

Abstract
Herpesvirus glycoprotein complex gH/gL provides a core entry function through interactions with the fusion protein gB and can also influence tropism through receptor interactions. The Epstein-Barr virus gH/gL and gH/gL/gp42 serve both functions for entry into epithelial and B cells, respectively. Human cytomegalovirus (HCMV) gH/gL can be bound by the UL128-131 proteins or gO. The phenotypes of gO and UL128-131 mutants suggest that gO-gH/gL interactions are necessary for the core entry function on all cell types, whereas the binding of UL128-131 to gH/gL likely relates to a distinct receptor-binding function for entry into some specific cell types (e.g., epithelial) but not others (e.g., fibroblasts and neurons). There are at least eight isoforms of gO that differ by 10 to 30% of amino acids, and previous analysis of two HCMV strains suggested that some isoforms of gO function like chaperones, disassociating during assembly to leave unbound gH/gL in the virion envelope, while others remain bound to gH/gL. For the current report, we analyzed the gH/gL complexes present in the virion envelope of several HCMV strains, each of which encodes a distinct gO isoform. Results indicate that all strains of HCMV contain stable gH/gL/gO trimers and gH/gL/UL128-131 pentamers and little, if any, unbound gH/gL. TR, TB40/e, AD169, and PH virions contained vastly more gH/gL/gO than gH/gL/UL128-131, whereas Merlin virions contained mostly gH/gL/UL128-131, despite abundant unbound gO remaining in the infected cells. Suppression of UL128-131 expression during Merlin replication dramatically shifted the ratio toward gH/gL/gO. These data suggest that Merlin gO is less efficient than other gO isoforms at competing with UL128-131 for binding to gH/gL. Thus, gO diversity may influence the pathogenesis of HCMV through effects on the assembly of the core versus tropism gH/gL complexes.
INTRODUCTION
Human cytomegalovirus (HCMV) is a ubiquitous human pathogen that contributes to significant disease throughout the world (1–4). Infections of immunocompetent adults are generally mild, but like other herpesviruses, HCMV establishes a persistent or latent infection. Immunosuppression resulting from HIV infection or posttransplantation chemotherapy can allow reactivation of latent HCMV, leading to a wide range of pathologies, including gastroenteritis, encephalitis, retinitis, pneumonitis, and, in the case of transplant recipients, graft rejection. In pregnant women, HCMV can cross the placental barrier and is associated with several types of birth defects. The pleomorphic manifestation of HCMV-associated disease likely relates to the ability of the virus to infect a diverse spectrum of cell types in the human body, including epithelial and endothelial cells, fibroblasts, monocyte/macrophages, dendritic cells, hepatocytes, neurons, and leukocytes (5, 6). The broad cell tropism of HCMV may reflect the abundance of distinct glycoprotein complexes in the virion envelope. Computer predictions and proteomics analyses of purified virions have suggested that HCMV envelope may contain up to 20 different glycoproteins (7–9).
Homologues of gB and gH/gL are found in all herpesviruses, and they represent the core entry machinery for the virus family. Extensive study of herpes simplex virus (HSV) and Epstein-Barr virus (EBV) has shown that gB is the fusion protein and that gH/gL provides a necessary regulatory role, likely involving direct interaction with gB (10–15). In addition, each of the different herpesviruses encodes glycoproteins that modify or regulate the activity of the gB-gH/gL core fusion machinery, such as HSV gD, EBV gp42, and human herpesvirus 6 (HHV-6) gQ1/gQ2. Each of these glycoproteins interacts transiently or stably with gH/gL, and each engages cell surface proteins as entry-mediating receptors (13, 16, 17). For EBV, the accessory protein gp42 has a well-characterized effect on tropism. Unmodified gH/gL can bind to αvβ5, αvβ6, and αvβ8 integrin molecules to mediate entry into epithelial cells (18, 19). When bound to gH/gL, gp42 likely blocks engagement of integrins by gH/gL, and instead gp42 engages major histocompatibility complex class II (MHC-II) molecules on the surfaces of B cells, thus acting like an adaptor molecule to switch viral tropism from epithelial cells to B cells (15, 20, 21). Importantly, entry of EBV into epithelial or B cells requires either gH/gL (epithelial) or gH/gL/gp42 (B cells), not both (22, 23). This indicates that, given the proper cell receptors, either complex can make the necessary interactions with gB to mediate fusion.
Because entry into all cell types requires membrane fusion mediated by gB, the regulation of gB through direct interactions may be considered the core entry function of the herpesvirus gH/gL. In a reduced system involving expression of HCMV proteins by replication-defective adenovirus (Ad) vectors, gH/gL and gB were sufficient to induce cell-cell fusion, indicating that the HCMV gH/gL provides the direct interaction surfaces involved in the regulation of gB fusion activity (24). However, like EBV, HCMV encodes a set of proteins that bind to gH/gL, i.e., gO and the UL128, UL130, and UL131 proteins (UL128-131) (25–28). Trafficking of HCMV gH/gL from the endoplasmic reticulum (ER) to the Golgi-derived viral envelopment compartment depends on interactions with either the UL128-131 proteins or gO, but the specific effects of these accessory proteins on the entry-mediating function(s) of gH/gL are not well understood (29, 30).
The HCMV UL128-131 proteins bind noncovalently to the ectodomain of HCMV gH/gL to form a pentameric complex, gH/gL/UL128-131 (25, 31, 32). UL128-131 mutants replicate well on fibroblasts but are severely impaired for entry into epithelial and endothelial cells (28, 31, 33). Ad vector expression of the gH/gL/UL128-131 complex rendered epithelial cells resistant to HCMV infection but had no effect on the susceptibility of fibroblasts (34). The observation of viral receptor interference, along with the restricted tropism phenotypes of UL128-131 mutants, suggests that gH/gL/UL128-131 facilitates entry by engaging molecules on the surfaces of specific cell types.
The role of gO as an interaction partner of HCMV gH/gL is complicated by the fact that it is among the most diverse proteins encoded by the virus. Rasmussen et al. performed a phylogenetic analysis of the predicted gO sequences of more than 40 clinical isolates of HCMV and found eight groups, or families (35). Within each group, the gO amino acid sequences were 98 to 100% identical, whereas differences between groups ranged from about 10 to 30%. Stanton et al. analyzed HCMV from clinical specimens collected from donors over 10 years and found gO sequences that fit within the families described by Rasmussen et al. Remarkably, the gO sequences within individual donors were stable over time (36). Similar results were also reported by Gorzer et al. (37). The tight grouping of gO sequences into families and their relative stability suggest that they represent alleles of the same gene, fixed in nature. Thus, the gO proteins encoded by different strains or isolates of HCMV may be considered different isoforms.
Only three isoforms of gO have been studied in detail. Huber et al. first characterized gO as an approximately 55-kDa protein encoded by the UL74 gene of the fibroblast-adapted strain AD169 (AD) (38). Posttranslational modifications, including extensive N-linked glycosylation, result in gO glycoforms that range from about 100 to 130 kDa, and interactions with gH/gL involve disulfide bonds such that a trimer of gH/gL/gO is present in the AD virion envelope (26, 27, 39). More recent analyses of the low-passage-number clinical strain TR failed to detect gO (TRgO) in the virion envelope, and several lines of evidence supported the conclusion that TRgO might act as a true chaperone, promoting trafficking of gH/gL lacking UL128-131 from the ER to the Golgi-derived envelopment compartment and then disassociating from the complex, leaving unmodified gH/gL in the virion envelope (29). Because HCMV strains encoding other gO isoforms were not analyzed, it was not clear if the observed difference between AD and TR reflected the diversity of gO or the fibroblast adaptation of AD. Regardless, gO mutants on the background of several strains have exhibited extensive replication defects in a wider range of cell types than were observed for UL128-131 mutants (40–43). Wille et al. found that a gO-null mutant TR released normal numbers of DNA-containing viral particles to the culture supernatants, but these particles were defective for entry into fibroblasts. Surprisingly, gO-null TR was also unable to enter epithelial or endothelial cells, despite elevated levels of the pentameric gH/gL/UL128-131 complex in the virion envelope. In sum, these observations suggest a model in which there are different requirements for entry of HCMV into different cells types. Entry into every cell type likely requires interactions between a gH/gL molecule and the fusion protein gB. The failure of gO-null TR to enter any cell type suggests that this function is performed by gH/gL/gO or gH/gL and not by the pentameric gH/gL/UL128-131. This is markedly different from the model of EBV tropism, in which either gH/gL or gH/gL/gp42 is sufficient as a “core gH/gL complex” for entry in epithelial or B cells, respectively.
The aim of the studies reported here was to further explore gO function(s) through comparative analysis of a wider set of gO isoforms. Experiments involving reconstruction of gH/gL-gO interactions by Ad vector expression suggested that, despite the sequence diversity, all isoforms of gO form stable disulfide-linked trimers with gH/gL. Analyses of extracellular HCMV virions indicated that, in contrast to previous reports, all HCMV strains, including TR, likely contain gH/gL/gO and gH/gL/UL128-131 and little, if any, unbound gH/gL. Furthermore, gO diversity may affect the ratio of gH/gL/gO to gH/gL/UL128-131 incorporated into the virion envelope, which may relate to the pleomorphic tropism and clinical pathology observed in HCMV infections.
MATERIALS AND METHODS
Cell lines.
