

Abstract
Bas-Congo virus (BASV) is a novel rhabdovirus recently identified from a patient with acute hemorrhagic fever in the Bas-Congo province of the Democratic Republic of Congo (DRC). Here we show that the BASV glycoprotein (BASV-G) can be successfully used to pseudotype glycoprotein-deficient vesicular stomatitis virus (VSV), allowing studies of BASV-G-driven membrane fusion and viral entry into target cells without replication-competent virus. BASV-G displayed broad tissue and species tropism in vitro, and BASV-G-mediated membrane fusion was pH dependent. The conformational changes induced in BASV-G by acidification were fully reversible and did not lead to inactivation of the viral fusion protein. Our data combined with comparative sequence similarity analyses suggest that BASV-G shares structural and functional features with other rhabdovirus glycoproteins and falls into the group of class III viral fusion proteins. However, activation of BASV-G-driven fusion required a lower pH and higher temperatures than did VSV-G-mediated fusion. Moreover, in contrast to VSV-G, mature BASV-G in VSV pseudotypes consists of a mixture of high-mannose and complex glycans that enables it to bind to certain C-type lectins, thereby enhancing its attachment to target cells. Taken together, the results presented in this study will facilitate future investigations of BASV-G-mediated cell entry and its inhibition in the absence of an infectious cell culture assay for BASV and at lower biosafety levels. Moreover, serology testing based on BASV-G pseudotype neutralization can be used to uncover the prevalence and importance of BASV as a potential novel human pathogen in the DRC and throughout Central Africa.
INTRODUCTION
The novel rhabdovirus Bas-Congo virus (BASV) was first identified in association with a small hemorrhagic fever outbreak in the Bas-Congo district of the Democratic Republic of Congo (DRC). A serum sample was obtained from one patient just before initiation of treatment, and the nearly complete viral sequence was recovered by deep sequencing and de novo genome assembly (1). Human BASV infection was subsequently confirmed by the detection of specific antibodies targeting the BASV glycoprotein (BASV-G) in the patient and an asymptomatic close contact (1).
Rhabdoviruses form a large family of enveloped, negative-sense, single-stranded RNA viruses (Rhabdoviridae) that infect a broad range of host species, including plants, invertebrate animals, and vertebrate animals, including humans (2). The neuroinvasive rabies virus (RABV) in the Lyssavirus genus causes acute encephalitis and represents the most pathogenic rhabdovirus in humans, with more than 55,000 deaths every year (3). The only members of the Rhabdoviridae associated with hemorrhagic disease are found in fish (4). The best-characterized rhabdovirus is vesicular stomatitis virus (VSV), which causes a mild but nevertheless economically important disease in cattle (5) and is often used as a model virus in laboratory settings. The rhabdovirus genome consists of at least 5 essential proteins: nucleoprotein (N), phosphoprotein (P), matrix protein (M), glycoprotein (G), and large protein or RNA-dependent RNA polymerase (L) (2). The viral glycoproteins of enveloped viruses make the first contact with the target cell and through a series of conformational changes bring the viral and cellular membranes into close proximity, which is required for membrane fusion and release of the viral genome into the target cell (6). Rhabdovirus glycoproteins belong to the group of class III viral fusion proteins and possess unique features that distinguish them from class I and II viral fusion proteins (7). Instead of the N-terminal fusion peptide observed in most class I and II viral fusion proteins, rhabdovirus glycoproteins display an internal fusion peptide that forms a bipartite fusion loop motif dominated by three aromatic amino acid residues (7). Moreover, the conformational changes that rhabdovirus glycoproteins undergo during the fusion process are fully reversible, unlike class I and II viral fusion proteins, which irreversibly collapse from their metastable prefusion state into their postfusion conformation (8, 9).
The structure and function of the viral glycoprotein is important for the initiation of the viral life cycle and the establishment of infection within a host. It is exposed to the host's immune system, thus presenting an important target for neutralizing antibodies. Antiviral drugs targeting the viral glycoprotein or the interaction with its cellular receptor(s) have successfully been identified for a number of pathogenic viruses and are based on detailed knowledge of the structure and function of the target protein (6). Here we sought to gain an understanding of the principal mechanism of BASV-G-mediated cell entry as well as information on its overall structure and possible modifications that could impact its susceptibility to therapeutic interference with its function.
MATERIALS AND METHODS
Cell lines.
The adherent human cell lines 293T (kidney), Huh-7.5 (liver), A549 (lung), HeLa (cervix), SW480 (colon), CaCo-2 (colon), HT1080 (connective tissue), and RD (muscle) as well as the adherent nonhuman cell lines Vero (African green monkey kidney), MC57 (mouse fibroblast), NIH 3T3 (mouse fibroblast), C6 (rat brain), NRK (rat kidney), BHK (hamster kidney), SK-RST (porcine kidney), MDBK (bovine kidney), and Tb1Lu (bat lung) were grown in Dulbecco's modified Eagle's medium (DMEM) (HyClone) supplemented with 10% fetal bovine serum (FBS; Gibco), the antibiotics penicillin and streptomycin (Gibco), l-glutamine (Gibco), and nonessential amino acids (Gibco) at 37°C and 5% CO2 in a humidified atmosphere. The insect cell lines C7/10 (mosquito) and C6/36 (mosquito) were grown in DMEM supplemented as described above but at 28°C and with 5% CO2 in a humidified atmosphere. The human suspension cell lines H9 (T lymphocyte), Jurkat (T lymphocyte), B-THP (B lymphocyte), THP-1 (monocyte), and HEL (erythroblast) were cultured in RPMI medium (Gibco) supplemented with 10% FBS, the antibiotics penicillin and streptomycin, l-glutamine, and nonessential amino acids at 37°C and 5% CO2 in a humidified atmosphere. The primary cell lines HUVEC (human umbilical vascular endothelium) and HUPEC (human pulmonary vascular endothelium) were maintained in complete EBM-2 medium with EGM-2 BulletKit supplement (Lonza) at 37°C and 5% CO2 in a humidified atmosphere. Stably transfected T-REx-293 cells (Invitrogen) were maintained in DMEM supplemented with 10% FBS, l-glutamine, nonessential amino acids, 200 μg/ml zeocin, and 5 μg/ml blasticidin. For induction of lectin expression, 1 μg/ml tetracycline was added to the culture medium, and cells were incubated for 24 h.
Plasmids.
The BASV-G sequence, as predicted by sequence alignments with other rhabdoviruses, was previously synthesized (Genscript) and subcloned into the mammalian expression plasmid pCAGGS (1). Furthermore, a tagged version of BASV-G (BASV-G.V5) was generated by addition of the V5 tag sequence to the C terminus of the BASV-G coding sequence. The plasmids for expression of VSV-G, chikungunya virus envelope (CHIKV env), Nipah virus (NiV) F protein, NiV G protein, and Ebola virus glycoprotein (EBOV-GP) (Zaire) were described previously (10–12). The fusion loop mutants of BASV-G were generated from the pCAGGS/BASV-G.V5 plasmid by overlap PCR using the following primers: forward outer primer 5′-GGCGGTACCACCATGACCCGC-3′, reverse outer primer 5′-GCCCTCGAGTCAGGTGCTATCCAGG-3′, W93A forward inner primer 5′-TGTGAAGAAACAGCTTATTTCACATCC-3′, W93A reverse inner primer 5′-GGATGTGAAATAAGCTGTTTCTTCACA-3′, Y94A forward inner primer 5′-AGAAACATGGGCTTTCACATCC-3′, Y94A reverse inner primer 5′-GGATGTGAAAGCCCATGTTTCT-3′, W137A forward inner primer 5′-AATGTAGACTGCTATGCTAACGCAATAAATAGT-3′, W137A reverse inner primer 5′-ACTATTTATTGCGTTAGCATAGCAGTCTACATT-3′, N138A forward inner primer 5′-TAGACTGCTATTGGGCTGCAATAAATAGT-3′, and N138A reverse inner primer 5′-ACTATTTATTGCAGCCCAATAGCAGTCTA-3′.
Viruses.
