

Abstract
Avoiding activation of immunity to vector-encoded proteins is critical to the safe and effective use of adeno-associated viral (AAV) vectors for gene therapy. While commonly used serotypes, such as AAV serotypes 1, 2, 7, 8, and 9, are often associated with minimal and/or dysfunctional CD8+ T cell responses in mice, the threshold for immune activation appears to be lower in higher-order species. We have modeled this discrepancy within the mouse by identifying two capsid variants with differential immune activation profiles: AAV serotype 8 (AAV8) and a hybrid between natural rhesus isolates AAVrh32 and AAVrh33 (AAVrh32.33). Here, we aimed to characterize the structural determinants of the AAVrh32.33 capsid that augment cellular immunity to vector-encoded proteins or those of AAV8 that may induce tolerance. We hypothesized that the structural domain responsible for differential immune activation could be mapped to surface-exposed regions of the capsid, such as hypervariable regions (HVRs) I to IX of VP3. To test this, a series of hybrid AAV capsids was constructed by swapping domains between AAV8 and AAVrh32.33. By comparing their ability to generate transgene-specific T cells in vivo versus the stability of transgene expression in the muscle, we confirmed that the functional domain lies within the VP3 portion of the capsid. Our studies were able to exclude the regions of VP3 which are not sufficient for augmenting the cellular immune response, notably, HVRs I, II, and V. We have also identified HVR IV as a region of interest in conferring the efficiency and stability of muscle transduction to AAVrh32.33.
INTRODUCTION
Adeno-associated virus (AAV) has been considered an ideal gene transfer vector due to its nonpathogenic, nonimmunogenic nature as well as its ability to transduce both dividing and nondividing cells and because it has a genome that persists over time to generate sustained, high-level expression (1). Since the discovery of the first AAV serotypes as contaminants in adenoviral preparations, 9 serotypes and over 120 capsid variants composing six phylogenetic clades have been described (2–11). The phylogenetic groups of capsids offer unique phenotypes in terms of transduction efficiency in target organs, tissue tropism, immunogenicity, and seroprevalence. In order to maximize the safety and efficacy of gene transfer, the ideal capsid would offer a low seroprevalence, a high transduction efficiency, and a lack of immunogenicity in vivo. As preclinical studies have begun identifying candidate capsids to progress toward clinical use, it has become apparent that the immunological sequelae of AAV vectors is quite complex. While many AAV capsid variants appear to be nonimmunogenic in murine models, they go on to generate robust cellular immune activation in higher-order species, which often threatens the stability of transduced cells (12).
In an attempt to investigate the mechanisms of activation of immunity to AAV, we recently identified two capsid variants that are functionally distinct in terms of their immune profiles in the muscle: AAV serotype 8 (AAV8) and a hybrid between natural rhesus isolates AAVrh32 and AAVrh33 (AAVrh32.33) (6, 7, 13, 14). AAV8 expressing nucleus-targeted LacZ (nLacZ) is associated with minimal CD8+ T cell priming, allowing stable β-galactosidase (β-Gal) expression in the muscle of C57BL/6 mice (14). In contrast, AAVrh32.33 generates a robust, polyfunctional CD8+ T cell response to both capsid and transgene antigens, resulting in a high degree of cellular infiltration in the muscle and a loss of β-Gal expression over time (14). While AAV8 provides the lack of cellular immunogenicity desired in a candidate gene therapy vector, a small population of humans has preexisting antibodies that interfere with vector administration (13). The seroprevalence of circulating neutralizing antibodies to AAVrh32.33 demonstrated naturally is very low; however, to ensure its safety and efficacy in gene transfer applications, one must first circumvent its inherent cellular immunogenicity.
These variants can be used to model differential immune activation in the mouse, by mimicking the discrepancy commonly seen between mice and nonhuman primates. From this model, we have learned that AAVrh32.33 augments transgene-specific CD8+ T cell activation by enhancing the activation and transduction of antigen-presenting cells (APCs) and, ultimately, CD4+ T cell help (14). Alternatively, AAV8 passively avoids the activation of innate and adaptive immunity while also inducing transgene-specific tolerance in the muscle of C57BL/6 mice (32). Overall, this model demonstrates that the structure of the AAV capsid is capable of dictating cellular immunity. By affecting critical vector-host cell interactions, such as receptor binding, endocytosis, endosomal escape, intracellular trafficking, nuclear entry, uncoating, or antigenicity (15), the capsid is influencing the generation of an immune response to vector-encoded proteins. This led us to hypothesize that the structural determinants that influence T cell activation or induction of tolerance to the transgene product could be mapped by creating capsid chimeras between AAV8 and AAVrh32.33.
