

Abstract
Alphaviruses are enveloped viruses with highly organized structures. The nucleocapsid (NC) core contains a capsid protein lattice enclosing the plus-sense RNA genome, and it is surrounded by a lipid bilayer containing a lattice of the E1 and E2 envelope glycoproteins. Capsid protein is synthesized in the cytoplasm and particle budding occurs at the plasma membrane (PM), but the traffic and assembly of viral components and the exit of virions from host cells are not well understood. To visualize the dynamics of capsid protein during infection, we developed a Sindbis virus infectious clone tagged with a tetracysteine motif. Tagged capsid protein could be fluorescently labeled with biarsenical dyes in living cells without effects on virus growth, morphology, or protein distribution. Live cell imaging and colocalization experiments defined distinct groups of capsid foci in infected cells. We observed highly motile internal puncta that colocalized with E2 protein, which may represent the transport machinery that capsid protein uses to reach the PM. Capsid was also found in larger nonmotile internal structures that colocalized with cellular G3BP and viral nsP3. Thus, capsid may play an unforeseen role in these previously observed G3BP-positive foci, such as regulation of cellular stress granules. Capsid puncta were also observed at the PM. These puncta colocalized with E2 and recruited newly synthesized capsid protein; thus, they may be sites of virus assembly and egress. Together, our studies provide the first dynamic views of the alphavirus capsid protein in living cells and a system to define detailed mechanisms during alphavirus infection.
INTRODUCTION
Enveloped virus budding reactions can take place at a variety of cellular membranes and may be dependent on the viral nucleocapsid, envelope proteins, and/or matrix proteins (reviewed in references 1 and 2). The alphaviruses are small enveloped plus-sense RNA viruses with highly organized structures (reviewed in references 3–5). Alphaviruses contain an internal core composed of the ∼11-kb RNA genome enclosed in an icosahedral capsid protein shell. This nucleocapsid (NC) is enveloped by the virus lipid bilayer containing a lattice of the E1 and E2 membrane glycoproteins. Alphavirus budding takes place at the plasma membrane (PM) and requires both the NC and the envelope proteins (6). The completed viral particle contains 240 copies of each of these structural proteins, with each capsid protein interacting 1:1 with the cytoplasmic domain of an E2 protein (7–9).
During infection, the alphavirus genomic RNA is translated to produce the four nonstructural proteins (nsP1 to nsP4) that mediate RNA replication, while the structural proteins are produced as a polyprotein from a subgenomic RNA (reviewed in references 3, 4, and 10). The N-terminal capsid protein contains a protease domain. Once it is translated it rapidly folds, autocleaves itself from the polypeptide, and is released into the cytoplasm. The rest of the polyprotein contains the viral membrane proteins, which are translocated into the endoplasmic reticulum and transported through the secretory pathway to the PM.
Two models have been proposed for alphavirus nucleocapsid assembly (reviewed in reference 11). One model predicts that the NC is preassembled in the cytoplasm and then drives virus budding by binding to the glycoproteins at the PM. This model is supported by the presence of abundant NC in the cytoplasm of infected cells (12) and by the efficient in vitro assembly of NC in the absence of glycoproteins (13). Microinjection of such preformed NCs into cells expressing the viral envelope proteins can generate infectious virus-like particles, albeit at a relatively low efficiency (14, 15).
An alternative model postulates that a capsid-RNA complex binds the E2 cytoplasmic domain at the PM, where the lateral interactions of the glycoproteins drive formation of the icosahedral NC and subsequent virus budding. In support of this model, particle production for capsid mutants defective in cytoplasmic NC formation is only mildly reduced compared to that of the wild type (WT), indicating that preformed NCs are not strictly required for virus budding (9, 16–18). A common feature of both models is that the cytoplasmic NC or the capsid-RNA complex must be transported to the PM. Based on its high protein concentration and extensive cytoskeletal network, the cytoplasmic milieu will greatly restrict the free diffusion of the capsid/NC (19), but potential transport mechanisms are undefined.
Early studies of the kinetics of alphavirus particle production indicated that only a fraction of the cellular pool of capsid protein is ultimately released in virus particles (20). Nascent capsid protein can associate at least transiently with ribosomes in infected cells (21–24). Later in infection, some capsid proteins associate with the cellular adaptor protein p62, which mediates capsid targeting to autophagosomes (25, 26). It is not clear how or where the remaining capsid proteins might accumulate in the host cell, whether they associate with specific cellular proteins or trigger host cell responses, or what other additional functions the capsid protein may have.
Here, we set out to characterize the dynamics of the alphavirus capsid protein during virus replication by visualizing the protein in its cellular context in live infected cells. We took advantage of the tetracysteine (TC) motif, a 12-amino-acid sequence that can be labeled with high affinity and specificity by biarsenical dyes such as FlAsH and ReAsH (27). Using a Sindbis virus (SINV) infectious clone, we defined a favorable insertion site for the TC motif on the viral capsid protein and optimized the system to monitor FlAsH- or ReAsH-labeled capsid in real time. Our studies identified three categories of capsid foci in SINV-infected Vero cells. One group was comprised of small, round, internal puncta that were highly motile and colocalized with the E2 protein, suggesting a role in transport. A second group consisted of larger, more irregular internal structures that colocalized with G3BP and nsP3, two proteins previously shown to interact and regulate stress granule formation. The third group was composed of capsid puncta that colocalized with the E2 envelope protein at the PM, suggesting their involvement in the assembly/exit of progeny viruses.
MATERIALS AND METHODS
Cells.
BHK-21 cells were cultured at 37°C in complete BHK medium (Dulbecco's modified Eagle's medium [DMEM] containing 5% fetal calf serum, 10% tryptose phosphate broth, 100 U penicillin/ml, and 100 μg streptomycin/ml). Vero cells from ATCC were kindly provided by Kartik Chandran and cultured in complete Vero medium (Dulbecco's modified Eagle's medium containing 10% fetal bovine serum, 100 U penicillin/ml, and 100 μg streptomycin/ml) at 37°C.
Construction of SINV infectious clones.
The WT SINV and mCherry SINV used in our studies were derived from the WT dsTE12Q infectious clone (28) and a dsTE12Q clone containing mCherry at the N terminus of the capsid (25), both kindly provided by Beth Levine. We constructed green fluorescent protein (GFP)-dsTE12Q and Toto1101 (29) clones containing superfolder GFP (sfGFP) at the N terminus of the capsid. This insertion was generated using a three-step overlap-extension PCR with sfGFP-C1 (30) (kindly provided by Erik Snapp) to amplify sfGFP and with dsTE12Q or Toto1101 to amplify the SINV capsid region. The fragment containing sfGFP-capsid was subcloned into the dsTE12Q or Toto1101 infectious clone using HpaI/AatII restriction endonuclease sites.
TC tag insertions in capsid protein were generated by overlapping PCR using primers encoding the optimized TC sequence FLNCCPGCCMEP (27). The fragment containing the TC insertion at the N terminus of the capsid was subcloned into the dsTE12Q infectious clone using the HpaI/AatII restriction sites, and fragments with TC inserted at other sites were subcloned using the HpaI/BclI sites. TC-LL (capsid Q94-TC plus capsid L108A/L110A) was constructed using overlapping PCR using Q94-TC dsTE12Q as a template and primers containing the capsid L108A and L110A mutations (9, 16). The fragment containing the Q94-TC insertion and capsid L108A/L110A mutation was subcloned into dsTE12Q using the HpaI/BclI sites. TC Y400K (capsid Q94-TC plus E2 Y400K) was constructed by subcloning the E2 Y400K-containing PmlI/BclI fragment from Y400K-dsTE12Q (G. Martinez, unpublished data) into Q94-TC dsTE12Q.
Sequence analysis (Genewiz Inc., North Brunswick, NJ) of the entire HpaI/AatII or HpaI/BclI PCR region confirmed that the TC insertions were in place without additional mutations introduced during PCR amplification. For each insertion mutant, two independent clones were tested to confirm the phenotype.
Production and titration of virus stocks.
Viral RNAs were in vitro transcribed from the XhoI-linearized SINV infectious clone and were electroporated into BHK-21 cells to generate virus stocks (31). All mutant virus stocks were prepared by incubation of electroporated cells for 12 h at 37°C to prevent possible generation of revertants during extended culture time.
Virus titers were measured as indicated by plaque titration on BHK-21 cells or by infectious center (IC) assays in BHK-21 cells or Vero cells. For infectious center assays, cells were infected with serial dilutions of SINV virus for 1.5 h at 37°C. Media were replaced with complete growth medium containing 20 mM NH4Cl to prevent secondary infection and were incubated at 28°C overnight to allow viral protein expression. Infected cells were quantitated by immunofluorescence (IF) using monoclonal antibodies (MAbs) R2 and R6 to the E1 and E2 proteins, respectively (kindly provided by William Klimstra) (32).
Virus assembly assay.
The assembly of mutant viruses was evaluated by pulse-chase analysis (33). The viral RNAs were introduced into BHK-21 cells by electroporation. After 6 h of incubation at 37°C, cells were pulse labeled with 50 μCi/ml [35S]methionine/cysteine (Express labeling mix; PerkinElmer Life and Analytical Sciences) at 37°C for 30 min and chased for 0 to 3 h at 37°C in the absence of radiolabel. The medium samples were collected, immunoprecipitated with MAb R6 against E2 (32) in the absence of detergent, and analyzed by SDS-PAGE. Cell lysates were collected in the presence of 40 μM N-ethylmaleimide to alkylate free cysteine in the TC tag, and aliquots of the lysates were analyzed directly by SDS-PAGE.
