolated B cells infected with rRABV or rRABV-UV or mock infected at the given time points, normalized to 1 μg of total input RNA. To compare two groups of data, we used an unpaired, two-tailed Student's t test (***, P < 0.001).
Live rRABV-based vaccines activate primary murine B cells.
Infection of B cells by rRABV carries a number of implications for RABV-based vaccine therapy. Previously, we observed prompt and potent B cell activation and antibody secretion by rRABV immunization (13). We hypothesized that direct infection of B cells with rRABV results in early B cell activation, providing additional stimulus to both T-independent and T-dependent antibody responses. Therefore, we measured expression of activation and costimulatory surface markers on in vitro-infected primary murine B cells by flow cytometry following the infection protocols described above. As noted above, rRABV-UV was included as a negative control to represent current inactivated RABV-based vaccines used in humans and to verify that the activation observed results from infection by live virus, as opposed to the activation that results only from viral uptake.
The C-type lectin CD69 is one of the earliest surface activation markers expressed by lymphocytes (42); therefore, it provides insight into the timing of B cell activation by rRABV. Figure 3A shows a representative gating strategy of B220+ B cells stained for CD69 and RABV-N. The number of RABV-N+ CD69hi-expressing B cells was significantly increased at all times postinfection in B cells infected with rRABV compared to B cells treated with rRABV-UV or PBS alone in both total splenocyte cultures (Fig. 3B) and isolated B cell cultures (Fig. 3C). To evaluate differences in levels of CD69 expression within the total infected B cell population, mean fluorescence intensities (MFI) were evaluated for shifts in fluorescence intensities between treatment groups, normalized to the CD69 MFI of B220+ cells from mock-infected cultures. Figure 3D shows a representative histogram of B220+ cells stained for CD69, displaying an upward shift in CD69 staining intensity of rRABV-infected and, to a lesser degree, rRABV-UV-treated B220+ RABV-N+ cells compared to that of mock-treated cultures. The CD69 staining intensity of B220+ RABV-N+ cells was significantly increased over mock infection intensity by day 1 postinfection with rRABV in both total splenocyte (Fig. 3E) and purified B cell (Fig. 3F) cultures. In splenocyte cultures, rRABV-UV stimulated CD69 surface expression on B220+ RABV-N+ cells above mock expression levels but to significantly lower intensities and at later time points postinfection than those of rRABV-infected cultures (Fig. 3E). In contrast, in purified B cell cultures, CD69 upregulation on RABV-N+ B cells in rRABV-UV-exposed cultures exceeded levels on RABV-N+ B cells from live rRABV-infected cultures (Fig. 3F). However, despite the greater intensity of CD69 expression on RABV-N+ B220+ cells from rRABV-UV-stimulated B cell cultures, the overall number of CD69hi cells was higher in rRABV-infected cultures (approximately 7,750 CD69 cells/100,000 live B220+ cells) than in rRABV-UV-treated cultures (approximately 3,500 CD69 cells/100,000 live B220+ cells) (Fig. 3C). Overall, the early upregulation of CD69 observed after exposure to rRABV suggests prompt activation of B cells, particularly in the presence of accessory splenocytes in culture.
Fig 3.

B220+ mouse B cells from total splenocyte and isolated B cell cultures infected with rRABV upregulate surface expression of the early activation marker CD69. Total mouse splenocytes and isolated splenic B cells, cultured and infected as described in the legend to Fig. 1, were stained for surface B220 and CD69, as well as intracellular RABV-N, for analysis by flow cytometry. Data shown are a combination of 5 (B and E) or 3 (C and F) independent cultures per treatment for each time point. (A) Representative flow cytometry gating strategy for live B220+ cells from total mouse splenocyte cultures gated on surface CD69 and intracellular RABV-N staining. (B) Numbers of live RABV-N+ CD69hi B220+ B cells normalized to the number of live cells from total splenocyte cultures infected with either rRABV or rRABV-UV, or mock infected, at the indicated time points. (C) Numbers of live RABV-N+ CD69hi B220+ B cells, normalized to live purified B220+ cells from isolated B cell cultures that were mock infected or infected with either rRABV or rRABV-UV at the indicated time points. (D) Representative histogram showing staining intensity of surface CD69 expression in live B220+ B cells from total splenocyte cultures that were mock infected or infected in vitro with either rRABV or rRABV-UV. (E) Mean fluorescence intensity (MFI) of CD69 staining on live RABV-N+ B220+ B cells from total splenocyte cultures infected in vitro with either rRABV (black bars) or rRABV-UV (gray bars), normalized to the CD69 MFI of B220+ B cells from mock-infected total splenocyte cultures. (F) MFI of CD69 staining on live RABV-N+ B220+ B cells from isolated splenic B cell cultures infected with either rRABV (black bars) or rRABV-UV (gray bars), normalized to the CD69 MFI of B220+ B cells from mock-infected total splenocyte cultures. To compare two groups of data, we used an unpaired, two-tailed Student's t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
