

Abstract
Most viruses possess strategies to circumvent host immune responses. The measles virus (MV) nonstructural C protein suppresses the interferon response, thereby allowing efficient viral growth, but its detailed mechanism has been unknown. We identified Shc Src homology 2 domain-binding protein 1 (SHCBP1) as one of the host proteins interacting with the C protein. Knockdown of SHCBP1 using a short-hairpin RNA greatly reduced MV growth. SHCBP1 was found to be required for viral RNA synthesis in the minigenome assay and to bind to the MV phosphoprotein, a subunit of the viral RNA polymerase. A stretch of 12 amino acid residues in the C protein were sufficient for SHCBP1 binding, and the peptide containing these 12 residues could suppress MV RNA synthesis, like the full-length C protein. The central region of SHCBP1 was found to bind to the C protein, as well as the phosphoprotein, but the two viral proteins did not compete for SHCBP1 binding. Our results indicate that the C protein modulates MV RNA polymerase activity by binding to the host protein SHCBP1. SHCBP1 may be exploited as a target of antiviral compounds.
INTRODUCTION
Measles, an acute human disease characterized by high fever and a generalized maculopapular rash, is still a major cause of morbidity and mortality of children worldwide (1). Measles virus (MV), the causative agent of the disease, is an enveloped virus classified as a member of the genus Morbillivirus in the family Paramyxoviridae. The virus contains a nonsegmented negative-strand RNA genome ∼16 kb in length, with six genes encoding the nucleocapsid (N), phospho- (P), matrix (M), fusion, hemagglutinin, and large (L) proteins, respectively. The P gene encodes two additional nonstructural proteins, the V and C proteins, via the RNA editing and alternative translational initiation in a different reading frame, respectively (2, 3).
The V and C proteins have been shown to counteract the host interferon (IFN) response by various mechanisms. The V protein interacts with several molecules involved in the induction or signal transduction of IFN-α and IFN-β (type I IFNs), including the RNA helicases melanoma differentiation-associated gene 5 (MDA5) and LGP2 (4), IκB kinase α (5), signal transducer and activator of transcription 1 (STAT1) (6), STAT2 (7, 8), and Janus kinase 1 (6), and interferes with their activity. The function of the C protein is less clear. A recombinant MV lacking the C protein (MVΔC) neither propagates nor causes symptoms such as Koplik spot and rash in experimentally infected nonhuman primates (9, 10). Furthermore, MVΔC exhibits attenuated growth in cells possessing the intact type I IFN system (11–14), partly through protein kinase PKR-mediated translation inhibition (13) and IFN-β induction (14). Since the C protein downregulates viral RNA synthesis (12, 15, 16), it was proposed that the C protein allows the virus to escape detection by the cytosolic RNA sensors retinoic acid-inducible gene I (RIG-I) and MDA5 and prevents IFN production (12). A recent study reported that the transfected C protein can interfere with IFN-β transcription in the nucleus (17). It remains to be determined whether this occurs in infected cells. The C protein has also been shown to affect the IFN-γ signaling by interfering with the dimer formation of STAT1 (18).
The MV genomic RNA is encapsidated by the N protein, forming the nucleocapsid. The MV RNA-dependent RNA polymerase, acting in both viral gene transcription and genome replication, is composed of two virally encoded subunits, the L and P proteins. The L protein has the enzymatic activities of nucleotide polymerization, 5′ capping, and polyadenylation, whereas the P protein acts as a cofactor, bridging the L protein and the nucleocapsid (19–22). The complex of the nucleocapsid and RNA polymerase is termed the ribonucleoprotein (RNP) complex. In MV-infected cells, the C protein and the RNP complex accumulate at the perinuclear area, forming small dots, which are thought to be the scaffold for viral transcription and/or replication (12, 23, 24). Despite its colocalization with the RNP complex, there is no evidence that the C protein directly interacts with any component of the RNP complex (23, 25). In contrast, the C protein of mouse Sendai virus inhibits viral RNA synthesis via its direct interaction with the viral RNA polymerase (26, 27). These observations suggest that the C protein of MV regulates viral RNA synthesis through its interaction with a host protein(s) involved in viral RNA synthesis.
In this study, we screened for host proteins that interact with the C protein, using the yeast two-hybrid system, and identified 12 binding proteins. By further analysis, Shc Src homology 2 (SH2) domain-binding protein 1 (SHCBP1) was identified as a host factor required for efficient MV growth. We also found that SHCBP1 interacts with the MV P protein and that the C protein inhibits viral RNA synthesis by interacting with SHCBP1 through its 12-mer peptide. Thus, the MV nonstructural C protein appears to fine-tune viral RNA synthesis so as not to induce type I IFNs, by modulating the host protein SHCBP1.
MATERIALS AND METHODS
Cells and viruses.
A549/hSLAM cells (28), which stably express a cellular receptor human signaling lymphocyte activation molecule (SLAM), were maintained in RPMI 1640 medium (Wako Pure Chemical Industries) supplemented with 10% fetal bovine serum (FBS) and 0.5 mg of G418 (Nacalai Tesque)/ml. VV5-4 cells (29) were maintained in RPMI 1640 medium supplemented with 10% FBS. Vero/hSLAM (30), L929, and HEK293T cells were maintained in Dulbecco modified Eagle medium (DMEM; Wako Pure Chemical Industries) supplemented with 10% FBS. PLAT-gp cells (a generous gift from M. Shimojima and T. Kitamura) expressing the gag-pol gene of Moloney murine leukemia virus (31) were maintained in DMEM supplemented with 10% FBS and 10 μg of blasticidin (Invitrogen)/ml. IC323-Luci, which is a recombinant MV based on the pathogenic IC-B strain (32), contains an additional transcription unit of the Renilla luciferase gene upstream of the N gene (33, 34). IC323/Ed-H-Luci is a derivative of IC323-Luci in which the H gene was replaced with that of the Edmonston tag strain (34). For the induction of SHCBP1, A549/hSLAM cells were mock treated, treated with 1,000 IU of IFN-αA/D/ml for 3 h or 18 h, treated with poly(I·C) for 24 h, or infected with IC323-Luci at a multiplicity of infection (MOI) of 0.05 for 24 h.
Plasmid constructions.
All mutations were introduced by PCR-based mutagenesis. Plasmids encoding the N gene (pCA7-IC-N), the P gene (pCA7-IC-PΔC) and enhanced green fluorescent protein (EGFP) (pCA7-EGFP) were described previously (35, 36). To construct expression plasmids, fragments encoding the influenza virus hemagglutinin (HA)-tagged P protein (pCA7-HA-IC-PΔC), the intact and mutated C proteins tagged with 6×His at the C terminus (pCA7-IC-C-His, pCA7-IC-C150-His, pCA7-IC-CΔ100-His, pCA7-IC-CΔ101-112-His, pCA7-IC-CΔ113-126-His, pCA7-IC-CΔ127-138-His, or pCA7-IC-CΔ139-150-His), Flag- or HA-tagged SHCBP1 (pCA7-Flag-SHCBP1 or pCA7-HA-SHCBP1), Flag-tagged truncated SHCBP1 (pCA7-Flag-SHCBP1-A, pCA7-Flag-SHCBP1-B or pCA7-Flag-SHCBP1-C), green fluorescent protein (GFP)-tagged C protein (pCA7-IC-C-GFP), and GFP and the 12-mer peptide comprised of amino acid residues at positions 127 to 138 of the C protein (pCA7-GFP-C12aa) were subcloned into the expression plasmid pCA7, a derivative of pCAGGS (37).
Gene knockdown by shRNA.