VSVΔG-green fluorescent protein (GFP)-based viruses, in which the glycoprotein (G) gene has been replaced with GFP, were produced by transient transfection of viral glycoprotein expression plasmids into 293T cells by calcium phosphate precipitation, as described previously (13). Briefly, cells were seeded into 10-cm culture dishes and allowed to attach for 24 h before transfection with 20 μg expression plasmid per plate. The transfection medium was changed at approximately 16 h posttransfection. The expression-enhancing reagent valproic acid (VPA) was added to a final concentration of 7.5 mM, and the cells were incubated for 3 to 4 h. The medium was changed again, and the cells were inoculated with VSVΔG-GFP virus pseudotyped with VSV-G at a multiplicity of infection (MOI) of 0.1 to 0.3 for 3 to 4 h before the medium was changed again. At about 24 h postinfection, the supernatants were collected and cleared of debris by filtration through a 0.45-μm syringe filter. To achieve higher titers, infectious supernatants were concentrated 10-fold by centrifugation in Amicon centrifugal filters with an exclusion size of 100,000 kDa (Millipore). The pseudotypes were titrated on different cell lines by infection of a known cell number, followed by fixation of infected cells with 2% paraformaldehyde (PFA) at 24 h postinfection and measurement of the number of GFP-expressing cells by flow cytometry using a Becton, Dickinson LSRII cytometer and FlowJo software. To avoid the inclusion of superinfection events, infectious rates of between 10 and 20% infected cells were used for calculations of viral titers, expressed as transducing units (TU)/ml. All experiments involving VSVΔG-GFP virus pseudotyped with BASV-G were conducted using biosafety level 3 (BSL-3) safety standards to address the risk of recombination.
Immunofluorescence staining.
BHK cells were seeded onto polylysine-coated 8-well culture slides (BD) and allowed to attach for 24 h. The cells were transfected with 2.5 μg pCAGGS plasmid expressing BASV-G with a C-terminal V5 tag or empty pCAGGS plasmid by using 7.5 μl TransIT2020 transfection reagent (Mirus) in 200 μl Opti-MEM medium (Gibco). The medium was changed at about 16 h posttransfection, and at 40 h posttransfection, the cells were washed 3 times with phosphate-buffered saline (PBS) and fixed by incubation with 2% paraformaldehyde (PFA) for 15 min at room temperature under mild rocking. The cells were then permeabilized with 200 μl binding buffer (3% bovine serum albumin [BSA], 0.5% Triton X-100, and 10% FBS in PBS) for 30 min at room temperature, followed by staining with 100 μl mouse anti-V5 monoclonal antibody (Invitrogen) diluted 1:200 in binding buffer for 1 h at room temperature under mild rocking. The slides were washed 3 times with PBS and incubated with 100 μl goat anti-mouse IgG conjugated with Alexa 488 (Invitrogen) at a 1:200 dilution for 1 h at room temperature under mild rocking. The slides were washed 2 times with PBS and 2 times with molecular-grade water to remove salts before the coverslips were mounted with Fluoroshield with 4′,6-diamidino-2-phenylindole (DAPI) mounting medium (Sigma-Aldrich). The samples were analyzed on a Leica DMI 6000B microscope, and pictures were taken with a Leica CTR6500 camera using iVision software (BioVision Technologies).
Western blotting.
Viruses were produced and processed as described above. Twenty-four hours after infection with VSVΔG-GFP/VSV-G, the supernatants were harvested and concentrated, and the producer cells were lysed by the addition of 500 μl radioimmunoprecipitation assay (RIPA) buffer (50 mM Tris [pH 8.0], 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) supplemented with protease inhibitors (Roche) per 10-cm culture dish. The cells were incubated for 10 min at room temperature, and the lysates were transferred into a 5-ml tube on ice. The lysates were sonicated for 20 s at 6 to 8 W on ice before the addition of lithium dodecyl sulfate (LDS) loading buffer and sample-reducing agent (Invitrogen). A milliliter each of the concentrated supernatants was overlaid onto 100 μl 20% sucrose in 1.5-ml tubes and centrifuged at 16,000 × g for 1 h. The complete liquid portion was removed to yield virus pellets that were each resuspended in 40 μl 1× LDS loading buffer–sample-reducing agent. All samples were denatured by incubation at 95°C for 10 min before gel electrophoresis on 10% NuPAGE Novex SDS gels with 1× morpholinepropanesulfonic acid (MOPS) buffer (Invitrogen) at 120 V for 1.5 h. The gel was transferred onto nitrocellulose membranes by using the iBlot system (Invitrogen), and the membranes were blocked with 2.5% dry milk in Tris-buffered saline (TBS; Fisher) plus 0.05% Tween 20 (Sigma) (TBS-T) for 1 h. The membranes were incubated overnight with a mouse anti-V5 monoclonal antibody (Invitrogen) diluted 1:5,000 in 2.5% dry milk in TBS-T. The membranes were washed 3 times for 10 min with TBS-T before incubation with a horseradish peroxidase (HRP)-conjugated goat anti-mouse IgG antibody (Thermo Scientific) diluted 1:5,000 in 2.5% dry milk in TBS-T for 1 h. The membranes were washed 4 times for 10 min with TBS-T and incubated with SuperSignal Femto ECL substrate (Pierce) for 5 min before exposure and chemiluminescence detection with an ImageQuant LAS4000 imaging system (GE Healthcare).
Glycosidase digest.
The VSVΔG-GFP pseudotypes were produced and harvested as described above. The concentrated infectious supernatants were centrifuged through a 20% sucrose cushion to pellet the virions. The pelleted viruses and the producer cells were lysed in RIPA buffer. Nine microliters of each sample was incubated with 1 μl denaturing buffer (NEB) at 100°C for 10 min. For the endoglycosidase H (Endo H) digest, 2 μl G5 reaction buffer (NEB), 7 μl water, and 1 μl enzyme were added, for a total volume of 20 μl. For the PNGase F digest, 2 μl G7 reaction buffer (NEB), 2 μl 10% NP-40 (NEB), 5 μl water, and 1 μl enzyme were added. The undigested control was treated with 2 μl G7 reaction buffer, 2 μl 10% NP-40, and 6 μl water. The samples were incubated at 37°C for 1.5 h before SDS gel electrophoresis and Western blot analysis, as described above.
Fusion assay.
For cell-to-cell fusion, 293T effector cells were seeded into 6-well plates and Vero target cells were seeded into 96-well plates and allowed to attach for 24 h. Subsequently, 293T cells were transfected with 3 μg β-galactosidase omega fragment expression plasmid and 1 μg (VSV-G, CHIKV env, and NiV F), 2 μg (NiV G), or 3 μg (pCAGGS, BASV-G, W93A, Y94A, W137A, and N138A) envelope DNA by using calcium phosphate precipitation, and Vero cells were transfected with 0.1 μg β-galactosidase alpha fragment per well by using 0.3 μl TransIT2020 transfection reagent and 10 μl Opti-MEM medium per well. At about 16 h posttransfection, the medium was replaced, and at 24 h posttransfection, VPA was added to a final concentration of 7.5 mM. At about 40 h posttransfection, the medium of the 293T cells was removed, and the cells were detached from the plates by incubation with 200 μl Versene (Gibco) per well at 37°C for 5 min. A volume of 800 μl DMEM was added per well, and the cells were carefully resuspended. The medium of the Vero target cells was replaced with fresh medium, and 40 μl of the 293T effector cells was added per well. The mixed cell populations were incubated for 1 h at 37°C. The plates were briefly centrifuged at 200 × g for 3 min, and the medium was carefully removed from the wells. Thirty microliters of pH-adjusted Earl's balanced salt solution with 20 mM morpholineethanesulfonic acid (MES) and 20 mM HEPES was then added per well, and the cells were incubated for 10 min at 37°C to induce pH-dependent fusion. To neutralize the pH, 150 μl DMEM and 20 mM HEPES (pH 7.4) were added per well, and the plates were returned to 37°C for an additional incubation for 5 h. The cells were lysed, and the β-galactosidase reporter activities in cell lysates were determined by using the chemiluminescence-based GalactoLight assay system (Applied Biosystems).
pH inactivation.
Vero cells were seeded into 48-well plates 24 h before infection. Per well, 10 μl virus was mixed with 40 μl pH-adjusted Earl's balanced salt solution with 20 mM MES and 20 mM HEPES (pH 4.5 to 7.0) and incubated for 30 min at 37°C. The pH was neutralized by the addition of 200 μl DMEM with 20 mM HEPES at pH 7.4 and incubation for 30 min. The medium was removed from the Vero cells and replaced with 250 μl pH-treated virus per well. The cells were incubated for 24 h, fixed with 2% PFA, and analyzed for GFP expression in a Becton, Dickinson LSRII cytometer and with FlowJo software.