Primary human foreskin fibroblasts (HFF; Life Technologies), and U373-MG human microglial cells (American Type Culture Collection; Manassas, VA, USA) were grown in Dulbecco's modified Eagle's medium (DMEM; Life Technologies) supplemented with 12% heat-inactivated fetal bovine serum (FBS; HyClone) or 10% bovine growth serum (BGS; HyClone). 239(IQ) cells (Microbix, Toronto, Canada) were grown in minimum essential medium (MEM; Life Technologies) plus 10% FBS. HFFtet cells, which express the tetracycline (Tet) repressor protein, were provided by Richard Stanton (Cardiff University, Cardiff, United Kingdom) and were propagated in DMEM supplemented with 12% FBS (44).
HCMVs.
All human cytomegaloviruses (HCMVs) were derived from bacterial artificial chromosome (BAC) clones. BAC clone TR was provided by Jay Nelson (Oregon Health and Sciences University, Portland OR, USA) (45). BAC clone TB40/e (BAC-4) (TB) was provided by Christian Sinzger (University of Tübingen, Tübingen, Germany) (46). BAC clones PH and AD169 (AD) were provided by Tom Shenk (Princeton University, Princeton, NJ, USA) (45). BAC clone Merlin (ME) (pAL1393), which harbors tetracycline operator sequences within the transcriptional promoter of UL130 and UL131, was provided by Richard Stanton (Cardiff University, Cardiff, United Kingdom) (44). Infectious HCMV was recovered by electroporation of BAC DNA into MRC-5 fibroblasts as described by Wille et al. (43). HCMV stocks were produced by infecting HFF (or HFFtet for ME) using 0.1 PFU per cell for 10 to 16 days. Cell-associated virus was prepared by sonic disruption of infected cells, and large cellular debris was cleared by centrifugation at 1,500 × g for 10 min. Extracellular virus was concentrated from culture supernatants by centrifugation at 50,000 × g for 1 h, and pellets were resuspended in DMEM plus 10% FBS. PFU counts were determined by plaque assay or the endpoint dilution method of Reed and Muench (47) on replicate HFF cultures.
Phylogenetic and sequence alignment analyses.
DNA sequences of the UL74 genes of clinical HCMV isolates and HCMV BAC clones have been reported previously along with GenBank accession numbers (35, 45, 48). Alignment and phylogenetic analyses were performed using the default parameters of the online MAFFT (version 7) tool offered by the Computational Biology Research Center (CBRC), National Institute of Advanced Industrial Science and Technology (AIST), Japan.
Antibodies.
Monoclonal antibodies (MAbs) specific for HCMV gH (14-4b) were provided by Bill Britt (University of Alabama, Birmingham, AL) (49). Anti-hemagglutinin (HA) MAbs were obtained from Sigma-Aldrich (St. Louis, MO, USA). Anti-UL128 MAb 4B10 was provided by Tom Shenk (32). Rabbit polyclonal antipeptide antibodies directed against HCMV gH, gL, TRgO, UL130, and UL131 were provided by David Johnson (Oregon Health and Sciences University, Portland, OR, USA) (29, 30). Additional rabbit polyclonal antisera were raised against synthetic peptides corresponding to residues 258 to 277 of MEgO, 250 to 269 of TBgO, 252 to 271 of ADgO, and 248 to 267 of TNgO. Peptide synthesis, rabbit immunization, and serum collection were performed by GenScript (Piscataway, NJ, USA.).
Replication-defective Ad vectors.
Nonreplicating (E1−) Ad vectors for expression of HCMV TR gH, gL, and gO, MEgO, TBgO, ADgO, and TNgO were generated using a commercial (Microbix) modification of the method of Matthews et al. (50). Briefly, HCMV genes (gH [UL75], gL [UL115], and gO [UL74]) were synthesized and codon optimized by GeneArt/Invitrogen (Regensburg, Germany). Coding sequence corresponding to the influenza HA epitope was added in frame with the C terminus of UL74 constructs. HCMV genes were ligated into shuttle plasmid pDC316(io) (Microbix), and shuttle plasmids were transfected into 293IQ cells along with the Ad genomic plasmid, pBHGloxΔE1,3Cre (Microbix). Cre-lox recombination resulted in Ad vectors that were subsequently propagated on 293IQ cells, which express the Lac repressor protein. The Lac repressor protein binds to lac operator sequences between the promoter and the HCMV transgene, reducing expression of the transgene in 293IQ cells. Ad vector PFU counts were determined using 293IQ cells. Multiplicities of infection (MOI) for experiments on U373 or HFF cells were determined empirically for each Ad vector to give appropriate expression, and ranged from 3 to 30 PFU/cell. There was little production of Ad proteins, and little or no cytopathic effect was observed.
Radiolabeling of Ad vector-transduced cells and IP analysis of expressed proteins.
Cells were transduced with Ad vectors for 12 to 24 h, washed extensively in labeling medium (DMEM lacking methionine and cysteine), and then incubated in this medium for 45 min at 37°C. Cells were then incubated for 5 min in labeling medium supplemented with [35S]methionine-cysteine (500 to 1,000 μCi/ml; PerkinElmer). Radioactivity was chased by incubating cells in DMEM containing a 10-fold excess of nonradioactive methionine and cysteine for the times indicated in the figure legends. Cell proteins were extracted with 1% Triton X-100 (TX100), 0.5% sodium deoxycholate (DOC) in 20 mM Tris (pH 6.8), and 100 mM NaCl (TBS-TX/DOC) supplemented with 0.5% bovine serum albumin (BSA) and 1 mM phenylmethylsulfonyl fluoride (PMSF). Extracts were clarified by centrifugation at 16,000 × g for 30 min and incubation with protein A-agarose beads (Invitrogen) for 1 to 2 h. Proteins were immunoprecipitated by the addition of MAbs, as indicated in the figure legends, for 2 h, followed by protein A-agarose for an additional 2 h. Agarose beads were collected by centrifugation and washed three times in TBS-TX/DOC. For endoglycosidase H (endo H) or peptide N-glycosidase F (PNGase F) treatment, immunoprecipitated proteins bound to protein A-agarose were incubated with endo H or PNGase F in the buffer specified by the enzyme manufacturer (New England BioLabs). Proteins were eluted from protein A-agarose by boiling in 2% SDS and 2% 2-mercaptoethanol (2-ME) and separated by electrophoresis using SDS-polyacrylamide gels. For sequential immunoprecipitation (IP) experiments, proteins were eluted from protein A-agarose beads by incubation in 2% SDS–30 mM dithiothreitol (DTT) in 100 mM Tris (pH 7.4)–100 mM NaCl at 95°C for 5 min. The samples were diluted 35-fold with a solution of 50 mM Tris (pH 7.4), 300 mM NaCl, 10 mM iodoacetamide, 1% TX100, 0.5% DOC, 5 mM EDTA, and 0.02% sodium azide. Then, the secondary IP antibody was added, and samples were analyzed as described above.
Western blot analysis of Ad vector- or HCMV-infected cells and HCMV extracellular virus particles.
HFF cells were transduced with Ad vectors for 12 to 24 days, and proteins were extracted in TBS-TX/DOC. For HCMV experiments, HFF or HFFtet cells were infected at an MOI of 3 with different strains of HCMV. At 5 to 10 days postinfection (depending on the strain), proteins were extracted from infected cells, and extracellular virus was collected from the culture supernatant by centrifugation, as described previously (29), in the same volume of TBS-TX/DOC. Extracts were clarified by centrifugation at 16,000 × g for 30 min, adjusted to 2% SDS with or without 2% 2-ME, and boiled for 10 min. Proteins were separated by SDS-PAGE and transferred to polyvinylidene fluoride membranes (Whatman) in a buffer containing 10 mM NaHCO3 and 3 mM Na2CO3 (pH 9.9), plus 10% methanol. Transferred proteins were probed with rabbit polyclonal antibodies or MAbs specific for HCMV proteins or the HA epitope, followed by horseradish peroxidase-conjugated secondary antibodies; chemiluminescence was detected using a Fujifilm LAS-3000 luminescent image analyzer.
RESULTS
Diversity of HCMV gO proteins among laboratory strains and clinical isolates of HCMV.
We performed a meta-analysis of the gO sequences reported by Rasmussen et al. using modern phylogeny software and included several commonly studied low-passage-number “clinical” BAC-cloned strains of HCMV that were not available at the time of the prior studies (35) (Fig. 1A). The gO isoforms encoded by HCMV TR, Merlin (ME), TB40/e (TB), and PH fit well into the groups described by Rasmussen et al. TRgO, MEgO, and TBgO are each identical to at least one of the other sequences from the same group, whereas PHgO differs from its closest family members (isolates 540, 1176, and 1736) by two amino acid substitutions (G280V and S398P). Strains AD169 (AD) and Towne (TN) were extensively propagated in cultured fibroblasts and are known to harbor many genetic mutations, deletions, and rearrangements compared to the low-passage-number strains, such as TR, ME, TB, and PH (45, 48, 51). However, the gO sequence of TN is identical to that of the clinical isolate DM13, and ADgO differs from the gO of clinical isolates 298, 6444, SW1, and SW3 by only one amino acid located within the predicted signal peptide. Thus, the gO isoforms encoded by these laboratory and clinical strains are reflective of the diversity of this protein in nature. It is important to note that, given the limited number of sequences available and the lack of understanding of gO function(s), no conclusions can be made concerning potential adaptive evolution of gO isoforms along lineages from a common ancestor. Despite this, there clearly exists extensive diversity in the gO proteins encoded by strains of HCMV in nature, and it is a compelling hypothesis that these differences affect the function(s) of gO and the biology of the virus.