Target sequences were designed using BLOCK-iT RNAi Designer (Invitrogen), and all transfections in the present study were carried out using Polyethylenimine-“Max” (Polysciences, Inc.). Duplexes of synthesized oligonucleotides containing the respective target sequences (5′-GCGATTCAGAGCCTATCAAGA-3′ for SHCBP1-1 and 5′-GCAAGGAAGGGATCCTCATTA-3′ for SHCBP1-2; target sequences of the other host proteins are provided upon request) were inserted into the retrovirus-based pRS-U6/puro vector (OriGene). To generate short hairpin RNA (shRNA)-transducing retroviruses, PLAT-gp cells plated on collagen-coated 10-cm dishes were transfected with 20 μg of the pRS-U6/puro vector expressing each shRNA and 2 μg of pCVSV-G, which encodes the G protein of vesicular stomatitis virus (38). Culture medium was replaced with fresh medium at 6 h after transfection, and supernatants containing retroviruses were harvested. To produce shRNA-expressing cells, A549/hSLAM cells were treated with retrovirus-containing culture media containing 8 μg of Polybrene per ml for 24 h. After a washing step with phosphate-buffered saline (PBS), the transduced cells were selected in the presence of 1 μg of puromycin (InvivoGen)/ml for 72 h and used for further experiments.
Virus infection and titration.
A549/hSLAM cells expressing shRNA were replated on six-well plate 1 day before infection, infected with IC323-Luci, encephalomyocarditis virus (EMCV), or herpes simplex virus 1 (HSV-1) at an MOI of 0.05 for 1 h at 37°C, washed with PBS, and cultured with complete medium. Both cells and culture medium was harvested at 48 h of infection for IC323-Luci and HSV-1 or at 24 h for EMCV. Monolayers of Vero/hSLAM cells on 12-well plates were incubated with serially diluted IC323-Luci or HSV-1 samples for 1 h at 37°C and then overlaid with DMEM containing 2% FBS and 0.75% agarose after the diluents were removed. At 5 days postinfection (p.i.), the plaques were counted. For EMCV titration, L929 cells were used and incubated for 2 days.
Minigenome assay.
A minigenome assay was performed as described previously (12). Briefly, monolayers of VV5-4 cells or A549/hSLAM cells expressing shRNA against SHCBP1 on 24-well plates were infected with vTF7-3 (39) or MVA-T7 (40), respectively, at an MOI of 0.5 for 1 h and then transfected with the minigenome plasmid p18MGFLuc01 encoding the firefly luciferase gene (41) and support plasmids comprised of pCA7-IC-N, pCA7-IC-PΔC and pGEMCR-9301B-L, with or without the expression plasmid encoding the protein to be examined. At 48 h posttransfection, enzymatic activities of the firefly luciferase were measured by the luciferase assay system (Promega) and luminometer Mithras LB 940 (Berthold Technologies). pGEMCR-9301B-L was omitted from the plasmid mixture of transfection for a negative control [indicated as L(−) in figures].
Yeast two-hybrid screening.
A yeast two-hybrid screening was performed as previously described with some modifications (23, 42). pDBLeu-C, a bait vector encoding the C protein downstream of the GAL4 DNA binding domain (23), and the pPC86 prey vector encoding a HeLa cDNA library (Life Technologies, catalog no. 11287-018) were introduced into the yeast strain MaV203 using polyethylene glycol in lithium acetate. The transformed cells were plated on synthetic complete (SC) medium plates lacking leucine, tryptophan, and histidine and supplemented with 10 mM 3-aminotriazole (SC/−Leu/−Trp/−His/3AT) to select cells that express the HIS3 reporter gene, and a small portion of the cells was plated on SC/−Leu/−Trp plates to determine the efficiency of the transformation by both the prey and the bait vectors. After 3 days of incubation, yeast colonies on the plates were further streaked on four different sets of selection plates (SC/−Leu/−Trp/−His/3AT plates, SC/−Leu/−Trp plates that lack uracil, SC/−Leu/−Trp plates supplemented with 0.2% 5-fluoroorotic acid, and YPAD medium [a rich medium for routine growth of yeast] plates supplemented with 5-bromo-5-chloro-3-indolyl-β-d-galactopyranoside [X-Gal]) (23). The grown cells on the second SC/−Leu/−Trp/−His/3AT plate, which expressed at least one of three reporter genes, were further tested on another set of the four selection plates by streaking for single colony isolation, to confirm reporter gene expression. Retention of both the bait and the prey vectors in yeast cells was confirmed by the growth on SC/−Leu/−Trp plates. Plasmids contained in colonies on the third SC/−Leu/−Trp/−His/3AT plates were extracted by phenol treatment and ethanol precipitation and then electroporated into E. coli DH10B strain. The transformed cells by prey plasmids containing cDNA were selected with ampicillin and the isolated prey plasmids from single colonies were analyzed by DNA sequencing. MaV203 cells transformed with respective prey plasmids in combination with either pDBLeu-C or pDBLeu empty vector were grown on the four different selection plates to reconfirm specific interactions and to test prey-independent activation of reporter gene, respectively.
Coimmunoprecipitation and Western blot analyses.
For immunoprecipitation, mouse anti-Flag (F1804; Sigma) monoclonal antibody (MAb) and mouse anti-HA (sc-7392; Santa Cruz) MAb were used. For Western blotting, we used rabbit anti-Flag (F7425; Sigma) polyclonal antibody (PAb), rabbit anti-His (PM032; MBL) PAb, mouse anti-GFP (632380; Clontech) MAb, rabbit anti-HA (sc-805; Santa Cruz) PAb, rabbit anti-SHCBP1 (12672-1-AP; Proteintech) PAb, mouse anti-β-actin (sc-8432; Santa Cruz) MAb, rabbit anti-P/V PAb (which recognizes the N terminus region shared between the V and P proteins) (11), mouse anti-C MAb (12), mouse anti-N MAb (ab9397; Abcam), horseradish peroxidase (HRP)-conjugated anti-rabbit IgG secondary PAb (Invitrogen), and HRP-conjugated anti-mouse IgG secondary PAb (Jackson ImmunoResearch). Subconfluent monolayers of HEK293T cells on six-well plates were transfected with an appropriate combination of expression plasmids. At 48 h posttransfection, the cells were washed with PBS and lysed in 1 ml of the whole-cell extract (WCE) buffer (50 mM Tris [pH 8.0], 280 mM NaCl, 0.5% Nonidet P-40, 0.2 mM EDTA, 2 mM EGTA, 10% glycerol, 1 mM dithiothreitol [DTT], and protease inhibitors cocktail [Sigma]) (43). The lysates were centrifuged at 20,630 × g for 5 min at 4°C to remove cell debris. A small amount (50 μl) of each supernatant was mixed with an equal volume of the sodium dodecyl sulfate (SDS) loading buffer (50 mM Tris [pH 6.8], 100 mM DTT, 2% SDS, 0.1% bromophenol blue, 10% glycerol), boiled for 5 min, and kept as the lysate sample. The rest of the supernatant was precleared with protein A/G-Sepharose (GE Healthcare) and normal murine serum and then incubated with Protein A/G-Sepharose and anti-HA MAb or anti-Flag MAb, depending on the target protein. In case of the assay analyzing the interaction between Flag-SHCBP1 and C protein mutants, protein A/G-Sepharose was replaced with Dynabeads Pan Mouse IgG (Invitrogen). After intensive washing with the WCE buffer without protease inhibitors, the polypeptides in the precipitated complexes were fractionated by SDS-PAGE and electroblotted onto polyvinylidene difluoride (PVDF) membranes (Hybond-P; GE Healthcare). Target proteins on the PVDF membranes were treated with a suitable combination of antibodies. The treated PVDF membranes were incubated with Chemi-Lumi One Super (Nacalai Tesque) to elicit chemiluminescent signals, and the signal were detected and visualized using a VersaDoc 5000 imager (Bio-Rad).
Immunofluorescence staining.
Procedures of the minigenome assay were carried out in VV5-4 or A549/hSLAM cells seeded on coverslips, with the expression plasmid encoding the protein to be examined. At 48 h posttransfection, cells were simultaneously fixed and permeabilized with PBS containing 2.5% formaldehyde and 0.5% Triton X-100. The cells were blocked with 1% normal donkey serum, and incubated with mouse anti-Flag MAb or mouse anti-C MAb, and rabbit anti-P/V PAb, followed by incubation with Alexa Fluor 488-conjugated donkey anti-mouse IgG (H+L) and Alexa Fluor 594-conjugated donkey anti-rabbit IgG (H+L) (Molecular Probes). The stained cells were observed under a confocal microscope (Radiance 2100; Bio-Rad).