Inhibition by lysosomotropic agents.
VSVΔG-GFP pseudotype infections were performed as described above. However, prior to infection, Vero cells were incubated with various concentrations of the lysosomotropic agents bafilomycin A, chloroquine, or ammonium chloride (Sigma-Aldrich) for 1 h at 37°C. The number of infected cells was determined by flow cytometric analysis of GFP expression.
Enhancement by lectin expression on target cells.
T-REx-293 cells (Invitrogen) were stably transfected to express the C-type lectins DC-SIGN, DC-SIGNR, SIGNR1, CLEC-1, CLEC-2, LSectin, or asialoglycoprotein receptor 1 (ASGPR1), as previously described (14, 15). The cells were grown in DMEM with 10% FBS, Glutamax, nonessential amino acids, 200 μg/ml zeocin, and 5 μg/ml blasticidin and seeded into 48-well plates 48 h before infection. Twenty-four hours before infection, 1 μg/ml tetracycline was added to induce lectin expression. The cells were infected with VSVΔG-GFP pseudotypes at a low MOI and incubated for 24 h. For mannan inhibition, cells were incubated with 200 μg/ml mannan for 1 h prior to infection. The cells were fixed with 2% PFA before flow cytometric analysis of GFP expression, as described above.
RESULTS
A multiple-sequence alignment of BASV-G with the glycoprotein sequences of 8 other rhabdovirus genomes, including RABV, Australian bat lyssavirus (ABLV), VSV, Chandipura virus (CHPV), Moussa virus (MOUV), viral hemorrhagic septicemia virus (VHSV), Tibrogargan virus (TIBV), and bovine ephemeral fever virus (BEFV), was performed. The alignment revealed structural similarities between the viral glycoproteins, such as the presence of an N-terminal signal peptide of 23 amino acids, two bipartite fusion loop motifs at positions 92/93 and 137/138, a single transmembrane domain of 23 amino acids, and a short intracellular C terminus of 33 amino acids (Fig. 1). N-glycosylation site prediction using NetNGlyc 1.0 detected four potential Asn sequons at residues 56, 194, 275, and 548 (Fig. 1A). This comparatively low level of glycosylation is typical for rhabdovirus glycoproteins such as RABV-G (3 N-glycosylation sites, of which only 2 are efficiently glycosylated [16]) and VSV-G (2 N-glycosylation sites [17]) and stands in contrast to other more extensively glycosylated viral glycoproteins, such as human immunodeficiency virus (HIV) gp120 (18 to 33 N-glycosylation sites [18]), EBOV-GP (17 N-glycosylation sites [19]), and severe acute respiratory syndrome coronavirus spike (SARS-S) (23 N-glycosylation sites [20]).
Fig 1.

BASV-G shares sequence and structure similarities with other rhabdovirus glycoproteins. The multiple-sequence alignment of the amino acid sequences of the glycoproteins of rabies virus (RABV), Australian bat lyssavirus (ABLV), vesicular stomatitis virus (VSV), Chandipura virus (CHPV), Moussa virus (MOUV), viral hemorrhagic septicemia virus (VHSV), Tibrogargan virus (TIBV), Bas-Congo virus (BASV), and bovine ephemeral fever virus (BEFV) was done by using the ClustalW multiple-sequence alignment tool (EMBL-EBI). The N-terminal signal peptide is shown in green, the bipartite fusion loop motif is shown in teal, and the transmembrane domain is shown in pink. Shades of blue indicate percent identity, with dark blue indicating 80 to 100%, medium blue indicating 60 to 80%, light blue indicating 40 to 60%, and no color indicating <40% identity.
The predicted BASV-G nucleotide sequence was previously synthesized (Genscript) and subcloned into the mammalian expression vector pCAGGS. For easy detection of the expressed protein, a V5 tag was included at the C terminus. To determine the expression level and localization of the V5-tagged BASV-G protein, BHK cells were transfected with the pCAGGS/BASV-G.V5 construct or the empty vector alone. The cells were fixed and permeabilized before staining with a mouse anti-V5 antibody and secondary detection with an Alexa 488-conjugated anti-mouse IgG antibody. The BASV-G protein was found in dot-like punctae throughout the cell, characteristic of a protein produced in the secretory pathway (Fig. 2A).
Fig 2.

BASV-G is expressed, glycosylated, and efficiently incorporated into virions. (A) Immunofluorescence staining of BHK cells transfected with V5-tagged BASV-G (left) or the empty vector control (right), permeabilized, and stained with a mouse anti-V5 antibody, Alexa 488-conjugated secondary antibody, and DAPI. A representative experiment of a total of two experiments is shown. (B) Western blot detection of V5-tagged BASV-G and BASV-G W93A, Y94A, W137A, and N138A fusion loop mutants in 293T virus producer cell lysates (left) and concentrated viral particles (right), using a mouse anti-V5 antibody and an HRP-conjugated secondary antibody. A representative experiment of a total of three experiments is shown. (C) Western blot detection of V5-tagged BASV-G in cell lysates (left) and concentrated viral particles (right) undigested or after digestion with Endo H or PNGase F. A representative experiment of a total of three experiments is shown.
To test for virion incorporation of V5-tagged BASV-G, cell lysates of pseudotype producer cells and cell-free pelleted viruses were subjected to Western blot analysis (Fig. 2B). In addition, alanine exchange mutants of the 4 amino acids in the putative bipartite fusion loop motif (W93, Y94, W137, and N138) were produced by overlap PCR and tested alongside the wild-type BASV-G protein. All proteins were expressed in the pseudotype producer cells, although at different levels, and could be detected in the lysates with an antibody targeted to the V5 tag (Fig. 2B). No size difference was observed for any of the mutants. Wild-type BASV-G could be clearly detected in pelleted VSV pseudoparticles (Fig. 2B), suggesting efficient incorporation. This was further confirmed by the fact that BASV-G conferred the ability to efficiently transduce target cells. Lower levels of incorporation were observed for the N138A and W93A mutants, while incorporation of the Y94A and W137A mutants was undetectable (Fig. 2B). Inoculation of Vero cells with wild-type and mutant pseudotype preparations resulted in reduced titers for the N138A mutant compared to wild-type BASV-G and no detectable infectivity for the remaining fusion loop mutants (data not shown). These results are in agreement with previous studies showing that mutation of the aromatic amino acids of the bipartite fusion loop motif of VSV-G abolishes fusion activity, while mutation of the nonaromatic amino acid merely reduces it (7). Mutation of the aromatic amino acids also markedly reduced the surface expression of the mutant VSV-G proteins at 37°C, which could be rescued by lowering the temperature to 32°C (7). Consistent with this, BASV-G-pseudotyped virions produced at lower temperatures were found to incorporate higher levels of mutant BASV-G proteins; however, this did not affect the infectivity of mutant viral particles (data not shown).
To experimentally confirm the presence of the predicted N-linked glycans, V5-tagged BASV-G protein from virus producer cells and cell-free pelleted virions was digested with the glycosidases endoglycosidase H (Endo H) and peptide-N-glycosidase F (PNGase F). While Endo H cleaves high-mannose and some hybrid oligosaccharides from N-linked glycoproteins, PNGase F removes almost all types of N-linked glycans, including high-mannose, hybrid, and bi-, tri-, and tetra-antennary glycans. Glycosidase digestion of BASV-G from cell lysates with both Endo H and PNGase F led to a reduction in molecular mass of approximately 10 kDa (Fig. 2C). However, BASV-G incorporated into virions migrated only slightly faster when digested with Endo H, while PNGase F digestion removed further glycans resistant to Endo H activity (Fig. 2C). These results reflect the state of glycan processing, with most of the cellular protein containing simple high-mannose glycans similarly sensitive to both glycosidases, while the viral protein comprises fully processed complex glycans only partially susceptible to Endo H digestion. The finding also confirms that posttranslational modification of BASV-G with N-linked glycans was relatively light compared to other viral glycoproteins such as HIV Env, contributing approximately 10 kDa to the total protein mass of 70 kDa.