Electron microscopy of virus-infected cells.
BHK-21 cells were electroporated with WT or mutant RNA, plated on 35-mm-diameter dishes, and incubated at 37°C for 12 h before fixing with 2.5% glutaraldehyde, 2% paraformaldehyde in 0.1 M sodium cacodylate buffer for 30 min at room temperature. Samples were then processed by the Einstein Analytical Imaging Facility by postfixing with 1% osmium tetroxide followed by 2% uranyl acetate, dehydration, and embedding in LX112 resin (LADD Research Industries, Burlington, VT). Thin sections were stained with uranyl acetate followed by lead citrate and examined on a JEOL 1200EX or a JEOL 100CXII electron microscope at 80 kV. Images were recorded at a magnification of ×20,000 and assembled with the software Adobe Photoshop CS.
Tetracysteine labeling with biarsenical dyes.
Vero cells were plated on 8-well Lab-TekTM II chambered cover glass (Thermo Scientific), incubated at 37°C overnight, and then infected with WT or TC-tagged SINV at a multiplicity of 0.5 IC/cell. After 2 h at 37°C, the medium was replaced with Vero complete medium and the incubation continued for another 5 h at 37°C to allow the expression of capsid proteins before labeling with biarsenical dyes. For the budding-negative Y400K mutant, Vero cells were transfected with viral RNA and incubated at 37°C for 7 h. Labeling protocols were modified from those published previously (34, 35), with additional helpful suggestions from Brett Lindenbach and Eric Freed. Cells were incubated in 2.5 μM FlAsH (or ReAsH) for 5 min at 37°C and washed three times at 37°C with DMEM containing 1 mM 2,3-dimercapto-1-propanol (British anti-Lewisite [BAL]; Sigma) (34). Live cell imaging was then performed as described below. Alternatively, cells were fixed with 3% paraformaldehyde for immunofluorescence experiments. The time course of infection of the Vero cell culture (and the cellular staining patterns) were not fully synchronous, even if the viral inoculum was prebound to the cells in the cold (data not shown). This is likely due to the effects of the local population context, including cell density (as described in reference 36).
For FlAsH/ReAsH pulse-chase labeling, cells were labeled with 5 μM FlAsH for 20 min at 37°C. After one wash with DMEM, cells were incubated with Vero growth media for 0 to 1 h at 37°C and then labeled with 2.5 μM ReAsH as described above.
Immunofluorescence analysis.
To stain for the capsid or cellular marker proteins, Vero cells were infected or transfected as described above, incubated at 37°C for 7 h, fixed with 3% formaldehyde, and permeabilized with 0.2% Triton X-100 for 5 min at room temperature. Antibodies used for immunofluorescence included mouse anti-capsid monoclonal antibody (MAb) C12-1 (kindly provided by Irene Greiser-Wilke) (37), rabbit antibodies to SINV nsP1 and nsP3 (kindly provided by Margaret MacDonald and Charles Rice) (38), rabbit anti-LC3 (PM036; MBL International Corporation), rabbit anti-G3BP (anti-G3BP1; G6046; Sigma), mouse anti-dsRNA MAb J2 (English & Science Consulting Kft.), and goat anti-eIF3 (sc-16377; Santa Cruz Biotechnology, Inc.). SINV E2 was detected using mouse anti-E2 MAb R6, and E1 was detected using MAb R2 (32). Preliminary studies showed that R6 recognized the ectodomain of the E2 protein on intact virus but not in cell lysates or detergent-solubilized viruses (data not shown), suggesting that the epitope correlates with trimer or lattice assembly. Secondary antibodies were Alexa 488 or 568 conjugated (Molecular Probes). For live cell staining, Vero cells were incubated for 30 min at 37°C with the R6 MAb, placed on ice, washed, incubated with secondary antibody on ice, and fixed with paraformaldehyde on ice for 30 min before imaging. Images were captured using a confocal microscope system (Duoscan; Carl Zeiss MicroImaging, Inc.) with a 63×, 1.4-numeric-aperture (NA) oil objective and a 489-nm, 100-milliwatt diode laser with a 500 to 550-nm band pass filter for FlAsH or Alexa 488 and a 40-milliwatt 561-nm diode laser with a 580-nm long pass filter for ReAsH or Alexa 568. All images were prepared with Adobe Photoshop CS4 software (Adobe System, San Jose, CA).
Live cell microscopy.
Live cells were imaged in imaging media (DMEM without phenol red or bicarbonate [D2902; Sigma], 10 mM HEPES, pH 7, 10% fetal bovine serum, 100 U penicillin/ml, and 100 μg streptomycin/ml) using the 37°C environmentally controlled chamber of the Duoscan confocal microscope system described above. Images were prepared with Volocity 3D image analysis software (PerkinElmer) and Adobe Photoshop CS4 and Illustrator CS4 software (Adobe System, San Jose, CA).
RESULTS
Development of functional SINV capsid protein with a fluorescent tag.
To visualize the movements of the alphavirus capsid protein during virus replication and assembly, we developed methods to fluorescently label and image capsid protein. SINV was used as a model alphavirus, and our constructs were based on the SINV infectious clone dsTE12Q.
We first tested a previously described virus containing mCherry at the N terminus of the capsid protein (mCherry-dsTE12Q) (25, 26). We also constructed a similar virus in which superfolder GFP was inserted at the capsid N terminus (GFP-dsTE12Q). BHK cells were electroporated with in vitro-transcribed viral RNAs, and progeny virus titers were measured. The titers of both mCherry-dsTE12Q and GFP-dsTE12Q were about 2 logs lower than those of the unlabeled WT virus at 24 h postelectroporation (data not shown). Transmission electron microscopy (EM) of virus-infected BHK cells showed WT SINV budding at the cell surface as spherical particles containing a dense core in the center (as shown below). In contrast, cells infected with GFP-dsTE12Q (as shown below) or mCherry-dsTE12Q (data not shown) produced larger aberrant particles containing multiple patches of dense core material. In addition, immunostaining with an antibody against the capsid protein showed that cells infected for 7 h with GFP- or mCherry-dsTE12Q virus contained unique cytoplasmic patches of fluorescence that were not observed in cells infected with WT SINV (data not shown). Given that the sizes of both GFP and mCherry are similar to those of the capsid protein (247, 250, and 264 amino acids, respectively), it seemed likely that GFP or mCherry interfered with the assembly of viral nucleocapsid and/or caused capsid protein aggregation.
As an alternative strategy, we tested insertion of the TC motif and its labeling with biarsenical dyes (27, 39). This system has been successfully used to study the trafficking of the human immunodeficiency virus (HIV) GAG protein, the hepatitis C virus core protein, and the vesicular stomatitis virus M protein (34, 35, 40–42). A TC motif introduced at the N terminus of the SINV capsid protein appeared to be rapidly lost during virus infection as detected by migration on SDS-PAGE (data not shown). Insertion of a flexible linker between the TC tag and the N terminus of the capsid protein stabilized the insertion, but the TC-tagged capsid protein was not successfully labeled by biarsenical dye treatment of infected cells (data not shown). This presumably reflects limited accessibility of the TC motif in the folded/assembled capsid protein. The alphavirus capsid protein assembles into NC, with its N-terminal region located internally in association with the viral RNA (43) and its C-terminal protease domain arrayed on the NC surface (44). We therefore narrowed our target site selection to the C-terminal domain and the region close to it in the hope of obtaining better dye accessibility. Within those regions of the capsid protein, we selected specific sites (Table 1) that appeared more likely to tolerate insertions based on the results of a whole-genome transposon-mediated insertion screen of the alphavirus Venezuelan equine encephalitis virus (45). Based on the structure of the SINV capsid protease domain (46), our selected sites were all localized at the tips of flexible loops, in keeping with their potential to tolerate a small insertion (data not shown). A series of TC-tagged SINV strains was constructed, and the viral growth kinetics were compared to those of WT SINV (Table 1). Most viruses with a TC tag inserted within the C-terminal capsid protease domain produced viral titers at least 2 logs lower than that of WT SINV. The single exception was D113-TC, which grew to a titer only 1 log lower than that of WT virus. D113 is located at the junction between the N-terminal and C-terminal regions of the capsid protein. Insertion sites just N-terminal to this junction (E89, Q94, K97, and G101) proved more amenable to the TC tag, with all of the resultant viruses showing growth comparable to that of WT SINV (Table 1). All of the TC-tagged viruses that grew to within 1 log of the WT titer (D113-TC, E89-TC, Q94-TC, K97-TC, and G101-TC) were then tested for their labeling intensity with the biarsenical dye FlAsH or ReAsH. Among these, K97- and Q94-TC showed the brightest signals. Q94-TC was selected for further studies.
Table 1.