B cells are professional APCs that can process and present antigen to CD4+ helper T cells alongside expression of costimulatory surface markers that serve as a second signal to activate T cells. MHC-II plays an essential role in this process as the vehicle for antigenic peptide presentation to the complementary specific T cell receptor (TCR). MHC-II can be upregulated in B cells, as in other professional APCs, by stimulation through a number of PAMP-sensitive pathways, such as TLR ligation or intracellular viral RNA sensors (43), as well as through the B cell receptor-mediated activation pathway (26). CD40 is a constitutively expressed B cell surface marker which, upon ligation to CD40L on activated T cells potently stimulates B cell maturation, antibody secretion, class switching, and division, as well as providing further positive feedback to the activated T cell (30, 35). Thus, the upregulation of MHC-II and CD40 reflects B cell activation to an antigen-presenting, costimulatory phenotype. Figures 4A and 5A show representative gating strategies for live B220+ cells gated for MHC-IIhi or CD40hi, respectively, and RABV-N. Notably, the number of RABV-N+ MHC-IIhi- or RABV-N+ CD40hi-expressing B cells was significantly higher in rRABV-infected cultures at all times postinfection than in rRABV-UV-infected or PBS-treated total splenocyte cultures (Fig. 4B and 5B) or isolated B cells (Fig. 4C and 5C). To evaluate differences in MHC-II or CD40 cell surface expression within the total infected B cell population, mean fluorescence intensities (MFI) (again, normalized to MFI levels of mock-infected B220+ cells) were evaluated for shifts by treatment. Figures 4D and 5D show representative histograms of B220+ cells stained for MHC-II and CD40, respectively. Compared to staining levels in mock-treated B220+ cells, the intensity of MHC-II and CD40 expression was significantly increased in B220+ RABV-N+ B cells by day 1 postinfection with rRABV in both total splenocyte (Fig. 4E and 5E) and purified B cell (Fig. 4F and 5F) cultures. Consistent with our observations of the expression of CD69 (Fig. 3), in total splenocyte cultures (Fig. 4E and 5E) surface expression levels of MHC-II or CD40 were upregulated more intensely in rRABV-infected RABV-N+ B cells than in rRABV-UV-exposed RABV-N+ B cells. In contrast, data from isolated B cell cultures generally indicated no significant differences in CD40 or MHC-II upregulation on RABV-N+ B cells between rRABV and rRABV-UV cultures. The MFI of MHC-II and CD40 on RABV-N+ B cells from both splenocyte and purified B cell cultures treated with rRABV-UV also increased compared to levels on mock-treated B cells (Fig. 4E and F and 5E and F). Activation occurs to a significantly lesser degree by treatment with rRABV-UV, as despite similar marker MFI upregulation to rRABV in isolated B cell cultures, significantly fewer RABV-N+ CD40hi/MHC-IIhi cells are detected from both splenocyte and isolated B cell cultures treated with rRABV-UV than from cultures treated with rRABV. However, even this level of activation by rRABV-UV was unexpected, as we included the inactive virus as a negative control to specifically check for the effects of live viral exposure on B cell activation. Overall, these data support a model of early activation of B cells to an antigen-presenting T-cell costimulatory phenotype by rRABV infection.
Fig 4.

B220+ mouse B cells from total splenocyte and isolated B cell cultures infected in vitro with rRABV upregulate surface expression of the antigen presentation molecule MHC-II. Total mouse splenocytes and isolated splenic B cells, cultured and infected as described in the legend to Fig. 1, were stained for surface B220 and MHC-II, as well as intracellular RABV-N, for analysis by flow cytometry. Data are from the experiments and number of replicates per treatment described in the legend to Fig. 3. (A) Representative gating strategy of cells, as described in the legend to Fig. 3A, gated on surface MHC-II and intracellular RABV-N staining. (B and C) Number of live RABV-N+ MHC-IIhi B220+ B cells normalized to the number of live cells from total splenocyte cultures (B) or live B220+ cells from isolated B cell cultures (C) that were mock infected or infected in vitro with either rRABV or rRABV-UV at the indicated time points. (D) Representative histogram showing staining intensity of surface MHC-II expression in live B220+ B cells from total splenocyte cultures either mock infected or infected in vitro with either rRABV or rRABV-UV. (E and F) MFI of MHC-II staining on live RABV-N+ B220+ B cells from total splenocyte cultures (E) or isolated B cell cultures (F) infected with either rRABV (black bars) or rRABV-UV (gray bars), normalized to the MHC-II MFI of B220+ B cells from corresponding mock-infected cultures. To compare two groups of data, we used an unpaired, two-tailed Student's t test (***, P < 0.001).
Fig 5.

B220+ mouse B cells from total splenocyte and isolated B cell cultures infected with rRABV upregulate surface expression of the survival and costimulatory molecule CD40. Cells cultured and infected as described in the legend to Fig. 1 were stained for surface B220 and CD40, as well as intracellular RABV-N, for analysis by flow cytometry. Data are from the same experiments and number of replicates per treatment described in the legend to Fig. 3. (A) Representative gating strategy of cells, as described in the legend to Fig. 3A, gated on surface CD40 and intracellular RABV-N staining. (B and C) Number of live RABV-N+ CD40hi B220+ B cells normalized to the number of live cells from total splenocyte cultures (B) or live B220+ cells from isolated B cell cultures (C) that were mock infected or infected in vitro with either rRABV or rRABV-UV at the indicated time points. (D) Representative histogram showing staining intensity of surface CD40 expression in live B220+ B cells from total splenocyte cultures infected in vitro with either rRABV or rRABV-UV or mock infected. (E and F) MFI of CD40 staining on live RABV-N+ B220+ B cells from total splenocyte cultures (E) or isolated splenic B cell cultures (F) infected in vitro with either rRABV (black bars) or rRABV-UV (gray bars), normalized to the MHC-II MFI of B220+ B cells from corresponding mock-infected cultures. To compare two groups of data, we used an unpaired, two-tailed Student's t test (*, P < 0.05; **, P < 0.01; ***, P < 0.001).
B cells infected with rRABV present antigen to CD4+ OT-II T cells, resulting in MHC-II-restricted T cell proliferation and IL-2 expression.