Fig 1.

Comparison of amino acid sequences of gO encoded by laboratory strains and clinical isolates of HCMV. (A) Phylogenetic relationships of predicted gO amino acid sequences of clinical HCMV isolates described in Rasmussen et al. (14) and the HCMV BAC clones used in the current studies (bold). Phylogeny was estimated using neighbor-joining with the Jones-Taylor-Thornton codon model implemented in MAFFT, version 7. Horizontal bars represent the expected number of amino acid substitutions per site according to the indicated scale. (B) MAFFT alignment of one representative gO sequence from each of the eight groups indicated in panel A is shown (top). Darker shading indicates sequence conservation. The approximate locations of six conserved cysteine (c) residues are indicated. The middle panel shows Kyte-Doolittle hydrophilicity analysis of TRgO. In the bottom panel are sequences of the synthetic peptides used to raise anti-gO antibodies. Asterisks indicate conserved residues, and shaded boxes highlight the position of a proline in each peptide sequence.
Sequence alignments of representative gO isoforms revealed regions of high diversity and regions of conservation (Fig. 1B). Sequence diversity was found in the N-terminal 100 residues, of which approximately 70 are predicted to remain following cleavage of the signal peptide. The region corresponding to approximately residues 270 to 340 of the alignment also harbored notable diversity. Six conserved cysteine (Cys) residues were identified at aligned positions 31, 152, 160, 178, 229, and 354. Cys 31 is located within the predicted signal peptide, suggesting that it is not relevant to the function of gO. The remaining five Cys residues are also found in the gO homologues encoded by other betaherpesviruses, such as HHV-6, rhesus CMV, and murine CMV, suggesting the importance of disulfide bonds for the structure and function of the gO interactions with the gH/gL complex (52–54; also data not shown).
Construction of replication-defective Ad vectors to express gO isoforms.
To study the effects of gO sequence diversity on interactions with gH/gL, we constructed Ad vectors to express the gO isoforms encoded by strains TR, ME, TB, AD, and TN, each with a C-terminal HA epitope tag (HA-gO). These Ad vectors represent the HCMV strains for which there are published mutational analyses of gO and/or the commonly studied fibroblast-adapted and low-passage-number clinical strains (40–43).
For initial characterization of gO isoforms, cells were transduced with Ad vectors, and proteins were pulse-radiolabeled and then chased for 0 or 180 min. Cell extracts were then analyzed by immunoprecipitation (IP) with anti-HA antibodies, followed by treatment with endoglycosidase H (endo H), which removes the high-mannose N-linked glycans added to proteins in the ER, or PNGase F, which can remove all types of N-linked glycans, including those that have been modified by enzymes present only in the Golgi complex. Proteins were then separated by SDS-PAGE under reducing conditions (Fig. 2). Each gO isoform migrated with an apparent molecular mass of approximately 100 kDa. Slight differences in mobility are likely related to differences in polypeptide lengths among these isoforms (predicted number of gO amino acids, including the signal peptide: TR, 462; ME, 472; TB, 464; AD, 466; TN, 456), as well as to differences in N-linked glycosylation or other posttranslation modifications. Faster-migrating bands were also observed in the untreated lanes of the 0-min chase samples. Since these bands were absent following glycosidase treatment and were not apparent after 3 h of chase, they were likely glycosylation intermediates of gO present in the cells after the pulse-labeling. The N-linked glycans on each gO isoform remained sensitive to endo H treatment for at least 3 h of chase time, as indicated by the decrease in apparent molecular mass to approximately 55 kDa. These data indicate that gO isoforms share the characteristics of being highly modified with N-linked glycans and are retained in the ER when they are expressed in the absence of other HCMV proteins.
Fig 2.
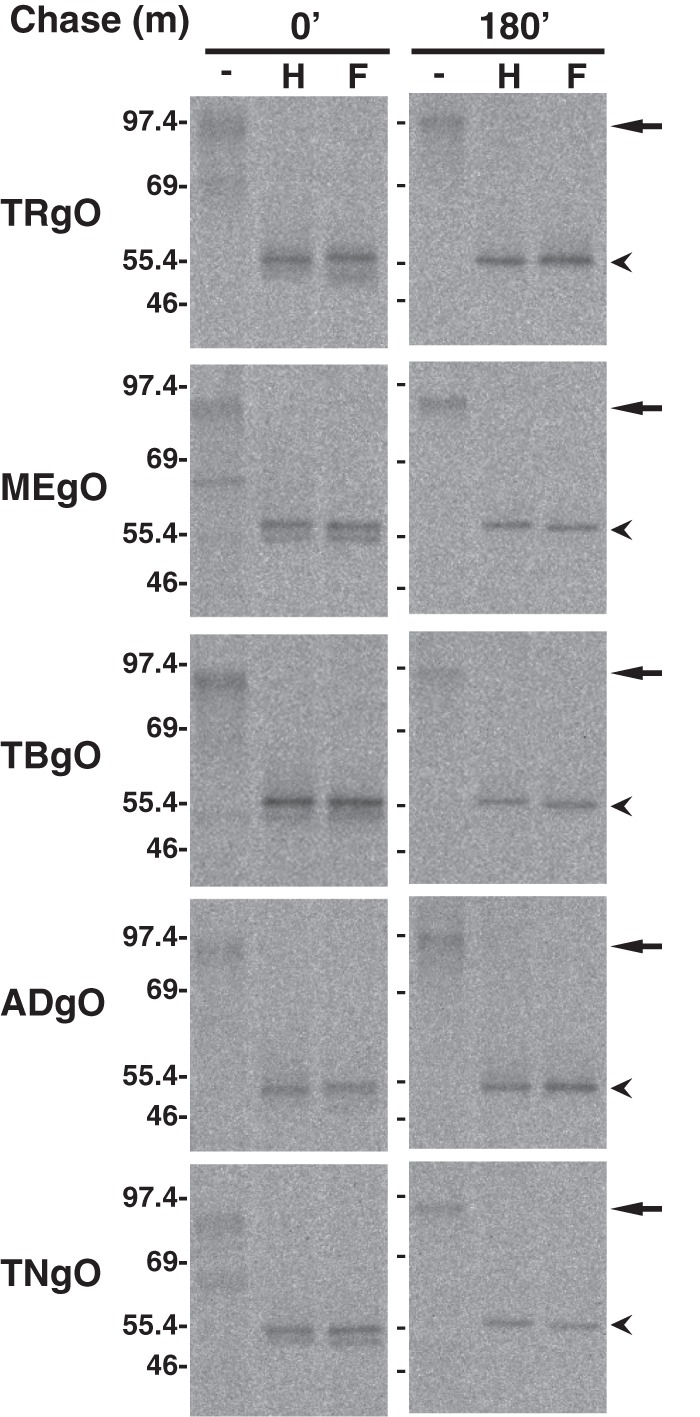
Expression of HCMV gO isoforms by replication-defective adenovirus (Ad) vectors. U373 cells were transduced with Ad vectors expressing gO isoforms (HA tagged) derived from HCMV TR (TRgO), Merlin (MEgO), TB40 (TBgO), AD169 (ADgO), and Towne (TNgO). Cells were labeled for 5 min with [35S]methionine-cysteine, and then the label was chased for 0 or 180 min. Proteins were immunoprecipitated with anti-HA antibodies, left untreated (−), or treated with endo H (H) or PNGase F (F), and separated by SDS-PAGE (8%) under reducing conditions. Arrows and arrowheads mark untreated and deglycosylated forms of gO, respectively. Mass markers (kDa) are shown on the left.
Cross-reactivity of antibodies raised against distinct gO isoforms.
Kyte-Doolittle analysis of TRgO indicated one prominent hydrophilic stretch including amino acids 245 to 271 (Fig. 1B). A synthetic peptide corresponding to these residues was used to raise in rabbits anti-TRgO antibodies that reacted well with TRgO but not other gO isoforms (29). Kyte-Doolittle analysis of other gO isoforms indicated the presence of a similar hydrophilic region at the aligned location (data not shown). However, there are some amino acid differences in this region that may explain the lack of cross-reactivity of the TRgO antiserum with other gO isoforms.
To allow more comprehensive comparative analyses of gO isoforms, antibodies were raised against peptides corresponding to the aligned hydrophilic regions of the gO isoforms encoded by strains ME, TB, AD, and TN and including conserved lysine and leucine residues (Fig. 1B). These antibodies were characterized by Western blot analysis using extracts from cells transduced with Ad vectors expressing the HA-tagged gO isoforms (Fig. 3A). The previously characterized anti-TRgO antibodies cross-reacted with TBgO but failed to detect MEgO or ADgO (Fig. 3B). Both the anti-TBgO and anti-ADgO antibodies showed overall stronger reactivity and unexpected cross-reactivity with other isoforms of gO, detecting TRgO better than the TRgO-specific antiserum (Fig. 3, compare B with D and E). In contrast, anti-MEgO antibodies were highly specific for MEgO (Fig. 3C). The specificity of the anti-MEgO antibodies may be related to the unique position of a proline residue in the aligned peptide sequences (Fig. 1B). The TNgO peptide sequence is less hydrophilic than the others (data not shown). Consistent with this, antibodies raised against TNgO peptides failed to react with any isoform of gO, and other antibodies failed to detect TNgO (data not shown). Thus, strain TN was not included in the comparative analyses of gO in the HCMV envelope described below. However, the TBgO and ADgO antibodies detected the gO isoform expressed by the low-passage-number strain PH (data not shown).
