

Abstract
Some cell types are more susceptible to viral gene transfer or virus infection than others, irrespective of the number of viral receptors or virus binding efficacy on their surfaces. In order to characterize the cell-line-specific features contributing to efficient virus entry, we studied two cell lines (Ea.hy926 and MG-63) that are nearly nonpermissive to insect-specific baculovirus (BV) and the human enterovirus echovirus 1 (EV1) and compared their characteristics with those of a highly permissive (HepG2) cell line. All the cell lines contained high levels of viral receptors on their surfaces, and virus binding was shown to be efficient. However, in nonpermissive cells, BV and its receptor, syndecan 1, were unable to internalize in the cells and formed large aggregates near the cell surface. Accordingly, EV1 had a low infection rate in nonpermissive cells but was still able to internalize the cells, suggesting that the postinternalization step of the virus was impaired. The nonpermissive and permissive cell lines showed differential expression of syntenin, filamentous actin, vimentin, and phosphorylated protein kinase C subtype α (pPKCα). The nonpermissive nature of the cells could be modulated by the choice of culture medium. RPMI medium could partially rescue infection/transduction and concomitantly showed lower syntenin expression, a modified vimentin network, and altered activities of PKC subtypes PKCα and PKCε. The observed changes in PKCα and PKCε activation caused alterations in the vimentin organization, leading to efficient BV transduction and EV1 infection. This study identifies PKCα, PKCε, and vimentin as key factors affecting efficient infection and transduction by EV1 and BV, respectively.
INTRODUCTION
Understanding mechanisms that regulate the cell entry of viruses, leading to efficient internalization, is equally important with pathogenic viruses and in the field of viral gene therapy. In order to understand the cellular mechanisms behind the cells' permissiveness to viruses, we studied the infection and transduction pathways of two viruses from distinct families, namely, an insect pathogen, baculovirus (BV), and a small human pathogen, echovirus 1 (EV1).
BV is a large, enveloped DNA virus that is nonpathogenic to humans and is considered a promising candidate for gene delivery applications (1–3). BV offers several advantages as a gene delivery vector compared to other viral vectors. They include high transgene capacity, easy production, and the nonreplicative nature of the virus. However, the development of baculovirus-based biomedical applications is hampered by a lack of knowledge about BV trafficking in human cells and a poor understanding of cellular factors affecting efficient gene transfer. Even though BV is able to internalize in and transduce several mammalian cell lines, the transduction efficiency varies among the cell types (4–8). We previously described BV capsid display as a novel tool for gene therapy that can be used to detect transduction efficiency (6). However, we also found cell lines, e.g., EA.hy926 and MG-63 cells, that were not able to efficiently express the targeted transgenes.
The factors affecting host cell permissiveness to BV transduction are still largely unknown. Cell lines such as HepG2 are commonly regarded as highly permissive (9, 10), whereas MG-63 and Ea.hy926 have been reported to be transduction-restricted cells (6, 11). In addition to the cell type, we showed previously that the cell culture medium affects the cells' permissiveness to viruses and may be used to enhance transgene delivery of BVs, adeno-associated viruses, adenoviruses, and lentiviruses (12).
EV1 is a small, nonenveloped RNA virus from the family Picornaviridae and genus Enterovirus. Enteroviruses form a large group of viruses containing known human pathogens, such as rhinoviruses, coxsackieviruses, and polioviruses (13). They cause a variety of illnesses, some of which may be severe, such as poliomyelitis, encephalitis, and hepatitis (14, 15). Although picornaviruses have a huge impact on human and animal health, the factors affecting their pathogenicity are not well understood (16). EV1 infects human cells from different origins, where its receptor, α2β1-integrin, is expressed. α2β1-Integrin is one of the major collagen binding integrins, and it is abundantly expressed in several cell types from endothelial and epithelial origins (17). We have previously shown that efficient EV1 internalization is dependent on Rac1, Pak1, PLC, and protein kinase C subtype α (PKCα) activation (18, 19).
We have recently reported that syndecan 1 acts as a receptor for BV in mammalian cells (K. E. Makkonen, P. Turkki, J. P. Laakkonen, S. Ylä-Herttuala, V. Marjomäki, and K. J. Airenne, submitted for publication). In addition to binding, our results indicate that syndecan 1 is also involved in the internalization of the virus (Makkonen et al., submitted). Syndecans are major cell membrane heparan sulfate proteoglycans (20, 21). Rather than being specific receptors for only one or two different ligands, syndecans can bind various ligands and multiple ligands at a time via their heparin sulfate side chains. Not much is known about syndecan internalization and trafficking. Syndecans internalize from membrane rafts by a clathrin- and caveolin-independent route, and the internalization is actin dependent. Clustering of syndecan 1 has been shown to induce interaction between its cytoplasmic tail and actin microfilaments (22), leading to actin reorganization (22, 23). EV1, instead, binds to its receptor, α2β1-integrin, which causes clustering and lateral movement of α2β1-integrin. This clustering is essential in the internalization of the virus and integrin complex (18). Syndecan 1 and α2β1-integrins are both cell surface receptors that are widely expressed in various cell types (17, 24, 25). Syndecans and integrins are transmembrane receptors that are capable of regulating both extracellular and intracellular activities. They play roles in various important processes in development and inflammation and in different diseases, such as cancer. Syndecans and integrins also act as receptors for various viruses (25–27).
In this study, we characterized nonpermissive and permissive cell lines for virus entry and identified the factors affecting viral infection and gene delivery. We show that in the nonpermissive cells, despite optimal levels of the respective viral receptors and virus binding on the cell surface, virus transduction and infection are halted. Additionally, our data demonstrate that PKC subtypes alpha and epsilon and intermediate filament vimentin dynamics are key regulators leading to successful virus internalization and cellular trafficking.
MATERIALS AND METHODS
Cells.
Human hepatocarcinoma (HepG2) cells, human umbilical vein endothelial hybridoma cells, A549 human lung carcinoma epithelial (Ea.hy926) cells, human embryonic kidney (293T) cells, human osteosarcoma (MG-63) cells, and mouse macrophage-like (RAW264) cells were grown in monolayers at 37°C in 5% CO2 in Dulbecco's modified Eagle's medium (DMEM) or RPMI 1640 medium supplemented with 10% inactivated fetal calf serum (FCS), l-glutamine, and penicillin-streptomycin (Gibco BRL, Paisley, United Kingdom). Between cell divisions, cells were kept in DMEM. During experiments, cells were kept in DMEM unless otherwise indicated.
Antibodies.
Antibodies directed against BV were as follows: monoclonal antibody against glycoprotein 64 (gp64) (B12D5), monoclonal antibody against capsid protein vp39 (p10C6), or polyclonal antibody against BV (L. Volkman, University of California, Berkeley, CA). For syndecan 1 detection, monoclonal (B-A38 and Ab27390; Abcam) and polyclonal (sc-5632; Santa Cruz) antibodies were used. For EV1 labeling, rabbit antisera for purified EV1 (28) was used. For the detection of α2β1-integrin, monoclonal antibody against α2-integrin (A2B111E10; from Fedor Berditchevski, Birmingham, United Kingdom) was used.
Monoclonal antibodies against Arf6 (Neomarker), CD81 (SC-166028; Santa Cruz), vimentin (NCL-VIM-V9; Leica Microsystems), syntenin (SC-100336), PKCε (sc-1681; Santa Cruz), PKCα (877-232-8995; Transduction Laboratories), and PIP2 (A-21327; Invitrogen) and polyclonal antibodies against phospho-PKCα (06-822; Upstate Biotechnology) and phospho-PKCε (06-821; Upstate Biotechnology) were used. For F-actin visualization, TRITC (tetramethyl rhodamine isocyanate)-conjugated phalloidin was used (Sigma). Fluorescence-conjugated goat secondary antibodies against mouse and rabbit (Alexa 488, 555, and 633; Invitrogen) were used. For the SDS-PAGE loading control, polyclonal actin antibody (Santa Cruz) was used.