The preceding data indicate that rRABV infects B cells, resulting in B cell activation and upregulated costimulatory marker expression. These observations suggest the potential for infected B cells to serve as rabies antigen-presenting cells to CD4+ T cells. To establish this capacity of RABV-infected murine B cells, we utilized a well-defined in vitro assay based on the ability of primary APCs to process and present chicken ovalbumin antigen to naive CD4+ T cells isolated from OT-II mice, which express a transgenic TCR recognizing OVA323-339 peptide in the context of MHC-II I-Ab (44). rRABV-encoded expression of OVA, rather than a natural RABV antigen, was chosen as the stimulus in these studies, as no equivalent rabies antigen-specific transgenic T cell model exists and because the assay using OT-II T cells is well established in the literature (40). An rRABV that expresses OVA (rRABV-OVA) was constructed, recovered, and characterized (Fig. 6A). OVA expression was verified by immunohistochemistry staining and Western blot analysis of in vitro-infected cells and culture supernatants using rabbit anti-chicken ovalbumin antibody (Fig. 6B), and the virus was confirmed to infect B cells similarly to rRABV by flow cytometry (Fig. 6C). On average, 11.9% of the OT-II CD4+ T cells stained positive for division after coculture with rRABV-OVA-infected B cells (Fig. 6D). T cell proliferation was not observed following coculture with B cells treated with UV-inactivated rRABV-OVA or with media alone. CD4+ T cell activation by rRABV-OVA-infected B cells was confirmed by staining for intracellular expression of IL-2, one of the earliest cytokines expressed by primed and activated T cells (45). On average, 14.2% of OT-II CD4+ T cells stained for IL-2 when cocultured with rRABV-OVA-infected B cells, compared to 2.5% background IL-2 staining observed in OT-II CD4+ T cells from rRABV-OVA-UV and mock-treated cocultures (Fig. 6E). Both OT-II CD4+ T cell proliferation and IL-2 expression were dependent on functional B cell MHC-II expression, since treatment of rRABV-OVA-stimulated cocultures with anti-MHC-II I-Ab blocking antibody significantly reduced OT-II CD4+ T cell IL-2 expression and reduced the percentage of divided cells to nonsignificance over background levels. To confirm the absence of effects from contaminating antigen-presenting cells in the isolated OT-II CD4+ T cell preparations used in these studies, magnetically isolated T cell suspensions were cultured and stimulated with either 25 μg/ml lipopolysaccharide purified from Escherichia coli strain O111:B4 (LPS-EB) and 150 μg/ml chicken ovalbumin or rRABV-OVA for 5 days. No significant levels of divided OT-II CD4+ T cells or IL-2 expression were observed above background levels from these cultures, from mock-treated OT-II CD4+ T cell cultures, or from B cell-T cell cocultures with rRABV not expressing OVA (data not shown). We conclude that naive primary C57BL/6 B cells infected with rRABV are able to process and present virally encoded antigens to naive CD4+ T cells, thereby priming and activating antigen-specific helper T cells in an MHC-II-restricted manner.
Fig 6.

Isolated naive mouse splenic B cells infected with rRABV-OVA stimulate proliferation and IL-2 secretion of naive OVA-specific OT-II CD4+ T cells in an in vitro coculture assay. Splenic B cells were isolated to a purity of >95% B220+ B cells and either mock infected with medium or infected at an MOI of 5 with rRABV-OVA, inactivated rRABV-UV-OVA, or rRABV-OVA combined with a I-Ab MHC-II blocking antibody. Infected B cells were cocultured with CellTrace-violet-stained isolated OT-II T cells (>92% CD3ε+ CD4+) for 120 h, and then harvested cells were stained for intracellular IL-2 and analyzed as described in Materials and Methods. (A) rRABV expressing ovalbumin (OVA) was constructed by cloning the chicken ovalbumin gene into the rRABV backbone, taking advantage of restriction enzyme sites inserted between the glycoprotein (G) and polymerase (L) genes. rRABV-OVA was recovered, characterized, and grown on baby hamster kidney cells. rRABV-UV-OVA was inactivated by UV light exposure for 10 min to <10 FFU/ml. (B) Western blot for ovalbumin from supernatants of neuroblastoma cell cultures infected with rRABV or rRABV-OVA. Purified chicken ovalbumin protein was included as a control. (C) Representative dot plots of B220+ RABV-N+-infected B cells from naive mouse splenocytes that were mock infected or infected at an MOI of 5 with rRABV-OVA or rRABV for 2 days. (D) Dividing CD4+ T cells, determined by diminution of CellTrace-violet staining intensity as a percentage of total CD4+ T cells from in vitro cocultures that were mock infected or exposed to rRABV-OVA, rRABV-OVA plus anti-MHC-II blocking antibody, or rRABV-UV-OVA for 120 h. (E) IL-2hi CD4+ T cells, determined by intracellular antibody staining as a percentage of total CD4+ T cells from in vitro cocultures exposed to rRABV-OVA, rRABV-OVA plus anti-MHC-II blocking antibody, rRABV-UV-OVA, or mock infection for 120 h. The inset shows a representative gating strategy histogram for IL-2hi CD4+ T cells. To compare two groups of data, we used an unpaired, two-tailed Student's t test (*, P < 0.05; ***, P < 0.001).
rRABV infects and activates primary human B cells.
Human lymphocytes were previously shown to have distinct responses and susceptibility to rabies infection compared to murine lymphocytes (20). To confirm that rRABV can also infect and activate primary human B cells, Ficoll-separated PBMCs from six healthy male donors were cultured with rRABV, rRABV-UV, or an equivalent volume of medium as a mock infection control, all in the absence of any additional mitogen in the culture medium. None of the donors had a history of rabies immunization, and all donors tested negative for serum titers of rabies VNA (data not shown). By staining for dead cells at the time of culture harvests using a fixable LIVE/DEAD stain, we found that, on average, 4.4% of live and 5.9% of dead B cells from PBMC cultures infected with rRABV were RABV-N+ by day 4 postinfection, which was significantly higher than the background level of RABV-N+ staining from PBMC cultures treated with rRABV-UV or media alone (Fig. 7C). In contrast to our data for murine B cells, rRABV-UV treatment of human PBMCs did not elevate the percentage of RABV-N+ B cells above the background level detected in mock-treated cultures.
Fig 7.

Human B and T cells from healthy nonimmunized donor PBMCs are infected by rRABV in vitro and upregulate the markers of antigen presentation and costimulation, HLA-DR, CD40, and CD28. Freshly isolated PBMCs were cultured at a density of 106 cells/ml and mock infected or infected with either rRABV or rRABV-UV at an MOI of 5 for 4 days. Cells were harvested and stained for B and T cell lineage marker expression, as well as costimulatory activation markers, for analysis by flow cytometry. (A) Representative gating strategy for live (LIVE/DEAD stain low) human cells stained for CD19 and RABV-N from mock-infected (left) or rRABV-infected (right) cultures. (B) Representative gating strategy for dead (LIVE/DEAD stain high) human cells stained with CD4 and RABV-N from mock-infected (left) or rRABV-infected (right) cultures. (C) Percentages of live and dead CD19+ B cells staining RABV-N+ 4 days postinfection in vitro from cultures infected with rRABV or rRABV-UV or mock infected. (D) Percentages of live and dead CD4+ T cells staining RABV-N+ 4 days postinfection in vitro from cultures infected with rRABV or rRABV-UV or mock infected. (E) MFI of HLA-DR and CD40 staining on days 2 and 4 postinfection of RABV-N+ CD19+ B cells from PBMC cultures infected with rRABV (black bars) or rRABV-UV (gray bars), normalized to the MFI of HLA-DR or CD40 on CD19+ B cells from mock-infected cultures. (F) MFI of CD28 staining on RABV-N+CD4+ T cells from PBMC cultures infected with rRABV (black bar) or rRABV-UV (gray bar), normalized to the MFI of CD28 on CD4+ T cells from mock-infected cultures. To compare two groups of data, we used an unpaired, two-tailed Student's t test (*, P < 0.05).