Fig 3.

Western blot detection of gO isoforms by antipeptide rabbit sera. Primary human fibroblasts were transduced with Ad vectors expressing the indicated gO isoforms (HA tagged). Cell extracts were analyzed by Western blotting using anti-HA antibodies (A) or antibodies raised against peptides derived from gO of HCMV TR (B), Merlin (C), TB40 (D), or AD169 (E). For detection with antipeptide antibodies (B to E), long and short chemiluminescent detections (top and bottom, respectively) were performed in parallel, allowing comparison of signal intensities. Mass markers (kDa) are shown on the left.
Reconstruction of gH/gL/gO complexes by Ad vector expression.
Unlike gO, gH/gL is highly conserved among strains of HCMV, differing by <5% of amino acids among strains, but it is possible that gO diversity reflects coevolution with specific gH/gL isoforms. To test whether distinct gO isoforms could bind to a common isoform of gH/gL, Ad vectors were used to express the gH/gL of strain TR (TRgH/gL) along with the different gO isoforms. Cells were pulse-chase-labeled and analyzed by IP with anti-gH antibodies, followed by treatment with endo H or PNGase F and separation by reducing SDS-PAGE (Fig. 4). In the absence of gO, gH/gL remained endo H sensitive for at least 3 h, indicative of ER retention. This was consistent with previous results (29, 30). Coexpression of any of the gO isoforms resulted in conversion of the majority of the labeled gH/gL to an endo H-resistant form, indicating transit from the ER to the trans-Golgi network (TGN).
Fig 4.
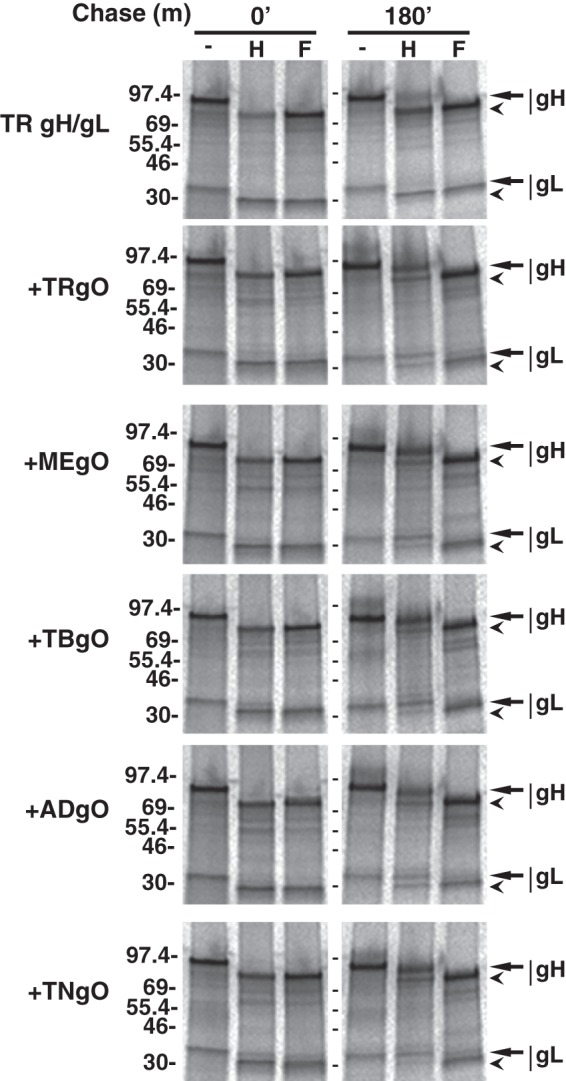
Effects of distinct gO isoforms on the intracellular trafficking of a common isoform of gH/gL. U373 cells were transduced with Ad vectors expressing gH/gL (derived from TR) alone or together (+) with Ad vectors expressing gO isoforms derived from TR (TRgO), Merlin (MEgO), TB40 (TBgO), AD169 (ADgO), and Towne (TNgO). Cells were pulse-labeled with [35S]methionine-cysteine, and then the label was chased for 0 or 180 min. Proteins were immunoprecipitated with anti-gH MAb 14-4b, left untreated (-), or treated with endo H (H) or PNGase F (F), and separated by SDS-PAGE under reducing conditions. Arrows mark untreated or endo H-resistant forms of gH and gL. Arrowheads mark deglycosylated forms of gH and gL. Mass markers (kDa) are shown on the left.
It was also of interest to determine whether gO isoforms remain bound to gH/gL during transit to the TGN. However, previous analyses of extracellular AD virions demonstrated that some of the N-linked glycans on gO remain endo H sensitive (i.e., high mannose content) despite transit through the TGN during virion morphogenesis (29, 55). This results in comigration of post-Golgi gO with ER-associated gH, precluding direct observation of gO glycoforms in the experiment described in the legend of Fig. 4. Thus, a modified version of the experiment was performed in which pulse-chase-labeled proteins were immunoprecipitated with anti-gH antibodies and heat denatured in the presence of SDS and reducing agents. The SDS and reducing agents in the samples were then diluted, and gO isoforms were analyzed by IP using anti-HA antibodies, treatment with endo H or PNGase F, and separation by reducing SDS-PAGE (Fig. 5). In the absence of endo H or PNGase F treatment, each gO isoform resolved into two bands migrating at approximately 100 and 120 kDa. After endo H treatment, these bands were replaced with a diffuse band of approximately 80 to 90 kDa that likely represented gO glycoforms bearing mostly endo H-resistant N-linked glycans (i.e., Golgi associated) and a tighter band of approximately 55 kDa that likely represented glycoforms bearing exclusively endo H-sensitive glycans (i.e., ER associated). PNGase F treatment yielded a similar 55-kDa band, as well as a slightly more slowly migrating, more diffuse band and a faint band at approximately 75 to 80 kDa. The presence of the latter band was inconsistent in these experiments, and given its apparently molecular mass, it likely represented residual gH-gO interactions recaptured in the second IP reaction. The 55-kDa band produced by PNGase F likely represented the same fraction of gO that resolved at 55 kDa following endo H treatment (i.e., ER associated), whereas the slower-migrating species produced by PNGase F likely corresponded to the fraction of gO that was endo H resistant (Golgi associated). This suggests the acquisition of modifications, in addition to N-linked oligosaccharides, such as O-linked glycans (39). Also, extracts from cells coexpressing gH/gL and any of the gO isoforms contained a protein species that migrated at approximately 240 to 260 kDa under nonreducing conditions and that was detected by anti-gL antibodies (Fig. 6A), anti-HA-gO antibodies (Fig. 6B), and anti-gH antibodies (data not shown). Together, these data strongly suggest that the assembly of all gH/gL/gO complexes involves disulfide interactions and the conserved sequences of gO. However, it is important to note that these Ad vector expression experiments were not well suited for comparisons of how efficiently each gO isoform interacts with gH/gL (i.e., binding affinity).
Fig 5.
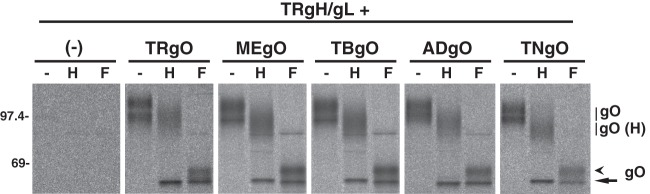
Detection of Golgi-associated glycoforms of gO isoforms during coexpression with gH/gL. U373 cells were transduced with Ad vectors expressing gH/gL (derived from HCMV TR) alone (left panel) or together with Ad vectors expressing gO isoforms derived from TR (TRgO), Merlin (MEgO), TB40 (TBgO), AD169 (ADgO), and Towne (TNgO). Cells were labeled for 5 min with [35S]methionine-cysteine, and then the label was chased for 180 min. Proteins were immunoprecipitated with anti-gH MAb 14-4b, denatured with SDS and reducing agents, and then gO isoforms were immunoprecipitated with anti-HA antibodies, left untreated (−) or treated with endo H (H) or PNGase F (F) as indicated, and analyzed by SDS-PAGE under reducing conditions. Diffuse bands corresponding to glycosylated gO and endo H-resistant gO glycoforms [gO (H)] are indicated with vertical lines. Forms of gO deglycosylated by endo H or PNGase F are indicted by the arrow. Forms of gO deglycosylated by PNGase F only are indicated by the arrowhead. Mass markers (kDa) are shown on the left.
Fig 6.