Cell viability assay.
A549/hSLAM cells expressing shRNA were replated on 96-well plate, and cell viability at 6 h and 48 h was analyzed using CellTiter 96 AQueous One Solution Cell Proliferation Assay (Promega), according to the manufacturer's instructions.
Statistics.
Data were analyzed for statistical significance by using a two-tailed Student t test. A P value of <0.05 were considered statistically significant.
RESULTS
Identification of cellular proteins interacting with the MV C protein.
To identify host proteins interacting with the MV C protein, we screened a cDNA library generated from HeLa cells using the yeast two-hybrid system with the full-length C protein as a bait. Approximately 6 × 106 cDNA clones were screened, and 209 positive clones were obtained after the selection by the expression of the HIS3 reporter gene. Those clones were further tested for the expression of HIS3, URA3, and lacZ reporter genes. A total of 161 clones expressed at least one of the three reporter genes, and they were assigned to 12 different cellular genes after sequence analysis (Table 1).
Table 1.
Results of yeast two-hybrid screening using the MV C protein as bait
| Gene ID | Symbol | Full name | No. of occurrences | Strength of the interaction |
|---|---|---|---|---|
| 5702 | PSMC3 | Proteasome (prosome, macropain) 26S subunit, ATPase, 3 | 107 | + |
| 9039 | UBA3 | Ubiquitin-like modifier activating enzyme 3 | 30 | +++ |
| 10615 | SPAG5 | Sperm-associated antigen 5 | 10 | + |
| 10241 | CALCOCO2 | Calcium binding and coiled-coil domain 2 | 4 | + |
| 80232 | WDR26 | WD repeat domain 26 | 2 | + |
| 87 | ACTN1 | Actinin, alpha 1 | 2 | + |
| 79801 | SHCBP1 | SHC SH2-domain binding protein 1 | 1 | ++ |
| 9240 | PNMA1 | Paraneoplastic Ma antigen 1 | 1 | + |
| 1039 | CDR2 | Cerebellar degeneration-related protein 2, 62 kDa | 1 | + |
| 84296 | GINS4 | GINS complex subunit 4 | 1 | + |
| 3858 | KRT10 | Keratin 10 | 1 | + |
| 3875 | KRT18 | Keratin 18 | 1 | + |
MV growth is suppressed in SHCBP1 knockdown cells.
We then examined whether these 12 genes identified by the yeast two-hybrid screening are involved in MV growth. A549/hSLAM cells, a human lung carcinoma cell line stably expressing human SLAM, a cellular receptor for MV, were transduced with the retroviruses expressing shRNAs that target respective host proteins. After brief selection with puromycin, the transduced A549/hSLAM cells were infected with IC323-Luci, the recombinant MV expressing the reporter Renilla luciferase, and enzymatic activities in MV-infected cells were measured. When SHCBP1 was knocked down, the luciferase activity was reduced to ∼10% of the control (the activity in cells expressing an shRNA against the EGFP) (Fig. 1A). Knockdown of eight other proteins also reduced the luciferase activity by 30 to 50%, whereas knockdown of WDR26 or CDR2 had an opposite effect on MV growth.
Fig 1.

Effect of SHCBP1 knockdown on MV growth. (A) A549/hSLAM cells expressing shRNA against individual target genes or EGFP gene were infected with IC323-Luci. At 48 h postinfection (p.i.), intracellular luciferase activity was measured. The average value for cells expressing shEGFP was set to 100%, and relative values are indicated. (B) A549/hSLAM cells were infected with IC323-Luci (MV) at an MOI of 0.05 or stimulated with 2 μg of poly(I·C)/ml for 24 h (left panel) or treated with 1,000 IU of IFN-αA/D/ml for the indicated times (right panel). The expression of SHCBP1 was examined by Western blotting. (C) A549/hSLAM cells expressing shSHCBP1-1, shSHCBP1-2, or shEGFP were stimulated with poly(I·C) for 24 h, and the SHCBP1 expression was examined. (D) Growth of MV in SHCBP1- or EGFP-knockdown cells. A549/hSLAM cells expressing shSHCBP1-1, shSHCBP1-2, or shEGFP were infected with IC323-Luci at an MOI of 0.05. At various times p.i., the intracellular luciferase activity was measured. (E) Cells were infected with IC323-Luci as for panel D. At 48 h p.i., the cells were scraped into culture medium, and virus titers were determined on Vero/hSLAM cells. (F) Viability of shRNA-expressing A549/hSLAM cells. After puromycin selection, the cells were replated on 96-well plate, and the cell viability was determined at 6 and 48 h. The data were indicated as the value at 48 h relative to that at 6 h. The relative value in shEGFP-expressing cells was set to 100%. (G) shRNA-expressing A549/hSLAM cells were infected with EMCV and HSV-1, and the virus titers at 24 and 48 h p.i., respectively, were determined. The data indicate means ± the standard deviations (SD) (A, D, E, F, and G). *, P < 0.05; **, P < 0.01; ***, P < 0.001. All of the results in this and other figures are representative of at least two independent experiments.
In the present study, we concentrated on SHCBP1 and examined its role in MV growth and interaction with the MV C protein. SHCBP1 was initially identified as a protein that interacts with the SH2 domain of the Shc adaptor protein (44). SHCBP1 is mainly expressed in the testis, spleen, and lung and is upregulated by growth factor stimulation, but its precise biological roles remain unknown (44).
We found that MV infection and poly(I·C) stimulation, like growth factor stimulation, enhance the expression of SHCBP1 (Fig. 1B, left panel). SHCBP1 was indeed strongly induced by IFN stimulation (Fig. 1B, right panel). To exclude an off-target effect of the first shRNA sequence selected (shSHCBP1-1) on MV growth, we generated another retrovirus vector that targets a different sequence of SHCBP1 (shSHCBP1-2). Both shRNAs expressed via the retrovirus vector strongly inhibited SHCBP1 expression even when its expression was enhanced by poly(I·C) stimulation (Fig. 1C). Using cells transduced with these retrovirus vectors, we examined the reporter gene expression at several time points after IC323-Luci infection. At 24, 48, and 72 h postinfection, enzymatic activities in shSHCBP1-1 or -2-expressing cells were reduced compared to those in shEGFP-expressing control cells, and the levels of reduction became more marked at later time points (Fig. 1D). Inhibition of MV growth was also observed when production of infectious progeny viruses was examined (Fig. 1E).
Since SHCBP1 is thought to be involved in cell growth signaling pathways (44), we tested the possibility that shSHCBP1 suppressed cell growth, which in turn led to inhibition of MV growth. Knockdown of SHCBP1 exhibited little, if any, effect on growth of A549/hSLAM cells, as measured by the cell proliferation/viability assay (Fig. 1F). It is also possible that knockdown of SHCBP1 suppresses the expression of the receptor SLAM, thereby affecting MV growth. However, the cell surface expression of SLAM was not affected by the shSHCBP1 treatment, as examined by flow cytometry (data not shown). Furthermore, SHCBP1 knockdown inhibited growth of IC323/Ed-H-Luci (34), which can use an alternative receptor CD46 ubiquitously expressed in all nucleated primate cells (data not shown).
To examine whether SHCBP1 plays a role in virus growth in general, we infected shSHCBP1-expressing cells with EMCV and HSV-1. Knockdown of SHCBP1 did not affect the growth of EMCV and HSV-1, unlike that of MV (Fig. 1G).
SHCBP1 is required for efficient MV RNA synthesis.
SHCBP1 allows efficient MV growth. On the other hand, the MV C protein binds to SHCBP1 and modulates viral RNA synthesis. These observations suggest that SHCBP1 is involved in MV RNA synthesis and that the MV C protein acts by affecting SHCBP1 activity. To test this hypothesis, we examined the effect of shSHCBP1 on MV RNA synthesis in the minigenome assay. Knockdown of SHCBP1 by either of the two independent shRNAs resulted in the expression level of the reporter gene 40 to 50% lower than that in control cells expressing shEGFP (Fig. 2A, middle). The RNA polymerase activities in SHCBP1-knockdown cells were further suppressed in the presence of the MV C protein (Fig. 2A, right), presumably because the C protein could suppress the remaining SHCBP1 activity after shRNA treatment, and/or because the C protein could inhibit the minigenome RNA synthesis through both SHCBP1-dependent and -independent mechanisms (see below).