Prior reports indicated that live-attenuated RABVs induce apoptosis in primary human T cells (20), so we ascertained the level of T cell death in our PBMC samples exposed to RABV. In contrast to B cells, of which both live and dead cells stained positive for RABV-N (Fig. 7A and C), only dead CD4+ T cells stained positive for RABV-N (Fig. 7D). Together, the data indicate that rRABV is able to infect B cells from both humans and mice, although the proportion of human B cells infected with rRABV was lower than that of the infection observed with murine B cells. Additionally, the data are consistent with previous reports that human CD4+ T cells infected by live rRABV in vitro undergo cell death, although the cause and timing of infected T cell death were not determined here. In contrast, however, only a subset of infected human B cells appeared to undergo cell death in response to rRABV infection. Additional research is required to investigate whether the cell death observed in infected primary human B cells is due to apoptosis and/or necrosis, as well as whether B cell-derived apoptotic bodies aid in promoting anti-RABV immunity similar to apoptotic bodies from mouse T cell lines expressing RABV-G (20). Nonetheless, our data indicate that live RABV-based vaccines infect primary RABV-naive human B cells.
Consistent with our activation data on primary murine B cells, rRABV also activated B cells derived from human PBMCs. Significantly elevated expression levels of HLA-DR and CD40 were detected in B cells from PBMC cultures exposed to rRABV compared to B cells from mock-infected controls (Fig. 7E). Furthermore, significantly elevated expression levels of CD28, which serves as the major early activating receptor on CD4+ T cells for the costimulatory CD80/CD86 pair on activated B cells, was detected in RABV-N+ CD4+ T cells from PBMC cultures infected with rRABV compared to T cells from mock-infected PBMC cultures (Fig. 7F). rRABV-UV-exposed B cells showed no upregulation of HLA-DR or CD40 expression, and rRABV-UV-exposed CD4+ T cells also showed no upregulation of CD28 expression compared to the mock-treated control. These data indicate that live but not UV-inactivated rRABV stimulates costimulatory marker expression on both human B and T cells, supporting the translatability of our results with murine live rRABV-infected and activated B cells to human live RABV-based vaccine immunization.
DISCUSSION
The data presented in this report show that live-attenuated rRABV-based vaccines infect primary murine and human B cells. Infection results in the expression of cell surface markers with the ability to induce primary naive murine T cells to proliferate and secrete IL-2, confirming the functional capabilities of B cells infected with rRABV. While it was previously shown that RABV infects primary T cells (20), DCs (46), and macrophages (24), the observation that attenuated rRABV specifically infects and activates primary mouse and human B lymphocytes is a novel finding.
Our data indicate that B cells in total splenocyte cultures are infected and activated more efficiently than isolated B cells. While the differences in percentage of infected B cells in total splenocyte cultures compared to purified B cells may be attributed to the overall poorer survival of isolated B cells in vitro, the data also suggest the existence of costimulatory effects by non-B cells in the culture that enhance infection and activation of B cells. This is consistent with previous findings that show that primary T cells cultured in the presence of mitogens are also more susceptible to RABV infection than are quiescent T cells (20). The lack of accessory costimulatory cells may also be a factor in the more even MFI levels of activation marker staining of rRABV- and rRABV-UV-treated isolated B cell cultures. Lacking cytokine or cell-associated survival signal support, cells of high activation states will undergo positive selection in the culture and be the only cells to survive out to 2 or 4 days postinfection. In any case, our central goal was not to compare activation induced by live versus inactivated virus but rather to discover novel interactions between live rRABV and B cells. The limited but significant activation of B cells by inactive rRABV-UV suggests that B cells are weakly sensitive to RABV antigens and/or RNA even in the absence of any live viral activity. However, our data incorporating both activated cell counts and activation marker MFIs make clear that a significant population of B cells, whether cultured in the presence of accessory cells or isolated, is infected and activated by rRABV. Future work is needed to identify any specific costimulatory molecules, such as cytokines or T cell-derived activation signals, which enhance B cell activation and APC function in the context of rRABV infection.
rRABV-infected and -activated B cells not only upregulate important molecules involved in efficient T cell-B cell interactions but also are functionally capable of directly priming and activating naive T cells to proliferate and secrete IL-2. This is consistent with the appreciation that B cell functions in an immune response include serving not only as precursors to antibody-secreting cells but also as prominent mediators of MHC-II antigen presentation to T cells. Prompt B cell encounters with T cells postimmunization was demonstrated in a model using hen egg lysozyme, in which antigen-specific B cell-T cell conjugates form in the spleen as early as 30 h postimmunization (31). This indicates that B cells have the ability to interact with T cells rapidly in secondary lymphoid organs; thus, they have the potential for mediating early priming and activation (26). Indeed, B cells have been shown to directly and independently prime T cells both in vitro (47) and in vivo (48, 49), although the importance of the contribution of B cells to antigen presentation to CD4+ T cells, compared to the contribution of DCs and macrophages, is highly antigen and model specific (50, 51). Thus, while the participation of B cells in initial T cell priming steps remains controversial, recent data suggest B cells are of importance to fine-tune the functional qualities of CD4+ helper T cells already primed by other APCs, since B cell-deficient mice show deficits in T cell cytokine secretion and B cell help provided by activated T cells postimmunization (34).