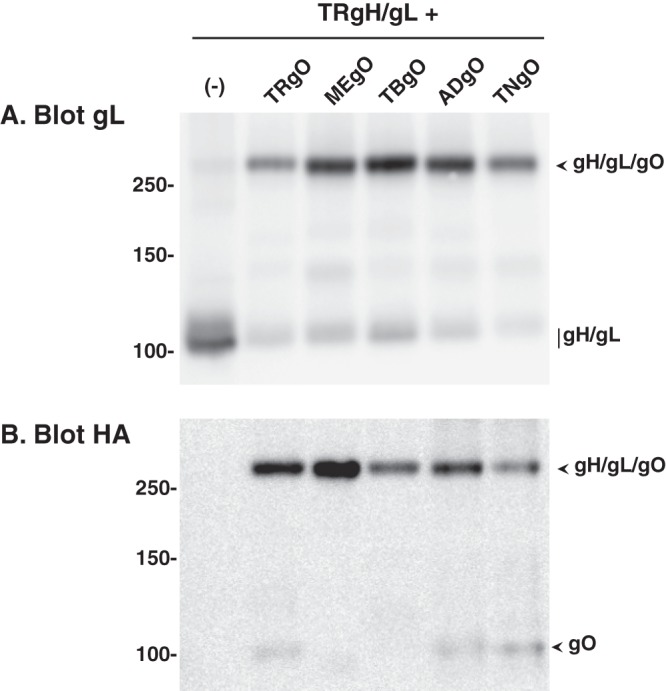
Analysis of disulfide bonds in the interactions between gH/gL and gO isoforms. U373 cells were transduced with Ad vectors expressing gH/gL (derived from TR) alone (−) or together with Ad vectors expressing gO isoforms derived from TR (TRgO), Merlin (MEgO), TB40 (TBgO), AD169 (ADgO), and Towne (TNgO). Cell extracts were separated under nonreducing conditions and analyzed by Western blotting using anti-gL (A) or anti-HA (B) antibodies. The predicted migrations of gH/gL/gO trimers, gH/gL dimers, and gO monomers are indicated to the right of both panels. Mass markers (kDa) are shown on the left.
HCMV strains differ in the ratio of gH/gL/gO and gH/gL/UL128-131 in the virion envelope.
The results of the Ad vector expression experiments described above indicated that all isoforms of gO can form a disulfide-linked complex with gH/gL. Previous comparative analysis of gO from strains TR and AD did not address disulfide interactions (29). Thus, using the new gO antibodies from the experiment shown in Fig. 3, we analyzed cell-free TR, ME, TB, AD, and PH virions and infected cells by Western blotting under reducing and nonreducing conditions (Fig. 7). Under reducing conditions and probing for gO, two bands were detected in TR-infected cell extracts, a prominent 100-kDa band and a fainter, more diffuse band of approximately 120 kDa (Fig. 7A). These bands likely corresponded to different glycoforms of gO, reflecting maturation of N-linked glycans and other modifications acquired during transit through the Golgi-derived, virion assembly compartment. Consistent with the previous report, gO was not detected in extracts of extracellular TR virions under reducing conditions (Fig. 7A) (29). However, when TR virions were analyzed under nonreducing conditions, a band with apparent molecular weight greater than 250 was detected in both cell and virion extracts, consistent with a gH/gL/gO complex (Fig. 7A). This species was also detected with anti-gL (Fig. 7B) and anti-gH (data not shown) antibodies, confirming its identity as gH/gL/gO. Analysis of four other strains of HCMV, each encoding a different gO isoform, yielded similar results (Fig. 7A and B). Since these experiments involved detection of gO isoforms using different antibodies and separate chemiluminescent detections, only the fraction of gO detected as gH/gL/gO versus gO monomer can be compared between strains. Note that on nonreducing gels, a greater fraction of the total gO migrated at the expected monomeric size in ME-infected cells than in cells infected with other strains (Fig. 7A).
Fig 7.

Comparative analysis of gH/gL/gO complexes from different strains of HCMV. Extracts of HFF cells (C) infected with BAC-derived HCMV TR, Merlin, TB40/e, AD169, or PH or virions (V) collected from the culture supernatant were separated by nonreducing or reducing SDS-PAGE, transferred to PVDF membranes, and probed with anti-gO (A) or anti-gL (B) antibodies. For gO blots, anti-TBgO antibodies were used for TR, TB, and PH samples, anti-MEgO antibodies were used for ME samples, and anti-ADgO antibodies were used for AD samples. Arrowheads mark bands corresponding to the disulfide-linked gH/gL/gO trimer, and vertical lines indicate bands that correspond to gO, gL, or the disulfide-linked gH/gL heterodimer. Mass markers (kDa) are shown on the left.
The detection of gO in TR virions when bound to gH/gL, but not when liberated from the complex by reduction of disulfide bonds, is difficult to explain since the antipeptide epitopes are generally more accessible following reduction of disulfide bonds. It is notable that similar results were observed for MEgO, ADgO (Fig. 7A), and gL (Fig. 7B) but not for TBgO and PHgO (Fig. 7A). While we cannot fully explain this phenomenon, the results of these experiments clearly indicate that the disulfide-linked gH/gL/gO complex is a conserved feature of the HCMV envelope.
The analyses of virion and infected cell extracts with anti-gL antibodies revealed a striking difference between ME and other strains (Fig. 7B). For each of the strains TR, TB, AD, and PH, the majority of the gL detected in virion extracts under nonreducing conditions was associated with gH/gL/gO, whereas for ME, most of the gL was detected at an apparent molecular mass consistent with gH/gL heterodimers, i.e., approximately 120 kDa (Fig. 7B, top row). Similar results were obtained using anti-gH antibodies (data not shown). Since efficient transport of HCMV gH/gL from the ER to the TGN-derived envelopment compartment depends on interactions with either gO or the UL128-131 proteins and since gO binds to gH/gL through disulfide bonds whereas UL128-131 associated with gH/gL through noncovalent interactions, it is likely that gH/gL dimers (approximately 120 kDa) detected in HCMV virions by nonreducing anti-gL Western blot assays represented gH/gL/UL128-131 (26, 27, 29, 30). In this context, these results suggest that TR, TB, AD, and PH virions contain vastly more gH/gL/gO than gH/gL/UL128-131, whereas ME virions contain more gH/gL/UL128-131.
The ratio of gH/gL/gO and gH/gL/UL128-131 in the HCMV virion envelope is influenced by competition between gO and the set of UL128-131 proteins for binding to gH/gL.
To confirm that the apparent gH/gL heterodimers detected in the ME virion envelope represented the gH/gL/UL128-131 complex and to test the hypotheses that the ratio of gH/gL complexes in the virion envelope is influenced by competition between gO and UL128-131 proteins, the tetracycline (Tet) expression-repression system described by Stanton et al. (44) was used. Briefly, ME is highly sensitive to mutational inactivation of the UL128-131 genes during serial propagation in cultured fibroblasts, making it difficult to maintain wild-type ME in culture (56, 57). To overcome this, Stanton et al. engineered an ME BAC with Tet operator sequences inserted within the promoter that regulates expression of UL130 and UL131 (UL130/131) and a fibroblast cell line expressing the Tet repressor protein (HFFtet). During replication in HFFtet cells, transcription from the UL130/131 promoter and, consequently, assembly of the gH/gL/UL128-131 complex are suppressed, reducing selective pressure against UL128-131.
Extracts of ME virions produced in HFF and HFFtet cells were analyzed by reducing and nonreducing Western blot assays (Fig. 8). Reducing blots indicated that virions produced by both cell types contained comparable amounts of total gL (Fig. 8A) and gB (data not shown). On nonreducing gels, the majority of gL detected in ME virions produced by HFF cells was in the form of gH/gL dimers, consistent with the results of the previous experiment (compare Fig. 7B and 8A). Strikingly, in virions produced under conditions of suppressed UL128-131 expression (HFFtet cells), the majority of the gL corresponded to the slower-migrating gH/gL/gO complex (Fig. 8A). Consistent with this, ME virions produced from HFFtet cells contained more gO (Fig. 7B) and less of the UL128-131 proteins (Fig. 7C). Note that due to the increased virion-associated MEgO under conditions of UL128-131 suppression, much shorter chemiluminescent exposures were needed for detection of MEgO in these experiments than in those described in the legend of Fig. 7A. Since unbound gH/gL dimers are poorly transported out of the ER and since the presence of UL128-131 in the virion exactly correlates with the detection of gH/gL dimers, these data strongly support the conclusion that the gH/gL heterodimers detected in ME virions represented the gH/gL/UL128-131 complex and that the ratio of the two gH/gL complexes in the virion envelope is influenced by competition between gO and the UL128-131 proteins for binding to gH/gL.
Fig 8.