Virus internalization, transduction, and infection experiments.
The previously described BVs Ba-CAG-EGFP/WPRE and p24Cherry were used in the experiments. Viruses were produced as described previously (8, 29). Different multiplicities of infection (MOIs) (100 to 1,000) were used, depending on the experimental setup. In BV internalization experiments, virus was first bound to cells for 1 h at 4°C, followed by washing off the unbound virus and internalization at 37°C for 5 min to 5 h. In transduction experiments, virus was added to the growth medium and kept until the cells were analyzed or fixed with 4% paraformaldehyde (PFA)–phosphate-buffered saline (PBS) at 24 to 48 h posttransduction (p.t.). After the transduction, the percentage of enhanced green fluorescent protein (EGFP)-expressing cells was analyzed with FACSCanto II and FACSDiva software (BD Biosciences) or with an Olympus confocal microscope IX81 or a Zeiss Cell Observer wide-field microscope.
The EV1 (strain Farouk; ATCC) used in the experiments was produced and purified as described previously (28). Virus dilutions from 3.2 × 106 to 8 × 107 PFU/ml were used, depending on the experimental setup. We used the smallest amount of viruses that usually resulted in 20 to 40% infection in our control cell cultures (human osteosarcoma cells) and thus did not saturate the entry pathways. In infection and internalization experiments, the cells were incubated with viruses for 1 h on ice in cell culture medium containing 1% fetal bovine serum (FBS) with gentle shaking. The unbound virus was then extensively removed by washing the cells with 0.5% bovine serum albumin (BSA)-PBS. The viruses were then allowed to internalize in the cells at 37°C in cell culture medium containing 10% FBS for 15 min to 2 h for internalization and for 6 to 7 h for infection experiments before fixation with 4% PFA-PBS.
α2β1-Integrin clustering experiment.
In α2β1 integrin clustering experiments, clusters of α2β1-integrins were allowed to form in the presence of α2-integrin antibody and Alexa 555 secondary antibody and were internalized at 37°C for 2 h. After internalization, the cells were cooled to 4°C and incubated with Alexa 488 secondary antibody, washed, and mounted with Prolong gold DAPI (4′,6-diamidino-2-phenylindole) (Invitrogen), leaving the internalized integrins red and the plasma membrane-associated integrin green or yellow as the channels were merged. Samples were then imaged, and the ratio of intracellular and extracellular integrin was analyzed.
SDS-PAGE and Western blotting.
Samples were separated in 12% SDS-polyacrylamide gels and electroblotted onto polyvinylidene difluoride membranes (Millipore). Appropriate primary antibodies, together with horseradish peroxidase-conjugated secondary antibodies, were used in immunoblotting. Bands were detected with a Supersignal chemiluminescence detection kit (Thermo Scientific). Cell lysates of 4 × 105 cells per lane were used. Actin was used as a loading control.
Immunofluorescence labeling and confocal microscopy.
After fixation, the cells were permeabilized with 0.2% Triton X-100. The cells were immunolabeled using standard protocols. The appropriate primary antibodies were used, together with fluorescence-conjugated goat secondary antibodies against mouse and rabbit antibodies (Alexa 488, 555, and 633; Molecular Probes, Inc.). The cells were then mounted with Prolong gold anti-fade reagent with DAPI (Invitrogen). Coverslips were imaged with an Olympus IX81 microscope with a Fluoview 1000 confocal setup or a Zeiss Cell Observer wide-field microscope. Appropriate excitation and emission settings were used (488-nm argon laser; 543-nm and 633-nm HeNe lasers), with a UPLSAPO 60× (numerical aperture [NA], 1.35) or Achroplan 20× (NA, 0.45) objective with a resolution of 512 by 512 or 640 by 640 pixels/image. Levels for the laser power, detector amplification, and optical sections were optimized for each channel before starting the imaging.
Differential labeling between the surface and intracellular ligands was performed as described previously (18, 19). Briefly, after the experiment, the cells were fixed with 3% PFA for 15 min. After fixation, the antigen present on the cell surface was labeled without permeabilization by using primary antibody, together with Alexa secondary antibody. After permeabilization with 0.2% Triton X-100, the same primary antibody was used again, but now with Alexa secondary antibody with a different fluorescent conjugate. Plasma membrane-associated antigen is stained with both Alexas, and the intracellular antigen is stained only with the latter Alexa antibody.
Microscopic data analysis.
Quantification of fluorescence intensities and colocalization analyses were performed with a free, open-source software package, BioImageXD (30). To quantify the level of colocalization, 30 cells from three separate experiments were randomly selected and optically sectioned using a confocal microscope. The colocalization thresholds were set manually so that background fluorescence and fluorescence from diffuse stain were eliminated.
For quantification of fluorescence intensities and thus the relative amount of immunolabeled antigen, at least 30 cells from three independent experiments were imaged. The threshold for each channel was manually adjusted to separate the signal from noise in BioImageXD. The total intensity was then divided by the DAPI-stained nucleus volume to obtain the relative amount of antigen per cell.
Transfection experiments.
A mixture of four small interfering RNAs (siRNAs) against human syntenin 1 (L-008270-00-0005) and vimentin (L-003551-00) was used. A nontargeting siRNA pool (Thermo Scientific Dharmacon; D-001810-10) was used for negative control. For siRNA transfections, JetPrime (Polyplus Transfections) reagent was used according to the manufacturer's instructions. With syntenin-targeted siRNA, cells were incubated in the presence of siRNA containing transfection reagent for 48 to 72 h, whereas with vimentin-targeted siRNA, the cells were incubated with siRNA for 72 h, after which the cells were split and cultured without transfection reagent or siRNA. A total of 120 h after transfection for vimentin and 48 h for syntenin, cells were treated with viruses, and phorbol myristate acetate (PMA) and samples were collected for analysis with SDS-PAGE or a confocal microscope. Western blotting confirmed 95% knockdown of syntenin in siRNA-transfected cells. Vimentin siRNA knockdown efficiency was determined from confocal images after vimentin immunolabeling. Vimentin siRNA showed 77% knockdown of vimentin.
DNA plasmid transfections were done using Trans-IT 2020 (Mirus) or JetPei (Polyplus transfections) transfection reagent according to the manufacturer's instructions. PH-GFP, Arf6 (wild type [wt] and constitutively active [CA]), and RhoA (wt, dominant negative [DN], and CA) constructs were used.
MTT assay.
An MTT [3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide] assay (Promega) was performed on PMA-treated cells (0, 0.125, 0.25, 0.5, and 1 μM) according to the manufacturer's instructions. The concentrations used in the experiments did not show any cellular cytotoxicity (P > 0.5).
Statistical testing.
Statistical pairwise comparison was done using the Student t test performed with Graphpad Prism software. For results reported as percentages, prior to statistical comparison, arcsine transformation was applied to convert results to follow a normal distribution. All data are presented as means and standard errors of the mean (SEM).
RESULTS
The Ea.hy926 and MG-63 cell lines are deficient for BV transduction and EV1 infection.
Five different mammalian cell lines, derived from different cell types, were characterized for the ability to be infected or transduced by EV1 or BV. Infection and transduction efficiencies were determined by immunofluorescence labeling of the newly synthesized viruses or by reporter gene expression analysis, respectively. With both viruses, HepG2 cells were efficiently transduced (85% ± 6%) and infected (100%), whereas the infection/transduction rates for Raw2647 cells were close to zero (0.5% ± 0% and 0% ± 0%). 293T cells showed moderate transduction (27% ± 5%) and infection (13% ± 1.5%) rates for both viruses. The infection/transduction efficiencies in Ea.hy926 (4% ± 0.7% and 5% ± 1.5%) and MG-63 (6% ± 1.5% and 5% ± 2%) cells were quite low. As the results show, BV transduction and EV1 infection levels were significantly similar between the viruses (Fig. 1A and B). In order to study the factors that lead to efficient virus entry, we studied more closely two cell lines that were almost nonpermissive to both viruses (Ea.hy926 and MG-63) and one highly permissive cell line (HepG2).