A stimulus of particular importance to B cell APC activity is signaling through CD40. The CD40-CD154 receptor/ligand pair on B and T cells maintains elevated levels of MHC-II and costimulatory molecules, such as CD80/CD86, on the surface of activated B cells, further promoting effective T cell-B cell interactions (35, 36, 52). Thus, in addition to BCR-mediated activation, B cells may be stimulated by a number of extracellular or intracellular signals, such as TLR ligation, BAFF-R or TACI signaling, or intracellular sensors of viral RNA, such as RIG-I-like receptors (28, 53). Which of these signals is of greatest importance for rRABV stimulation of B cells is a mechanistic question beyond the scope of the current study; however, it seems that B cell activation is not solely mediated by specific binding to antigen recognition regions of the BCR based on the proportion of B cells from RABV naive mice being infected and activated by rRABV. Overall, the independent upregulation of activation markers on polyclonal B cells following infection by rRABV may help to promote effective antigen presentation to naive T cells as well as effective postprimed T cell-B cell interactions in secondary lymphoid organs, questions which require further in vivo study.
The ability of rRABV to infect and activate B cells can be compared to other select viruses that also infect B cells, inducing various outcomes for both the host and virus. These include, most notably and classically, Epstein-Barr virus, which polyclonally infects B cells, resulting in broad B cell activation, antibody secretion, and lymphomagenesis (54). In contrast, a subset of in vitro-activated human B cells expressing the C-type lectin DC-SIGN are infected by human herpesvirus 8 (HHV-8), resulting in downregulation of CD20 and MHC-I on the B cell surface that aids immunoevasion by the virus (55). A human B cell line infected with hepatitis C virus was established using non-Hodgkin's B cell lymphoma tissue from an infected patient, indicating that HCV is capable of infecting B cells both in vivo and in vitro (56). Examples also exist of virally infected and activated B cells functioning as APCs, as we found in this study. The retrovirus mouse mammary tumor virus (MMTV) infects and activates B cells through the TLR4 receptor and uses the interaction of these infected B cells with antigen-specific T cells to further viral spread (57). The negative-sense single-stranded RNA respiratory syncytial virus (RSV), which is tropic for lower respiratory tract epithelial cells, was found to persistently infect bovine airway-resident B cells in vivo and continuing under ex vivo culture (58). Human RSV was found to infect and stimulate in vitro-cultured mouse B cells, causing prompt upregulation of markers of activation and costimulation (59). Thus, B cells are infected by a limited range of viruses, which can influence in diverse ways the pathogenicity of the virus or immunity of the host, based on the conditions of infection and phenotype of the B cells. As such, while we have established that rRABV infection of naive primary B cells results in an antigen-presenting phenotype that can directly prime naive T cells, it is possible that B cells at different stages of development or activation respond differently to rRABV infection, including by apoptosis, as was observed for T lymphocytes. Similar to other systems where viral infection was shown to activate B cells, the importance of live RABV infection in our system is demonstrated by the observation that equivalent levels of B cell activation were not observed with exposure to inactivated rRABV compared to an equivalent initial dose of live virus. Interestingly, our inactivated RABV-based vaccine activates primary murine B cells (albeit to lower levels than live RABV-based vaccines, as just described), potentially through B cell-intrinsic signaling events, such as TLR ligation. Our finding that inactivated RABV can activate B cells is consistent with observations of other inactivated viruses, such as influenza virus (60) or Chandipura virus (61). However, despite demonstrating limited activation of primary B cells by inactivated RABV-based vaccines, our results support the superiority of live RABV-based vaccines for promoting effective B cell responses.
While our present studies suggest a role for rRABV-infected B cells as APCs for helper T cells, DCs typically are recognized as more important initiators of the T cell-dependent immune response against a wide range of viral infections (62). This includes RABV infection, as human immature DCs and monocytes were found to be infected by both attenuated and pathogenic RABV in vitro, leading to upregulation of costimulatory markers CD80/CD86 and maturation of infected cells (23). Mouse bone marrow-derived DCs are also infected and activated by RABV, leading to interferon production and costimulatory marker expression by RIG-I-like receptor-mediated mechanisms (22). Thus, the contribution of DCs, being more specialized for surveillance of the initial site of infection in peripheral tissues, cannot be discounted, and we hypothesize that B cell infection and activation serve alongside DC stimulation to generate the prompt and potent antibody responses observed following immunization with live rRABV-based vaccines. Thus, while the role of DCs as APCs centers on their ability to detect antigen from barrier surfaces and undergo independent activation in peripheral tissues through a suite of PAMP sensors (63), the APC capabilities of B cells may result from their activation in secondary lymphoid organs containing concentrated T cell populations, putting them in prime position to fine-tune the helper T cell response toward the development of an appropriate antibody response and cytokine profile.
To translate our findings from the murine system to humans, we also show that rRABV is able to infect and activate B cells derived from primary human PBMCs, although the level of infection of human PBMC B cells is reduced compared to that for mouse B cells. The lower proportion of B cells in human PBMCs (6 to 8%) compared to the proportion in mouse secondary lymphoid organs (20 to 60%) may contribute to differences of infection rates between mice and humans. The phenotype of recirculating B cells contained in PBMCs differs from that of B cells residing in secondary lymphoid organs, which also may account for discrepancies (64, 65). Activation marker upregulation by RABV-infected human B cells was also diminished compared to that of murine B cells. Human and mouse B cells differ in their expression of and sensitivity to pathogen sensors, such as TLRs. In particular, the murine and human TLR7 and TLR8, while both sensing single-stranded RNA, differ in their sensitivity to pharmacological activators (66), and with age, mice acquire a B cell subset that is refractory to BCR-mediated activation but uniquely sensitive to TLR7 stimulation (67). Thus, a combination of diminished infection and differing sensitivity to activation by rabies may be responsible for the weaker impact of in vitro RABV infection on human B cell activation and for the lack of any response by human B cells to rRABV-UV. Nonetheless, rRABV was able to significantly infect and activate primary human B cells, supporting translation of our murine findings to humans.