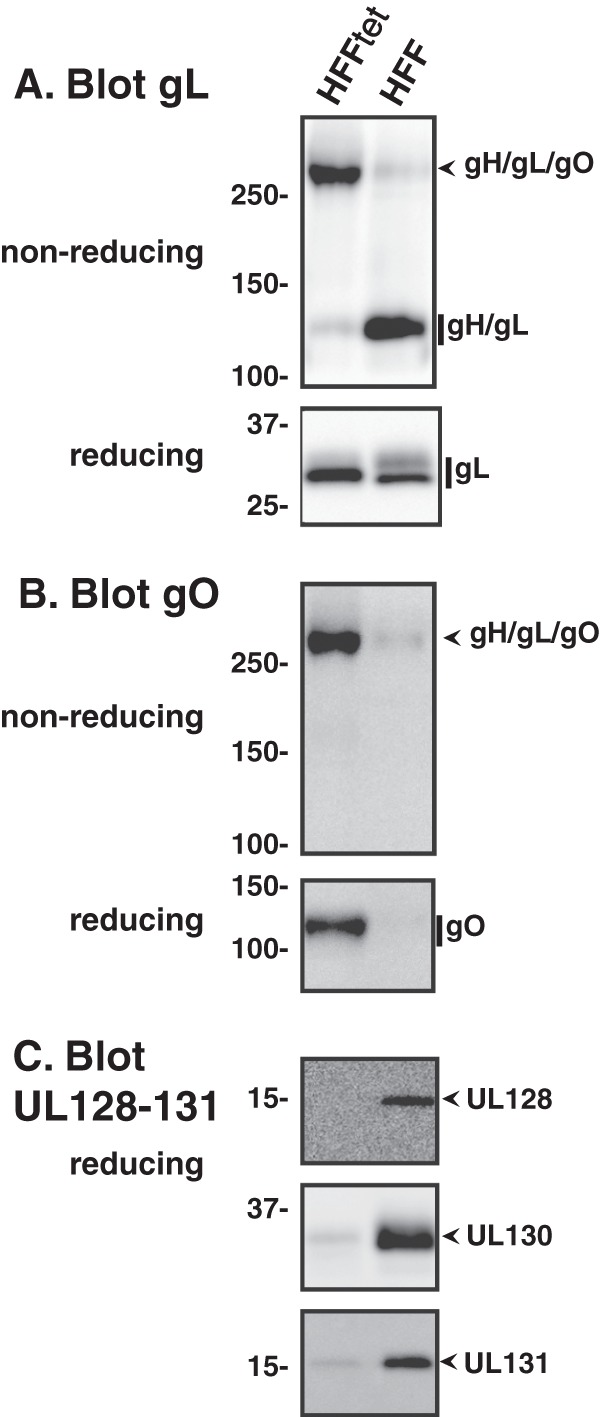
Suppression of UL128-131 expression enhances incorporation of gH/gL/gO into the Merlin virion envelope. The BAC-derived Merlin clone containing Tet operator sequences in the UL130/131 promoter was used to infect HFF cells or HFF cells expressing the Tet repressor protein (HFFtet). Extracellular virions collected from culture supernatants were analyzed by reducing and/or nonreducing Western blotting using anti-gL (A), anti-gO (B), or anti-UL128, anti-UL130, and UL131 (C) antibodies, as indicated. Mass markers (kDa) are shown on the left.
DISCUSSION
Previous analyses suggested that some gO isoforms remain bound to gH/gL in the virion envelope and that others behave more like chaperones to promote assembly of unbound gH/gL dimers into the virion envelope (29). The chaperone model was based largely on the failure to detect gO in TR virions by reducing Western blot assay. Here, we used nonreducing gel systems to compare several strains of HCMV. In all cases, gO was clearly detected as part of a disulfide-linked complex with gH/gL in the virion envelope.
The discrepancy between the results with nonreducing and reducing Western blot assays is puzzling. As noted, antipeptide antibodies are generally thought to react better with proteins that have been separated under denaturing-reducing conditions because continuous epitopes should be more accessible. It is notable that virion-associated ADgO was clearly detected in the reducing Western blot experiments reported in Ryckman et al. (29) but not in the similar experiments reported here. This difference might be explained by the fact that in the previous studies, ADgO was detected with the polyclonal serum described by Huber and Compton (38), which was raised against full-length gO, whereas in the current studies, antipeptide antibodies were used for detection of all gO isoforms. This observation points toward specific modification of gO, such as proteolysis, at or near the antipeptide antibody target site (the conserved hydrophilic domain shown in Fig. 1B). If this were the case, then perhaps the disulfide nature of the gH/gL/gO complex might preserve epitope recognition in Western blot experiments. Potential modifications are likely coincident with virion morphogenesis and release because intracellular gO was easily detected with the antipeptide antibodies in reducing Western blot assays. Also, since detection of some gO isoforms was less affected by disulfide reduction, sequence differences might affect these potential modifications. It is also important to note that a similar phenomenon was observed during analysis of gL, and it is interesting that the anti-gL antibodies used were also raised against synthetic peptides. In order to efficiently elicit antibodies, synthetic peptides are conjugated to a carrier protein (such as keyhole limpet hemocyanin [KLH]) via a terminal Cys residue that either represents a Cys in the protein of interest or one that is added to the synthetic peptide for the sole purpose of conjugation to the carrier. Thus, it is tempting to suggest that the presence of a Cys in the peptides used to generate the antibodies might contribute to the observed phenomenon. However, for all antibodies described, the Cys used for conjugation was extraneous and not present in the actual protein. Finally, it is possible that the observed effects relate to differences between cell-associated and virion-associated proteins in the transfer to blotting membranes and that these differences are less apparent during transfer of the intact gH/gL/gO complex. Again, this explanation would suggest posttranslational modifications of proteins coincident with virion release. Regardless, the analyses of TR and of other HCMV strains reported here clearly demonstrate that disulfide-linked gH/gL/gO complexes are a conserved feature of the HCMV virion envelope.
Regulation of gB can be considered the core entry function of the herpesvirus gH/gL. HCMV gH/gL, without UL128-131 or gO, can regulate gB-mediated cell-cell fusion, indicating that the necessary surfaces for gB interaction are comprised of amino acids contained within gH/gL alone (24). However, data presented here indicate that the HCMV envelope contains a mixture of gH/gL/UL128-131 and gH/gL/gO and little or no unbound gH/gL. Thus, for HCMV, the core function involving interactions with gB must be provided by either the pentameric gH/gL/UL128-131 or the trimeric gH/gL/gO. EBV also has different types of gH/gL, unmodified gH/gL or a trimeric gH/gL/gp42, and either can perform this function depending on the cell type involved (22, 23). In contrast, the phenotypes of HCMV gO and UL128-131 mutants strongly suggest that gH/gL/gO performs the core gB-interaction function during entry into all cell types, whereas gH/gL/UL128-131 serves a distinct, receptor-binding function that is an additional necessity for entry into some specific cell types, such as epithelial, endothelial, and dendritic cells, but not others, such as fibroblasts or neurons (25, 28, 31, 40–43, 58–60). The receptors for gH/gL/UL128-131 are unknown, but engagement of these molecules might trigger endocytosis, actin rearrangement, or other cell surface functions that then allow for the universally required gH/gL/gO-gB interactions that result in membrane fusion and entry. An important prediction of this model is that the surfaces on gH/gL that interact with gB are inaccessible on gH/gL/UL128-131 but not on gH/gL/gO, and studies to test aspects of this model are in progress.
The sequence diversity of gO adds a layer of complexity to our understanding of HCMV envelope composition and models of entry and tropism that have not been previously addressed. In the analyses presented here, all isoforms of gO bound to a common isoform of gH/gL to form a disulfide-linked trimer that was transported from the ER to the TGN. This suggests that gH/gL/gO complexes share a similar structure involving the conserved sequences of gO. This conclusion is consistent with the model of gH/gL/gO providing a core entry function since all gO isoforms must leave critical surfaces on gH/gL available for interaction with gB. These results are also consistent with those of Rasmussen et al., who investigated genetic linkages of polymorphic loci in clinical HCMV isolates and found evidence of several combinations of gH/gL and gO isoforms (61).
The finding that TR, TB, AD, and PH virions contained vastly more gH/gL/gO than gH/gL/UL128-131 whereas ME virions contained more gH/gL/UL128-131 than gH/gL/gO was unexpected and has a number of important implications. First, the observation that UL128-131 suppression during ME replication dramatically shifted the ratio of gH/gL complexes in the virion envelope toward gH/gL/gO indicates that gO and UL128-131 compete for binding of gH/gL to form the two complexes, and the relative abundance of unbound gO in ME-infected cells suggests that MEgO may be less able to compete than other gO isoforms. Thus, the variable sequences of gO might affect the ratio of the core entry factor gH/gL/gO to the tropism factor gH/gL/UL128-131. However, these results do not exclude the possibility that differences in protein expression levels or small sequence differences in gH/gL or UL128-131 also contribute to strain variation. Wille et al. showed that a TR mutant that could not express gO produced virions that contained elevated amounts of gH/gL/UL128-131 but drastically lower total amounts of gH/gL, indicating that UL128-131 was unable to bind a significant fraction of the remaining gH/gL (43). This might indicate that TR expresses more gH/gL or less UL128-131 than ME. Second, it is well appreciated that there is a selective pressure against UL128-131 during replication in fibroblast cultures and that ME is especially sensitive to this selection (56, 57; also R. Stanton, personal communication). If the assembly of gH/gL/gO and gH/gL/UL128-131 comes at the expense of one another and if the gH/gL/UL128-131 cannot participate in the interactions with gB that are necessary for fusion, then mutation of UL128-131 would be expected to result in a greater fibroblast replication fitness advantage for a virus like ME than for a virus like TR. This model predicts an evolutionary balancing act requiring the virus to express enough gH/gL/UL128-131 for broad tropism while retaining enough gH/gL/gO to promote fusion on all cell types. Clearly, the amounts of each complex in the ME virion are sufficient to allow infection of both fibroblasts and epithelial/endothelial cells, but the relative efficiency (i.e., per virion) compared to other HCMV strains with different amounts of the complexes is difficult to directly measure. As noted above, it is possible that HCMV strains differ in the total amount of gH/gL, and this could either lessen or magnify the importance of the ratio of the two complexes. Clearly, suppression of UL128-131 enhances the replication of ME, but the effects of manipulation of the expression of gO or gH/gL have not been addressed. Finally, infection of neuronal cells is likely independent of gH/gL/UL128-131 (60). Thus, it is conceivable that the ratio of the two distinct gH/gL complexes might be subject to similar selective pressures in neuronal or other tissues, as occurs in fibroblast cultures. If so, then the gO genotype of HCMV might influence viral pathology and the ability of the virus to spread to other hosts, which likely involves replication in epithelial and endothelial tissues.