Fig 1.

A block in infection/transduction is not due to lack of receptor or deficient binding of virus. (A) Cells were incubated with baculovirus (MOI, 800) and analyzed at 48 h p.t. with a confocal microscope. The percentages of cells expressing the fluorescent reporter gene are shown. (B) Echovirus infection (8 × 107 PFU/ml) was analyzed at 6 h p.i. Cells were immunolabeled with antibody against EV1, and the percentages of cells showing cytoplasmic expression of newly synthesized virus particles are shown. (C to F) Cell lines showed no statistical difference in virus binding or numbers of viral receptors. The amounts of bound viruses, syndecan 1, and α2β1-integrin were determined by immunolabeling with antibodies against the virus, syndecan 1, and α2-integrin. Cells were imaged by confocal microscopy, and the relative amounts of viruses and their receptors are shown as total intensity of antibody per nucleus volume. Analysis was performed using BioImageXD. The error bars indicate SEM.
We first wanted to assess whether expression of the viral receptors on different cell types is the determining factor for successful viral entry. All three cell lines showed high expression levels of the BV binding receptor syndecan 1 on the cell surface, and there were no significant differences between the cell lines (Fig. 1C). BV binding to cells was in accordance with the expressed receptor amounts (Fig. 1D). Corresponding experiments were performed for EV1 and its receptor, α2β1-integrin. As with BV and syndecan 1, α2β1-integrin was abundantly expressed on the cell surface and EV1 was able to bind to all cell types (Fig. 1E and F).
Next, we determined the transduction and infection levels in the nonpermissive cells. With BV transduction, only 6% transduction efficiency for MG-63 and 5% for Ea.hy926 cells were achieved in the nonpermissive cells, even when a large amount (MOI, 1,000) of BV was used. With EV1, we could reach 24% infected cells when 25-times-larger amounts of virus were used (data not shown). Altogether, these results show that Ea.hy926 and MG-63 cells are deficient for BV transduction and EV1 infection, even though these cells possess large numbers of viral receptors and virus binds efficiently to their surfaces. In HepG2 cells, both viruses show exceptionally high transduction and infection efficiencies, despite the similar expression levels of viral receptors in nonpermissive cells.
In nonpermissive Ea.hy926 and MG-63 cells, trafficking of BV and syndecan 1 is restricted on the plasma membrane.
Next, we wanted to assess at which stage the viruses are blocked along their internalization pathway. An internalization assay with BV was performed in Ea.hy926 and MG-63 cells, using a differential labeling method (19). With this assay, we can determine whether the antibody, and thus the protein of interest, is in the cytosol or on the cell surface. The results showed that, in nonpermissive cells, even after 5 h, the majority of BVs had not entered the cell but remained mainly on the cell surface in atypically large clusters (Fig. 2A). This was evident, as the colocalization of the total BV signal with the surface signal was high.
Fig 2.

BV aggregates syndecan 1 on the plasma membrane in Ea.hy-926 and MG-63 cells. EV-1 internalization is not blocked. (A and B) With differential labeling before and after permeabilization, we were able to differentiate antigens on the cell surface from the intracellular antigens. After baculovirus (MOI, 400) internalization (5 h p.t), cells were immunolabeled and imaged by confocal microscopy. (C and D) Baculovirus internalization was studied in Ea.hy926 cells. The cells were treated with baculovirus (MOI, 500), fixed at different time points, immunolabeled with BV and sdc-1 antibodies, and imaged with a confocal microscope. Colocalization and particle analysis were performed with BioImageXD. (E) Internalization of clustered α2β1-integrin. α2β1-integrin was clustered with antibodies in the presence of fluorescence-conjugated secondary antibodies (Alexa 555). After 2 h of internalization, α2β1-integrin remaining on the cell surface was labeled with secondary antibody conjugated with a different fluorescent dye (Alexa 488), showing clustered intracellular α2β1-integrin in red and extracellular α2β1-integrin in green. (F) Quantification of images with BioImageXD showed no statistical difference between the cell lines. Scale bars, 20 μm. The error bars indicate SEM. **, P < 0.01; ***, P < 0.001.
As the viruses were able to bind to the cells but unable to enter, we wanted to study the possibility that viruses attach to a decoy receptor on the nonpermissive cells, e.g., another heparan sulfate proteoglycan (HSPG) molecule showing optimal sulfation. However, microscopy analysis showed that BV associated with syndecan 1, but not with other syndecan family members on the cell surface. Moreover, the viruses were predominantly associated with the cell surface syndecan 1 rather than the intracellular protein (Fig. 2B), confirming that the deficient internalization is not due to binding to another HSPG. In HepG2 cells at 5 h p.t., the majority of the viruses had already uncoated, and large extracellular-virus–syndecan 1 clusters were not observed, as expected (Fig. 2B). Further colocalization analysis performed with Ea.hy926 cells showed that the colocalization of BV with syndecan 1 stayed approximately the same during the 4-h time course, whereas the colocalization of syndecan 1 with BV grew over time (Fig. 2C), suggesting that these BV clusters actually gathered more syndecan 1 over time. When we evaluated the colocalization of syndecan 1 with BV, we observed that the colocalization increased from 10% at 5 min to 30% at 2 h, suggesting that more syndecan was trapped with BV on the cell surface (Fig. 2C). Furthermore, detailed size measurements from confocal images showed that the aggregates grew bigger by over 4-fold within 2 h. The average size of syndecan 1 aggregates at 2 h postinternalization was 1.1 (±0.4) μm, while 4 h later, the average size was 4.8 (±1.1) μm (Fig. 2D).
Previously, we showed that antibody-clustered α2β1-integrin follows the same path as EV1-clustered α2β1-integrin (18, 19, 31). To study whether the trafficking of α2β1-integrin, and thus EV1, was also blocked on the surfaces of nonpermissive cells, we monitored the entry of antibody-clustered α2β1-integrin. In contrast to BV and syndecan 1, we could not detect any block in integrin internalization. After 2 h of internalization of the antibody-clustered integrin, clear intracellular clusters were observed in all tested cell lines (Fig. 2E). With careful quantification of confocal images, we confirmed that the amounts of internalized and cell surface integrin did not significantly differ between the cell lines (Fig. 2F), suggesting that the internalization of α2β1-integrin was not affected in the nonpermissive cells. As Ea.hy926 and MG-63 cells had a poor infection rate despite the internalization of the receptor, it seems probable that these cells have postinternalization defects in the infection pathway.
In conclusion, we show that in nonpermissive cells, BV clusters syndecan 1 on the cell membrane into large aggregates that are not able to internalize. EV1 internalization is not arrested in nonpermissive cells, indicating that the block is localized after the initial internalization step.
Cells show differences in cellular distribution and expression levels of F-actin, syntenin, vimentin, and phosphorylated PKCα (pPKCα).
In order to define the factors affecting BV and EV1 trafficking, we identified features that differ between the permissive and nonpermissive cell lines. Proteins that have previously been associated with BV, EV1, syndecan 1, or α2β1-integrin trafficking were studied by confocal microscopy.
The trafficking of BV (32), as well as syndecan (33, 34), is actin dependent, suggesting that actin could be one factor likely to contribute to deficient internalization. We thus tested the involvement of F-actin in the nonpermissive cell phenotype. Permissive HepG2 cells contained high levels of F-actin, whereas in nonpermissive cells, F-actin levels were substantially lower (Fig. 3A). As Rho GTPases regulate actin dynamics and we have previously shown that RhoA regulates BV uptake (29), we wanted to test the involvement of RhoA in the nonpermissive cell phenotype. However, our results using RhoA wt, DN, and CA constructs showed that RhoA had no apparent effect on BV transduction in Ea.hy926 cells (data not shown), suggesting that even though the F-actin levels in the nonpermissive cells were low, changing actin dynamics with RhoA is not the determining factor in the nonpermissive phenotype.
Fig 3.