We next sought to narrow down the cellular and tissue tropism of BASV-G-driven infection. A spectrum of adherent and nonadherent human cell lines was infected with both VSV-G- and BASV-G-pseudotyped VSVΔG-GFP viruses. As expected, high viral titers of between 1.0E+06 and 1.0E+08 transducing units (TU)/ml were achieved with VSV-G on all adherent human cell lines (Fig. 3A and Table 1). Interestingly, very similar results were obtained with BASV-G pseudotypes. Titers in human colon SW480 cells as well as human hepatoma Huh-7 cells were roughly identical for both glycoproteins, while titers in human kidney 293T cells, human alveolar epithelial A549 cells, and human cervix HeLa cells were slightly lower for BASV-G than for VSV-G (Fig. 3A and Table 1). In contrast, BASV-G titers were slightly increased over VSV-G titers in human muscle RD cells and human intestinal CaCo-2 cells (Fig. 3A and Table 1). The greatest variance in titers for adherent cells, however, was found in human fibrosarcoma HT1080 cells, with 9.0E+07 TU/ml for VSV-G and 2.0E+07 TU/ml for BASV-G (a 4.5-fold difference) (Fig. 3A and Table 1). Distinctively, infection experiments with human H9 and Jurkat T cells, human B-THP/Raji B-cells, human THP-1 monocytes, and human erythroleukemia HEL cells resulted in 1- to 2-log-lower titers for BASV-G than for VSV-G, without exception (Fig. 3B and Table 1). However, all tested human cell lines were susceptible to BASV-G-driven infection, suggesting a broad tissue tropism, including lymphocytes.
Fig 3.

BASV-G pseudotypes are able to infect a broad range of human and nonhuman cell lines. (A) Human adherent cell lines HT1080, SW480, RD, 293T, CaCo-2, Huh-7.5, A549, and HeLa were infected with VSV-G (left) or BASV-G (right) pseudotypes, and titers were calculated based on the percentage of GFP reporter-expressing cells at 24 h postinfection. Green, fibrosarcoma; blue, colon adenocarcinoma; yellow, muscle cells. (B) Human suspension cell lines B-THP, HEL, Jurkat, H9, and THP-1 were infected with VSV-G (left) or BASV-G (right) pseudotypes, and titers were calculated based on the percentage of GFP reporter-expressing cells at 24 h postinfection. Red, T lymphocytes. (C) Nonhuman adherent cell lines C6/36, C7/10, 3T3, NRK, BHK, SK-RST, Tb1Lu, C6, MC57, Vero, and MDBK were infected with VSV-G (left) or BASV-G (right) pseudotypes, and titers were calculated based on the percentage of GFP reporter-expressing cells at 24 h postinfection. Red, mosquito cells; green, mouse cells; blue, rat cells; yellow, bat cells. (D) Human primary cell lines HUVEC and HUPEC were infected with VSV-G (left) or BASV-G (right) pseudotypes, and titers were calculated based on the percentage of GFP reporter-expressing cells at 24 h postinfection. Background levels for bald particles lacking the glycoprotein were below 1.0E+04 TU/ml. Infections were performed in duplicate and were repeated at least three times for each cell line.
Table 1.
Actual and relative titers of VSV-G and BASV-G pseudotype infections in various cell lines and primary cellsa
| Cell line | Mean VSV-G titer (TU/ml) ± SD | Mean BASV-G titer (TU/ml) ± SD | Mean relative titer ± SDb |
|---|---|---|---|
| Human adherent cell lines | |||
| 293T | 1.2E+07 ± 3.7E+06 | 9.0E+06 ± 1.2E+05 | 74.5 ± 1.1 |
| Huh-7 | 4.8E+06 ± 5.5E+05 | 4.3E+06 ± 2.1E+04 | 89.8 ± 0.4 |
| A549 | 4.6E+06 ± 8.8E+04 | 3.2E+06 ± 3.1E+04 | 69.3 ± 0.7 |
| HeLa | 4.1E+06 ± 3.4E+05 | 2.8E+06 ± 1.8E+05 | 68.2 ± 4.4 |
| SW480 | 2.9E+07 ± 5.8E+05 | 2.8E+07 ± 4.7E+05 | 95.1 ± 1.6 |
| CaCo-2 | 7.2E+06 ± 8.1E+04 | 8.4E+06 ± 4.9E+05 | 116.7 ± 6.7 |
| HT1080 | 8.8E+07 ± 2.1E+06 | 1.9E+07 ± 4.5E+06 | 21.6 ± 5.1 |
| RD | 1.3E+07 ± 7.4E+05 | 2.0E+07 ± 1.0E+04 | 153.9 ± 0.1 |
| Human suspension cell lines | |||
| H9 | 1.4E+06 ± 3.7E+04 | 1.9E+05 ± 1.1E+04 | 13.1 ± 0.8 |
| Jurkat | 1.2E+07 ± 3.2E+05 | 6.1E+05 ± 1.8E+05 | 5.1 ± 1.5 |
| B-THP | 5.1E+07 ± 2.9E+06 | 5.7E+05 ± 2.1E+05 | 1.1 ± 0.4 |
| THP-1 | 1.0E+06 ± 2.1E+05 | 5.8E+04 ± 3.6E+03 | 5.6 ± 0.3 |
| HEL | 1.4E+07 ± 1.8E+06 | 2.7E+06 ± 2.5E+05 | 19.3 ± 1.7 |
| Vertebrate cell lines | |||
| Vero | 3.9E+06 ± 7.9E+05 | 8.7E+05 ± 1.5E+05 | 22.5 ± 3.8 |
| MC57 | 4.6E+06 ± 1.4E+06 | 2.4E+06 ± 1.5E+05 | 53.5 ± 3.2 |
| NIH 3T3 | 3.2E+07 ± 7.8E+05 | 2.8E+05 ± 1.8E+04 | 0.9 ± 0.1 |
| C6 | 4.8E+06 ± 3.3E+05 | 3.6E+05 ± 1.5E+05 | 7.5 ± 3.2 |
| NRK | 1.5E+07 ± 8.7E+04 | 2.8E+06 ± 1.2E+04 | 18.5 ± 0.1 |
| BHK | 7.0E+06 ± 2.2E+05 | 3.0E+06 ± 1.1E+04 | 42.7 ± 0.2 |
| SK-RST | 5.2E+06 ± 1.1E+04 | 2.3E+05 ± 5.5E+04 | 4.4 ± 1.0 |
| MDBK | 3.0E+06 ± 3.9E+05 | 4.0E+05 ± 2.9E+04 | 13.5 ± 1.0 |
| Tb1Lu | 5.0E+06 ± 4.8E+05 | 3.9E+05 ± 9.8E+04 | 7.8 ± 2.0 |
| Invertebrate cell lines | |||
| C7/10 | 6.9E+07 ± 1.0E+07 | 7.4E+06 ± 5.7E+04 | 10.7 ± 0.1 |
| C6/36 | 8.4E+07 ± 2.8E+05 | 6.1E+06 ± 1.3E+05 | 7.2 ± 0.2 |
| Human primary cells | |||
| HUVEC | 1.5E+07 ± 1.4E+06 | 6.5E+06 ± 6.1E+05 | 43.3 ± 6.1 |
| HUPEC | 1.1E+07 ± 1.1E+06 | 2.6E+06 ± 4.4E+05 | 23.6 ± 4.0 |
For the negative control, the background level of infectivity of bald particles was 5.6E+03 ± 1.4E+03 TU/ml.
Relative titer is the BASV-G/VSV-G ratio as a percentage of VSV-G.
Since the natural reservoir and the initial route of BASV transmission to humans are unknown, BASV-G pseudotype infection of cell lines from various animals was performed to give an indication to the susceptibilities of different species to BASV-G-driven infection. Interestingly, the highest titers for both VSV-G and BASV-G were observed in the mosquito cell lines C6/36 and C7/10, with about 10-fold-lower titers for BASV-G than for VSV-G (Fig. 3C and Table 1). BASV-G-driven infection of other adherent cell lines, including several rodent cell lines as well as bovine, porcine, monkey, and bat cells, yielded lower titers than did VSV-G-driven infection. However, all tested cell lines were readily susceptible to BASV-G-mediated infection (Fig. 3C and Table 1). The greatest difference in titers was observed for the murine fibroblast cell line NIH 3T3, with 100-fold-lower titers for BASV-G than for VSV-G. These results suggest that a number of species could potentially act as a natural host reservoir for BASV, including rodents, bats, or mosquitoes.
Finally, to test BASV-G pseudotype infection in primary cells, the human endothelial cell lines HUVEC and HUPEC were subjected to VSV-G- and BASV-G-mediated infection. Both cell types were susceptible to BASV-G-driven infection, again with slightly lower titers for BASV-G than for VSV-G (Fig. 3D and Table 1). Other primary cells, such as PBMCs and fetal hepatocytes, could not be tested due to low titers even with VSV-G, likely attributable to the sensitivity of the VSVΔG-GFP viral backbone to interferon responses more potently induced by primary cells.