Host cell characteristics may also influence the protein composition of the HCMV envelope. Wang et al. found that HCMV produced by epithelial cells entered epithelial cells through fusion at the plasma membrane rather than via fusion following endocytosis, as had been described for fibroblast-produced virus. They suggested that this correlated with an approximately 2-fold increase in the ratio of gH/gL/UL128-131 versus gH/gL/gO in epithelial cell-produced virions (62). Scrivano et al. similarly compared HCMV virions produced by fibroblasts with those produced by endothelial cells and suggested that the two cell types produce virion populations that are heterogeneous in the relative levels of gH/gL/gO and gH/gL/UL128-131 (63). The notion of heterogeneity in the amounts of gH/gL complexes incorporated into the HCMV envelope had been previously suggested by Li et al. (64). Our results linking gO diversity with the ratio of the two gH/gL complexes raises the question of whether the envelopes of all strains are similarly heterogeneous or affected by the producer cell.
In summary, the data presented in this report clearly indicate that the gH/gL/gO complex is an envelope component of all strains of HCMV and suggest that the sequence differences between gO isoforms may affect the ratio of gH/gL complexes incorporated in the virion envelope. The diverse sequences of gO may also affect other functions and aspects of the gH/gL/gO complex. For example, Vanarsdall et al. provided evidence from receptor interference experiments that suggested that gH/gL/gO might act as a receptor-binding complex and that the gO isoform might participate in these interactions or modulate the activity (65). Also, Jiang et al. recently suggested that the extensive array of N-linked glycans on gO might act to shield the virus from the effects of neutralizing antibodies (66). Current efforts are directed at further characterizing these and other possible significances of gO diversity by analyzing interstrain gO recombinant HCMVs.
ACKNOWLEDGMENTS
We are grateful to Bill Britt, David Johnson, Jay Nelson, Tom Shenk, Christian Sinzger, and Richard Stanton for generously supplying HCMV BAC clones, antibodies, and cell lines as indicated in Materials and Methods. We thank John McCutcheon and Dan Vanderpool (University of Montana) for their expert advice with phylogenetic analyses. We thank Sherry Colman for important early characterizations of anti-gO peptide antibodies, Jean-Marc Lanchy for manuscript editing, and members of the Ryckman lab for support and insightful discussions.
B.J.R. is supported by grants from the National Institutes of Health (5-R01-AI097274-02 and PG20GM103546).
Experiments were designed by B.J.R. and M.Z. and performed by M.Z., Q.Y., and A.W. Data were analyzed and the manuscript was prepared by B.J.R. and M.Z.
Footnotes
Published ahead of print 26 June 2013
REFERENCES
- 1.Britt WJ. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr. Top. Microbiol. Immunol. 325:417–470 [DOI] [PubMed] [Google Scholar]
- 2.Landolfo S, Gariglio M, Gribaudo G, Lembo D. 2003. The human cytomegalovirus. Pharmacol. Ther. 98:269–297 [DOI] [PubMed] [Google Scholar]
- 3.Pass R, Fowler K, Boppana S, Britt W, Stagno S. 2006. Congenital cytomegalovirus infection following first trimester maternal infection: symptoms at birth and outcome. J. Clin. Virol. 35:216–220 [DOI] [PubMed] [Google Scholar]
- 4.Streblow D, Orloff S, Nelson J. 2007. Acceleration of allograft failure by cytomegalovirus. Curr. Opin. Immunol. 19:577–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Plachter B, Sinzger C, Jahn G. 1996. Cell types involved in replication and distribution of human cytomegalovirus. Adv. Virus Res. 46:195–261 [DOI] [PubMed] [Google Scholar]
- 6.Sinzger C, Grefte A, Plachter B, Gouw A, The T, Jahn G. 1995. Fibroblasts, epithelial cells, endothelial cells and smooth muscle cells are major targets of human cytomegalovirus infection in lung and gastrointestinal tissues. J. Gen. Virol. 76:741–750 [DOI] [PubMed] [Google Scholar]
- 7.Murphy E, Rigoutsos I, Shibuya T, Shenk T. 2003. Reevaluation of human cytomegalovirus coding potential. Proc. Natl. Acad. Sci. U. S. A. 100:13585–13590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rigoutsos I, Novotny J, Huynh T, Chin-Bow ST, Parida L, Platt D, Coleman D, Shenk T. 2003. In silico pattern-based analysis of the human cytomegalovirus genome. J. Virol. 77:4326–4344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Varnum S, Streblow D, Monroe M, Smith P, Auberry K, Pasa-Tolic L, Wang D, Camp DN, Rodland K, Wiley S, Britt W, Shenk T, Smith R, Nelson J. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. J. Virol. 78:10960–10966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Atanasiu D, Saw W, Cohen G, Eisenberg R. 2010. Cascade of events governing cell-cell fusion induced by herpes simplex virus glycoproteins gD, gH/gL, and gB. J. Virol. 84:12292–12299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Backovic M, Longnecker R, Jardetzky T. 2009. Structure of a trimeric variant of the Epstein-Barr virus glycoprotein B. Proc. Natl. Acad. Sci. U. S. A. 106:2880–2885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chowdary T, Cairns T, Atanasiu D, Cohen G, Eisenberg R, Heldwein E. 2010. Crystal structure of the conserved herpesvirus fusion regulator complex gH-gL. Nat. Struct. Mol. Biol. 17:882–888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Connolly SA, Jackson JO, Jardetzky TS, Longnecker R. 2011. Fusing structure and function: a structural view of the herpesvirus entry machinery. Nat. Rev. Microbiol. 9:369–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heldwein E, Lou H, Bender F, Cohen G, Eisenberg R, Harrison S. 2006. Crystal structure of glycoprotein B from herpes simplex virus 1. Science 313:217–220 [DOI] [PubMed] [Google Scholar]
- 15.Matsuura H, Kirschner A, Longnecker R, Jardetzky T. 2010. Crystal structure of the Epstein-Barr virus (EBV) glycoprotein H/glycoprotein L (gH/gL) complex. Proc. Natl. Acad. Sci. U. S. A. 107:22641–22646 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hutt-Fletcher LM. 2007. Epstein-Barr virus entry. J. Virol. 81:7825–7832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mori Y, Akkapaiboon P, Yonemoto S, Koike M, Takemoto M, Sadaoka T, Sasamoto Y, Konishi S, Uchiyama Y, Yamanishi K. 2004. Discovery of a second form of tripartite complex containing gH-gL of human herpesvirus 6 and observations on CD46. J. Virol. 78:4609–4616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chesnokova L, Hutt-Fletcher LM. 2011. Fusion of Epstein-Barr virus with epithelial cells can be triggered by αvβ5 in addition to αvβ6 and αvβ8, and integrin binding triggers a conformational change in glycoproteins gHgL. J. Virol. 85:13214–13223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chesnokova L, Nishimura S, Hutt-Fletcher LM. 2009. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins αvβ6 or αvβ8. Proc. Natl. Acad. Sci. U. S. A. 106:20464–20469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Borza C, Morgan A, Turk S, Hutt-Fletcher LM. 2004. Use of gHgL for attachment of Epstein-Barr virus to epithelial cells compromises infection. J. Virol. 78:5007–5014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kirschner A, Lowrey A, Longnecker R, Jardetzky T. 2007. Binding-site interactions between Epstein-Barr virus fusion proteins gp42 and gH/gL reveal a peptide that inhibits both epithelial and B-cell membrane fusion. J. Virol. 81:9216–9229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Borza C, Hutt-Fletcher LM. 2002. Alternate replication in B cells and epithelial cells switches tropism of Epstein-Barr virus. Nat. Med. 8:594–599 [DOI] [PubMed] [Google Scholar]
- 23.Wang X, Kenyon W, Li Q, Mullberg J, Hutt-Fletcher LM. 1998. Epstein-Barr virus uses different complexes of glycoproteins gH and gL to infect B lymphocytes and epithelial cells. J. Virol. 72:5552–5558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Vanarsdall A, Ryckman B, Chase M, Johnson D. 2008. Human cytomegalovirus glycoproteins gB and gH/gL mediate epithelial cell-cell fusion when expressed either in cis or in trans. J. Virol. 82:11837–11850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Adler B, Scrivano L, Ruzcics Z, Rupp B, Sinzger C, Koszinowski U. 2006. Role of human cytomegalovirus UL131A in cell type-specific virus entry and release. J. Gen. Virol. 87:2451–2460 [DOI] [PubMed] [Google Scholar]