Fig 2.
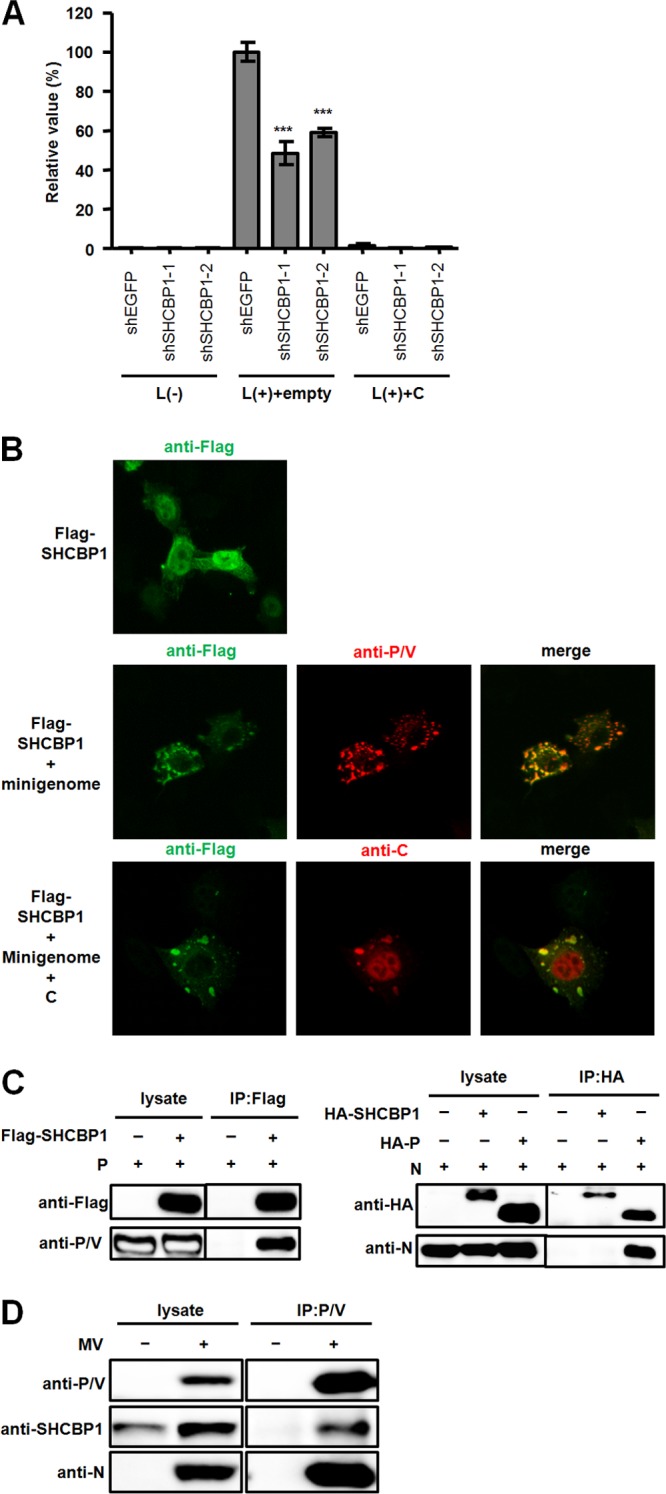
Role of SHCBP1 in MV RNA synthesis. (A) Effect of SHCBP1 knockdown on MV RNA synthesis. A minigenome assay was performed in shRNA-expressing A549/hSLAM cells. The reporter activities were determined 48 h after transfection. The minigenome and support plasmids were supplemented with empty vector or the expression plasmid encoding the C protein. L(−), the plasmid encoding the L protein, was omitted. (B) Intracellular localization of proteins. The upper panel shows VV5-4 cells expressing Flag-tagged SHCBP1 (green). The middle panels show VV5-4 cells expressing the minigenome, the N, P (red), and L proteins, and Flag-tagged SHCBP1 (green). The lower panels show VV5-4 cells expressing the minigenome, the N, P, and L proteins, Flag-tagged SHCBP1 (green), and the C protein (red). (C) Interaction of SHCBP1 with the RNP complex. For the left panel, a coimmunoprecipitation (IP) assay was performed with lysates from HEK293T cells expressing the MV P protein with or without Flag-tagged SHCBP1. At 48 h after transfection, cell lysates were prepared, and proteins were precipitated with anti-Flag antibody and examined by Western blotting. For the right panel, a coimmunoprecipitation (IP) assay with lysates from cells expressing the MV N protein and HA-tagged-SHCBP1 or P protein was performed. Proteins were precipitated with anti-HA antibody. (D) A coimmunoprecipitation (IP) assay was performed with lysates from MV-infected A549/SLAM cells. Protein complexes were precipitated with anti-P/V antibody.
We next examined whether SHCBP1 supports MV RNA synthesis by interacting with the MV RNP complex comprised of the RNA genome and N, P, and L proteins. Previous studies indicated that the MV RNP complex forms small dot-like structures at the perinuclear area (12, 23, 24). This small compartment is thought to be a kind of scaffold where viral RNA synthesis takes place (24). Similar structures containing RNA replication complexes have also been found in cells infected with other viruses (45, 46). When expressed alone, SHCBP1 was localized throughout the cell, but the majority was found in the nucleus (Fig. 2B, upper panel). In contrast, when expressed together with minimum elements for the MV minigenome system (the minigenome and N, P, and L proteins), SHCBP1 was redistributed to the small perinuclear foci and colocalized with the MV P protein (Fig. 2B, middle panel). The MV C protein was also colocalized with SHCBP1 in the foci, when included in the transfection (Fig. 2B, lower panel).
In coimmunoprecipitation experiments, SHCBP1 interacted with the P protein, but not with the N protein under the condition where the N-P interaction could be detected (19) (Fig. 2C). We could not obtain enough expression of the L protein to detect by Western blotting, so the interaction of SHCBP1 with the L protein remains to be determined. The interaction of endogenous SHCBP1 with the P protein was also detected in MV-infected cells (Fig. 2D). These data support the idea that SHCBP1 stimulates MV polymerase activity by binding to the P protein.
The MV C protein inhibits viral polymerase activity via its interaction with SHCBP1.
We next examined whether the interaction of the C protein with SHCBP1 is indeed involved in its regulation of viral RNA synthesis. We first verified their binding in human cells, by coimmunoprecipitation of the histidine (His)-tagged C protein (wt-C) and Flag-tagged SHCBP1 (Fig. 3A, lanes 1, 2, 9, and 10). Then, to determine the binding site for SHCBP1 in the C protein, we prepared a panel of its deletion mutants (Fig. 3B) and performed coimmunoprecipitation experiments (Fig. 3A). All of the C protein mutants, except the one missing the residues at positions 127 to 138 (CΔ127-138), were coprecipitated by SHCBP1 (Fig. 3A), suggesting that these 12 residues contain the SHCBP1 binding site.
Fig 3.