We next sought to determine the principal route of entry into target cells for BASV-G-pseudotyped virions. The fusion activity of VSV-G and other rhabdovirus glycoproteins is induced by a low-pH trigger in the endosome (8, 21). The same is true for alphavirus type I fusion proteins like CHIKV env (22), while membrane fusion mediated by the F and G proteins of the paramyxovirus NiV does not require acidification (23). Utilizing this knowledge, we adapted a cell-cell fusion assay based on the α-complementation of β-galactosidase (24–26) to investigate the pH optimum of BASV-G-mediated fusion. Effector 293T cells were transfected to express NiV F/G, VSV-G, CHIKV env, BASV-G, or the four fusion loop mutants along with the omega fragment of β-galactosidase and were mixed with target Vero cells transfected with the β-galactosidase alpha fragment and known to be susceptible to infection with the respective viruses (7, 27, 28). Lowering the pH to values between 4.5 and 7.0 induced fusion. After neutralization of the acidic pH and additional incubation time, the reporter activity in the cell lysates could be determined as a measure of fusion activity (Fig. 4A). The fusion activity of NiV F/G was not influenced by the pH but yielded consistently high measurements, as expected from the highly fusogenic NiV F protein (Fig. 4A). The pH-sensitive fusion proteins VSV-G and CHIKV env showed some residual fusion activity at neutral pH, as anticipated based on previous studies (29, 30), but demonstrated a clear increase in fusogenicity at pH values of ≤6.0 (Fig. 4A). In contrast, the fusion properties of BASV-G were activated only at pH values of ≤5.5, indicating a tighter pH barrier for the induction of the fusogenic conformation of BASV-G (Fig. 4A). Of the BASV-G fusion loop mutants, only the N138A mutant was able to induce cell-cell fusion although at reduced levels, while alanine replacement of the aromatic amino acids completely abolished the ability of BASV-G to function in the fusion assay (Fig. 4A). This observation is consistent with those of a similar study performed for VSV-G, which found that mutation of the aromatic fusion loop residues led to a loss of fusion activity (7).
Fig 4.
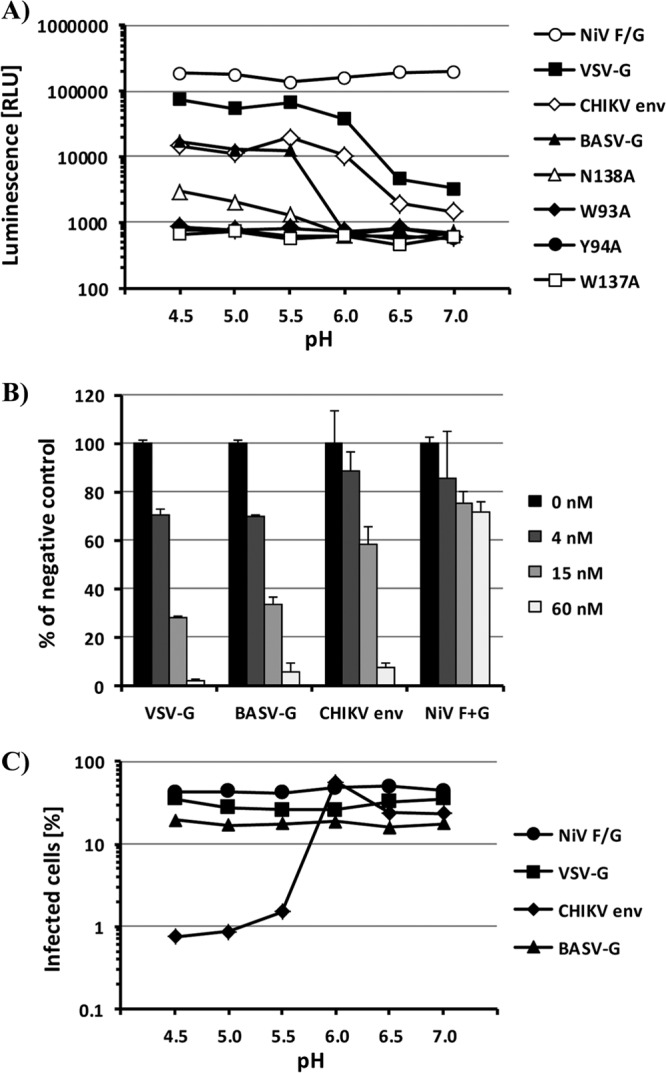
Activation of BASV-G fusion activity is pH dependent and reversible and occurs in the late endosome. (A) Cell-cell fusion mediated by the viral glycoproteins NiV F/G, CHIKV env, VSV-G, BASV-G, and the BASV-G W93A, Y94A, W137A, and N138A fusion loop mutants. Vero cells expressing the alpha subunit of β-galactosidase and 293T cells expressing the omega subunit of β-galactosidase and one of the glycoproteins were mixed and allowed to fuse at the indicated pH values. β-Galactosidase activities in cell lysates were determined at 5 h postfusion by using the chemiluminescence-based GalactoLight assay system. The experiment was performed in triplicate, and a representative experiment of a total of five experiments is shown. RLU, relative light units. (B) Vero cells were incubated with the indicated concentrations of bafilomycin A prior to infection with VSVΔG-GFP pseudotypes bearing VSV-G, BASV-G, CHIKV env, or NiV F/G. The level of infection was determined as the percentage of GFP-expressing cells at 24 h postinfection and is shown as a percentage of infection in the absence of bafilomycin A. The experiment was performed in duplicate, and a representative experiment of a total of four experiments is shown. Error bars indicate standard deviations. (C) VSVΔG-GFP pseudotypes bearing VSV-G, BASV-G, CHIKV env, or NiV F/G were exposed to the indicated pH values for 30 min before neutralization and infection of Vero cells. The level of infection was determined as a percentage of GFP-expressing cells at 24 h postinfection. The experiment was performed in duplicate, and a representative experiment of a total of three experiments is shown.
The induction of BASV-G-mediated fusion by acidic pH suggests endosomal uptake as the principal route of target cell entry. To confirm the requirement of endosomal acidification for BASV-G-driven infection, Vero cells were treated with bafilomycin A prior to infection with pseudotyped viruses. Bafilomycin A belongs to a group of specific inhibitors of vacuolar-type H+-ATPases which are found in endosomes, lysosomes, and secretory vesicles and play a crucial role in the acidification of these organelles (31). As expected, NiV F/G-mediated entry was only slightly affected by bafilomycin A treatment of target cells, even at a concentration of 60 nM (Fig. 4B). The observed reduction of titers is likely due to a general effect of bafilomycin A on cell proliferation and viability. In contrast, VSV-G- and CHIKV env-pseudotyped viruses, both known to require endosomal acidification for productive cell entry (8, 22), were markedly inhibited by bafilomycin treatment of target cells (Fig. 4B). Similarly, BASV-G-mediated cell entry was reduced in a dose-dependent manner by preincubation of cells with increasing concentrations of bafilomycin A (Fig. 4B). Comparable effects were obtained with other lysosomotropic agents such as ammonium chloride and chloroquine (data not shown). These results confirm the requirement of endosomal acidification for BASV-G-driven cell entry.
A striking feature of VSV-G and other rhabdovirus glycoproteins, in comparison to viral class I and II fusion proteins, is the full reversibility of the conformational changes induced by the low-pH trigger (8). To test if BASV-G shares the reversible fusion properties of the related rhabdovirus glycoproteins, pseudotypes harboring NiV F/G, VSV-G, CHIKV env, or BASV-G were incubated at pH values between 7.0 and 4.5 and brought back to neutral pH before infection of target cells. Virions pseudotyped with the pH-independent NiV F/G or the reversible VSV-G entered the cells unhindered (Fig. 4C). In contrast, CHIKV env pseudotypes exhibited markedly reduced infectivity levels after transient exposure to pH values of <6.0, consistent with a pH optimum of CHIKV env-mediated fusion of about pH 5.5 (Fig. 4C). Interestingly, BASV-G-driven infection of target cells was not affected by preincubation at acidic pH (Fig. 4C), supporting its addition to the group of reversible class III fusion proteins.