Previously we described the early, protective, extrafollicular antibody response to immunization with replication-deficient rRABV-ΔM as containing both T-independent and T-dependent antibody production components (13). Our discovery that B cells infected with rRABV are activated to a costimulatory antigen-presenting phenotype suggests a novel means of initiation and support of the early T-dependent component of this antibody response. We suggest a model whereby polyclonal draining lymph node B cells infected by rRABV in the hours or days immediately following immunization serve as antigen-presenting cells for rabies-specific CD4+ T cells in the B cell-T cell border of the follicle. The localized T cell activation may then in turn quickly provide help to RABV-specific B cells, driving them toward early IgG class-switched antibody production. This sequence of events is consistent with our previous data showing the rapid induction of T cell-dependent IgG isotype antibodies within 5 days of immunization and could contribute to the efficacy of a live, single-dose RABV-based vaccine (10, 13). Possible T-independent effects on the RABV-specific antibody response, such as the stimulation of antibody secretion or class-switching in RABV-infected B cells, were not examined in the current study but remain to be elucidated, as T-independent antibody production is an important mechanism contributing to the rapid antibody responses needed in postexposure settings (13).
In summary, we discovered and characterized the in vitro infection and activation of naive mouse and human B cells by rRABV and found that rRABV-infected mouse B cells directly prime and activate naive CD4+ T cells in vitro in an antigen-specific, MHC-II-dependent manner. While it is unlikely that a live replication-competent RABV-based vaccine would replace currently licensed human rabies vaccines due to safety concerns, these findings lay the groundwork for future studies of B cell-RABV interactions, as well as for the design of a single-dose RABV-based vaccine that specifically targets B cells for infection and activation.
ACKNOWLEDGMENTS
This work was supported by NIH/NIAID grant R01AI079211 to J.P.M.
We thank Ran You for recovering the rRABV-OVA virus used in the study and Matthew Farabaugh of the Kimmel Cancer Center Flow Cytometry Facility for assistance.
Footnotes
Published ahead of print 12 June 2013
REFERENCES
- 1. Dietzschold B, Rupprecht CE, Fu ZF, Koprowski H. 1996. Rhabdoviruses, p 1137–1152 In Fields BN. (ed), Fields virology. Lippincott-Raven Publishers, Philadelphia, PA [Google Scholar]
- 2. WHO 2013. WHO rabies fact sheet no. 99. WHO, Geneva, Switzerland [Google Scholar]
- 3. Plotkin SA. 2008. Vaccines: correlates of vaccine-induced immunity. Clin. Infect. Dis. 47:401–409 [DOI] [PubMed] [Google Scholar]
- 4. Turner GS. 1978. Immunoglobulin (IgG) and (IgM) antibody responses to rabies vaccine. J. Gen. Virol. 40:595–604 [DOI] [PubMed] [Google Scholar]
- 5. WHO 1997. WHO recommendations on rabies post-exposure treatment and the correct technique of intradermal immunization against rabies. WHO, Geneva, Switzerland [Google Scholar]
- 6. Moore SM, Hanlon CA. 2010. Rabies-specific antibodies: measuring surrogates of protection against a fatal disease. PLoS Negl. Trop. Dis. 4:e595. 10.1371/journal.pntd.0000595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. WHO 2010. WHO recommendations for active rabies immunization after exposure where rabies vaccines are in short supply. WHO, Geneva, Switzerland [Google Scholar]
- 8. Rupprecht CE, Briggs D, Brown CM, Franka R, Katz SL, Kerr HD, Lett SM, Levis R, Meltzer MI, Schaffner W, Cieslak PR, Centers for Disease Control and Prevention 2010. Use of a reduced (4-dose) vaccine schedule for postexposure prophylaxis to prevent human rabies: recommendations of the advisory committee on immunization practices. MMWR Recomm. Rep. 59:1–9 [PubMed] [Google Scholar]
- 9. McGettigan JP. 2010. Experimental rabies vaccines for humans. Expert Rev. Vaccines 9:1177–1186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cenna J, Hunter M, Tan GS, Papaneri AB, Ribka EP, Schnell MJ, Marx PA, McGettigan JP. 2009. Replication-deficient rabies virus-based vaccines are safe and immunogenic in mice and nonhuman primates. J. Infect. Dis. 200:1251–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cenna J, Tan GS, Papaneri AB, Dietzschold B, Schnell MJ, McGettigan JP. 2008. Immune modulating effect by a phosphoprotein-deleted rabies virus vaccine vector expressing two copies of the rabies virus glycoprotein gene. Vaccine 26:6405–6414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mebatsion T, Weiland F, Conzelmann KK. 1999. Matrix protein of rabies virus is responsible for the assembly and budding of bullet-shaped particles and interacts with the transmembrane spike glycoprotein G. J. Virol. 73:242–250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dorfmeier CL, Lytle AG, Dunkel AL, Gatt A, McGettigan JP. 2012. Protective vaccine-induced CD4+ T cell-independent B cell responses against rabies Infection. J. Virol. 86:11533–11540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jackson AC. 2008. Rabies. Neurol. Clin. 26:717–726 [DOI] [PubMed] [Google Scholar]
- 15. Turner GS. 1976. Thymus dependence of rabies vaccine. J. Gen. Virol. 33:535–538 [DOI] [PubMed] [Google Scholar]
- 16. Mifune K, Takeuchi E, Napiorkowski PA, Yamada A, Sakamoto K. 1981. Essential role of T cells in the postexposure prophylaxis of rabies in mice. Microbiol. Immunol. 25:895–904 [DOI] [PubMed] [Google Scholar]
- 17. Bunschoten H, Dietzschold B, Claassen I, Klapmuts R, Uytdehaag F, Osterhaus A. 1990. Rabies virus cross-reactive murine T cell clones: analysis of helper and delayed-type hypersensitivity function. Viral Immunol. 3:41–53 [DOI] [PubMed] [Google Scholar]