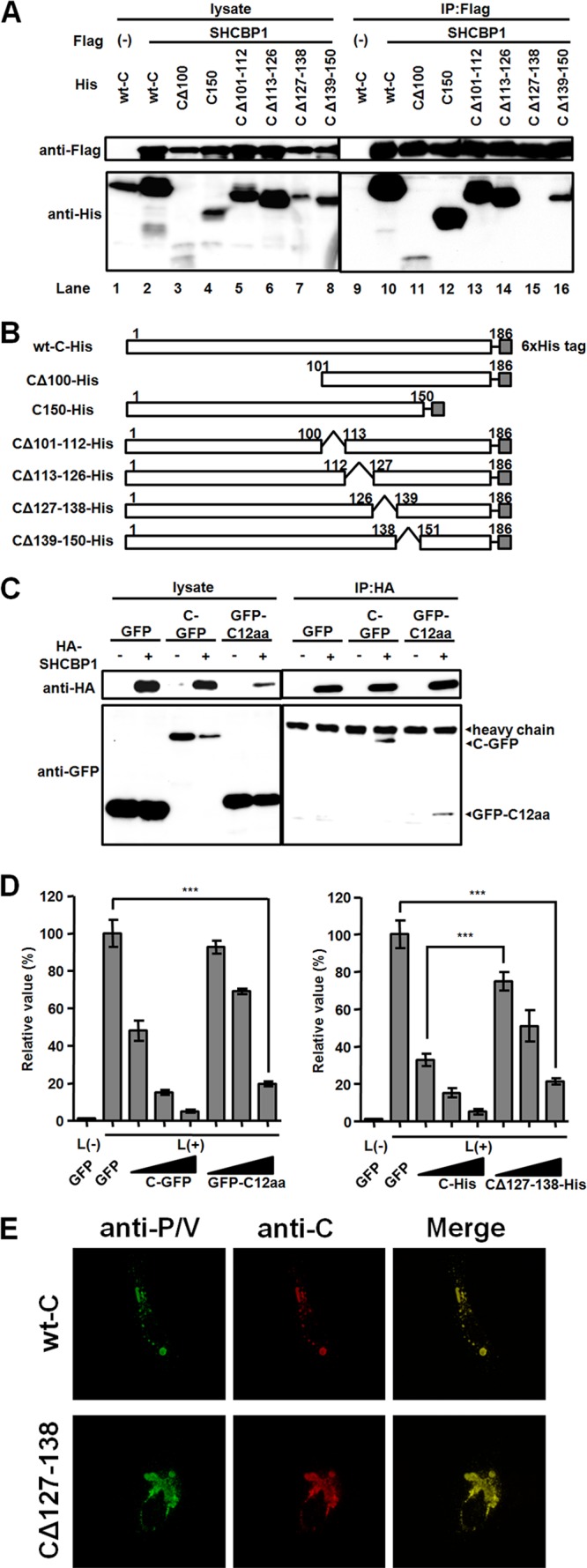
The interaction of the C protein with SHCBP1. (A) A coimmunoprecipitation (IP) assay was carried out with lysates from HEK293T cells expressing indicated combinations of Flag-tagged SHCBP1 and His-tagged wild type (wt) or mutant C protein. At 48 h after transfection, protein complexes were precipitated with anti-Flag antibody and examined by Western blotting. (B) Diagram of His-tagged wild-type (wt) and mutant C proteins. (C) Lysates from HEK 293T cells expressing indicated combinations of GFP-fused proteins and HA-tagged SHCBP1 were used for an immunoprecipitation assay. Proteins were precipitated with anti-HA antibody and examined by Western blotting. (D) Expression plasmids encoding the indicated proteins were included in the minigenome assay. Triangles below the lanes indicate increasing concentrations of the plasmids transfected (12.5, 50, and 200 ng). The average value in the sample expressing GFP was set to 100%. The data indicate means ± the SD. ***, P < 0.001. (E) Intracellular localization of CΔ127-138 and P proteins. A549/hSLAM cells were transfected with expression plasmids encoding P, N, and C proteins. At 48 h after transfection, the C (red) and P proteins (green) were immunostained.
To determine whether the peptide corresponding to the residues at positions 127 to 138 of the C protein can bind to SHCBP1, the 12-mer peptide was fused to the C terminus of GFP (GFP-C12aa) and used for coimmunoprecipitation. Both GFP-C12aa and the full-length C protein fused to GFP (C-GFP) were coprecipitated with SHCBP1, whereas GFP alone was not (Fig. 3C), indicating that the 12-mer peptide is sufficient for the interaction with SHCBP1. GFP-C12aa was also found to inhibit, although not as strong as C-GFP, the expression of the reporter gene in the MV minigenome assay (Fig. 3D, left panel), implying that the interaction of the C protein with SHCBP1 through these 12 residues is involved in the inhibition of MV RNA synthesis. On the other hand, CΔ127-138 still suppressed viral RNA synthesis especially at high concentrations, although not as strong as the full-length C protein (Fig. 3D, right panel). The results suggest that other host factor(s) than SHCBP1 may also bridge the C protein and the RNP complex and that the C protein regulates viral RNA synthesis via SHCBP1-dependent (through its 12 residues at positions 127 to 138) and -independent mechanisms. In fact, CΔ127–138 was found to still colocalize with the P protein at the perinuclear region of cells (Fig. 3E).
The central region of SHCBP1 is involved in its interaction with the P protein, as well as with the C protein.
In order to determine how SHCBP1 interacts with the P protein to facilitate efficient MV RNA synthesis, we constructed truncated forms of SHCBP1 (Fig. 4A) and examined whether they can interact with the P protein. SHCBP1-A, comprised of residues at positions 1 to 129 of the protein, could not be detected by Western blotting (Fig. 4B, left panel, lanes 3 and 8). Whereas SHCBP1-B, an N- and C-terminus-truncated mutant containing residues at positions 130 to 488, was successfully coprecipitated with the P protein (Fig. 4B, left panel, lanes 4 and 9), SHCBP1-C, comprised of residues at positions 489 to 672, was not (Fig. 4B, left panel, lanes 5 and 10). SHCBP1-B, but not SHCBP1-C, also bound to the C protein (Fig. 4B, right panel). Since both the P and the C proteins interacted with SHCBP1-B, we performed a competition assay to examine whether these two MV proteins bind to the same or overlapping site in SHCBP1. The P protein did not compete with the C protein and did coprecipitate with SHCBP1 even more efficiently in the presence of the C protein (Fig. 4C).
Fig 4.
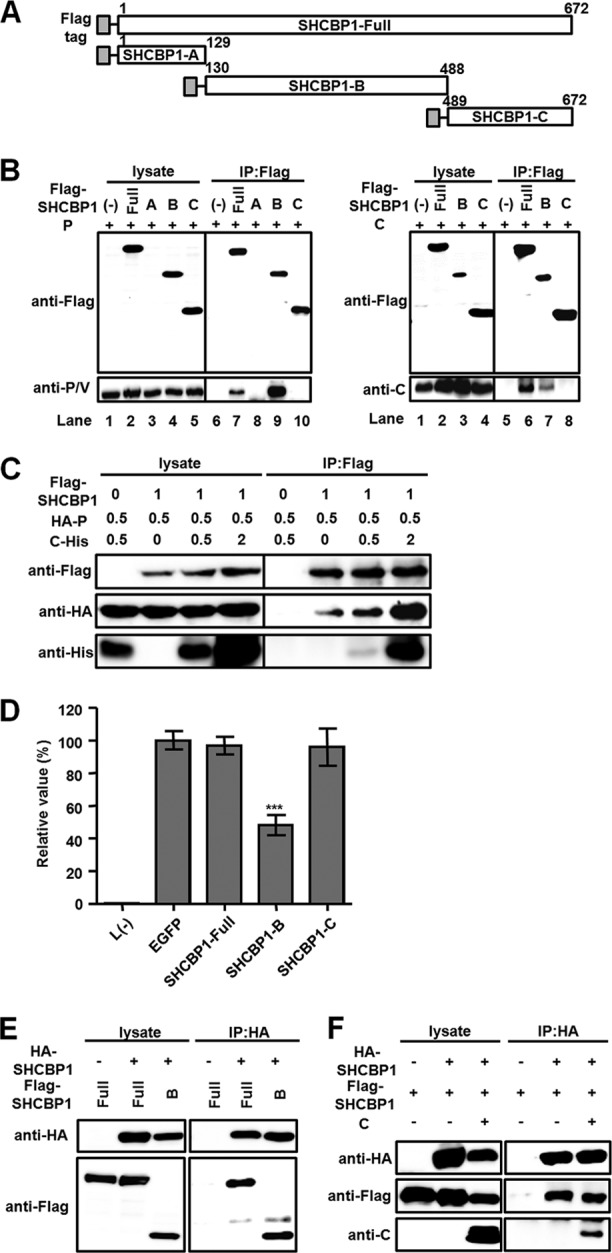
Interaction of the central region of SHCBP1 with the P and C proteins. (A) Diagram of Flag-tagged SHCBP1 and its truncated mutants. (B) Cell lysates containing indicated Flag-tagged SHCBP1 constructs and the P (left panel) or C protein (right panel) were precipitated with anti-Flag antibody and examined by Western blotting. (C) Competition for SHCBP1 binding between the C and P proteins was examined. The indicated amounts (μg) of plasmids encoding Flag-tagged SHCBP1, HA-tagged P protein, and His-tagged C protein were introduced into HEK293T cells. The total amounts of transfected DNA were kept constant by using pCA7-EGFP. At 48 h after transfection, proteins were precipitated with anti-Flag antibody and examined by Western blotting. (D) SHCBP1 and its truncated mutants were included in the minigenome assay. The average value in the sample expressing GFP was set to 100%. The data indicate means ± the SD. ***, P < 0.001. (E) Multimer formation of SHCBP1. A coimmunoprecipitation assay was performed with lysates containing indicated HA- and Flag-tagged proteins. Proteins were precipitated with anti-HA antibody and examined by Western blotting. (F) A coimmunoprecipitation (IP) assay was performed with lysates containing indicated HA- and Flag-tagged SHCBP1 and MV C protein. Proteins were precipitated with anti-HA antibody and examined by Western blotting.