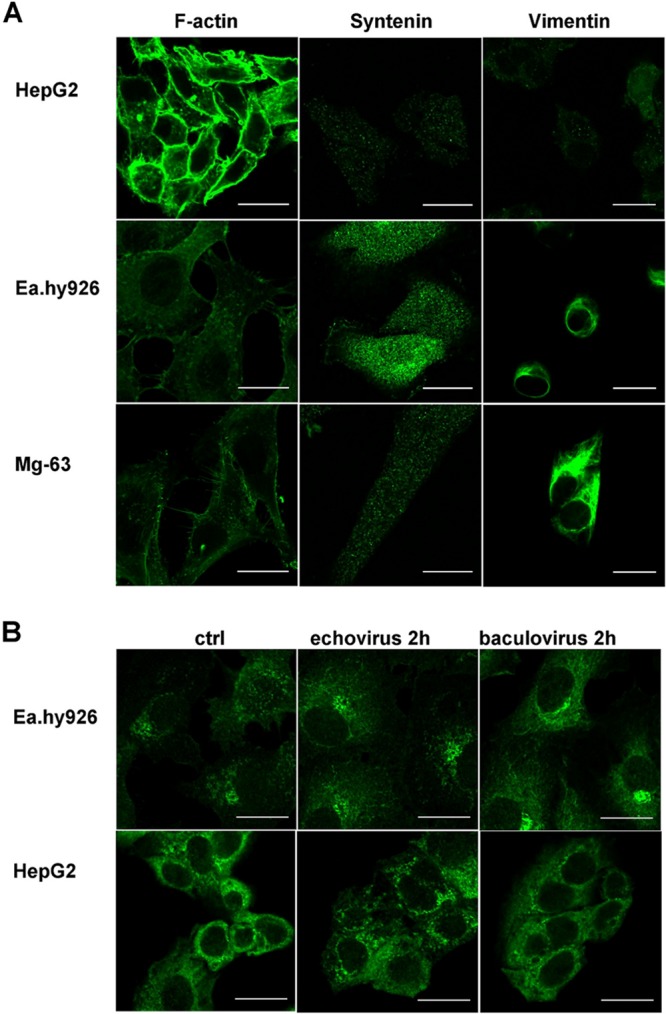
Nonpermissive and permissive cell lines show differences in expression patterns of F-actin, syntenin, vimentin, and pPKCα. (A) Immunolabeling of syntenin and vimentin in different cell types. Actin was labeled using TRITC-conjugated phalloidin. (B) pPKCα immunolabeled from untreated (control [ctrl]) or virus-treated (virus 2 h p.i.) Ea.hy926 and HepG2 cells. Scale bars, 20 μm.
Syntenin is a syndecan binding protein that is involved in syndecan recycling (35). Thus, the role of syntenin was studied in detail. The results revealed that high syntenin expression levels correlated negatively with effective virus transduction and infection. Syntenin was highly overexpressed in Ea.hy926 cells. In the other nonpermissive cell line, MG-63 cells, the expressed syntenin levels were lower than in Ea.hy926 cells but still higher than in permissive HepG2 cells (Fig. 3A). The results thus suggested that syntenin might be one of the key factors contributing to the nonpermissive cell phenotype.
Considering our previous results indicating that vimentin could be involved in effective BV transduction (12), we studied vimentin distribution in permissive and nonpermissive cells further. We noticed considerable differences between the cell lines. First, the overall level of vimentin expression was notably higher in nonpermissive cell lines than in the permissive cell line. Second, a clear difference was seen in the form of vimentin in the cells, with nonpermissive cells showing complex and abundant vimentin networks whereas the permissive HepG2 cell line showed no clear network and vimentin was seen in vesicle-like structures. This vesicle-like distribution further suggested that in HepG2 cells vimentin is more often in monomeric rather than in filamentous form (Fig. 3A).
PKCα is a protein kinase that has been shown to regulate β1-integrin trafficking (36) and EV1 internalization (18). Here, we studied both the phosphorylated form and the total pool of PKCα in the cells. We did not observe any significant differences in the total PKCα pool between the cell lines (data not shown), whereas the phosphorylated form showed distinctive distribution patterns in the nonpermissive cell lines, where it was mostly cytoplasmic and generally aggregated in the perinuclear compartment. In the permissive HepG2 cells, pPKCα was evenly distributed in the cell, and no perinuclear aggregates were observed (Fig. 3B). Interestingly, EV1 internalization was observed to lead to a reorganization of the pPKCα pool in permissive HepG2 cells, where 2 h postinternalization, pPKCα was relocated from the plasma membrane to the cytosol. This is in accordance with our previous results, where we found pPKCα activation during virus internalization and after antibody-induced integrin clustering (18). In contrast, in Ea.hy926 cells, there was no difference between the control and virus-treated cells, suggesting that although EV1 is able to internalize in these cells, due to the different pPKCα function/response in the cells, internalization does not lead to efficient infection.
The results revealed that F-actin, syntenin, vimentin, and pPKCα showed clear differences in permissive versus nonpermissive cell lines and prompted us to study these factors in more detail.
Change of cell culture medium from DMEM to RPMI induces BV transduction and EV1 infection.
Previously, we showed that BV transduction efficiency can be induced by using RPMI medium, in contrast to other commonly used media (12). BV transduction efficiency was also assessed here, and the results were in accordance with our previous results (Fig. 4A). To study whether the medium change would have an effect on EV1 infection as well, we performed an EV1 infection assay in different cell lines kept in either DMEM or RPMI medium for 24 h prior to the experiment. In all the cell lines studied, we were able to at least double the infection efficiency with RPMI medium, in contrast to DMEM (Fig. 4A). In order to determine if the medium change affected EV1 internalization or merely boosted the binding of the viruses on the cell surface, we measured the binding of EV1 onto the cells. There was no difference in virus binding between different media (data not shown), suggesting that RPMI medium induces virus trafficking in the cells, rather than virus binding.
Fig 4.

Vimentin, syntenin, and pPKCα expression levels are changed in optimal medium. (A) Baculovirus transduction (MOI, 1,000 in nonpermissive cells and 400 in permissive cells) or echovirus 1 infection (a proportional amount of cells highly expressing viral capsid proteins after 6 h p.i.; 8 × 107 PFU/ml in nonpermissive cells and 3.2 × 106 PFU/ml in permissive cells) determined from cells cultured in DMEM or RPMI medium. (B) Confocal images of cells immunolabeled with antibodies against vimentin, syntenin, or pPKCα and, in the case of F-actin, fluorescence-conjugated phalloidin. (C) Western blot showing the effect of syntenin siRNA transfection on syntenin levels in cells. (D) Syntenin knockdown could not rescue EV1 infection or BV transduction in nonpermissive Ea.hy926 cells. (E) Western blots showing the level of pPKCα in nonpermissive cells cultured in different media. Scale bars, 20 μm. The error bars indicate SEM. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We then wanted to characterize whether the previously identified proteins that were differentially expressed in the nonpermissive and permissive cell lines were affected by the medium change. No difference in the expression and distribution of F-actin was observed in cells cultured in different media (Fig. 4B). In contrast to F-actin, syntenin levels changed substantially in response to the culture medium. In HepG2 cells, where the expression level was fairly low to begin with, the effect was less obvious, whereas in the nonpermissive cell lines, an apparent drop in syntenin levels was observed in RPMI medium. The change in syntenin expression was especially drastic in Ea.hy926 cells (Fig. 4B). To further assess the role of syntenin, we performed a knockdown experiment with a syntenin-targeted siRNA smart pool. The syntenin level was downregulated efficiently (Fig. 4C), but to our surprise, siRNA treatment had no effect on BV transduction or EV1 infection, suggesting that syntenin was not a determining factor for cell susceptibility (Fig. 4D).
Since pPKCα showed a distinct distribution in the nonpermissive and permissive cells, the role of pPKCα in the medium phenomenon was characterized, as well. Strikingly, change of culture medium affected pPKCα distribution and also its phosphorylation status in the cells. In nonpermissive cells cultured in RPMI medium, the phospho-PKCα was translocated from the cytosol to the plasma membrane, and the overall level of pPKCα expression was higher (Fig. 4B). The induction in the phosphorylation levels seen from the confocal images was also confirmed by Western blotting (Fig. 4E).