There is speculation about the possible animal reservoir and/or vector harboring BASV (1), and cell lines from different species, including mosquito cells, were found to be susceptible to BASV-G-driven infection (Fig. 3C). The need for additional data to facilitate the identification of the reservoir/vector species led us to investigate the temperature range of effective BASV-G fusion to provide potential insight into its natural reservoir in the wild. The cell-cell fusion assay described above was modified by moving the cells, media, and buffers to a range of different temperatures shortly before, during, and after the induction of fusion at pH 5.0. At 8°C, no, or very limited, fusion was observed for any of the tested glycoproteins, consistent with a largely reduced membrane fluidity at lower temperatures (Fig. 5). Interestingly, at 22°C, fusion activity was high for CHIKV env and already maximal for VSV-G, while BASV-G fusion activity was only slightly above background levels (Fig. 5). CHIKV env fusion activity continued to increase at 28°C and 37°C, while VSV-G maintained high levels of fusion activity throughout this temperature range (Fig. 5). The activity of both fusion proteins was slightly reduced at 40°C, indicating a temperature optimum for VSV-G- and CHIKV env-mediated fusion of about 37°C (Fig. 5). In contrast, BASV-G fusion activity increased slowly and steadily between 22°C and 37°C and continued to increase at 40°C (Fig. 5). Notably, the BASV-G fusion loop N138A mutant with a reduced overall fusion capacity displayed even stronger temperature dependence, with a 4-fold increase of fusion activity between 28°C and 37°C and another 3-fold increase between 37°C and 40°C (Fig. 5). This finding is surprising given the high titers of BASV-G pseudotypes that were obtained in mosquito cells grown at 28°C (Fig. 3C).
Fig 5.
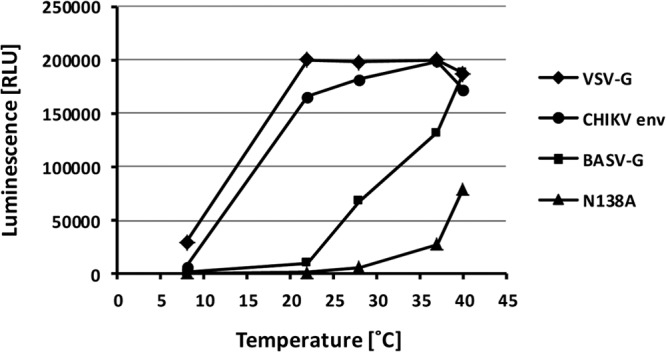
BASV-G-mediated fusion is heat activated and highly temperature dependent. Cell-cell fusion mediated by the viral glycoproteins VSV-G, CHIKV env, BASV-G, and the BASV-G N138A fusion loop mutant induced at pH 5.0 was measured at a range of different temperatures, as described in the legend of Fig. 4A. The experiment was performed in triplicate, and a representative experiment of a total of three experiments is shown.
Finally, we sought to investigate the importance of cellular lectins as attachment factors for BASV-G. C-type lectins such as DC-SIGN and DC-SIGNR have been found to bind to the glycoproteins of and enhance infection with a number of different viruses (32, 33). The interaction is based on high-mannose carbohydrate modifications of the viral glycoproteins acting as DC-SIGN ligands (34). While infections with viruses bearing highly glycosylated viral glycoproteins, such as HIV-1 gp120, EBOV-GP, and SARS-S, are markedly enhanced in the presence of C-type lectins, no such effect was observed for glycoproteins with lower glycan content, such as VSV-G (33). However, glycosidase digests of BASV-G with Endo H and PNGase F strongly suggested the presence of a mixture of high-mannose and complex carbohydrates (Fig. 2C). Tetracycline-inducible T-REx-293 cells expressing the lectins DC-SIGN, DC-SIGNR, or SIGNR1 were infected with VSV-G, BASV-G, or EBOV-GP pseudotypes, and the resulting titers were compared to those of infections of regular 293T cells devoid of lectins. As expected, EBOV-GP-driven infection of DC-SIGN- and DC-SIGNR-expressing T-REx-293 cells was enhanced 10-fold and 12-fold, respectively, compared to infection of regular 293T cells (Fig. 6). The effect of SIGNR1 expression on EBOV-GP pseudotype infection was less pronounced and led to a 3-fold increase. In contrast, none of the lectins enhanced VSV-G-driven infection to >1.5-fold (Fig. 6). Interestingly, expression of DC-SIGN, DC-SIGNR, and SIGNR1 enhanced BASV-G-mediated infection 6-, 5-, and 8-fold, respectively, compared to infection of regular 293T cells (Fig. 6). Finally, all enhancing effects of lectin expression on EBOV-GP and BASV-G pseudotype infection could be counteracted by incubation of cells with mannan prior to infection, thereby blocking the carbohydrate recognition domains of the lectins (Fig. 6). The observed effect seemed to be selective for DC-SIGN, DC-SIGNR, and SIGNR1, since CLEC-1, CLEC-2, LSectin, and ASGPR1 (asialoglycoprotein receptor 1) were not found to have enhancing effects on BASV-G-mediated infection (data not shown).
Fig 6.
The lectins DC-SIGN, DC-SIGNR, and SIGNR1 act as attachment factors and enhance BASV-G-mediated infection. Expression of DC-SIGN, DC-SIGNR, and SIGNR1 was induced in T-REx-293 cells 24 h prior to infection with VSV-G, BASV-G, or EBOV-GP pseudotypes. To block lectin-mediated enhancement of infection, cells were pretreated with 200 μg/ml mannan. The level of infection was determined as a percentage of GFP-expressing cells at 24 h postinfection and is shown as a percentage of infection of 293T control cells. The experiment was performed in duplicate, and a representative experiment of a total of three experiments is shown. Error bars indicate standard deviations.
DISCUSSION
Emerging viruses pose a substantial threat to global public health, and the number of events of zoonotic transmission of previously unknown pathogens to the human population has risen significantly over time (35). Novel viruses like the SARS coronavirus, pandemic influenza virus strains, and human immunodeficiency virus (HIV) have demonstrated the potential of emerging viruses to rapidly spread among the population, more rapidly than public health measures, reliable diagnostic tools, and potential therapeutics or vaccines can be deployed. Therefore, it is crucial to detect and characterize emerging viruses that have recently been introduced into the human population, to enable us to predict future transmission events and the dissemination potential of novel pathogens.
In this study, we performed a detailed characterization of the glycoprotein of BASV, a novel rhabdovirus recently discovered in association with a small hemorrhagic fever outbreak in Central Africa, and its role in cell entry. Serological tests will determine the prevalence of BASV infection in the Bas-Congo province and throughout Central Africa, and a comprehensive study of the structural and functional features of the BASV glycoprotein will provide critical information for the prevention and therapy of BASV infection. However, it is currently not possible to culture the virus from patient samples, and a reverse genetics system to culture the virus has yet to be established. Furthermore, conducting these studies with a replication-competent and potentially highly pathogenic virus may eventually require containment in a biosafety level 4 laboratory. Thus, we resorted to working with a VSVΔG/BASV-G pseudotype system, allowing a comprehensive analysis of the functional features of the BASV glycoprotein, as well as serological testing for neutralizing antibodies in human sera, at lower biosafety levels.
Amino acid sequence alignments of BASV-G with 8 other rhabdovirus glycoproteins revealed conserved structures, such as the internal bipartite fusion loop motif that was previously described for a number of animal and plant rhabdoviruses (7). The most striking difference of BASV-G and its phylogenetically closest relatives, TIBV-G and BEFV-G, compared to VSV-G and other rhabdovirus glycoproteins is the additional 100 to 150 amino acids in length, with the longest stretch unique to this group between amino acids 400 and 460. The function of these additional sequence elements is unknown at present. However, functionally, BASV-G and VSV-G appear to be similar in many ways. Mutations of the three aromatic amino acids in the fusion loops previously shown to be critical for VSV-G-mediated fusion (7) also led to a loss of function of BASV-G. For VSV-G, this observation has been attributed to a defect in protein transport to the cell surface at 37°C, which could be partially rescued at lower temperatures. However, the fusion activity of VSV-G fusion loop mutants could not be restored by improved surface expression (7). Consistent with this, our experiments showed no infectivity of BASV-G W93A, Y94A, or W137A mutants when produced at either 37°C or 28°C. More likely, the aromatic amino acid residues of the fusion loops are involved directly in the insertion of the protein into the target membrane that initiates membrane fusion. As for VSV-G, mutation of the fourth, nonaromatic fusion loop residue in BASV-G led to an overall reduction of fusion activity and a shift to a lower pH optimum without complete loss of function.