- 18. Perry LL, Lodmell DL. 1991. Role of CD4+ and CD8+ T cells in murine resistance to street rabies virus. J. Virol. 65:3429–3434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dorfmeier CL, Tzvetkov EP, Gatt A, McGettigan JP. 2013. Investigating the role for IL-21 in rabies virus vaccine-induced immunity. PLoS Negl. Trop. Dis. 3:e2129. 10.1371/journal.pntd.0002129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Thoulouze MI, Lafage M, Montano-Hirose JA, Lafon M. 1997. Rabies virus infects mouse and human lymphocytes and induces apoptosis. J. Virol. 71:7372–7380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wanjalla CN, Faul EJ, Gomme EA, Schnell MJ. 2010. Dendritic cells infected by recombinant rabies virus vaccine vector expressing HIV-1 Gag are immunogenic even in the presence of vector-specific immunity. Vaccine 29:130–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Faul EJ, Wanjalla CN, Suthar MS, Gale M, Wirblich C, Schnell MJ. 2010. Rabies virus infection induces type I interferon production in an IPS-1 dependent manner while dendritic cell activation relies on IFNAR signaling. PLoS Pathog. 6:e1001016. 10.1371/journal.ppat.1001016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li J, McGettigan JP, Faber M, Schnell MJ, Dietzschold B. 2008. Infection of monocytes or immature dendritic cells (DCs) with an attenuated rabies virus results in DC maturation and a strong activation of the NFkappaB signaling pathway. Vaccine 26:419–426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ray NB, Ewalt LC, Lodmell DL. 1995. Rabies virus replication in primary murine bone marrow macrophages and in human and murine macrophage-like cell lines: implications for viral persistence. J. Virol. 69:764–772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Rodriguez-Pinto D, Moreno J. 2005. B cells can prime naive CD4+ T cells in vivo in the absence of other professional antigen-presenting cells in a CD154-CD40-dependent manner. Eur. J. Immunol. 35:1097–1105 [DOI] [PubMed] [Google Scholar]
- 26. Rodriguez-Pinto D. 2005. B cells as antigen presenting cells. Cell. Immunol. 238:67–75 [DOI] [PubMed] [Google Scholar]
- 27. Hon H, Oran A, Brocker T, Jacob J. 2005. B lymphocytes participate in cross-presentation of antigen following gene gun vaccination. J. Immunol. 174:5233–5242 [DOI] [PubMed] [Google Scholar]
- 28. Pone EJ, Zan H, Zhang J, Al-Qahtani A, Xu Z, Casali P. 2010. Toll-like receptors and B-cell receptors synergize to induce immunoglobulin class-switch DNA recombination: relevance to microbial antibody responses. Crit. Rev. Immunol. 30:1–29 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Oliver AM, Martin F, Gartland GL, Carter RH, Kearney JF. 1997. Marginal zone B cells exhibit unique activation, proliferative and immunoglobulin secretory responses. Eur. J. Immunol. 27:2366–2374 [DOI] [PubMed] [Google Scholar]
- 30. Lee BO, Moyron-Quiroz J, Rangel-Moreno J, Kusser KL, Hartson L, Sprague F, Lund FE, Randall TD. 2003. CD40, but not CD154, expression on B cells is necessary for optimal primary B cell responses. J. Immunol. 171:5707–5717 [DOI] [PubMed] [Google Scholar]
- 31. Okada T, Miller MJ, Parker I, Krummel MF, Neighbors M, Hartley SB, O'Garra A, Cahalan MD, Cyster JG. 2005. Antigen-engaged B cells undergo chemotaxis toward the T zone and form motile conjugates with helper T cells. PLoS Biol. 3:e150. 10.1371/journal.pbio.0030150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Epstein MM, Di Rosa F, Jankovic D, Sher A, Matzinger P. 1995. Successful T cell priming in B cell-deficient mice. J. Exp. Med. 182:915–922 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kleindienst P, Brocker T. 2005. Concerted antigen presentation by dendritic cells and B cells is necessary for optimal CD4 T-cell immunity in vivo. Immunology 115:556–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Macaulay AE, DeKruyff RH, Umetsu DT. 1998. Antigen-primed T cells from B cell-deficient JHD mice fail to provide B cell help. J. Immunol. 160:1694–1700 [PubMed] [Google Scholar]
- 35. Faassen AE, Dalke DP, Berton MT, Warren WD, Pierce SK. 1995. CD40-CD40 ligand interactions stimulate B cell antigen processing. Eur. J. Immunol. 25:3249–3255 [DOI] [PubMed] [Google Scholar]
- 36. Evans DE, Munks MW, Purkerson JM, Parker DC. 2000. Resting B lymphocytes as APC for naive T lymphocytes: dependence on CD40 ligand/CD40. J. Immunol. 164:688–697 [DOI] [PubMed] [Google Scholar]
- 37. Conzelmann KK, Cox JH, Schneider LG, Thiel HJ. 1990. Molecular cloning and complete nucleotide sequence of the attenuated rabies virus SAD B19. Virology 175:485–499 [DOI] [PubMed] [Google Scholar]
- 38. McGettigan JP, Naper K, Orenstein J, Koser M, McKenna PM, Schnell MJ. 2003. Functional human immunodeficiency virus type 1 (HIV-1) Gag-Pol or HIV-1 Gag-Pol and env expressed from a single rhabdovirus-based vaccine vector genome. J. Virol. 77:10889–10899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Gomme EA, Wirblich C, Addya S, Rall GF, Schnell MJ. 2012. Immune clearance of attenuated rabies virus results in neuronal survival with altered gene expression. PLoS Pathog. 8:e1002971. 10.1371/journal.ppat.1002971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Quah BJ, Warren HS, Parish CR. 2007. Monitoring lymphocyte proliferation in vitro and in vivo with the intracellular fluorescent dye carboxyfluorescein diacetate succinimidyl ester. Nat. Protoc. 2:2049–2056 [DOI] [PubMed] [Google Scholar]
- 41. Finke S, Conzelmann KK. 1997. Ambisense gene expression from recombinant rabies virus: random packaging of positive- and negative-strand ribonucleoprotein complexes into rabies virions. J. Virol. 71:7281–7288 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sancho D, Gomez M, Sanchez-Madrid F. 2005. CD69 is an immunoregulatory molecule induced following activation. Trends Immunol. 26:136–140 [DOI] [PubMed] [Google Scholar]