We also observed that the vimentin network was modified in response to the culture medium in nonpermissive cell lines. In DMEM, the vimentin network was tight and compact, whereas in RPMI-cultured cells, the vimentin network was looser. In addition to vimentin network changes, it was also evident that, as the network was more compact, there was clearly more of the filamentous vimentin in the cells cultured in DMEM than there was in the cells cultured in RPMI. In permissive HepG2 cells, we could not detect any changes (Fig. 4B). Since the culture medium was shown to affect vimentin distribution in nonpermissive cells, we wanted to test the involvement of PKCε, which is the kinase that affects both vimentin dynamics and β1-integrin trafficking (37). However, we were not able to observe any difference in the amount of total PKCε between the media used. In contrast, in cells that were cultured in RPMI, a clear reduction in the phosphorylated form of PKCε was observed (Fig. 5A). In addition, when pPKCε distribution was monitored after BV internalization, we noticed that pPKCε associated with the BV-syndecan 1 aggregate on the cell surface (Fig. 5B). With EV1, no association between the virus and pPKCε was seen (data not shown). However, Western blotting of virus-treated Ea.hy926 cells showed clear downregulation of pPKCε in response to EV1 internalization (15 min postinfection [p.i.]) (Fig. 5C), suggesting that changes in PKCε phosphorylation may be a key factor regulating EV1 internalization. However, there was no difference in the amount of pPKCε in response to BV (Fig. 5C), further suggesting that lack of pPKCε downregulation could lead to deficient BV internalization. As blockage of β1-integrin recycling and its entrapment in CD81-positive vesicles upon PKCε inhibition have been reported by Ivaska et al. (38), we wanted to study whether CD81 was associated with the BV aggregates. However, we could not find CD81 in our aggregates (data not shown), suggesting that they are not the same structures previously reported by Ivaska et al.
Fig 5.

pPKCε is associated with BV and syndecan 1 aggregate on the plasma membrane in nonpermissive cells cultured in DMEM, with downregulation of pPKCε by RPMI medium and EV1 internalization. (A) Amounts of pPKCε in cells cultured in different media. pPKCε was immunolabeled with anti-pPKCε antibody, and the amount of pPKCε in cells was determined from confocal images and analyzed with BioImageXD. (B) Confocal images of pPKCε associated with baculovirus aggregate 5 h p.i. Scale bars, 20 μm. (C) pPKCε labeled in Ea.hy926 cells after SDS-PAGE and blotting after BV or EV1 treatments for 15 min to 2 h. The error bars indicate SEM. **, P < 0.01.
Altogether, these results indicate that PKC subtypes α and ε, together with vimentin reorganization, are the key factors affecting viral gene transfer and virus infection in different culture media.
PMA causes opposite effects on BV and EV1 entry in permissive versus nonpermissive cells.
PMA is a commonly used PKC activator. PMA can lead to the activation of both classical and novel PKC isoforms and thus to the activation of both PKCα and pPKCε. PKC's activity is regulated by phosphorylation and subcellular localization (39, 40). Since PKCα and PKCε were both shown to play a role in the culture medium phenomenon, we wanted to study the effects of PMA treatment on BV transduction and EV1 infection efficiency. The response to PMA treatment was studied in both cell media, as well.
With a flow cytometer, we first monitored the changes in BV transduction efficiency in response to PMA treatment in different media. As the results show, the effects of PMA treatment differed radically between the permissive and nonpermissive cell lines. In the permissive HepG2 cells, PMA was able to induce transduction efficiency to the level of RPMI medium, whereas in nonpermissive cells, strikingly, the transduction efficiency was actually reduced (Fig. 6A). EV1 infection levels during PMA treatments were determined by confocal microscopy. The results were again similar to those with BV; in permissive cells, PMA induced infection in both media, and in nonpermissive cells, PMA reduced infection (Fig. 6B).
Fig 6.

PMA treatment induces EV-1 infection and BV transduction in permissive cells, but not in nonpermissive cells. (A) Baculovirus transduction efficiency (MOI, 1,000 in nonpermissive cells and 400 in permissive cells) after transduction was determined from similar samples with fluorescence-activated cell sorter (FACS) analysis by monitoring reporter gene expression. (B) Echovirus 1 (8 × 107 PFU/ml in nonpermissive cells and 3.2 × 106 PFU/ml in permissive cells) infection efficiencies were determined in different media after PMA (30-min treatment) by immunolabeling and confocal microscopy. (C) Syntenin-targeted siRNA treatment did not have an effect on pPKCα in Ea.hy926 cells. The error bars indicate SEM. *, P < 0.05; **, P < 0.01.
To further control the effects of PMA treatment in the cells, we determined its effect on syntenin. Syntenin and PKCα activation were previously reported to be regulated interdependently in response to fibronectin (41). To study whether we had analogous interdependent regulation between syntenin and PKC, we tested the effects of PKC activation on syntenin expression and, conversely, the effects of syntenin knockdown on PKCα activation. However, in our studies, no interdependent regulation was observed (Fig. 6C).
Taken together, these results showed that PMA treatment had opposite effects on virus transduction and infection in nonpermissive versus permissive cell lines. In permissive cells, PMA treatment could further boost transduction and infection, due to PKCα activation. In contrast, in nonpermissive cells, PMA treatment activated inhibitory factors that blocked the beneficial effect of the RPMI medium.
Vimentin organization regulates BV transduction and EV1 infection in nonpermissive cells.
As PKCε is a subtype of PKC that is activated by PMA, and its downregulation was shown by RPMI change and by EV1 internalization, we next wanted to monitor changes in the vimentin network upon PMA treatment.
With a confocal microscope, we monitored changes in the vimentin organization in response to the culture medium and PMA treatment. With thorough analysis of multiple cells, we were able to observe subtle but consistent changes in the vimentin network. In nonpermissive Ea.hy926 cells, the vimentin network was more loosely packed in RPMI-cultured cells (Fig. 7A), where virus transduction and infection were induced. In both media, PMA treatment made the vimentin network even more compact. In permissive HepG2 cells, neither the culture medium nor PMA treatment had any effect on vimentin (Fig. 7B), suggesting that changes in the vimentin dynamics were responsible for the reduction in transduction and infection seen after PMA treatment in nonpermissive cells. As RPMI medium was shown to downregulate pPKCε in the nonpermissive cell lines, these results suggest that PKCε activation by PMA treatment leads to changes in vimentin organization in nonpermissive cells. These changes are then able to overcome the positive effect of PKCα activation on virus internalization, resulting in reduction in BV transduction and EV1 infection.
Fig 7.

PMA treatment modulates cellular distribution of vimentin to a more restrictive phenotype in Ea.hy926 cells. (A) Vimentin was immunolabeled from Ea.hy926 after PMA treatment (30 min) and imaged with a confocal microscope. Cell outlines are shown. (B) Vimentin in HepG2 cells after similar treatments. (C) EV1 infection (8 × 107 PFU/ml) and BV transduction (MOI, 500) were determined after siRNA transfection and subsequent PMA treatment (30 min). The error bars indicate SEM. (D) Vimentin knockdown efficiency after siRNA transfection (120 h) in Ea.hy926 cells was evaluated by immunolabeling. Scale bars, 20 μm. The error bars indicate SEM. *, P < 0.05; **, P < 0.01.
In addition to filamentous versus monomeric forms of vimentin observed in the nonpermissive and permissive cell types, respectively, nonpermissive cells also contained notably higher levels of vimentin than the permissive cells. We thus wanted to study whether vimentin knockdown would have an effect on BV transduction and EV1 infection. For this purpose, we used a vimentin-targeted siRNA smart pool and monitored virus transduction and infection efficiency with a confocal microscope. Vimentin knockdown did not have an apparent effect on EV1 infection or BV transduction in nonpermissive cells (Fig. 7C). In order to confirm that vimentin overexpression was not the restricting factor in PMA-treated cells, vimentin siRNA treatment was performed, with simultaneous PMA treatment (Fig. 7B), which proved negative. Knockdown of vimentin due to the siRNA treatment was confirmed by confocal microscopy. Although the siRNA treatment did not reduce vimentin expression to zero (77% knockdown), it was able to significantly diminish vimentin expression (Fig. 7D).