Class I and II fusion proteins exist in a metastable prefusion state that, upon triggering, irreversibly transitions into the postfusion state, leading to virus-cell membrane fusion. Triggering in the absence of a target membrane leads to the inactivation of the fusion machinery (9, 36). In contrast, class III fusion proteins exist in a dynamic equilibrium of pre- and postfusion conformations that is driven toward the active state at low pH but allows the protein to return to its inactive state upon neutralization of the ambient pH (8). This observation led to the classification of VSV-G and certain herpesvirus glycoproteins as novel class III viral fusion proteins (7). The finding that pseudotypes bearing BASV-G are not inactivated by pretreatment at pH 5.0 along with the presence of an internal bipartite fusion loop motif in BASV-G and other similarities found in multiple-sequence alignments with other rhabdovirus glycoproteins suggest that BASV-G is a class III fusion protein.
A number of rhabdoviruses, including BEFV, TIBV, and VSV, are arboviruses that are transmitted to their animal hosts via biting flies, while other rhabdoviruses like RABV are zoonotic viruses transmitted directly between mammals (37). Bat and rodent species in particular have been associated with the transmission of hemorrhagic fever viruses to humans and are considered natural reservoirs for such viruses in the Filoviridae and Arenaviridae families (38, 39). Although no direct conclusions on animal reservoirs and transmission routes can be drawn from cell culture-derived virus titers, in vitro testing of BASV-G pseudotype infectivity on a number of cell lines from different species and tissues revealed a broad tropism of BASV-G similar to that of VSV-G, which is well known for its broad tropism and exceptionally high efficiency in viral cell entry. This suggests that various cell types from different species are susceptible to BASV-G-mediated infection and therefore likely express a conserved receptor (or group of receptors) used by BASV-G to initiate target cell entry.
Like VSV-G, CHIKV env, and many other viral glycoproteins, BASV-G requires endosomal acidification in order to achieve efficient entry. The threshold pH to induce BASV-G fusion activity was found to be <5.5, lower than those observed for VSV-G or CHIKV env, both of which fuse at pH <6.0 (40, 41). This suggests that higher activation energy is required for BASV-G to adopt its fusion-active conformation. Similarly, the temperature optimum of BASV-G-mediated fusion was found to differ from those of VSV-G and CHIKV env. The fusion activity of BASV-G increased more slowly with higher temperatures and, unlike VSV-G and CHIKV env, kept increasing at temperatures above 37°C. This observation was even more striking for the N138A fusion loop mutant, suggesting a heat-activated mechanism for BASV-G-mediated fusion and an even higher energy barrier for the N138A mutant. The relatively low fusion activity of BASV-G at 28°C appears at odds with the high titers obtained with BASV-G pseudotypes in mosquito cells. However, temperature dependence was measured in a cell-cell fusion assay, whereas titers were calculated from pseudotype infection of cell lines, which involves additional steps such as endosomal uptake, acidification, and, downstream of membrane fusion, reporter gene expression. Multiple aspects differing between the two experimental systems, such as lipid compositions of viral and cellular membranes, surface availability of the glycoprotein, spatial constraints, and endosomal milieu versus extracellular space, could account for the observed differences. Nevertheless, taken together, our investigation of BASV-G fusion activity implies that, while exhibiting broad species and tissue tropism and likely using a conserved and ubiquitously expressed receptor, BASV-G may be less fusogenic at lower temperatures than the envelope proteins of the two arboviruses VSV and CHIKV. More investigation will be needed to determine whether insects are a potential vector for BASV.
Enveloped viruses have evolved to mask their envelope proteins by glycosylation. The addition of glycans not only protects the virus from recognition by the host's immune system as intruding nonself structures but also assists in the interaction between viruses and their target cells. This can have important implications for the route of transmission used by a virus to enter a new host. For example, our current understanding of arthropod-borne virus transmission suggests a potentially important role of C-type lectins expressed on the surface of dendritic cells (DCs) and reticuloendothelial cells in the establishment of infection in a new host (32). These cell types are likely to be one of the first to be encountered by the virus following its injection into the skin of a new host by the arthropod vector. The attachment of the virus to these cells may lead to either direct infection of DCs in cis or transport of the virus by DCs to other tissues and infection of more suitable target cells in trans (32). The finding that BASV-G, in contrast to VSV-G and other rhabdovirus glycoproteins, contains high-mannose glycans and interacts with C-type lectins like DC-SIGN, DC-SIGNR, and SIGNR1 to enhance infection of lectin-expressing target cells could therefore have important consequences with respect to the infectivity and pathogenesis of BASV-G. Thus, despite the similarities shared with VSV-G and other rhabdovirus glycoproteins, BASV-G possesses some distinctive features that distinguish it from the rest of the viral family and could explain its suspected clinical manifestation of acute hemorrhagic fever in humans.
The pseudotype system described in this study will be a practical tool to determine the seroprevalence and clinical relevance of BASV infection and to allow the development of prophylactic and therapeutic intervention strategies for BASV infection.
ACKNOWLEDGMENTS
This work was supported by grant R01AI074986 from the National Institute of Allergy and Infectious Diseases. Metabiota is graciously supported by the U.S. Department of Defense Armed Forces Health Surveillance Center, Division of Global Emerging Infections, Surveillance Operations (AFHSC GEIS); the Defense Threat Reduction Agency Cooperative Biological Engagement Program (DTRA-CBEP), the Department of Defense HIV/AIDS Prevention Program (DHAPP); Google.org; the Skoll Foundation; and the U.S. Agency for International Development (USAID) Emerging Pandemic Threats Program PREDICT project, under the terms of cooperative agreement number GHN-A-OO-09-00010-00.
Footnotes
Published ahead of print 19 June 2013
REFERENCES
- 1.Grard G, Fair JN, Lee D, Slikas E, Steffen I, Muyembe JJ, Sittler T, Veeraraghavan N, Ruby JG, Wang C, Makuwa M, Mulembakani P, Tesh RB, Mazet J, Rimoin AW, Taylor T, Schneider BS, Simmons G, Delwart E, Wolfe ND, Chiu CY, Leroy EM. 2012. A novel rhabdovirus associated with acute hemorrhagic fever in central Africa. PLoS Pathog. 8:e1002924. 10.1371/journal.ppat.1002924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kuzmin IV, Novella IS, Dietzgen RG, Padhi A, Rupprecht CE. 2009. The rhabdoviruses: biodiversity, phylogenetics, and evolution. Infect. Genet. Evol. 9:541–553 [DOI] [PubMed] [Google Scholar]
- 3.WHO 2013. Rabies fact sheet n°99. WHO, Geneva, Switzerland [Google Scholar]
- 4.Hoffmann B, Beer M, Schutze H, Mettenleiter TC. 2005. Fish rhabdoviruses: molecular epidemiology and evolution. Curr. Top. Microbiol. Immunol. 292:81–117 [DOI] [PubMed] [Google Scholar]
- 5.Rodriguez LL. 2002. Emergence and re-emergence of vesicular stomatitis in the United States. Virus Res. 85:211–219 [DOI] [PubMed] [Google Scholar]
- 6.Dimitrov DS. 2004. Virus entry: molecular mechanisms and biomedical applications. Nat. Rev. Microbiol. 2:109–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sun X, Belouzard S, Whittaker GR. 2008. Molecular architecture of the bipartite fusion loops of vesicular stomatitis virus glycoprotein G, a class III viral fusion protein. J. Biol. Chem. 283:6418–6427 [DOI] [PubMed] [Google Scholar]