Summary of TC motif insertion sites tested in SINV capsid protein
| Insert region on capsid protein and insertion | Virus growthd | Labeling efficiencye |
|---|---|---|
| Capsid N terminus | ||
| (−1)-TCa | WT | Loses the TC tag |
| (−1)-TC-Lb | WT | + |
| Protease domainc | ||
| D113-TC | 1 log lower | ++ |
| E176-TC | 2 logs lower | NA |
| S182-TC | 5 logs lower | NA |
| E186-TC | 6 logs lower | ++++ |
| S199-TC | >7 logs lower | NA |
| E259-TC | >7 logs lower | NA |
| N terminal to protease domainc | ||
| E89-TC | WT | ++ |
| Q94-TC | WT | +++ |
| K97-TC | WT | +++ |
| G101-TC | WT | ++ |
The TC motif plus an N-terminal ATG codon was introduced just before the first amino acid (methionine) of the capsid protein.
The TC motif plus an N-terminal ATG codon and a C-terminal GSSGGSSGGSSG flexible linker region was introduced before the first amino acid of the capsid protein.
The TC motif was introduced directly after the indicated residue.
The growth of all TC insertion mutants was compared to that of WT SINV at 24 h postelectroporation (see Materials and Methods). The log of the WT SINV titer was ∼8.6 as measured by plaque assay on BHK-21 cells. The titer of all mutants designated WT was >8.
The labeling efficiency of virus-infected Vero cells with biarsenical dyes. NA, not applicable; +, barely detectable fluorescence; ++, low fluorescence intensity; +++, strong fluorescence intensity; ++++, very bright fluorescence intensity.
Characterization of Q94-TC virus.
BHK-21 cells were electroporated with WT or Q94-TC viral RNA and the kinetics of progeny virus production determined (Fig. 1A). Efficient production of both viruses was observed by 6 h postelectroporation, and titers reached maximal levels by ∼14 h. Similar results were observed when the titers of progeny viruses were determined using an infectious center assay on Vero cells, the cell line used for our imaging studies (data not shown). Therefore, the growth kinetics of Q94-TC were comparable to those of WT SINV.
Fig 1.
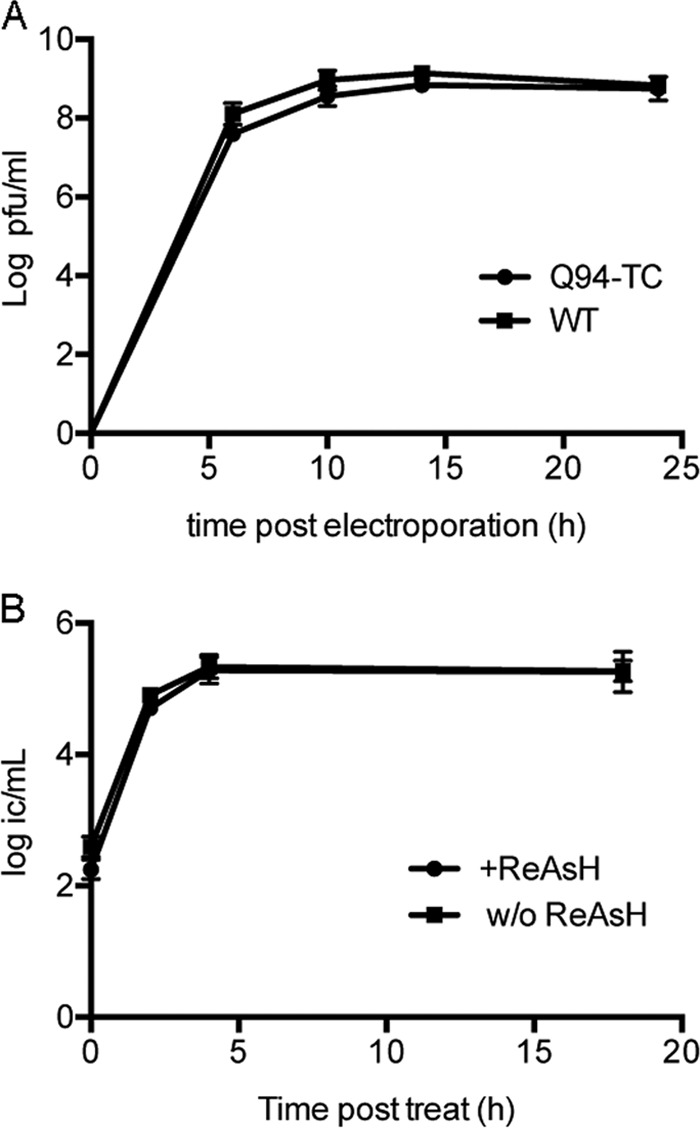
Growth properties of Q94-TC SINV. (A) Growth kinetics of Q94-TC versus WT virus. BHK-21 cells were electroporated with WT or mutant virus RNA and incubated at 37°C for the indicated times. Media were collected, and titers of progeny viruses were determined by plaque assay. (B) The effect of ReAsH labeling on Q94-TC virus production. Vero cells were infected with Q94-TC at a multiplicity of 0.5 IC/cell, cultured for 7 h at 37°C, and mock treated or treated with ReAsH using the conditions described in Materials and Methods. The incubation was continued at 37°C for the indicated times, and progeny virus production was quantitated by infectious center assays on Vero cells. Data are averages from two independent experiments with ranges indicated.
The assembly of virus particles was evaluated by pulse-chase analysis of BHK-21 cells 6 h after RNA electroporation. Cells infected with Q94-TC or WT virus produced comparable amounts of viral proteins (Fig. 2, lysate samples). The E2 precursor pE2 was clearly detected at the 0-h chase time, and maturation comparable to that of E2 was observed in WT- versus Q94-TC-infected cells during a 3-h chase. Analysis of medium samples indicated that the budding efficiencies of Q94-TC and WT SINV were comparable. The capsid protein in both Q94-TC media and lysate samples migrated as a single band that ran slightly above the position of the WT SINV capsid (Fig. 2), consistent with the stable incorporation of the 12-amino-acid TC motif in Q94-TC.
Fig 2.
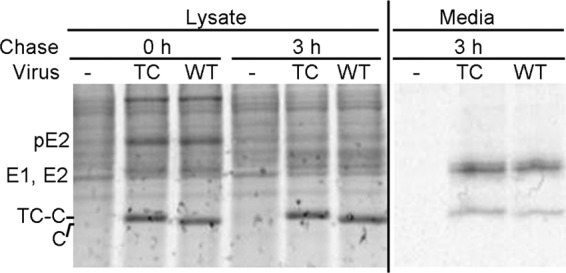
Assembly properties of Q94-TC SINV. BHK-21 cells were electroporated with Q94-TC (TC) or WT virus RNA, incubated at 37°C for 6 h, pulse labeled with [35S]methionine/cysteine, and chased for 0 or 3 h at 37°C. (Left) Cell lysates were collected and an aliquot analyzed directly by SDS-PAGE. The positions of TC-capsid (TC-C) and WT capsid (C) proteins are indicated, with TC-C migrating slightly slower than WT C. The pE2 protein is visible in the 0-h lysate sample, but E1 and E2 are obscured by the host cell proteins. (Right) Samples of the chase media were immunoprecipitated with an antibody against SINV E2 in the absence of detergent to recover intact virus particles and were analyzed by SDS-PAGE. The E1 and E2 proteins migrate as a doublet.
The morphology of Q94-TC virus particles was evaluated by transmission EM of infected BHK cells at 12 h postelectroporation (Fig. 3). Abundant Q94-TC virus particles were observed budding at the plasma membrane. Their morphology was comparable to that of WT virus, with a particle diameter of about 70 nm and a dense NC core in the center (compare Fig. 3A and C). In contrast, the GFP- or mCherry-capsid viruses both produced aberrant particles (Fig. 3E and data not shown). Typical membranous replication structures, termed CPVI (3, 47), were detected within WT- and Q94-TC-infected cells (data not shown). In addition, both WT- and Q94-TC-infected cells contained CPVII with associated NC (Fig. 3B and D). Together, all of the evidence indicated that the introduction of a TC tag at the capsid Q94 position did not cause detectable effects on virus replication or progeny virus production.
Fig 3.

Electron microscopy of WT- and mutant-infected cells. BHK-21 cells were electroporated with Q94-TC, WT SINV, or GFP-dsTE12Q viral RNA, incubated at 37°C for 12 h, and processed for electron microscopy. Panels A, C, and E show representative examples of the morphology of budding virus particles (indicated by arrows). Panels B and D show representative examples of cytopathic vacuole type II (CPVII) in WT (B)- or Q94-TC (D)-infected cells. Note that the CPVII-associated nucleocapsids are larger, denser, and more regular in shape than adjacent ribosomes in the field, differentiating these structures from the endoplasmic reticulum. All images were acquired at a magnification of 20,000×, with the scale bar representing 200 nM.
ReAsH labeling specificity and effects of ReAsH labeling on virus assembly.
We next tested the specificity of ReAsH labeling and its effects on virus production. Vero cells were infected with Q94-TC or WT virus for 7 h and stained with ReAsH as described in Materials and Methods. Vero cells were chosen for imaging, given their efficient infection by SINV and flat morphology. After labeling, the cells were fixed, permeabilized, and stained with a MAb against the capsid protein (Fig. 4). No ReAsH signal was detected in WT SINV-infected cells, while in Q94-TC-infected cells clear intracellular staining was observed. The distribution pattern of the ReAsH staining in Q94-infected cells colocalized with the signal from the MAb against capsid protein. Thus, the Q94-TC motif was specifically labeled by the ReAsH dye with minimal background and accurately reflected the distribution of capsid protein in infected cells. Similar results were obtained with FlAsH dye labeling (data not shown).