- 43. Barr TA, Brown S, Ryan G, Zhao J, Gray D. 2007. TLR-mediated stimulation of APC: Distinct cytokine responses of B cells and dendritic cells. Eur. J. Immunol. 37:3040–3053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Robertson JM, Jensen PE, Evavold BD. 2000. DO11.10 and OT-II T cells recognize a C-terminal ovalbumin 323-339 epitope. J. Immunol. 164:4706–4712 [DOI] [PubMed] [Google Scholar]
- 45. Bachmann MF, Oxenius A. 2007. Interleukin 2: from immunostimulation to immunoregulation and back again. EMBO Rep. 8:1142–1148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Faul EJ, Wanjalla CN, Suthar MS, Gale M, Wirblich C, Schnell MJ. 2010. Rabies virus infection induces type I interferon production in an IPS-1 dependent manner while dendritic cell activation relies on IFNAR signaling. PLoS Pathog. 6:e1001016. 10.1371/journal.ppat.1001016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cassell DJ, Schwartz RH. 1994. A quantitative analysis of antigen-presenting cell function: activated B cells stimulate naive CD4 T cells but are inferior to dendritic cells in providing costimulation. J. Exp. Med. 180:1829–1840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Constant S, Schweitzer N, West J, Ranney P, Bottomly K. 1995. B lymphocytes can be competent antigen-presenting cells for priming CD4+ T cells to protein antigens in vivo. J. Immunol. 155:3734–3741 [PubMed] [Google Scholar]
- 49. Morris SC, Lees A, Finkelman FD. 1994. In vivo activation of naive T cells by antigen-presenting B cells. J. Immunol. 152:3777–3785 [PubMed] [Google Scholar]
- 50. Lyons JA, San M, Happ MP, Cross AH. 1999. B cells are critical to induction of experimental allergic encephalomyelitis by protein but not by a short encephalitogenic peptide. Eur. J. Immunol. 29:3432–3439 [DOI] [PubMed] [Google Scholar]
- 51. Byersdorfer CA, Dipaolo RJ, Petzold SJ, Unanue ER. 2004. Following immunization antigen becomes concentrated in a limited number of APCs including B cells. J. Immunol. 173:6627–6634 [DOI] [PubMed] [Google Scholar]
- 52. Clatza A, Bonifaz LC, Vignali DA, Moreno J. 2003. CD40-induced aggregation of MHC class II and CD80 on the cell surface leads to an early enhancement in antigen presentation. J. Immunol. 171:6478–6487 [DOI] [PubMed] [Google Scholar]
- 53. Xu LG, Jin L, Zhang BC, Akerlund LJ, Shu HB, Cambier JC. 2012. VISA is required for B cell expression of TLR7. J. Immunol. 188:248–258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Heath E, Begue-Pastor N, Chaganti S, Croom-Carter D, Shannon-Lowe C, Kube D, Feederle R, Delecluse HJ, Rickinson AB, Bell AI. 2012. Epstein-Barr virus infection of naive B cells in vitro frequently selects clones with mutated immunoglobulin genotypes: implications for virus biology. PLoS Pathog. 8:e1002697. 10.1371/journal.ppat.1002697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rappocciolo G, Hensler HR, Jais M, Reinhart TA, Pegu A, Jenkins FJ, Rinaldo CR. 2008. Human herpesvirus 8 infects and replicates in primary cultures of activated B lymphocytes through DC-SIGN. J. Virol. 82:4793–4806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sung VM, Shimodaira S, Doughty AL, Picchio GR, Can H, Yen TS, Lindsay KL, Levine AM, Lai MM. 2003. Establishment of B-cell lymphoma cell lines persistently infected with hepatitis C virus in vivo and in vitro: the apoptotic effects of virus infection. J. Virol. 77:2134–2146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rassa JC, Meyers JL, Zhang Y, Kudaravalli R, Ross SR. 2002. Murine retroviruses activate B cells via interaction with toll-like receptor 4. Proc. Natl. Acad. Sci. U. S. A. 99:2281–2286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Valarcher JF, Bourhy H, Lavenu A, Bourges-Abella N, Roth M, Andreoletti O, Ave P, Schelcher F. 2001. Persistent infection of B lymphocytes by bovine respiratory syncytial virus. Virology 291:55–67 [DOI] [PubMed] [Google Scholar]
- 59. Angel Rico M, Trento A, Ramos M, Johnstone C, Del Val M, Melero JA, Lopez D. 2009. Human respiratory syncytial virus infects and induces activation markers in mouse B lymphocytes. Immunol. Cell Biol. 87:344–350 [DOI] [PubMed] [Google Scholar]
- 60. Sha Z, Compans RW. 2000. Induction of CD4(+) T-cell-independent immunoglobulin responses by inactivated influenza virus. J. Virol. 74:4999–5005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Balakrishnan A, Shahir P. 2012. Early IgM antibody response in chandipuri virus infection: T cell-independent activation of B cells. J. Clin. Cell Immunol. 3:123 [Google Scholar]
- 62. Belz G, Mount A, Masson F. 2009. Dendritic cells in viral infections. Handb. Exp. Pharmacol. 188:51–77 [DOI] [PubMed] [Google Scholar]
- 63. Diebold SS. 2009. Activation of dendritic cells by toll-like receptors and C-type lectins. Handb. Exp. Pharmacol. 188:3–30 [DOI] [PubMed] [Google Scholar]
- 64. Stein JV, Nombela-Arrieta C. 2005. Chemokine control of lymphocyte trafficking: a general overview. Immunology 116:1–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Gray D, MacLennan IC, Bazin H, Khan M. 1982. Migrant mu+ delta+ and static mu+ delta-B lymphocyte subsets. Eur. J. Immunol. 12:564–569 [DOI] [PubMed] [Google Scholar]
- 66. Heil F, Hemmi H, Hochrein H, Ampenberger F, Kirschning C, Akira S, Lipford G, Wagner H, Bauer S. 2004. Species-specific recognition of single-stranded RNA via toll-like receptor 7 and 8. Science 303:1526–1529 [DOI] [PubMed] [Google Scholar]
- 67. Hao Y, O'Neill P, Naradikian MS, Scholz JL, Cancro MP. 2011. A B-cell subset uniquely responsive to innate stimuli accumulates in aged mice. Blood 118:1294–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]