- 8.Roche S, Albertini AA, Lepault J, Bressanelli S, Gaudin Y. 2008. Structures of vesicular stomatitis virus glycoprotein: membrane fusion revisited. Cell. Mol. Life Sci. 65:1716–1728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schibli DJ, Weissenhorn W. 2004. Class I and class II viral fusion protein structures reveal similar principles in membrane fusion. Mol. Membr. Biol. 21:361–371 [DOI] [PubMed] [Google Scholar]
- 10.Negrete OA, Levroney EL, Aguilar HC, Bertolotti-Ciarlet A, Nazarian R, Tajyar S, Lee B. 2005. EphrinB2 is the entry receptor for Nipah virus, an emergent deadly paramyxovirus. Nature 436:401–405 [DOI] [PubMed] [Google Scholar]
- 11.Salvador B, Zhou Y, Michault A, Muench MO, Simmons G. 2009. Characterization of Chikungunya pseudotyped viruses: identification of refractory cell lines and demonstration of cellular tropism differences mediated by mutations in E1 glycoprotein. Virology 393:33–41 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Salvador B, Sexton NR, Carrion R, Jr, Nunneley J, Patterson JL, Steffen I, Lu K, Muench MO, Lembo D, Simmons G. 2013. Filoviruses utilize glycosaminoglycans for their attachment to target cells. J. Virol. 87:3295–3304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Whitt MA. 2010. Generation of VSV pseudotypes using recombinant DeltaG-VSV for studies on virus entry, identification of entry inhibitors, and immune responses to vaccines. J. Virol. Methods 169:365–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pohlmann S, Baribaud F, Lee B, Leslie GJ, Sanchez MD, Hiebenthal-Millow K, Munch J, Kirchhoff F, Doms RW. 2001. DC-SIGN interactions with human immunodeficiency virus type 1 and 2 and simian immunodeficiency virus. J. Virol. 75:4664–4672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pohlmann S, Zhang J, Baribaud F, Chen Z, Leslie GJ, Lin G, Granelli-Piperno A, Doms RW, Rice CM, McKeating JA. 2003. Hepatitis C virus glycoproteins interact with DC-SIGN and DC-SIGNR. J. Virol. 77:4070–4080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shakin-Eshleman SH, Remaley AT, Eshleman JR, Wunner WH, Spitalnik SL. 1992. N-linked glycosylation of rabies virus glycoprotein. Individual sequons differ in their glycosylation efficiencies and influence on cell surface expression. J. Biol. Chem. 267:10690–10698 [PubMed] [Google Scholar]
- 17.Robertson JS, Etchison JR, Summers DF. 1976. Glycosylation sites of vesicular stomatitis virus glycoprotein. J. Virol. 19:871–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vigerust DJ, Shepherd VL. 2007. Virus glycosylation: role in virulence and immune interactions. Trends Microbiol. 15:211–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feldmann H, Volchkov VE, Volchkova VA, Stroher U, Klenk HD. 2001. Biosynthesis and role of filoviral glycoproteins. J. Gen. Virol. 82:2839–2848 [DOI] [PubMed] [Google Scholar]
- 20.Rota PA, Oberste MS, Monroe SS, Nix WA, Campagnoli R, Icenogle JP, Penaranda S, Bankamp B, Maher K, Chen MH, Tong S, Tamin A, Lowe L, Frace M, DeRisi JL, Chen Q, Wang D, Erdman DD, Peret TC, Burns C, Ksiazek TG, Rollin PE, Sanchez A, Liffick S, Holloway B, Limor J, McCaustland K, Olsen-Rasmussen M, Fouchier R, Gunther S, Osterhaus AD, Drosten C, Pallansch MA, Anderson LJ, Bellini WJ. 2003. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300:1394–1399 [DOI] [PubMed] [Google Scholar]
- 21.Florkiewicz RZ, Rose JK. 1984. A cell line expressing vesicular stomatitis virus glycoprotein fuses at low pH. Science 225:721–723 [DOI] [PubMed] [Google Scholar]
- 22.Sourisseau M, Schilte C, Casartelli N, Trouillet C, Guivel-Benhassine F, Rudnicka D, Sol-Foulon N, Le Roux K, Prevost MC, Fsihi H, Frenkiel MP, Blanchet F, Afonso PV, Ceccaldi PE, Ozden S, Gessain A, Schuffenecker I, Verhasselt B, Zamborlini A, Saib A, Rey FA, Arenzana-Seisdedos F, Despres P, Michault A, Albert ML, Schwartz O. 2007. Characterization of reemerging chikungunya virus. PLoS Pathog. 3:e89. 10.1371/journal.ppat.0030089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Diederich S, Thiel L, Maisner A. 2008. Role of endocytosis and cathepsin-mediated activation in Nipah virus entry. Virology 375:391–400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Holland AU, Munk C, Lucero GR, Nguyen LD, Landau NR. 2004. Alpha-complementation assay for HIV envelope glycoprotein-mediated fusion. Virology 319:343–352 [DOI] [PubMed] [Google Scholar]
- 25.Moosmann P, Rusconi S. 1996. Alpha complementation of LacZ in mammalian cells. Nucleic Acids Res. 24:1171–1172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ullmann A, Jacob F, Monod J. 1967. Characterization by in vitro complementation of a peptide corresponding to an operator-proximal segment of the beta-galactosidase structural gene of Escherichia coli. J. Mol. Biol. 24:339–343 [DOI] [PubMed] [Google Scholar]
- 27.Aljofan M, Saubern S, Meyer AG, Marsh G, Meers J, Mungall BA. 2009. Characteristics of Nipah virus and Hendra virus replication in different cell lines and their suitability for antiviral screening. Virus Res. 142:92–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wikan N, Sakoonwatanyoo P, Ubol S, Yoksan S, Smith DR. 2012. Chikungunya virus infection of cell lines: analysis of the East, Central and South African lineage. PLoS One 7:e31102. 10.1371/journal.pone.0031102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kononchik JP, Jr, Hernandez R, Brown DT. 2011. An alternative pathway for alphavirus entry. Virol. J. 8:304. 10.1186/1743-422X-8-304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roberts PC, Kipperman T, Compans RW. 1999. Vesicular stomatitis virus G protein acquires pH-independent fusion activity during transport in a polarized endometrial cell line. J. Virol. 73:10447–10457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yoshimori T, Yamamoto A, Moriyama Y, Futai M, Tashiro Y. 1991. Bafilomycin A1, a specific inhibitor of vacuolar-type H(+)-ATPase, inhibits acidification and protein degradation in lysosomes of cultured cells. J. Biol. Chem. 266:17707–17712 [PubMed] [Google Scholar]
- 32.Klimstra WB, Nangle EM, Smith MS, Yurochko AD, Ryman KD. 2003. DC-SIGN and L-SIGN can act as attachment receptors for alphaviruses and distinguish between mosquito cell- and mammalian cell-derived viruses. J. Virol. 77:12022–12032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gramberg T, Soilleux E, Fisch T, Lalor PF, Hofmann H, Wheeldon S, Cotterill A, Wegele A, Winkler T, Adams DH, Pohlmann S. 2008. Interactions of LSECtin and DC-SIGN/DC-SIGNR with viral ligands: differential pH dependence, internalization and virion binding. Virology 373:189–201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Feinberg H, Mitchell DA, Drickamer K, Weis WI. 2001. Structural basis for selective recognition of oligosaccharides by DC-SIGN and DC-SIGNR. Science 294:2163–2166 [DOI] [PubMed] [Google Scholar]
- 35.Jones KE, Patel NG, Levy MA, Storeygard A, Balk D, Gittleman JL, Daszak P. 2008. Global trends in emerging infectious diseases. Nature 451:990–993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Colman PM, Lawrence MC. 2003. The structural biology of type I viral membrane fusion. Nat. Rev. Mol. Cell Biol. 4:309–319 [DOI] [PubMed] [Google Scholar]
- 37.Bourhy H, Cowley JA, Larrous F, Holmes EC, Walker PJ. 2005. Phylogenetic relationships among rhabdoviruses inferred using the L polymerase gene. J. Gen. Virol. 86:2849–2858 [DOI] [PubMed] [Google Scholar]
- 38.Ogbu O, Ajuluchukwu E, Uneke CJ. 2007. Lassa fever in West African sub-region: an overview. J. Vector Borne Dis. 44:1–11 [PubMed] [Google Scholar]
- 39.Smith I, Wang LF. 2013. Bats and their virome: an important source of emerging viruses capable of infecting humans. Curr. Opin. Virol. 3:84–91 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fredericksen BL, Whitt MA. 1996. Mutations at two conserved acidic amino acids in the glycoprotein of vesicular stomatitis virus affect pH-dependent conformational changes and reduce the pH threshold for membrane fusion. Virology 217:49–57 [DOI] [PubMed] [Google Scholar]
- 41.Tsetsarkin KA, McGee CE, Higgs S. 2011. Chikungunya virus adaptation to Aedes albopictus mosquitoes does not correlate with acquisition of cholesterol dependence or decreased pH threshold for fusion reaction. Virol. J. 8:376. 10.1186/1743-422X-8-376 [DOI] [PMC free article] [PubMed] [Google Scholar]