Fig 4.
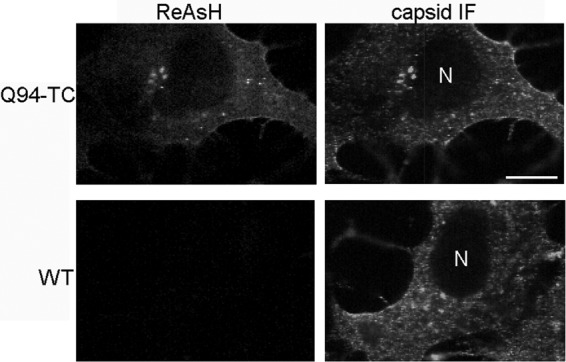
Specificity of biarsenical dye labeling. Vero cells were infected with Q94-TC or WT SINV at a multiplicity of 0.5 IC/cell, cultured for 7 h at 37°C, and labeled with ReAsH as described in Materials and Methods. Cells were then fixed, permeabilized, and stained with antibody against the capsid protein. Images are single internal z sections and are representative of two independent experiments. Bar, 10 μM.
We then measured the titers of Q94-TC progeny viruses collected at different time points following ReAsH labeling or mock treatment. Similar titers were detected in the ReAsH- versus mock-treated samples (Fig. 1B), indicating that the ReAsH treatment and the binding of ReAsH dye to the capsid protein had minimal effects on the production of infectious virus particles.
Distribution of the capsid protein in infected cells.
We then used dye labeling to monitor the distribution of the capsid protein after various times of infection. No significant staining of Q94-TC cells was detectable during the first ∼5 h of infection (data not shown). After 5.5 to 6 h of infection, Q94-TC-infected cells showed relatively weak ReAsH staining that was diffusely distributed in the cytoplasm (data not shown). By 7 to 8 h postinfection, some cells still showed weak ReAsH staining, but the majority of the cells contained a much brighter ReAsH capsid signal. This staining was distributed diffusely in the cytoplasm as before but also showed distinct foci, as discussed in detail below and depicted in Fig. 5. The absence of clear capsid foci at earlier times of infection could reflect their formation at a certain point in the infection cycle or could simply be due to their intensities lying below the detection limit compared to the surrounding cytoplasmic capsid signal.
Fig 5.

Examples of the three types of capsid foci. Vero cells were infected with Q94-TC and labeled with ReAsH at 7 h postinfection. Cells were then fixed, permeabilized, and stained with a MAb against the SINV E2 protein. The images illustrate three groups of capsid foci. (A) Small internal capsid puncta that colocalize with the E2 protein (arrows point to representative examples). (B) Irregular internal capsid structures that did not colocalize with the E2 protein (arrows point to representative examples). (C) PM-proximal capsid puncta that colocalize with E2 protein. The inset shows a zoomed view of the boxed region (3× magnification). All images are single z sections, where panels A and B are internal sections and C is a PM-proximal section. All are representative examples from three experiments. Bar, 10 μM.
We performed a detailed analysis of the intracellular capsid foci by confocal microscopy of Vero cells infected for 7 to 8 h, approximately at the start of exponential virus production (as shown in Fig. 1). Q94-TC-infected cells were labeled with ReAsH at 7 h postinfection, fixed, permeabilized, and stained with MAb R6 against the SINV E2 protein (32). The cytoplasmic capsid foci were separated into three groups based on their size, shape, approximate cellular location, and colocalization with E2. The first category contained capsid foci (termed “small internal capsid puncta”) (Fig. 5A) that colocalized with E2 in internal regions of the cell and were small (0.1 to 0.5 μm3) and round. In live cells, these small internal capsid puncta were highly motile (data not shown). As they contain both capsid and E2 protein, we hypothesize that these puncta are vesicles that transport both capsid and envelope proteins. Detailed studies using a virus labeled in both the capsid and E2 protein are currently in progress to characterize the role of these puncta in virus biogenesis.
A second category contained capsid foci in internal regions of the cell (termed “irregular internal capsid structures”) (Fig. 5B) that were larger (0.6 to 3.0 μm3) and more irregularly shaped than the first group and showed no detectable E2 protein. Most of the cells that were positive for capsid foci contained both the small internal puncta and the irregular internal structures. The frequency of small internal capsid puncta was 5.2 per cell (standard deviations [SD], 5.3; n = 20 cells), which was not significantly different (P > 0.05 by Student's t test) from the frequency of the irregular internal capsid structures (8.0 per cell; SD, 5.0; n = 20 cells). Given their frequencies, these classes of capsid foci were not always in the same confocal z section. The third group of capsid foci (Fig. 5C) was observed as many discrete puncta at a focal plane that appeared close to the PM by confocal microscopy. This class of foci (termed “PM-proximal capsid puncta”) also colocalized with E2 and was detected in all capsid focus-containing cells.
The irregular internal capsid structures colocalize with G3BP and nsP3.
Immunofluorescence analysis demonstrated that, unlike the other two classes of capsid foci, the irregular internal capsid structures did not colocalize with either the E2 or E1 envelope protein (Fig. 5B and data not shown). We therefore evaluated cellular markers that might be characteristic of this group of capsid foci. The structures did not show colocalization with p115, a marker of the Golgi complex, or with LC3, a marker of autophagosomes (data not shown). However, our studies showed that these irregular internal capsid foci were strongly positive for the Ras-GAP SH3 domain binding protein G3BP (Fig. 6A). G3BP, a multifunctional RNA binding protein, has been reported to be a key factor in the nucleation of cellular stress granules (SGs) and is activated by dephosphorylation during SG formation (48). To determine if the irregular internal capsid structures also contained other SG markers, such as eukaryotic translation initiation factor 3 (eIF3), we tested the colocalization of ReAsH-labeled capsid protein and MAb-stained eIF3. Control cells in which SGs were chemically induced by sodium arsenite treatment (49, 50) displayed eIF3-positive puncta (data not shown). In contrast, eIF3 was diffusely distributed in the cytoplasm of SINV-infected cells and did not colocalize with any of the three types of capsid structures (Fig. 6A, lower). Consistent with this result, when Q94-TC- or WT-infected cells were stained with the anti-capsid MAb, the irregular internal capsid structures colocalized with G3BP but not with eIF3 (Fig. 6C and data not shown). Together, our marker analysis suggested that the capsid-G3BP foci are not typical SGs or autophagosomes. We do observe autophagosome induction and LC3-Q94 capsid colocalization at later times of infection (data not shown), in agreement with the studies of Orvedahl et al. (25).
Fig 6.
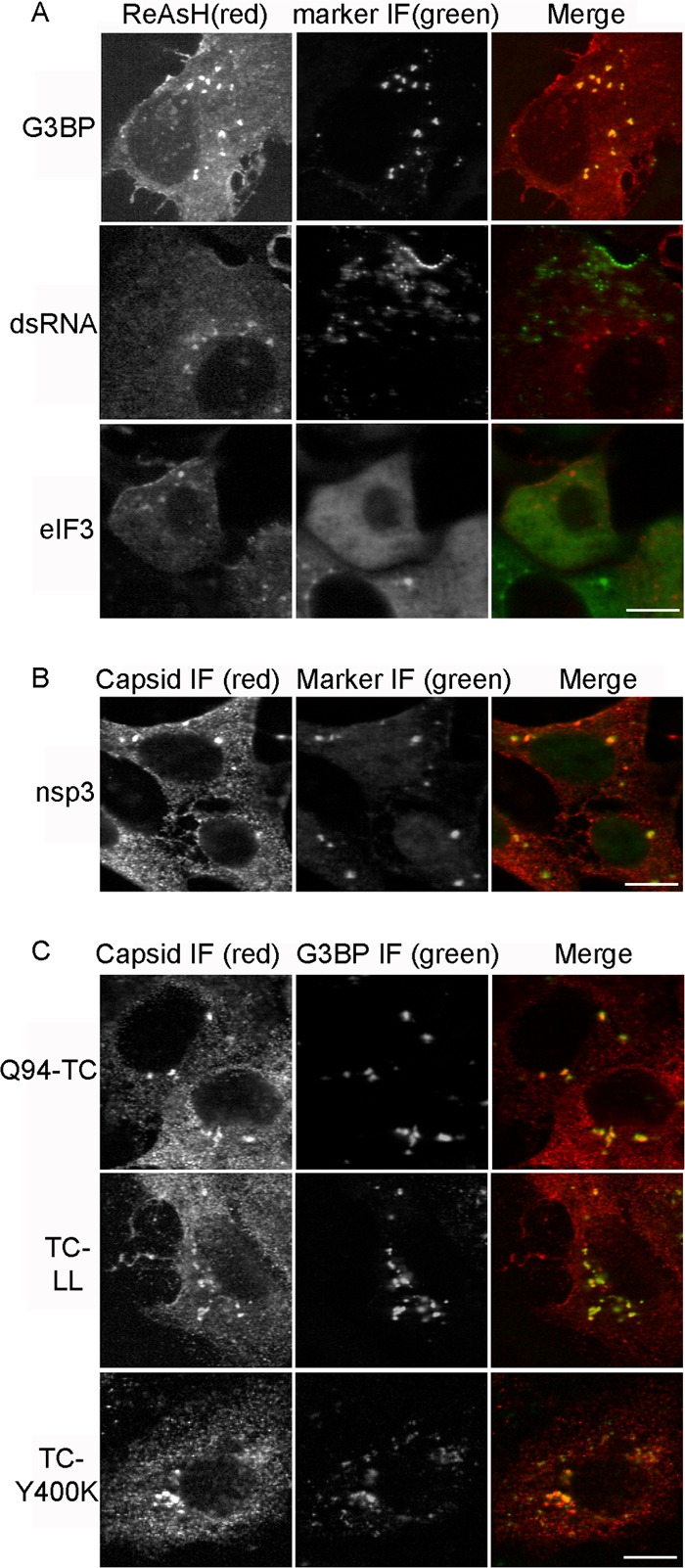
Irregular internal capsid structures colocalize with G3BP and nsP3. (A) Vero cells were infected with Q94-TC and labeled with ReAsH (left) at 7 h postinfection. Cells were then fixed, permeabilized, and stained with antibodies against G3BP, dsRNA, or eIF3 (middle panels). (B) Vero cells were infected with Q94-TC virus, incubated for 7 h, fixed, and costained with MAb recognizing the capsid protein and rabbit antibody against nsP3. (C) Vero cells were infected with either Q94-TC or Q94-TC plus L108A/L110A (indicated as TC-LL). Alternatively, cells were transfected with RNA for the budding-defective mutant Q94-TC plus E2Y400K (indicated as TC-Y400K). Cells were incubated for 7 h, fixed, and costained with MAb to capsid and rabbit antibody against G3BP. Images are representative of two independent experiments. Scale bar, 10 μM.