We also analyzed the effects of these SHCBP1 truncated mutants on the MV minigenome activity (Fig. 4D). The addition of the full-length SHCBP1 did not increase the reporter gene expression, suggesting that endogenous SHCBP1 provides a saturated amount of support activity for the MV minigenome system. Whereas SHCBP1-C did not affect MV RNA synthesis, SHCBP1-B significantly inhibited the reporter gene expression. Presumably, SHCBP1-B did so by disrupting the interaction between the full-length SHCBP1 and the P protein. It is also possible that SHCBP1-B acted in a dominant-negative manner. Indeed, SHCBP1 was found to form multimers, and SHCBP1-B could form multimers with the full-length SHCBP1 (Fig. 4E). However, the C protein did not disrupt the multimer formation of SHCBP1 (Fig. 4F).
DISCUSSION
In this study, we show that the host protein SHCBP1 plays an important role in the activity of the MV RNA polymerase (Fig. 5). SHCBP1 was originally identified as a binding partner of Shc, a molecule that acts as an adaptor of numerous cell surface receptors and is involved in cell growth, apoptosis, and development (44, 47–49). Upon knockdown of SHCBP1, MV growth was greatly suppressed, but the multiplication of HSV-1 or EMCV was not affected. Thus, this host protein appears to enhance virus growth in an MV-specific manner. Importantly, knockdown of Shc did not affect MV replication (data not shown), indicating that Shc signaling pathways are not related to the regulation of MV growth. Furthermore, knockdown of SHCBP1 did not affect cell proliferation and viability, suggesting that disruption of cell growth is not the cause of the inhibition of MV replication. The minigenome assay revealed that SHCBP1 is required for the full activity of the MV RNA polymerase. Consistent with this, SHCBP1 was redistributed to the perinuclear foci and colocalized with the MV P protein after transfection or MV infection, whereas it was located in both the cytoplasm and the nuclei of untreated and uninfected cells. Its binding to the P protein was confirmed by coprecipitation experiments.
Fig 5.
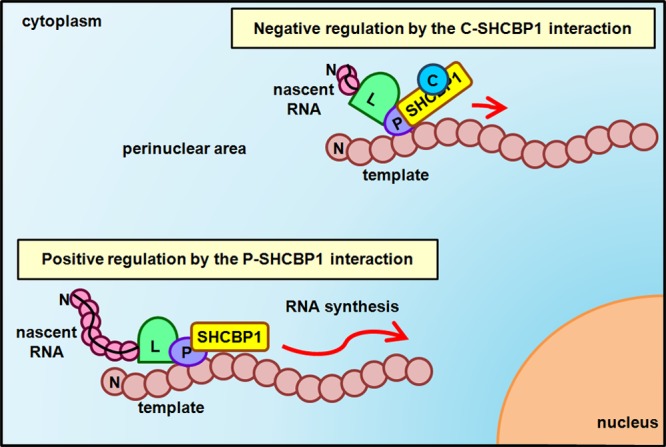
Model for the role of SHCBP1 in MV RNA synthesis and its modulation by the C protein.
Unlike the minigenome assay, the effect of SHCBP1 knockdown on viral RNA levels, as determined by quantitative reverse transcription-PCR, was not evident in the context of MV infection of A549/hSLAM cells (data not shown). The reason for the discrepancy is unknown, but viral RNA levels in infected cells may also be influenced by other factors than MV RNA polymerase activity. Furthermore, it is possible that SHCBP1 affects MV growth not only in viral RNA synthesis but also in other unknown steps. SHCBP1 knockdown did not elevate IFN-β mRNA levels in infected cells (data not shown), indicating that SHCBP1 does not affect MV growth through its direct effect on IFN induction.
We first identified SHCBP1 as a host protein binding to the MV C protein. The C protein has been shown to regulate viral RNA synthesis so as not to stimulate intracellular RNA sensors such as RIG-I and MDA5 (12). Although the C protein does not associate with any of the N, P, and L proteins (23, 25), it colocalizes with the RNP complex in the perinuclear areas of infected cells (12, 23, 24). The fragment containing amino acids at positions 127 to 138 of the C protein could bind to SHCBP1 and exhibited an inhibitory function in the MV minigenome assay. These results suggested that the C protein interacts with the MV RNP complex through SHCBP1 and thereby exerts its regulatory activity in MV RNA synthesis.
How does the C protein regulate the MV RNA polymerase? Since both the C and the P proteins can bind to SHCBP1, we first thought that the C protein inhibits the activity of the P protein by competing with the latter for SHCBP1 binding. However, the results showed that these two proteins do not compete with each other. We then found that SHCBP1 forms homomultimers and tested the possibility that the C protein disrupts the multimer formation of SHCBP1, thereby inhibiting its function. However, our results disproved the possibility. Thus, it is likely that the binding of the C protein somehow affects the structure and/or function of SHCBP1 so that SHCBP1 can no longer support MV RNA polymerase activity, even though it retains the ability to interact with the P protein (Fig. 5).
It should be noted that the interaction with SHCBP1 may not be the only mechanism by which the C protein regulates MV RNA synthesis, because the C protein lacking the SHCBP1-binding site still possesses the inhibitory function. Considering that knockdown of several other host proteins also reduced MV growth, albeit not as strongly as SHCBP1, there may be other host proteins that support MV RNA polymerase activity, and bridge the C protein and the RNP complex.
Heat shock protein 72 (50, 51) and peroxiredoxin 1 (52) have been reported to enhance MV RNA polymerase activity by interacting with the P protein-binding motif of the N protein at its C-terminal region. The MV M protein binds to the C-terminal region of the N protein and wraps the genomic RNA-N protein complex (23, 53, 54). This M protein coat might prevent the binding and/or moving of the P protein, thereby inhibiting RNA polymerase activity. Since neither the C protein nor SHCBP1 associates with the N protein, the inhibitory mechanism of the C protein should be different from that of the M protein. Casein kinase II, another host factor, has been reported to phosphorylate the P protein (55), and the activity of the P protein is differently influenced by the phosphorylation status in different paramyxoviruses (56, 57). A recent study reported that the phosphorylation of the MV P protein at S86 and/or S151 diminishes its polymerase activity (58). Thus, it is possible that SHCBP1 controls the phosphorylation status of the P protein to promote the polymerase activity and that the C protein inhibits the process by binding to SHCBP1.
It has been reported that SHCBP1 is expressed in actively proliferating cells and that its expression increases after growth factor stimulation (44). We found that expression of SHCBP1 is also induced by MV infection, poly(I·C) stimulation, and IFN treatment, indicating that SHCBP1 is an IFN-inducible gene. It is surprising that MV utilizes one of the IFN-inducible genes for its efficient growth, whereas the V and P proteins strongly prevent the expression of these genes (4, 7, 59). At the early stage of its replication cycle, MV probably induces a small amount of type I IFNs, which in turn induce the SHCBP1 required for efficient MV replication. Once enough amounts of SHCBP1 are produced, the V and P proteins inhibit the IFN induction and signal transduction, thereby inhibiting the expression of IFN-inducible genes, including SHCBP1. The C protein also inhibits induction of IFNs, further reducing expression of IFN-inducible genes.