These results together suggest that the unfavorable effects of the vimentin network on cellular entry and intracellular trafficking of viruses is not due to the vimentin levels, but rather, is due to changes in vimentin organization, which are regulated by PKCε.
DISCUSSION
Identification of cell-type-specific characteristics affecting the permissiveness of cells to virus transduction or infection is important in order to understand virus-host interactions, as well as to identify factors that need to be considered in developing virus-based therapies. In nonpermissive cells, virus-mediated gene delivery and virus infection may be arrested in several steps from the initial binding to the eventual gene transcription and new virion budding. Here, we have characterized cell lines that are known to be restricted in virus vector transduction and determined the cell-line-specific features contributing to efficient virus entry. Two distinct model viruses, BV and EV1, were used.
BV has gained interest as a vector for biomedical applications, such as vaccine therapy, immune therapy, and gene therapy. In order to further define its use in biomedicine, factors affecting the permissiveness of the target cells must be identified. BV can transduce several mammalian cell types, but the efficiency varies between cell lines. Our previous studies on nonpermissive Ea.hy926 and MG-63 cells suggested that the block in transduction has similarities in different nonpermissive cells (6). Additionally we showed that medium change from DMEM to RPMI makes cells more permissive to BV transduction (8). However, the factors mediating this nonpermissive-to-permissive phenotype change remained undefined. Therefore, in this study, we monitored human cell lines derived from different cell types in more detail. In order to further define the cell-type-specific features mediating a nonpermissive phenotype and thus leading to reduced virulence, we also studied human picornavirus EV1 infection in these cells. In this study, we show that despite the fact that BV and EV1 have very different origins, the transduction and infection levels were highly similar in all of the mammalian cell lines tested. Furthermore, transduction and infection efficiencies were independent of the number of viral receptors or virus binding on the cell surface.
The cell lines under study, referred to as nonpermissive (Ea.hy926 and MG-63) and permissive (HepG2) cells, showed differential trafficking of BV and syndecan 1. In Ea.hy926 and MG-63 cells, transduction and infection levels were extremely low, whereas in HepG2 cells, they were exceptionally high. In the nonpermissive cells, BV and syndecan 1 formed large clusters on the plasma membrane that actively gathered more syndecan 1, leading to a block in internalization. This was surprising, as syndecan 1 clustering normally leads to efficient internalization of its ligand (34, 42). Even though EV1 infection was deficient in these cells as well, the mechanism of restriction seemed to differ between the viruses. EV1 internalization was not blocked, suggesting that restriction for EV1 was in intracellular trafficking. Arf6 and PIP2 are known regulators of syndecan and/or integrin recycling (35, 43). We have previously shown that the GTP binding protein Arf6 regulates BV uptake in permissive cells (29). In nonpermissive Ea.hy926 cells, however, Arf6 wt or CA overexpression could not induce BV transduction. Additionally, no apparent changes were seen in PIP2 distribution monitored by a PH-GFP-expressing construct in either nonpermissive or permissive cell lines. Furthermore, no colocalization was observed with BV or EV1 and Arf6 or PIP2 (data not shown). These results indicate that neither Arf6 nor PIP2 is associated with poor viral internalization.
By comparing the permissive and nonpermissive cell lines, several proteins that are associated with the nonpermissive cell phenotype were identified. The permissive cell line contained a substantially larger amount of F-actin and lower levels of syntenin and vimentin and showed differences in the cellular distribution of pPKCα. In addition to lower expression levels of vimentin, the organization of vimentin also appeared to differ: in nonpermissive cell lines, vimentin was arranged into a tight filamentous network, whereas in the permissive HepG2 cell line, the network was hardly detectable.
In all studied cell lines, infection and transduction could be induced by culturing cells in RPMI medium. This rescuing medium effect has been previously shown for BV, lentivirus, adeno-associated virus, and adenovirus (12). Here, we showed that EV1 infection could also be induced with a culture medium change from DMEM to RPMI. In RPMI medium, a reduction in vimentin expression and rearrangements in the vimentin network were observed. Previously, several viruses have been reported to depend on or to be responsible for vimentin rearrangements during virus infection, suggesting that vimentin could be one of the common regulators associated with effective virus entry. These viruses include, e.g., African swine fever virus (44), vaccinia virus (45), retrovirus (46), adenovirus (47), parvovirus (minute virus of mice [MVM]) (48), dengue virus (49), Japanese encephalitis virus (50, 51), herpesvirus (52), hepatitis C virus (53), and foot and mouth disease virus and bovine enterovirus (54). In this study, knockdown of vimentin could not rescue the transduction or infection of the viruses in nonpermissive cells, suggesting that it is not the overall vimentin level, but rather, the cellular organization of vimentin that promotes virus transduction and infection.
Vimentin dynamics are closely regulated by PKC subtypes (55). Interestingly, we found different effects of two interesting PKC subtypes upon RPMI medium change, namely, PKCα and PKCε. An increase in the level of phosphorylated PKCα was associated with its relocation from the cytosol to the plasma membrane. PKCα activation has previously been associated with efficient internalization of α2β1-integrin and EV1 infection (18), as well as syndecan 4 (56–58). Even though its association with syndecans has thus far been specifically only with syndecan 4 (59), our results indicate that syndecan 1 trafficking could also be regulated by PKCα. The structure and assembly of intermediate filaments, such as vimentin, are regulated by their phosphorylation status (55, 60). PKCε is one of the kinases that phosphorylate vimentin (37). Upon RPMI change, we observed downregulation of the phosphorylated form of PKCε, suggesting that the activation of PKCε could be inhibitory to cellular entry of viruses. This explains well the complex responses of permissive and nonpermissive cells to PMA treatment, which activates all PKC subtypes. If a tight filamentous vimentin network was the main inhibitory factor in nonpermissive cells, then the phosphorylation status of filamentous vimentin into a looser soluble network could be crucial to successful virus entry. In line with this idea, upon BV and EV1 treatment of the nonpermissive cells, pPKCε was effectively downregulated with EV1, but not with BV, leading to efficient internalization of EV1, but not of BV, which stayed on the plasma membrane. Interestingly, PKCε-mediated vimentin phosphorylation was shown earlier to regulate β1-intergin trafficking (37). In our study, RPMI medium was associated with high pPKCα and low pPKCε expression levels. Whether they both contribute to vimentin depolymerization directly or indirectly remains to be shown.
Taken together, this study demonstrates that the nonpermissive cell phenotype can be associated with low levels of F-actin, high expression levels of syntenin, a filamentous vimentin network, and differential regulation of PKCα and PKCε. Upon rescuing virus entry with RPMI medium, no change in F-actin organization was observed, suggesting that F-actin is not crucially involved. Even though syntenin expression was lowered by RPMI medium change, knockdown of its expression could not rescue the infection/transduction. Our results clearly indicated that differential activation of PKC subtypes α and ε, together with the status of intermediate filament vimentin, regulated BV transduction and EV1 infection (Fig. 8). The exact mechanism through which these molecules interact in the internalization pathways needs to be further studied.
Fig 8.

Factors associated with permissive and nonpermissive cell phenotypes affecting BV transduction and EV1 infection efficiency.
ACKNOWLEDGMENTS
For technical support, we thank Laura Pitkänen and Arja Mansikkaviita from the University of Jyväskylä, Tarja Taskinen from Ark Therapeutics Oy, and Anneli Miettinen and Joonas Malinen from the University of Eastern Finland. We are grateful to Loy Volkman (University of California, Berkley) for the BV antibodies.
This work was supported by the Academy of Finland and the Alfred Kordelin Foundation.