Previous studies reported that typical SGs are induced at early times (∼2 to 4 h) of Semliki Forest virus (SFV) infection and are disassembled at later times of infection when the cells become positive for viral gene expression (49). Studies with SINV, Chikungunya virus (CHIKV), and SFV showed that the viral protein nsP3 interacts with G3BP, sequesters it into cytoplasmic foci, and inhibits the formation of bona fide SGs (38, 50–52). We therefore tested the colocalization of the irregular internal capsid structures with nsP3 and dsRNA, a marker for the viral replication complex. Approximately 80% of the dsRNA foci in SINV-infected Vero cells were associated with the PM at this infection time, with some small dsRNA puncta in the cytoplasm (Fig. 6A). However, none of the dsRNA foci colocalized with the capsid protein. The irregular internal capsid structures were also negative for nsP1 (data not shown). Together, these results suggest the structures were not associated with the viral replication complex. In contrast, the nsP3 protein was detected in the irregular internal capsid foci (Fig. 6B), suggesting that these structures represent the previously observed nsP3/G3BP foci reported to function in inhibiting formation of bona fide SG (50, 52).
The role of NC assembly and E2 interactions in the generation of capsid protein-G3BP structures was determined by engineering additional mutations into SINV Q94-TC. The capsid substitutions L108A/L110A inhibit the formation of the cytoplasmic NC and CPVII but still allow virus budding at the PM (9, 16). Vero cells infected with TC-LL showed somewhat more irregular internal capsid structures than Q94-TC (33.0 per cell; SD, 8.2; n = 20 cells), but clear colocalization with G3BP was observed (Fig. 6C). Thus, cytoplasmic NC formation is not required for formation of these structures. The substitution Y400K in the cytoplasmic tail of E2 blocks E2-capsid interaction, formation of CPVII, NC localization at the PM, and virus budding (7). However, cells infected with TC-Y400K showed irregular capsid structures that colocalized with G3BP (Fig. 6C), demonstrating that interaction with the E2 protein was not necessary for formation of the capsid/G3BP foci. Vero cells infected with the alphaviruses SFV and CHIKV also express capsid/G3BP foci in the cytoplasm (data not shown), suggesting that formation of these structures is conserved among alphaviruses.
Dynamics of the irregular internal capsid structures.
The TC-tagged virus made it possible to track the movement of the irregular internal capsid structures by ReAsH labeling of Q94-TC-infected cells and live cell imaging. As shown by a representative time series (Fig. 7A), this population of capsid foci was relatively immobile. Similar results were observed in cells infected with the TC-LL or TC-Y400K mutant (data not shown).
Fig 7.
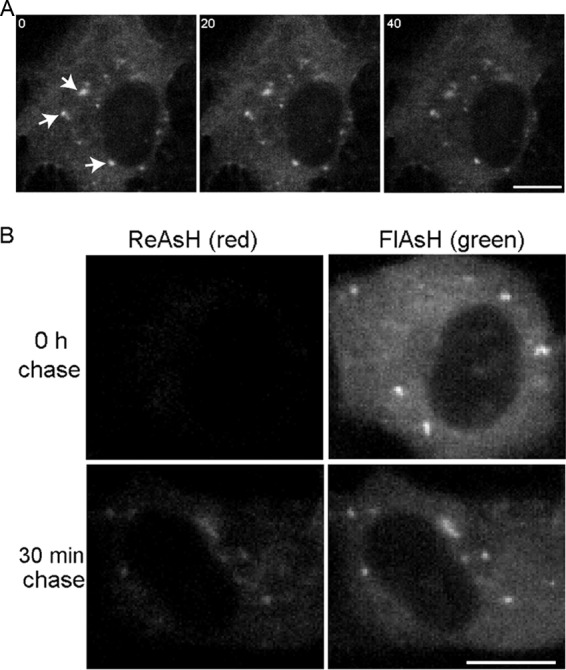
Dynamics of irregular internal capsid structures. (A) Time series of internal capsid structures. Vero cells were infected with Q94-TC and labeled with ReAsH at 7 h postinfection. The positions of specific foci (indicated by arrows) were tracked by images acquired every second. Images collected at 0, 20, and 40 s are shown, documenting the relative immobility of these structures. (B) Recruitment of newly synthesized capsid protein to preexisting irregular internal capsid foci. Vero cells were infected with Q94-TC and labeled with FlAsH at 7 h postinfection (right column). At the indicated chase time, the cells were labeled with ReAsH (left column). Images all were acquired at the same gain and are representative of two independent experiments. Bar, 10 μM.
We then wished to address whether these immobile structures can still actively recruit new capsid proteins. This question could be addressed by testing for an increase of fluorescence intensity with time. However, the ReAsH dye is not very photostable, and the gradual bleaching of fluorescence, even using low laser power and short exposure times, made it complicated to directly quantify changes in fluorescence intensity (data not shown). Alternatively, fluorescence recovery after photobleaching (FRAP) experiments could be used to monitor delivery of labeled capsid protein. This system requires that the high-power laser photobleach is irreversible, allowing fluorescence recovery exclusively from the unbleached capsid pool. In our system, bleaching of the ReAsH dye was reversible and ReAsH signal rapidly reappeared after a complete photobleach of the whole cell (data not shown).
Therefore, we used FlAsH/ReAsH pulse-chase experiments to monitor the newly synthesized capsid protein. Cells were infected for 6.5 h with Q94-TC and pulse labeled with FlAsH to saturate the available TC sites. After incubation in growth media at 37°C for 30 min (chase), the cells were treated with ReAsH to label the newly synthesized capsid protein. No ReAsH labeling was detected in the absence of chase, confirming that the FlAsH labeling is complete (Fig. 7B, upper). In contrast, ReAsH labeling was readily detected in the cells following a 30-min chase (Fig. 7B, lower). In the chase samples, the irregular internal capsid structures were labeled with both FlAsH and ReAsH, suggesting that newly synthesized capsid was being delivered into preexisting capsid/G3BP/nsP3 structures. Thus, although these structures were relatively immobile, they were dynamic in acting as delivery sites for newly synthesized capsid protein.
Characterization of capsid puncta at the plasma membrane.
As shown in Fig. 5C, distinct capsid puncta that colocalized with E2 were detected close to the PM and were particularly clear when focusing at the basolateral region of the cells. To address whether these PM-proximal capsid puncta are actually localized to the PM, we labeled Q94-TC-infected Vero cells with ReAsH and then performed immunostaining on ice to specifically detect the PM-localized pool of E2. Under these conditions, E2 was not detected inside the cells (Fig. 8A), confirming that only the PM pool was visualized. Strong colocalization of capsid ReAsH signal and E2 protein was observed at the surface of infected cells. We will refer to this group of capsid structures as “PM capsid puncta” from this point on in the text. Live cell imaging showed that most of the PM capsid puncta were relatively immobile (Fig. 8B).
Fig 8.
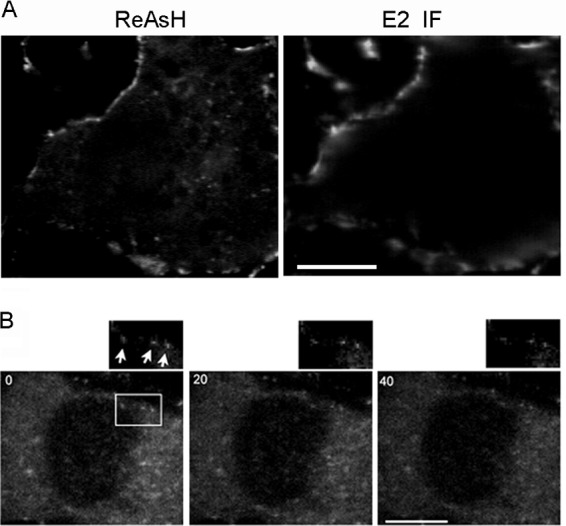
Localization and dynamics of the PM-proximal capsid puncta. Vero cells were infected with Q94-TC and labeled with ReAsH at 7 h postinfection. (A) Localization of the PM-proximal capsid puncta. Following ReAsH labeling, cells were immunolabeled on ice to detect the cell surface E2 protein as described in Materials and Methods. A middle section in the z direction is shown. (B) Time series of PM capsid puncta. The positions of specific puncta (indicated by arrows) were tracked by images acquired every second during a 37°C incubation. Images of a z section at the PM collected at 0, 20, and 40 s are shown, with a zoomed view of the boxed region on the upper-right side, documenting the relative immobility of this puncta type. Images are representative examples from two independent experiments. Bar, 10 μM.