Despite the availability of efficient vaccine, measles is still prevalent in developing countries. Measles is an acute febrile disease and generally does not cause persistent infection among immunocompetent hosts. An exception is subacute sclerosing panencephalitis (SSPE), which is caused by persistent infection of MV in the brain and is characterized by progressive cognitive and motor disability (1). There is currently no effective treatment for the disease. We identified here a 12-mer peptide derived from the C protein that is able to inhibit MV RNA polymerase activity by interacting with SHCBP1. This peptide could be a good lead compound to treat SSPE and other MV-related conditions. Considering the high error rate of the viral RNA polymerase and resulting escape mutations, the use of compounds targeting a host protein rather than viral proteins may be effective to treat MV infection.
ACKNOWLEDGMENTS
We thank the staff of the Research Support Center, Graduate School of Medical Sciences, Kyushu University, for technical support. We also thank M. Shimojima, T. Kitamura, W. Chang, and K. Komase for providing reagents.
This study was supported by MEXT KAKENHI grant 24115005 and the Uehara Memorial Foundation.
Footnotes
Published ahead of print 26 June 2013
REFERENCES
- 1.Griffin DE. 2007. Measles virus, p 1551–1585 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott/The Williams & Wilkins Co, Philadelphia, PA [Google Scholar]
- 2.Bellini WJ, Englund G, Rozenblatt S, Arnheiter H, Richardson CD. 1985. Measles virus P gene codes for two proteins. J. Virol. 53:908–919 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cattaneo R, Kaelin K, Baczko K, Billeter MA. 1989. Measles virus editing provides an additional cysteine-rich protein. Cell 56:759–764 [DOI] [PubMed] [Google Scholar]
- 4.Parisien JP, Bamming D, Komuro A, Ramachandran A, Rodriguez JJ, Barber G, Wojahn RD, Horvath CM. 2009. A shared interface mediates paramyxovirus interference with antiviral RNA helicases MDA5 and LGP2. J. Virol. 83:7252–7260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pfaller CK, Conzelmann KK. 2008. Measles virus V protein is a decoy substrate for IκB kinase alpha and prevents Toll-like receptor 7/9-mediated interferon induction. J. Virol. 82:12365–12373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Caignard G, Guerbois M, Labernardiere JL, Jacob Y, Jones LM, Wild F, Tangy F, Vidalain PO. 2007. Measles virus V protein blocks Jak1-mediated phosphorylation of STAT1 to escape IFN-alpha/beta signaling. Virology 368:351–362 [DOI] [PubMed] [Google Scholar]
- 7.Palosaari H, Parisien JP, Rodriguez JJ, Ulane CM, Horvath CM. 2003. STAT protein interference and suppression of cytokine signal transduction by measles virus V protein. J. Virol. 77:7635–7644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ramachandran A, Parisien JP, Horvath CM. 2008. STAT2 is a primary target for measles virus V protein-mediated alpha/beta interferon signaling inhibition. J. Virol. 82:8330–8338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Devaux P, Hodge G, McChesney MB, Cattaneo R. 2008. Attenuation of V- or C-defective measles viruses: infection control by the inflammatory and interferon responses of rhesus monkeys. J. Virol. 82:5359–5367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Takeuchi K, Takeda M, Miyajima N, Ami Y, Nagata N, Suzaki Y, Shahnewaz J, Kadota S, Nagata K. 2005. Stringent requirement for the C protein of wild-type measles virus for growth both in vitro and in macaques. J. Virol. 79:7838–7844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nakatsu Y, Takeda M, Ohno S, Koga R, Yanagi Y. 2006. Translational inhibition and increased interferon induction in cells infected with C protein-deficient measles virus. J. Virol. 80:11861–11867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nakatsu Y, Takeda M, Ohno S, Shirogane Y, Iwasaki M, Yanagi Y. 2008. Measles virus circumvents the host interferon response by different actions of the C and V proteins. J. Virol. 82:8296–8306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toth AM, Li Z, Cattaneo R, Samuel CE. 2009. RNA-specific adenosine deaminase ADAR1 suppresses measles virus-induced apoptosis and activation of protein kinase PKR. J. Biol. Chem. 284:29350–29356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McAllister CS, Toth AM, Zhang P, Devaux P, Cattaneo R, Samuel CE. 2010. Mechanisms of protein kinase PKR-mediated amplification of beta interferon induction by C protein-deficient measles virus. J. Virol. 84:380–386 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bankamp B, Wilson J, Bellini WJ, Rota PA. 2005. Identification of naturally occurring amino acid variations that affect the ability of the measles virus C protein to regulate genome replication and transcription. Virology 336:120–129 [DOI] [PubMed] [Google Scholar]
- 16.Reutter GL, Cortese-Grogan C, Wilson J, Moyer SA. 2001. Mutations in the measles virus C protein that upregulate viral RNA synthesis. Virology 285:100–109 [DOI] [PubMed] [Google Scholar]
- 17.Sparrer KM, Pfaller CK, Conzelmann KK. 2012. Measles virus C protein interferes with beta interferon transcription in the nucleus. J. Virol. 86:796–805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yokota S, Okabayashi T, Fujii N. 2011. Measles virus C protein suppresses gamma-activated factor formation and virus-induced cell growth arrest. Virology 414:74–82 [DOI] [PubMed] [Google Scholar]
- 19.Bankamp B, Horikami SM, Thompson PD, Huber M, Billeter M, Moyer SA. 1996. Domains of the measles virus N protein required for binding to P protein and self-assembly. Virology 216:272–277 [DOI] [PubMed] [Google Scholar]
- 20.Cevik B, Holmes DE, Vrotsos E, Feller JA, Smallwood S, Moyer SA. 2004. The phosphoprotein (P) and L binding sites reside in the N terminus of the L subunit of the measles virus RNA polymerase. Virology 327:297–306 [DOI] [PubMed] [Google Scholar]
- 21.Harty RN, Palese P. 1995. Measles virus phosphoprotein (P) requires the NH2- and COOH-terminal domains for interactions with the nucleoprotein (N) but only the COOH terminus for interactions with itself. J. Gen. Virol. 76:2863–2867 [DOI] [PubMed] [Google Scholar]
- 22.Horikami SM, Smallwood S, Bankamp B, Moyer SA. 1994. An amino-proximal domain of the L protein binds to the P protein in the measles virus RNA polymerase complex. Virology 205:540–545 [DOI] [PubMed] [Google Scholar]
- 23.Iwasaki M, Takeda M, Shirogane Y, Nakatsu Y, Nakamura T, Yanagi Y. 2009. The matrix protein of measles virus regulates viral RNA synthesis and assembly by interacting with the nucleocapsid protein. J. Virol. 83:10374–10383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nakatsu Y, Ma X, Seki F, Suzuki T, Iwasaki M, Yanagi Y, Komase K, Takeda M. 2013. Intracellular transport of the measles virus RNP complex is mediated by Rab11A-positive recycling endosomes and drives virus release from the apical membrane of polarized epithelial cells. J. Virol. 87:4683–4693 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liston P, DiFlumeri C, Briedis DJ. 1995. Protein interactions entered into by the measles virus P, V, and C proteins. Virus Res. 38:241–259 [DOI] [PubMed] [Google Scholar]
- 26.Cadd T, Garcin D, Tapparel C, Itoh M, Homma M, Roux L, Curran J, Kolakofsky D. 1996. The Sendai paramyxovirus accessory C proteins inhibit viral genome amplification in a promoter-specific fashion. J. Virol. 70:5067–5074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grogan CC, Moyer SA. 2001. Sendai virus wild-type and mutant C proteins show a direct correlation between L polymerase binding and inhibition of viral RNA synthesis. Virology 288:96–108 [DOI] [PubMed] [Google Scholar]