Footnotes
Published ahead of print 3 July 2013
REFERENCES
- 1.Chen CY, Lin CY, Chen GY, Hu YC. 2011. Baculovirus as a gene delivery vector: recent understandings of molecular alterations in transduced cells and latest applications. Biotechnol. Adv. 29:618–631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Airenne KJ, Hu YC, Kost TA, Smith RH, Kotin RM, Ono C, Matsuura Y, Wang S, Yla-Herttuala S. 2013. Baculovirus: an insect-derived vector for diverse gene transfer applications. Mol. Ther. 10.1038/mt.2012.286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Airenne KJ, Makkonen KE, Mahonen AJ, Yla-Herttuala S. 2010. In vivo application and tracking of baculovirus. Curr. Gene Ther. 10:187–194 [DOI] [PubMed] [Google Scholar]
- 4.Kost TA, Condreay JP. 2002. Recombinant baculoviruses as mammalian cell gene-delivery vectors. Trends Biotechnol. 20:173–180 [DOI] [PubMed] [Google Scholar]
- 5.Kost TA, Condreay JP, Jarvis DL. 2005. Baculovirus as versatile vectors for protein expression in insect and mammalian cells. Nat. Biotechnol. 23:567–575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kukkonen SP, Airenne KJ, Marjomaki V, Laitinen OH, Lehtolainen P, Kankaanpaa P, Mahonen AJ, Raty JK, Nordlund HR, Oker-Blom C, Kulomaa MS, Yla-Herttuala S. 2003. Baculovirus capsid display: a novel tool for transduction imaging. Mol. Ther. 8:853–862 [DOI] [PubMed] [Google Scholar]
- 7.Airenne KJ, Laitinen OH, Mahonen AJ, Yla-Herttuala S. 2009. Transduction of vertebrate cells with recombinant baculovirus. Cold Spring Harb. Protoc. 2009:pdb.prot5182. 10.1101/pdb.prot5182 [DOI] [PubMed] [Google Scholar]
- 8.Mahonen AJ, Airenne KJ, Purola S, Peltomaa E, Kaikkonen MU, Riekkinen MS, Heikura T, Kinnunen K, Roschier MM, Wirth T, Yla-Herttuala S. 2007. Post-transcriptional regulatory element boosts baculovirus-mediated gene expression in vertebrate cells. J. Biotechnol. 131:1–8 [DOI] [PubMed] [Google Scholar]
- 9.Boyce FM, Bucher NL. 1996. Baculovirus-mediated gene transfer into mammalian cells. Proc. Natl. Acad. Sci. U. S. A. 93:2348–2352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hofmann C, Sandig V, Jennings G, Rudolph M, Schlag P, Strauss M. 1995. Efficient gene transfer into human hepatocytes by baculovirus vectors. Proc. Natl. Acad. Sci. U. S. A. 92:10099–10103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song SU, Shin SH, Kim SK, Choi GS, Kim WC, Lee MH, Kim SJ, Kim IH, Choi MS, Hong YJ, Lee KH. 2003. Effective transduction of osteogenic sarcoma cells by a baculovirus vector. J. Gen. Virol. 84:697–703 [DOI] [PubMed] [Google Scholar]
- 12.Mahonen AJ, Makkonen KE, Laakkonen JP, Ihalainen TO, Kukkonen SP, Kaikkonen MU, Vihinen-Ranta M, Yla-Herttuala S, Airenne KJ. 2010. Culture medium induced vimentin reorganization associates with enhanced baculovirus-mediated gene delivery. J. Biotechnol. 145:111–119 [DOI] [PubMed] [Google Scholar]
- 13.Tuthill TJ, Groppelli E, Hogle JM, Rowlands DJ. 2010. Picornaviruses. Curr. Top. Microbiol. Immunol. 343:43–89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sawyer MH. 2002. Enterovirus infections: diagnosis and treatment. Semin. Pediatr. Infect. Dis. 13:40–47 [DOI] [PubMed] [Google Scholar]
- 15.Muir P, Kammerer U, Korn K, Mulders MN, Poyry T, Weissbrich B, Kandolf R, Cleator GM, van Loon AM. 1998. Molecular typing of enteroviruses: current status and future requirements. The European Union Concerted Action on Virus Meningitis and Encephalitis. Clin. Microbiol. Rev. 11:202–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Whitton JL, Cornell CT, Feuer R. 2005. Host and virus determinants of picornavirus pathogenesis and tropism. Nat. Rev. Microbiol. 3:765–776 [DOI] [PubMed] [Google Scholar]
- 17.Mizejewski GJ. 1999. Role of integrins in cancer: survey of expression patterns. Proc. Soc. Exp. Biol. Med. 222:124–138 [DOI] [PubMed] [Google Scholar]
- 18.Upla P, Marjomaki V, Kankaanpaa P, Ivaska J, Hyypia T, Van Der Goot FG, Heino J. 2004. Clustering induces a lateral redistribution of alpha 2 beta 1 integrin from membrane rafts to caveolae and subsequent protein kinase C-dependent internalization. Mol. Biol. Cell 15:625–636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Karjalainen M, Kakkonen E, Upla P, Paloranta H, Kankaanpaa P, Liberali P, Renkema GH, Hyypia T, Heino J, Marjomaki V. 2008. A Raft-derived, Pak1-regulated entry participates in alpha2beta1 integrin-dependent sorting to caveosomes. Mol. Biol. Cell 19:2857–2869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teng YH, Aquino RS, Park PW. 2012. Molecular functions of syndecan-1 in disease. Matrix Biol. 31:3–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Couchman JR. 2010. Transmembrane signaling proteoglycans. Annu. Rev. Cell Dev. Biol. 26:89–114 [DOI] [PubMed] [Google Scholar]
- 22.Rapraeger A, Jalkanen M, Bernfield M. 1986. Cell surface proteoglycan associates with the cytoskeleton at the basolateral cell surface of mouse mammary epithelial cells. J. Cell Biol. 103:2683–2696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carey DJ, Stahl RC, Tucker B, Bendt KA, Cizmeci-Smith G. 1994. Aggregation-induced association of syndecan-1 with microfilaments mediated by the cytoplasmic domain. Exp. Cell Res. 214:12–21 [DOI] [PubMed] [Google Scholar]
- 24.Choi Y, Chung H, Jung H, Couchman JR, Oh ES. 2011. Syndecans as cell surface receptors: unique structure equates with functional diversity. Matrix Biol. 30:93–99 [DOI] [PubMed] [Google Scholar]
- 25.Chen Y, Gotte M, Liu J, Park PW. 2008. Microbial subversion of heparan sulfate proteoglycans. Mol. Cells 26:415–426 [PubMed] [Google Scholar]
- 26.Stewart PL, Nemerow GR. 2007. Cell integrins: commonly used receptors for diverse viral pathogens. Trends Microbiol. 15:500–507 [DOI] [PubMed] [Google Scholar]
- 27.Fears CY, Woods A. 2006. The role of syndecans in disease and wound healing. Matrix Biol. 25:443–456 [DOI] [PubMed] [Google Scholar]
- 28.Marjomaki V, Pietiainen V, Matilainen H, Upla P, Ivaska J, Nissinen L, Reunanen Huttunen HP, Hyypia T, Heino J. 2002. Internalization of echovirus 1 in caveolae. J. Virol. 76:1856–1865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laakkonen JP, Makela AR, Kakkonen E, Turkki P, Kukkonen S, Peranen J, Yla-Herttuala S, Airenne KJ, Oker-Blom C, Vihinen-Ranta M, Marjomaki V. 2009. Clathrin-independent entry of baculovirus triggers uptake of E. coli in non-phagocytic human cells. PLoS One 4:e5093. 10.1371/journal.pone.0005093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kankaanpaa P, Paavolainen L, Tiitta S, Karjalainen M, Paivarinne J, Nieminen J, Marjomaki V, Heino J, White DJ. 2012. BioImageXD: an open, general-purpose and high-throughput image-processing platform. Nat. Methods 9:683–689 [DOI] [PubMed] [Google Scholar]