- 26.Huber M, Compton T. 1997. Characterization of a novel third member of the human cytomegalovirus glycoprotein H-glycoprotein L complex. J. Virol. 71:5391–5398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li L, Nelson J, Britt W. 1997. Glycoprotein H-related complexes of human cytomegalovirus: identification of a third protein in the gCIII complex. J. Virol. 71:3090–3097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang D, Shenk T. 2005. Human cytomegalovirus UL131 open reading frame is required for epithelial cell tropism. J. Virol. 79:10330–10338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ryckman B, Chase M, Johnson D. 2010. Human cytomegalovirus TR strain glycoprotein O acts as a chaperone promoting gH/gL incorporation into virions but is not present in virions. J. Virol. 84:2597–2609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ryckman B, Rainish B, Chase M, Borton J, Nelson J, Jarvis M, Johnson D. 2008. Characterization of the human cytomegalovirus gH/gL/UL128-131 complex that mediates entry into epithelial and endothelial cells. J. Virol. 82:60–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ryckman B, Jarvis M, Drummond D, Nelson J, Johnson D. 2006. Human cytomegalovirus entry into epithelial and endothelial cells depends on genes UL128 to UL150 and occurs by endocytosis and low-pH fusion. J. Virol. 80:710–722 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang D, Shenk T. 2005. Human cytomegalovirus virion protein complex required for epithelial and endothelial cell tropism. Proc. Natl. Acad. Sci. U. S. A. 102:18153–18158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hahn G, Revello M, Patrone M, Percivalle E, Campanini G, Sarasini A, Wagner M, Gallina A, Milanesi G, Koszinowski U, Baldanti F, Gerna G. 2004. Human cytomegalovirus UL131-128 genes are indispensable for virus growth in endothelial cells and virus transfer to leukocytes. J. Virol. 78:10023–10033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ryckman B, Chase M, Johnson D. 2008. HCMV gH/gL/UL128-131 interferes with virus entry into epithelial cells: evidence for cell type-specific receptors. Proc. Natl. Acad. Sci. U. S. A. 105:14118–14123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Rasmussen L, Geissler A, Cowan C, Chase A, Winters M. 2002. The genes encoding the gCIII complex of human cytomegalovirus exist in highly diverse combinations in clinical isolates. J. Virol. 76:10841–10848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Stanton R, Westmoreland D, Fox J, Davison A, Wilkinson G. 2005. Stability of human cytomegalovirus genotypes in persistently infected renal transplant recipients. J. Med. Virol. 75:42–46 [DOI] [PubMed] [Google Scholar]
- 37.Gorzer I, Guelly C, Trajanoski S, Puchhammer-Stockl E. 2010. Deep sequencing reveals highly complex dynamics of human cytomegalovirus genotypes in transplant patients over time. J. Virol. 84:7195–7203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Huber M, Compton T. 1998. The human cytomegalovirus UL74 gene encodes the third component of the glycoprotein H-glycoprotein L-containing envelope complex. J. Virol. 72:8191–8197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Huber M, Compton T. 1999. Intracellular formation and processing of the heterotrimeric gH-gL-gO (gCIII) glycoprotein envelope complex of human cytomegalovirus. J. Virol. 73:3886–3892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Dunn W, Chou C, Li H, Hai R, Patterson D, Stolc V, Zhu H, Liu F. 2003. Functional profiling of a human cytomegalovirus genome. Proc. Natl. Acad. Sci. U. S. A. 100:14223–14228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hobom U, Brune W, Messerle M, Hahn G, Koszinowski U. 2000. Fast screening procedures for random transposon libraries of cloned herpesvirus genomes: mutational analysis of human cytomegalovirus envelope glycoprotein genes. J. Virol. 74:7720–7729 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jiang X, Adler B, Sampaio K, Digel M, Jahn G, Ettischer N, Stierhof Y, Scrivano L, Koszinowski U, Mach M, Sinzger C. 2008. UL74 of human cytomegalovirus contributes to virus release by promoting secondary envelopment of virions. J. Virol. 82:2802–2812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wille P, Knoche A, Nelson J, Jarvis M, Johnson D. 2010. A human cytomegalovirus gO-null mutant fails to incorporate gH/gL into the virion envelope and is unable to enter fibroblasts and epithelial and endothelial cells. J. Virol. 84:2585–2596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Stanton R, Baluchova K, Dargan D, Cunningham C, Sheehy O, Seirafian S, McSharry B, Neale M, Davies J, Tomasec P, Davison A, Wilkinson G. 2010. Reconstruction of the complete human cytomegalovirus genome in a BAC reveals RL13 to be a potent inhibitor of replication. J. Clin. Invest. 120:3191–3208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Murphy E, Yu D, Grimwood J, Schmutz J, Dickson M, Jarvis M, Hahn G, Nelson J, Myers R, Shenk T. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. U. S. A. 100:14976–14981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sinzger C, Hahn G, Digel M, Katona R, Sampaio K, Messerle M, Hengel H, Koszinowski U, Brune W, Adler B. 2008. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J. Gen. Virol. 89:359–368 [DOI] [PubMed] [Google Scholar]
- 47.Reed LJ, Muench H. 1938. A simple method of estimating fifty per cent endpoints. Am. J. Hyg. 27:493–497 [Google Scholar]
- 48.Dolan A, Cunningham C, Hector R, Hassan-Walker AF, Lee L, Addison C, Dargan D, McGeoch D, Gatherer D, Emery V, Griffiths P, Sinzger C, McSharry B, Wilkinson G, Davison A. 2004. Genetic content of wild-type human cytomegalovirus. J. Gen. Virol. 85:1301–1312 [DOI] [PubMed] [Google Scholar]
- 49.Bogner E, Reschke M, Reis B, Reis E, Britt W, Radsak K. 1992. Recognition of compartmentalized intracellular analogs of glycoprotein H of human cytomegalovirus. Arch. Virol. 126:67–80 [DOI] [PubMed] [Google Scholar]
- 50.Matthews D, Cummings D, Evelegh C, Graham F, Prevec L. 1999. Development and use of a 293 cell line expressing lac repressor for the rescue of recombinant adenoviruses expressing high levels of rabies virus glycoprotein. J. Gen. Virol. 80:345–353 [DOI] [PubMed] [Google Scholar]
- 51.Cha T, Tom E, Kemble G, Duke G, Mocarski E, Spaete R. 1996. Human cytomegalovirus clinical isolates carry at least 19 genes not found in laboratory strains. J. Virol. 70:78–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gompels U, Nicholas J, Lawrence G, Jones M, Thomson B, Martin M, Efstathiou S, Craxton M, Macaulay H. 1995. The DNA sequence of human herpesvirus-6: structure, coding content, and genome evolution. Virology 209:29–51 [DOI] [PubMed] [Google Scholar]
- 53.Rivailler P, Kaur A, Johnson R, Wang F. 2006. Genomic sequence of rhesus cytomegalovirus 180.92: insights into the coding potential of rhesus cytomegalovirus. J. Virol. 80:4179–4182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith L, McWhorter A, Masters L, Shellam G, Redwood A. 2008. Laboratory strains of murine cytomegalovirus are genetically similar to but phenotypically distinct from wild strains of virus. J. Virol. 82:6689–6696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Theiler R, Compton T. 2002. Distinct glycoprotein O complexes arise in a post-Golgi compartment of cytomegalovirus-infected cells. J. Virol. 76:2890–2898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Akter P, Cunningham C, McSharry B, Dolan A, Addison C, Dargan D, Hassan-Walker AF, Emery V, Griffiths P, Wilkinson G, Davison A. 2003. Two novel spliced genes in human cytomegalovirus. J. Gen. Virol. 84:1117–1122 [DOI] [PubMed] [Google Scholar]
- 57.Dargan D, Douglas E, Cunningham C, Jamieson F, Stanton R, Baluchova K, McSharry B, Tomasec P, Emery V, Percivalle E, Sarasini A, Gerna G, Wilkinson G, Davison A. 2010. Sequential mutations associated with adaptation of human cytomegalovirus to growth in cell culture. J. Gen. Virol. 91:1535–1546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gerna G, Percivalle E, Lilleri D, Lozza L, Fornara C, Hahn G, Baldanti F, Revello M. 2005. Dendritic-cell infection by human cytomegalovirus is restricted to strains carrying functional UL131-128 genes and mediates efficient viral antigen presentation to CD8+ T cells. J. Gen. Virol. 86:275–284 [DOI] [PubMed] [Google Scholar]
- 59.Hahn G, Rose D, Wagner M, Rhiel S, McVoy M. 2003. Cloning of the genomes of human cytomegalovirus strains Toledo, TownevarRIT3, and Townelong as BACs and site-directed mutagenesis using a PCR-based technique. Virology 307:164–177 [DOI] [PubMed] [Google Scholar]
- 60.Luo M, Schwartz P, Fortunato E. 2008. Neonatal neural progenitor cells and their neuronal and glial cell derivatives are fully permissive for human cytomegalovirus infection. J. Virol. 82:9994–10007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Rasmussen L, Geissler A, Winters M. 2003. Inter- and intragenic variations complicate the molecular epidemiology of human cytomegalovirus. J. Infect. Dis. 187:809–819 [DOI] [PubMed] [Google Scholar]
- 62.Wang D, Yu Q, Schroer J, Murphy E, Shenk T. 2007. Human cytomegalovirus uses two distinct pathways to enter retinal pigmented epithelial cells. Proc. Natl. Acad. Sci. U. S. A. 104:20037–20042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Scrivano L, Sinzger C, Nitschko H, Koszinowski U, Adler B. 2011. HCMV spread and cell tropism are determined by distinct virus populations. PLoS Pathog. 7:e1001256. 10.1371/journal.ppat.1001256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li L, Coelingh K, Britt W. 1995. Human cytomegalovirus neutralizing antibody-resistant phenotype is associated with reduced expression of glycoprotein H. J. Virol. 69:6047–6053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Vanarsdall A, Chase M, Johnson D. 2011. Human cytomegalovirus glycoprotein gO complexes with gH/gL, promoting interference with viral entry into human fibroblasts but not entry into epithelial cells. J. Virol. 85:11638–11645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Jiang X, Sampaio K, Ettischer N, Stierhof Y, Jahn G, Kropff B, Mach M, Sinzger C. 2011. UL74 of human cytomegalovirus reduces the inhibitory effect of gH-specific and gB-specific antibodies. Arch. Virol. 156:2145–2155 [DOI] [PubMed] [Google Scholar]