Earlier studies have utilized domain-swapping methods to identify regions of the AAV capsid important for obtaining desired phenotypes. For instance, the improved muscle transduction observed in AAV1 and AAV6 in comparison to that observed in AAV2 has been mapped to a specific region of VP1 (amino acids 350 to 430) using this method (16). In this case, due to the low primary sequence homology between AAV8 and AAVrh32.33, the creation of viable capsid chimeras may be structurally limited, as swapping domains between divergent sequences could affect the proper folding and assembly of the capsid proteins. Knowledge of capsid structure is important in designing compatible swaps where interactions contributing to symmetry axes and secondary structure are maintained. The AAV capsid is encoded by 3 cap genes, VP1, VP2, and VP3. VP3 monomers comprise ∼90% of the capsid secondary structure and consist of a highly conserved eight-stranded β-barrel motif (βB to βI) (17). Due to this conservation, the basic architecture of the icosahedron, including critical protein interactions between each symmetry axis, is maintained between AAV8 and AAVrh32.33, despite differences in primary sequence (18; unpublished data). The majority of sequence variation falls within the surface loops linking these β strands, referred to as hypervariable regions (HVRs) I to IX. HVRs I to IX are the most surface-exposed loops of the AAV capsid and have been reported to dictate receptor binding, transduction efficiency, and antigenicity in AAV2 (which shares 83% sequence identity with AAV8) and AAV4 (which is a close relative of AAVrh32.33) (18–21). Thus, we further hypothesized that the ability of each capsid to augment or downregulate cellular immunity could be mapped to the specific domains of VP3 associated with these properties, a subset of hypervariable regions I to IX.
In this study, we aimed to characterize the structural determinants of the capsid responsible for driving differential activation of immunity to vector-encoded proteins. To do so, a series of hybrid AAV capsids was constructed by swapping domains between AAV8 and AAVrh32.33. By comparing their ability to generate transgene-specific T cells in vivo with the stability of transgene expression in the muscle, we were able to confirm that the functional domain lies within the VP3 portion of the capsid. Our studies were also able to exclude several regions of VP3 which are not sufficient for augmenting the cellular immune response, notably, HVRs I, II, and V. This work demonstrates the importance of structural analysis in the design of structurally viable hybrids between two capsid variants with low primary amino acid sequence identity. We have also identified HVR IV to be a region of interest in conferring the efficiency and stability of muscle transduction to AAVrh32.33 by generating an AAVrh32.33-based vector with the combined properties of low seroprevalence and robust, stable transgene expression.
MATERIALS AND METHODS
Cloning of hybrid AAV capsid-packaging plasmids.
The PCR splicing by overlap extension (SOE) technique was employed for the construction of AAV8-AAVrh32.33 hybrid capsids (22). In order to swap two domains, individual fragments were first generated by PCR and then combined in the presence of external primers to splice overlapping sequences together by SOE. This concept was used to swap single or multiple domains at a time to generate hybrid AAV cap genes, which were then cloned onto a packaging plasmid containing AAV2 rep using the restriction endonucleases HindIII and EcoRV for digestion, followed by subsequent ligation. Packaging plasmids were used to transform Escherichia coli, from which a single colony was amplified by a Qiagen Megaprep kit. The full-length hybrid cap gene sequences were then confirmed by DNA sequencing (Qiagen).
Production and purification of AAV vectors.
Recombinant AAV vectors with viral capsids from AAV8 or AAVrh32.33 expressing firefly Luciferase (ffLuc; small-scale transfections) or nucleus-targeted β-galactosidase (nLacZ; large-scale transfections) were manufactured as previously described (23) by PennVector at the University of Pennsylvania (Philadelphia, PA). Briefly, an AAV cis-plasmid containing transgene cDNA driven by either the cytomegalovirus (CMV; for ffLuc [CMV.ffLuc]) or the chicken β-actin (CB; for nLacZ [CB.nLacZ]) promoter and flanked by AAV2 inverted terminal repeats was packaged by triple transfection of human embryonic kidney 293 (HEK293) cells using an adenovirus helper plasmid (pAdΔF6) and a chimeric packaging construct containing the AAV2 rep gene and the AAV8-AAVrh32.33 wild-type or hybrid cap genes. Small-scale transfections using CaCl2 and HEPES-buffered saline (HBS) were performed on 6-well plates with cells at ∼80 to 90% confluence at the time of transfection, 3.25 μg of both packaging plasmid and cis-plasmid, and 6.5 μg of adenovirus helper. Both supernatant and cell lysate were harvested at day 2 following three freeze-thaw cycles in a final volume of 3 ml per preparation. Large-scale transfections were performed by two methods: 40 transfections on 15-cm plates using CaCl2 and HBS for harvesting of the cell pellet only or a single-cell stack transfection using polyethylenimine (PEI; PEI-MAX; Polysciences Inc., Warrington, PA) for harvesting of total lysate at day 5. Large-scale vector preparations were purified by two rounds of cesium chloride gradient centrifugation. The genome titer (number of genome copies [GCs] per milliliter) of the AAV vectors was determined by real-time PCR.