- 28.Takeda M, Ohno S, Seki F, Nakatsu Y, Tahara M, Yanagi Y. 2005. Long untranslated regions of the measles virus M and F genes control virus replication and cytopathogenicity. J. Virol. 79:14346–14354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bair CH, Chung CS, Vasilevskaya IA, Chang W. 1996. Isolation and characterization of a Chinese hamster ovary mutant cell line with altered sensitivity to vaccinia virus killing. J. Virol. 70:4655–4666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ono N, Tatsuo H, Hidaka Y, Aoki T, Minagawa H, Yanagi Y. 2001. Measles viruses on throat swabs from measles patients use signaling lymphocytic activation molecule (CDw150) but not CD46 as a cellular receptor. J. Virol. 75:4399–4401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morita S, Kojima T, Kitamura T. 2000. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther. 7:1063–1066 [DOI] [PubMed] [Google Scholar]
- 32.Takeda M, Takeuchi K, Miyajima N, Kobune F, Ami Y, Nagata N, Suzaki Y, Nagai Y, Tashiro M. 2000. Recovery of pathogenic measles virus from cloned cDNA. J. Virol. 74:6643–6647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hashimoto K, Ono N, Tatsuo H, Minagawa H, Takeda M, Takeuchi K, Yanagi Y. 2002. SLAM (CD150)-independent measles virus entry as revealed by recombinant virus expressing green fluorescent protein. J. Virol. 76:6743–6749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takeda M, Tahara M, Hashiguchi T, Sato TA, Jinnouchi F, Ueki S, Ohno S, Yanagi Y. 2007. A human lung carcinoma cell line supports efficient measles virus growth and syncytium formation via a SLAM- and CD46-independent mechanism. J. Virol. 81:12091–12096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Takeda M, Ohno S, Seki F, Hashimoto K, Miyajima N, Takeuchi K, Yanagi Y. 2005. Efficient rescue of measles virus from cloned cDNA using SLAM-expressing Chinese hamster ovary cells. Virus Res. 108:161–165 [DOI] [PubMed] [Google Scholar]
- 36.Komune N, Ichinohe T, Ito M, Yanagi Y. 2011. Measles virus V protein inhibits NLRP3 inflammasome-mediated interleukin-1β secretion. J. Virol. 85:13019–13026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Niwa H, Yamamura K, Miyazaki J. 1991. Efficient selection for high-expression transfectants with a novel eukaryotic vector. Gene 108:193–199 [DOI] [PubMed] [Google Scholar]
- 38.Shirogane Y, Takeda M, Tahara M, Ikegame S, Nakamura T, Yanagi Y. 2010. Epithelial-mesenchymal transition abolishes the susceptibility of polarized epithelial cell lines to measles virus. J. Biol. Chem. 285:20882–20890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Fuerst TR, Niles EG, Studier FW, Moss B. 1986. Eukaryotic transient-expression system based on recombinant vaccinia virus that synthesizes bacteriophage T7 RNA polymerase. Proc. Natl. Acad. Sci. U. S. A. 83:8122–8126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schneider H, Spielhofer P, Kaelin K, Dotsch C, Radecke F, Sutter G, Billeter MA. 1997. Rescue of measles virus using a replication-deficient vaccinia-T7 vector. J. Virol. Methods 64:57–64 [DOI] [PubMed] [Google Scholar]
- 41.Komase K, Nakayama T, Iijima M, Miki K, Kawanishi R, Uejima H. 2006. The phosphoprotein of attenuated measles AIK-C vaccine strain contributes to its temperature-sensitive phenotype. Vaccine 24:826–834 [DOI] [PubMed] [Google Scholar]
- 42.Ashizuka M, Fukuda T, Nakamura T, Shirasuna K, Iwai K, Izumi H, Kohno K, Kuwano M, Uchiumi T. 2002. Novel translational control through an iron-responsive element by interaction of multifunctional protein YB-1 and IRP2. Mol. Cell. Biol. 22:6375–6383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kraus TA, Garza L, Horvath CM. 2008. Enabled interferon signaling evasion in an immune-competent transgenic mouse model of parainfluenza virus 5 infection. Virology 371:196–205 [DOI] [PubMed] [Google Scholar]
- 44.Schmandt R, Liu SK, McGlade CJ. 1999. Cloning and characterization of mPAL, a novel Shc SH2 domain-binding protein expressed in proliferating cells. Oncogene 18:1867–1879 [DOI] [PubMed] [Google Scholar]
- 45.Gosert R, Egger D, Lohmann V, Bartenschlager R, Blum HE, Bienz K, Moradpour D. 2003. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring subgenomic replicons. J. Virol. 77:5487–5492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Westaway EG, Khromykh AA, Mackenzie JM. 1999. Nascent flavivirus RNA colocalized in situ with double-stranded RNA in stable replication complexes. Virology 258:108–117 [DOI] [PubMed] [Google Scholar]
- 47.Lai KM, Pawson T. 2000. The ShcA phosphotyrosine docking protein sensitizes cardiovascular signaling in the mouse embryo. Genes Dev. 14:1132–1145 [PMC free article] [PubMed] [Google Scholar]
- 48.Gotoh N, Tojo A, Shibuya M. 1996. A novel pathway from phosphorylation of tyrosine residues 239/240 of Shc, contributing to suppress apoptosis by IL-3. EMBO J. 15:6197–6204 [PMC free article] [PubMed] [Google Scholar]
- 49.Salcini AE, McGlade J, Pelicci G, Nicoletti I, Pawson T, Pelicci PG. 1994. Formation of Shc-Grb2 complexes is necessary to induce neoplastic transformation by overexpression of Shc proteins. Oncogene 9:2827–2836 [PubMed] [Google Scholar]
- 50.Zhang X, Glendening C, Linke H, Parks CL, Brooks C, Udem SA, Oglesbee M. 2002. Identification and characterization of a regulatory domain on the carboxyl terminus of the measles virus nucleocapsid protein. J. Virol. 76:8737–8746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang X, Oglesbee M. 2003. Use of surface plasmon resonance for the measurement of low affinity binding interactions between HSP72 and measles virus nucleocapsid protein. Biol. Proc. Online 5:170–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Watanabe A, Yoneda M, Ikeda F, Sugai A, Sato H, Kai C. 2011. Peroxiredoxin 1 is required for efficient transcription and replication of measles virus. J. Virol. 85:2247–2253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Brown HR, Goller N, Thormar H, Norrby E. 1987. Fuzzy material surrounding measles virus nucleocapsids identified as matrix protein. Arch. Virol. 94:163–168 [DOI] [PubMed] [Google Scholar]
- 54.Liljeroos L, Huiskonen JT, Ora A, Susi P, Butcher SJ. 2011. Electron cryotomography of measles virus reveals how matrix protein coats the ribonucleocapsid within intact virions. Proc. Natl. Acad. Sci. U. S. A. 108:18085–18090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Das T, Schuster A, Schneider-Schaulies S, Banerjee AK. 1995. Involvement of cellular casein kinase II in the phosphorylation of measles virus P protein: identification of phosphorylation sites. Virology 211:218–226 [DOI] [PubMed] [Google Scholar]
- 56.Hu CJ, Kato A, Bowman MC, Kiyotani K, Yoshida T, Moyer SA, Nagai Y, Gupta KC. 1999. Role of primary constitutive phosphorylation of Sendai virus P and V proteins in viral replication and pathogenesis. Virology 263:195–208 [DOI] [PubMed] [Google Scholar]
- 57.Villanueva N, Hardy R, Asenjo A, Yu Q, Wertz G. 2000. The bulk of the phosphorylation of human respiratory syncytial virus phosphoprotein is not essential but modulates viral RNA transcription and replication. J. Gen. Virol. 81:129–133 [DOI] [PubMed] [Google Scholar]
- 58.Sugai A, Sato H, Yoneda M, Kai C. 2012. Phosphorylation of measles virus phosphoprotein at S86 and/or S151 downregulates viral transcriptional activity. FEBS Lett. 586:3900–3907 [DOI] [PubMed] [Google Scholar]
- 59.Ohno S, Ono N, Takeda M, Takeuchi K, Yanagi Y. 2004. Dissection of measles virus V protein in relation to its ability to block alpha/beta interferon signal transduction. J. Gen. Virol. 85:2991–2999 [DOI] [PubMed] [Google Scholar]