Tests of TC-LL-infected cells (Fig. 9A) showed that the formation of the PM capsid puncta did not require cytoplasmic NCs. In contrast, PM capsid puncta were not detected in TC-Y400K-infected cells, indicating that the E2-capsid interaction was critical (data not shown). Our earlier data showed that at this time of infection in Vero cells, the majority of dsRNA-positive puncta were associated with the PM (Fig. 6A). While we readily detected both dsRNA puncta and PM capsid puncta at the basolateral membrane of infected cells, no colocalization was observed (Fig. 9B). Thus, the capsid puncta at the PM were not associated with the viral RNA replication complex.
Fig 9.

Properties of PM capsid puncta. (A) Generation of PM capsid puncta does not require cytoplasmic NC formation. Vero cells infected with TC-LL for 7 h were labeled with ReAsH and then fixed, permeabilized, and stained with E2 MAb R6. (B) PM capsid puncta do not colocalize with dsRNA. Vero cells were infected with Q94-TC for 7 h, labeled with ReAsH, and then fixed, permeabilized, and stained with MAb against dsRNA. (C) Newly synthesized capsid protein was delivered to preexisting PM capsid puncta. Vero cells were infected with Q94-TC for 7 h and stained with FlAsH to label the existing capsid protein pool (second column). At the indicated chase time, the cells were labeled with ReAsH (first column). After 1 h of chase, ReAsH-labeled capsid protein was detected in PM capsid puncta containing the FlAsH signal. The inset is a zoomed view of the boxed region (2.5× magnification). Images are representative of two independent experiments. Bar, 10 μM.
We then used FlAsH/ReAsH pulse-chase experiments as described above to address whether the PM capsid puncta can actively recruit newly synthesized capsid protein. As early as 1 h after chase, newly synthesized capsid proteins were detected in PM puncta that were also labeled with the FlAsH signal (Fig. 9C). Based on their enrichment for viral structural proteins and recruitment of newly synthesized capsid protein, our results suggest that the PM capsid puncta are sites of virus assembly and budding.
DISCUSSION
In summary, we developed a TC-based system to image the movements of the SINV capsid protein in live infected cells without affecting its biological activity. At early times of infection, labeling of the TC-capsid produced diffuse cytoplasmic staining that may include ribosome-bound capsid proteins. By the beginning of exponential virus production, three distinct types of intracellular capsid foci were detected. Based on properties such as motility and colocalization with viral and cellular proteins, we hypothesize that these foci are involved in capsid protein delivery to the PM, in the regulation of cellular SGs, and in virus assembly/exit from the PM.
Fluorescent labeling of the alphavirus capsid protein.
The alphavirus NC has a diameter of ∼400 Å and contains 240 copies of the ∼30-kDa capsid protein arranged in a T=4 icosahedral lattice (53, 54). Given this organized NC structure, introducing the ∼27-kDa, 42-Å-long β-barrel structure of GFP or mCherry (55) into the complete capsid protein is spatially challenging. Insertion of GFP or mCherry at the N terminus of the capsid protein did produce infectious virus, but its growth was reduced by ∼2 logs, and both constructs produced aberrant particle morphology and capsid protein distribution. Aberrant budding of HIV GFP-Gag is rescued by coexpression of WT-Gag (56, 57). We tested an analogous complementation strategy for rescue of the budding defects in SINV GFP-capsid. We coexpressed the GFP- or mCherry-SINV infectious clone with the WT capsid protein, reasoning that the incorporation of WT capsid might reduce the spatial hindrance in NC formation. However, even though WT capsid protein was well expressed and displayed a normal cellular distribution pattern, it was not efficiently recruited into the GFP- or mCherry-labeled virus (data not shown). The highly symmetric structure of the alphavirus NC and/or the possible importance of cis expression of capsid and envelope proteins may limit such complementation approaches. We conclude that the GFP- or mCherry-capsid virus does not accurately reflect the complete WT capsid pathway and is not optimal for imaging studies of capsid dynamics. However, such GFP- or mCherry-capsid viruses have clearly been very useful tools to identify specific alphavirus-host protein interactions that confirm using WT SINV (25, 26).
We chose the small TC motif as an alternative strategy for live cell imaging of the alphavirus capsid protein. Virus particles in the medium of ReAsH-labeled WT- or Q94-TC-infected cells could be captured on poly-l-lysine-coated culture slides (data not shown). Both the WT and Q94-TC produced particles that labeled with mixtures of MAbs against the E1 and E2 proteins. ReAsH-labeled particles were detected only in the Q94-TC sample, and the number of particles was dependent on the multiplicity of infection. This result indicates that the Q94-TC virus supports imaging of both intracellular and particle-assembled capsid protein. Q94-TC virus particles did not show labeling when stained with ReAsH after adsorption to coverslips (data not shown), presumably reflecting inhibition of free diffusion of the biarsenical dye into the Q94-TC site in assembled virus.
Capsid protein traffic and NC production.
While the alphavirus envelope proteins use the endogenous cellular secretory pathway for delivery to the PM, the traffic of the capsid protein is not clear. We observed a diffuse distribution of capsid in the cytoplasm that could provide a local source during virus budding. Our labeling studies also identified small, highly motile capsid puncta, which we hypothesize are capsid transport vehicles. Immunofluorescence experiments showed that this class of capsid foci colocalized with the E2 protein, suggesting their vesicular nature. Further characterization is necessary to determine if these puncta deliver the capsid/NC to the PM and what mediates their rapid movement. We are currently analyzing their properties using a virus containing both the TC-94 capsid and a GFP-E2 protein, which permits live cell imaging of both proteins. If the capsid/NC does travel with the E2 protein, it will be interesting to explore the cellular location where this E2-capsid interaction initiates and the specific mechanisms of transport.
Labeling of Q94-TC virus-infected cells also identified a group of capsid puncta at the PM. Based on the coenrichment of the viral envelope proteins at these sites and the absence of such PM capsid puncta in the budding-defective Y400K mutant (data not shown), we propose that they represent assembly/budding sites for alphavirus particles. Further experiments will address whether the capsid protein is delivered to these sites as preassembled NCs or as capsid-RNA complexes. FlAsH-based superresolution microscopy (58) and/or correlative electron microscopy of ReAsH-labeled capsid (27, 59) may provide the spatial resolution to differentiate between these two states of the capsid protein.
G3BP/nsP3-positive capsid structures.
Labeling of Q94-TC virus-infected cells revealed a population of large internal capsid structures that colocalize with G3BP and nsP3. Immunofluorescence analysis of WT-infected cells demonstrated similar capsid foci that were positive for G3BP and nsP3. To our knowledge, while the G3BP/nsP3 foci have been quite extensively studied, this is the first report that these foci can contain the alphavirus capsid protein. Further colocalization studies showed that this group of intracellular capsid foci was negative for viral envelope proteins, nsP1, dsRNA, and eIF3.
dsRNA is a hallmark of plus-strand RNA virus replication complexes (60) and can be detected by staining with the J2 MAb. In SFV-infected BHK cells, replication complexes are first localized on regions of the PM (1 to 3 h postinfection), then in small and scattered cytoplasmic vesicles, and finally (∼8 h postinfection) in large perinuclear CPVI vacuolar structures (52, 61). We observed cell line differences in this immunofluorescence pattern, detecting large perinuclear dsRNA structures at 8 h postinfection in SFV-infected BHK cells but not in SFV-infected Vero cells (data not shown). Similarly, at ∼6 to 8 h postinfection, SINV-infected Vero cells showed MAb J2 staining on specific regions of the PM or on small vesicles scattered in the cytoplasm but no large perinuclear dsRNA structures then or at later times of infection. The dsRNA structures in WT- or Q94-TC-infected Vero cells did not colocalize with capsid MAb or capsid ReAsH staining (data not shown). Thus, it appears that the internal capsid/G3BP foci are not viral replication complexes.
As part of the host defense against virus infection, cells can trigger the formation of canonical SGs that sequester translation factors and inhibit protein synthesis (reviewed in references 62–64). Some viruses have evolved strategies to inhibit or reverse the formation of cellular SGs at later times of infection. For example, the poliovirus 3C protease cleaves G3BP (65), while West Nile virus sequesters the SG protein TIAR (66). Alphaviruses such as SFV and CHIKV have been reported to reduce the SG response late in infection by sequestering the G3BP protein into cytoplasmic foci (49, 50, 52). These G3BP foci do not contain other SG markers, such as eIF3 or TIAR; thus, they are not bona fide cellular SGs (50, 52). G3BP sequestration into these cytoplasmic structures is mediated by the alphavirus nsP3 protein, which contains a short sequence at its C terminus that binds G3BP (50, 52, 67).