- 31.Karjalainen M, Rintanen N, Lehkonen M, Kallio K, Maki A, Hellstrom K, Siljamaki V, Upla P, Marjomaki V. 2011. Echovirus 1 infection depends on biogenesis of novel multivesicular bodies. Cell. Microbiol. 13:1975–1995 [DOI] [PubMed] [Google Scholar]
- 32.Ohkawa T, Volkman LE, Welch MD. 2010. Actin-based motility drives baculovirus transit to the nucleus and cell surface. J. Cell Biol. 190:187–195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tkachenko E, Lutgens E, Stan RV, Simons M. 2004. Fibroblast growth factor 2 endocytosis in endothelial cells proceed via syndecan-4-dependent activation of Rac1 and a Cdc42-dependent macropinocytic pathway. J. Cell Sci. 117:3189–3199 [DOI] [PubMed] [Google Scholar]
- 34.Fuki IV, Kuhn KM, Lomazov IR, Rothman VL, Tuszynski GP, Iozzo RV, Swenson TL, Fisher EA, Williams KJ. 1997. The syndecan family of proteoglycans. Novel receptors mediating internalization of atherogenic lipoproteins in vitro. J. Clin. Invest. 100:1611–1622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zimmermann P, Zhang Z, Degeest G, Mortier E, Leenaerts I, Coomans C, Schulz J, N′Kuli F, Courtoy PJ, David G. 2005. Syndecan recycling is controlled by syntenin-PIP2 interaction and Arf. Dev. Cell 9:377–388 [DOI] [PubMed] [Google Scholar]
- 36.Ng T, Shima D, Squire A, Bastiaens PI, Gschmeissner S, Humphries MJ, Parker PJ. 1999. PKCalpha regulates beta1 integrin-dependent cell motility through association and control of integrin traffic. EMBO J. 18:3909–3923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ivaska J, Vuoriluoto K, Huovinen T, Izawa I, Inagaki M, Parker PJ. 2005. PKCepsilon-mediated phosphorylation of vimentin controls integrin recycling and motility. EMBO J. 24:3834–3845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ivaska J, Whelan RD, Watson R, Parker PJ.2002. PKCϵ controls the traffic of β1 integrins in motile cells. EMBO J. 21:3608–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Freeley M, Kelleher D, Long A. 2011. Regulation of protein kinase C function by phosphorylation on conserved and non-conserved sites. Cell. Signal. 23:753–762 [DOI] [PubMed] [Google Scholar]
- 40.Steinberg SF. 2008. Structural basis of protein kinase C isoform function. Physiol. Rev. 88:1341–1378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hwangbo C, Kim J, Lee JJ, Lee JH. 2010. Activation of the integrin effector kinase focal adhesion kinase in cancer cells is regulated by crosstalk between protein kinase Calpha and the PDZ adapter protein mda-9/Syntenin. Cancer Res. 70:1645–1655 [DOI] [PubMed] [Google Scholar]
- 42.Fuki IV, Meyer ME, Williams KJ. 2000. Transmembrane and cytoplasmic domains of syndecan mediate a multi-step endocytic pathway involving detergent-insoluble membrane rafts. Biochem. J. 351:607–612 [PMC free article] [PubMed] [Google Scholar]
- 43.Powelka AM, Sun J, Li J, Gao M, Shaw LM, Sonnenberg A, Hsu VW. 2004. Stimulation-dependent recycling of integrin beta1 regulated by ARF6 and Rab11. Traffic 5:20–36 [DOI] [PubMed] [Google Scholar]
- 44.Stefanovic S, Windsor M, Nagata KI, Inagaki M, Wileman T. 2005. Vimentin rearrangement during African swine fever virus infection involves retrograde transport along microtubules and phosphorylation of vimentin by calcium calmodulin kinase II. J. Virol. 79:11766–11775 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferreira LR, Moussatche N, Moura Neto V. 1994. Rearrangement of intermediate filament network of BHK-21 cells infected with vaccinia virus. Arch. Virol. 138:273–285 [DOI] [PubMed] [Google Scholar]
- 46.Snasel J, Shoeman R, Horejsi M, Hruskova-Heidingsfeldova O, Sedlacek J, Ruml T, Pichova I. 2000. Cleavage of vimentin by different retroviral proteases. Arch. Biochem. Biophys. 377:241–245 [DOI] [PubMed] [Google Scholar]
- 47.Belin MT, Boulanger P. 1987. Processing of vimentin occurs during the early stages of adenovirus infection. J. Virol. 61:2559–2566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nuesch JP, Lachmann S, Rommelaere J. 2005. Selective alterations of the host cell architecture upon infection with parvovirus minute virus of mice. Virology 331:159–174 [DOI] [PubMed] [Google Scholar]
- 49.Kanlaya R, Pattanakitsakul SN, Sinchaikul S, Chen ST, Thongboonkerd V. 2010. Vimentin interacts with heterogeneous nuclear ribonucleoproteins and dengue nonstructural protein 1 and is important for viral replication and release. Mol. Biosyst. 6:795–806 [DOI] [PubMed] [Google Scholar]
- 50.Das S, Ravi V, Desai A. 2011. Japanese encephalitis virus interacts with vimentin to facilitate its entry into porcine kidney cell line. Virus Res. 160:404–408 [DOI] [PubMed] [Google Scholar]
- 51.Liang JJ, Yu CY, Liao CL, Lin YL. 2011. Vimentin binding is critical for infection by the virulent strain of Japanese encephalitis virus. Cell. Microbiol. 13:1358–1370 [DOI] [PubMed] [Google Scholar]
- 52.Miller MS, Hertel L. 2009. Onset of human cytomegalovirus replication in fibroblasts requires the presence of an intact vimentin cytoskeleton. J. Virol. 83:7015–7028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ghosh S, Ahrens WA, Phatak SU, Hwang S, Schrum LW, Bonkovsky HL. 2011. Association of filamin A and vimentin with hepatitis C virus proteins in infected human hepatocytes. J. Viral Hepat. 18:e568–77. 10.1111/j.1365-2893.2011.01487.x [DOI] [PubMed] [Google Scholar]
- 54.Armer H, Moffat K, Wileman T, Belsham GJ, Jackson T, Duprex WP, Ryan M, Monaghan P. 2008. Foot-and-mouth disease virus, but not bovine enterovirus, targets the host cell cytoskeleton via the nonstructural protein 3Cpro. J. Virol. 82:10556–10566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eriksson JE, Brautigan DL, Vallee R, Olmsted J, Fujiki H, Goldman RD. 1992. Cytoskeletal integrity in interphase cells requires protein phosphatase activity. Proc. Natl. Acad. Sci. U. S. A. 89:11093–11097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Keum E, Kim Y, Kim J, Kwon S, Lim Y, Han I, Oh ES. 2004. Syndecan-4 regulates localization, activity and stability of protein kinase C-alpha. Biochem. J. 378:1007–1014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lim ST, Longley RL, Couchman JR, Woods A. 2003. Direct binding of syndecan-4 cytoplasmic domain to the catalytic domain of protein kinase C alpha (PKC alpha) increases focal adhesion localization of PKC alpha. J. Biol. Chem. 278:13795–13802 [DOI] [PubMed] [Google Scholar]
- 58.Horowitz A, Simons M. 1998. Phosphorylation of the cytoplasmic tail of syndecan-4 regulates activation of protein kinase Calpha. J. Biol. Chem. 273:25548–25551 [DOI] [PubMed] [Google Scholar]
- 59.Simons M, Horowit A. 2001. Syndecan-4-mediated signalling. Cell. Signal. 13:855–862 [DOI] [PubMed] [Google Scholar]
- 60.Eriksson JE, He T, Trejo-Skalli AV, Harmala-Brasken AS, Hellman J, Chou YH, Goldman RD. 2004. Specific in vivo phosphorylation sites determine the assembly dynamics of vimentin intermediate filaments. J. Cell Sci. 117:919–932 [DOI] [PubMed] [Google Scholar]