At late times of infection a number of RNA viruses, including hepatitis C virus, SFV, and dengue virus, are observed to induce the dynamic assembly and disassembly of cytoplasmic SGs over a time span of hours (68). Such oscillations allow cells to cycle between translational activity and arrest, potentially promoting cell survival and chronic infection (68). Alphaviruses inhibit the formation of bona fide SGs at about 6 to 8 h postinfection, and the G3BP/nsP3-mediated SG inhibition might reflect the disassembly phase of SG oscillation. As the time-lapse experiments we performed to track the movement of the GBP3/capsid foci were relatively short, it is not clear if there are oscillations in these capsid structures that influence the oscillations in cellular SGs. The overall process of SG disassembly has been shown to occur in cells infected with recombinant alphaviruses lacking the capsid protein (50, 52).
Although G3BP depletion by short interfering RNA produces a small enhancement of SINV virus production (69), the effect is relatively modest. This might be explained by the important roles G3BP plays in mediating both initial SG assembly and subsequent SG disassembly. In contrast, an SINV nsP3 mutant that fails to form nsP3/G3BP cytoplasmic foci or to dissociate SG displays a relatively strong (2 log) decrease in virus titer (67). In this case, the robust inhibition of virus growth could be due to initial SG formation in the absence of subsequent SG disassembly. We speculate that the capsid protein in the nsP3/G3BP foci plays a role in the complex biology controlling viral replication. Clearly, further studies will be needed to determine the mechanism of capsid protein recruitment to G3BP/nsP3 foci, the potential interactions of capsid protein with the components of these foci, and the role(s) of capsid in SG regulation and oscillation.
ACKNOWLEDGMENTS
We thank all members of our laboratory for their helpful discussions and experimental suggestions and Mathieu Dube, Guadalupe Martinez, and Claudia Sánchez-San Martín for critical readings of the manuscript. We thank Erik Snapp and Guadalupe Martinez for many helpful discussions and input on imaging, Youqing Xiang for her excellent technical assistance, and the staff of the Einstein Analytical Imaging Facility for their support.
The data in this paper are from a thesis submitted by Y.Z. in partial fulfillment of the requirements for the Degree of Doctor of Philosophy in the Graduate Division of Medical Sciences, Albert Einstein College of Medicine, Yeshiva University.
This work was supported by a grant to M.K. from the National Institute of General Medicine (R01-GM057454) and by Cancer Center Core Support Grant NIH/NCI P30-CA13330.
The content of this work is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of General Medicine or the National Institutes of Health.
Footnotes
Published ahead of print 19 June 2013
REFERENCES
- 1.Garoff H, Hewson R, Opstelten D-JE. 1998. Virus maturation by budding. Microbiol. Mol. Biol. Rev. 62:1171–1190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weissenhorn W, Poudevigne E, Effantin G, Bassereau P. 2013. How to get out: ssRNA enveloped viruses and membrane fission. Curr. Opin. Virol. 3:159–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kuhn RJ. 2007. Togaviridae: the viruses and their replication, p 1001–1022 In Knipe DM, Howley PM. (ed), Fields virology, Fifth ed, vol 1 Lippincott, Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 4.Jose J, Snyder JE, Kuhn RJ. 2009. A structural and functional perspective of alphavirus replication and assembly. Future Microbiol. 4:837–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vaney MC, Duquerroy S, Rey FA. 2013. Alphavirus structure: activation for entry at the target cell surface. Curr. Opin. Virol. 3:151–158 [DOI] [PubMed] [Google Scholar]
- 6.Suomalainen M, Liljeström P, Garoff H. 1992. Spike protein-nucleocapsid interactions drive the budding of alphaviruses. J. Virol. 66:4737–4747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhao H, Lindqvist B, Garoff H, von Bonsdorff C-H, Liljeström P. 1994. A tyrosine-based motif in the cytoplasmic domain of the alphavirus envelope protein is essential for budding. EMBO J. 13:4204–4211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skoging U, Vihinen M, Nilsson L, Liljeström P. 1996. Aromatic interactions define the binding of the alphavirus spike to its nucleocapsid. Structure 4:519–529 [DOI] [PubMed] [Google Scholar]
- 9.Lee S, Owen KE, Choi H-K, Lee H, Lu G, Wengler G, Brown DT, Rossmann MG, Kuhn RJ. 1996. Identification of a protein binding site on the surface of the alphavirus nucleocapsid and its implication in virus assembly. Structure 4:531–541 [DOI] [PubMed] [Google Scholar]
- 10.Strauss JH, Strauss EG. 1994. The alphaviruses: gene expression, replication, and evolution. Microbiol. Rev. 58:491–562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Garoff H, Sjoberg M, Cheng RH. 2004. Budding of alphaviruses. Virus Res. 106:103–116 [DOI] [PubMed] [Google Scholar]
- 12.Acheson NH, Tamm I. 1967. Replication of Semliki Forest virus: an electron microscopic study. Virology 32:128–143 [DOI] [PubMed] [Google Scholar]
- 13.Tellinghuisen TL, Hamburger AE, Fisher BR, Ostendorp R, Kuhn RJ. 1999. In vitro assembly of alphavirus cores by using nucleocapsid protein expressed in Escherichia coli. J. Virol. 73:5309–5319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Snyder JE, Azizgolshani O, Wu B, He Y, Lee AC, Jose J, Suter DM, Knobler CM, Gelbart WM, Kuhn RJ. 2011. Rescue of infectious particles from preassembled alphavirus nucleocapsid cores. J. Virol. 85:5773–5781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheng F, Mukhopadhyay S. 2011. Generating enveloped virus-like particles with in vitro assembled cores. Virology 413:153–160 [DOI] [PubMed] [Google Scholar]
- 16.Skoging-Nyberg U, Liljestrom P. 2001. M-X-I motif of semliki forest virus capsid protein affects nucleocapsid assembly. J. Virol. 75:4625–4632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Forsell K, Griffiths G, Garoff H. 1996. Preformed cytoplasmic nucleocapsids are not necessary for alphavirus budding. EMBO J. 15:6495–6505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Forsell K, Xing L, Kozlovska T, Cheng RH, Garoff H. 2000. Membrane proteins organize a symmetrical virus. EMBO J. 19:5081–5091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sodeik B. 2000. Mechanisms of viral transport in the cytoplasm. Trends Microbiol. 8:465–472 [DOI] [PubMed] [Google Scholar]
- 20.Scheele CM, Pfefferkorn ER. 1969. Kinetics of incorporation of structural proteins into Sindbis virions. J. Virol. 3:369–375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Soderlund H, Ulmanen I. 1977. Transient association of Semliki Forest virus capsid protein with ribosomes. J. Virol. 24:907–909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ulmanen I, Soderlund H, Kaariainen L. 1976. Semliki Forest virus capsid protein associates with the 60S ribosomal subunit in infected cells. J. Virol. 20:203–210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ulmanen I, Soderlund H, Kaariainen L. 1979. Role of protein synthesis in the assembly of Semliki forest virus nucleocapsid. Virology 99:265–276 [DOI] [PubMed] [Google Scholar]
- 24.Wengler G, Gros C. 1996. Analyses of the role of structural changes in the regulation of uncoating and assembly of alphavirus cores. Virology 222:123–132 [DOI] [PubMed] [Google Scholar]
- 25.Orvedahl A, MacPherson S, Sumpter R, Jr, Talloczy Z, Zou Z, Levine B. 2010. Autophagy protects against Sindbis virus infection of the central nervous system. Cell Host Microbe 7:115–127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Orvedahl A, Sumpter R, Jr, Xiao G, Ng A, Zou Z, Tang Y, Narimatsu M, Gilpin C, Sun Q, Roth M, Forst CV, Wrana JL, Zhang YE, Luby-Phelps K, Xavier RJ, Xie Y, Levine B. 2011. Image-based genome-wide siRNA screen identifies selective autophagy factors. Nature 480:113–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Adams SR, Campbell RE, Gross LA, Martin BR, Walkup GK, Yao Y, Llopis J, Tsien RY. 2002. New biarsenical ligands and tetracysteine motifs for protein labeling in vitro and in vivo: synthesis and biological applications. J. Am. Chem. Soc. 124:6063–6076 [DOI] [PubMed] [Google Scholar]
- 28.Hardwick JM, Levine B. 2000. Sindbis virus vector system for functional analysis of apoptosis regulators. Methods Enzymol. 322:492–508 [DOI] [PubMed] [Google Scholar]
- 29.Rice CM, Levis R, Strauss JH, Huang HV. 1987. Production of infectious RNA transcripts from Sindbis virus cDNA clones: Mapping of lethal mutations, rescue of a temperature-sensitive marker, and in vitro mutagenesis to generate defined mutants. J. Virol. 61:3809-3838l9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pedelacq JD, Cabantous S, Tran T, Terwilliger TC, Waldo GS. 2006. Engineering and characterization of a superfolder green fluorescent protein. Nat. Biotechnol. 24:79–88 [DOI] [PubMed] [Google Scholar]
- 31.Liljeström P, Lusa S, Huylebroeck D, Garoff H. 1991. In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release. J. Virol. 65:4107–4113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Meyer WJ, Johnston RE. 1993. Structural rearrangement of infecting Sindbis virions at the cell surface: Mapping of newly accessible epitopes. J. Virol. 67:5117–5125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chanel-Vos C, Kielian M. 2004. A conserved histidine in the ij loop of the Semliki Forest virus E1 protein plays an important role in membrane fusion. J. Virol. 78:13543–13552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Counihan NA, Rawlinson SM, Lindenbach BD. 2011. Trafficking of hepatitis C virus core protein during virus particle assembly. PLoS Pathog. 7:e1002302. 10.1371/journal.ppat.1002302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gousset K, Ablan SD, Coren LV, Ono A, Soheilian F, Nagashima K, Ott DE, Freed EO. 2008. Real-time visualization of HIV-1 GAG trafficking in infected macrophages. PLoS Pathog. 4:e1000015. 10.1371/journal.ppat.1000015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Snijder B, Sacher R, Ramo P, Damm EM, Liberali P, Pelkmans L. 2009. Population context determines cell-to-cell variability in endocytosis and virus infection. Nature 461:520–523 [DOI] [PubMed] [Google Scholar]
- 37.Greiser-Wilke I, Moennig V, Kaaden O-R, Figueiredo LTM. 1989. Most alphaviruses share a conserved epitopic region on their nucleocapsid protein. J. Gen. Virol. 70:743–748 [DOI] [PubMed] [Google Scholar]
- 38.Cristea IM, Carroll JW, Rout MP, Rice CM, Chait BT, MacDonald MR. 2006. Tracking and elucidating alphavirus-host protein interactions. J. Biol. Chem. 281:30269–30278 [DOI] [PubMed] [Google Scholar]
- 39.Griffin BA, Adams SR, Tsien RY. 1998. Specific covalent labeling of recombinant protein molecules inside live cells. Science 281:269–272 [DOI] [PubMed] [Google Scholar]
- 40.Rudner L, Nydegger S, Coren LV, Nagashima K, Thali M, Ott DE. 2005. Dynamic fluorescent imaging of human immunodeficiency virus type 1 gag in live cells by biarsenical labeling. J. Virol. 79:4055–4065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Coller KE, Heaton NS, Berger KL, Cooper JD, Saunders JL, Randall G. 2012. Molecular determinants and dynamics of hepatitis C virus secretion. PLoS Pathog. 8:e1002466. 10.1371/journal.ppat.1002466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Das SC, Panda D, Nayak D, Pattnaik AK. 2009. Biarsenical labeling of vesicular stomatitis virus encoding tetracysteine-tagged m protein allows dynamic imaging of m protein and virus uncoating in infected cells. J. Virol. 83:2611–2622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Coombs KM, Brown DT. 1989. Form-determining functions in Sindbis virus nucleocapsids: nucleo somelike organization of the nucleocapsid. J. Virol. 63:883–891 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mukhopadhyay S, Zhang W, Gabler S, Chipman PR, Strauss EG, Strauss JH, Baker TS, Kuhn RJ, Rossmann MG. 2006. Mapping the structure and function of the E1 and E2 glycoproteins in alphaviruses. Structure 14:63–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beitzel BF, Bakken RR, Smith JM, Schmaljohn CS. 2010. High-resolution functional mapping of the venezuelan equine encephalitis virus genome by insertional mutagenesis and massively parallel sequencing. PLoS Pathog. 6:e1001146. 10.1371/journal.ppat.1001146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Choi H-K, Tong L, Minor W, Dumas P, Boege U, Rossman MG, Wengler G. 1991. Structure of Sindbis virus core protein reveals a chymotrypsin-like serine proteinase and the organization of the virion. Nature 354:37–43 [DOI] [PubMed] [Google Scholar]
- 47.Salonen A, Ahola T, Kaariainen L. 2005. Viral RNA replication in association with cellular membranes. Curr. Topics Microbiol. Immunol. 285:139–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tourriere H, Chebli K, Zekri L, Courselaud B, Blanchard JM, Bertrand E, Tazi J. 2003. The RasGAP-associated endoribonuclease G3BP assembles stress granules. J. Cell Biol. 160:823–831 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 49.McInerney GM, Kedersha NL, Kaufman RJ, Anderson P, Liljestrom P. 2005. Importance of eIF2alpha phosphorylation and stress granule assembly in alphavirus translation regulation. Mol. Biol. Cell 16:3753–3763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fros JJ, Domeradzka NE, Baggen J, Geertsema C, Flipse J, Vlak JM, Pijlman GP. 2012. Chikungunya Virus nsP3 Blocks Stress Granule Assembly by Recruitment of G3BP into Cytoplasmic Foci. J. Virol. 86:10873–10879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frolova E, Gorchakov R, Garmashova N, Atasheva S, Vergara LA, Frolov I. 2006. Formation of nsP3-specific protein complexes during Sindbis virus replication. J. Virol. 80:4122–4134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Panas MD, Varjak M, Lulla A, Er Eng K, Merits A, Karlsson Hedestam GB, McInerney GM. 2012. Sequestration of G3BP coupled with efficient translation inhibits stress granules in Semliki Forest virus infection. Mol. Biol. Cell 23:4701–4712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cheng RH, Kuhn RJ, Olson NH, Rossman MG, Choi H-K, Smith TJ, Baker TS. 1995. Nucleocapsid and glycoprotein organization in an enveloped virus. Cell 80:621–630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Paredes AM, Brown DT, Rothnagel R, Chiu W, Schoepp RJ, Johnston RE, Prasad BVV. 1993. Three-dimensional structure of a membrane-containing virus. Proc. Natl. Acad. Sci. U. S. A. 90:9095–9099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ormo M, Cubitt AB, Kallio K, Gross LA, Tsien RY, Remington SJ. 1996. Crystal structure of the Aequorea victoria green fluorescent protein. Science 273:1392–1395 [DOI] [PubMed] [Google Scholar]
- 56.Larson DR, Johnson MC, Webb WW, Vogt VM. 2005. Visualization of retrovirus budding with correlated light and electron microscopy. Proc. Natl. Acad. Sci. U. S. A. 102:15453–15458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Jouvenet N, Bieniasz PD, Simon SM. 2008. Imaging the biogenesis of individual HIV-1 virions in live cells. Nature 454:236–240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lelek M, Di Nunzio F, Henriques R, Charneau P, Arhel N, Zimmer C. 2012. Superresolution imaging of HIV in infected cells with FlAsH-PALM. Proc. Natl. Acad. Sci. U. S. A. 109:8564–8569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gaietta G, Deerinck TJ, Adams SR, Bouwer J, Tour O, Laird DW, Sosinsky GE, Tsien RY, Ellisman MH. 2002. Multicolor and electron microscopic imaging of connexin trafficking. Science 296:503–507 [DOI] [PubMed] [Google Scholar]
- 60.Weber F, Wagner V, Rasmussen SB, Hartmann R, Paludan SR. 2006. Double-stranded RNA is produced by positive-strand RNA viruses and DNA viruses but not in detectable amounts by negative-strand RNA viruses. J. Virol. 80:5059–5064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Spuul P, Balistreri G, Hellstrom K, Golubtsov AV, Jokitalo E, Ahola T. 2011. Assembly of alphavirus replication complexes from RNA and protein components in a novel trans-replication system in mammalian cells. J. Virol. 85:4739–4751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Reineke LC, Lloyd RE. 2013. Diversion of stress granules and P-bodies during viral infection. Virology 436:255–267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Valiente-Echeverria F, Melnychuk L, Mouland AJ. 2012. Viral modulation of stress granules. Virus Res. 169:430–437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Beckham CJ, Parker R. 2008. P bodies, stress granules, and viral life cycles. Cell Host Microbe 3:206–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.White JP, Cardenas AM, Marissen WE, Lloyd RE. 2007. Inhibition of cytoplasmic mRNA stress granule formation by a viral proteinase. Cell Host Microbe 2:295–305 [DOI] [PubMed] [Google Scholar]
- 66.Emara MM, Brinton MA. 2007. Interaction of TIA-1/TIAR with West Nile and dengue virus products in infected cells interferes with stress granule formation and processing body assembly. Proc. Natl. Acad. Sci. U. S. A. 104:9041–9046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Foy NJ, Akhrymuk M, Akhrymuk I, Atasheva S, Bopda-Waffo A, Frolov I, Frolova EI. 2013. Hypervariable domains of nsP3 proteins of New World and Old World alphaviruses mediate formation of distinct, virus-specific protein complexes. J. Virol. 87:1997–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ruggieri A, Dazert E, Metz P, Hofmann S, Bergeest JP, Mazur J, Bankhead P, Hiet MS, Kallis S, Alvisi G, Samuel CE, Lohmann V, Kaderali L, Rohr K, Frese M, Stoecklin G, Bartenschlager R. 2012. Dynamic oscillation of translation and stress granule formation mark the cellular response to virus infection. Cell Host Microbe 12:71–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Cristea IM, Rozjabek H, Molloy KR, Karki S, White LL, Rice CM, Rout MP, Chait BT, MacDonald MR. 2010. Host factors associated with the Sindbis virus RNA-dependent RNA polymerase: role for G3BP1 and G3BP2 in virus replication. J. Virol. 84:6720–6732 [DOI] [PMC free article] [PubMed] [Google Scholar]