In vitro transduction assays.
Small-scale vector preparations of AAV hybrid capsids encoding CMV.ffLuc were used to transduce either HEK293 cells or the U937 human monocyte-derived cell line in vitro. Cells (1 × 105) were plated in a 96-well plate, and vector was added at a multiplicity of infection (MOI) of 104 GCs/cell. The MOI was calculated as follows: desired GC number/cell × number of cells/well = number of GCs needed/well. A wild-type adenoviral helper construct was used to cotransduce cells at an MOI of 102. Two negative controls were performed: one in which no packaging plasmid was added to the transfection mixture and a second one of completely untransfected cells. Small-scale transfections were performed in triplicate; all three samples were tested for in vitro transduction in duplicate.
Structural alignment of the AAV8 and AAVrh32.33 VP3 monomers.
The VP3 coordinates of the AAVrh32.33 capsid crystal structure (Protein Data Bank [PDB] accession no. 4IOV; M. Agbandje-McKenna et al., unpublished data) were superimposed onto the AAV8 crystal structure (PDB accession no. 2QA0) (http://www.rcsb.org/pdb/home/home.do) (18) using the secondary structure matching option (24) of the Coot program (25). The program also generates a list of atomic distances (in Å) between each C-α position aligned. This list was used to identify amino acid residues that are structurally equivalent versus those with differences (defined as C-α atoms that are >1.0 Å apart). Stretches of two or more C-α positions that were greater than 1 Å apart between the two structures were identified as variable loop regions.
Animals.
Male C57BL/6 mice (6 to 8 weeks old) were purchased from The Jackson Laboratory. Mice were maintained in the Animal Facility of Translational Research Laboratories. All experimental procedures involving the use of mice were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania.
Animal procedures.
Mice were anesthetized intraperitoneally (i.p.) with ketamine (70 mg/kg of body weight) and xylazine (7 mg/kg of body weight). A total of 5 × 1010 or 1 × 1011 GCs of recombinant AAV vector were injected into the anterior tibialis muscle in a volume of 50 μl of sterile phosphate-buffered saline (PBS). Where indicated, whole blood was extracted by retro-orbital bleeds, using a heparinized capillary tube, of the lateral canthus of the eye of lightly anesthetized animals (ketamine [35 mg/kg] and xylazine [5 mg/kg] i.p.). At day 28 postinjection, mice were sacrificed by CO2 inhalation, followed by cervical dislocation to harvest muscle for histochemistry.
MHC-I tetramer stain.
Phycoerythrin (PE)-conjugated major histocompatibility complex (MHC) class I (MHC-1) H2-Kb-ICPMYARV tetramer complex was obtained from Beckman Coulter (Miami, FL). At various time points after vector injection, tetramer staining was performed on heparinized whole blood cells isolated by retro-orbital bleeds. Cells were costained for 30 min at room temperature (RT) with PE-conjugated tetramer and fluorescein isothiocyanate-conjugated anti-CD8α (Ly-2) antibody (BD Pharmingen). Red blood cells were then lysed, and cells were fixed with iTAg MHC tetramer lysing solution supplemented with fix solution (Beckman Coulter, Miami, FL) for 15 min at RT. The cells were then washed three times in PBS and resuspended in 0.01% BD CytoFix fixation buffer (BD Biosciences). Data were gathered with an FC500 flow cytometer (Beckman Coulter, Miami, FL) and were analyzed with FlowJo analysis software (TreeStar, San Carlos, CA). In the analysis, lymphocytes were selected on the basis of forward and side scatter characteristics, followed by selection of CD8+ cells and then the tetramer-positive CD8+ T cell population.
Intracellular cytokine staining.
Spleens harvested from treated mice were transferred into Liebowitz's 15 (L-15) medium (Cellgro; Mediatech, Herndon, VA) at RT. The tissue was then homogenized and passed through a 70-μm-pore-size nylon cell strainer (Fisher Scientific, Pittsburgh, PA) to remove cell clumps and undissociated tissue. The cells were centrifuged for 5 min at 1,600 rpm and RT, resuspended in fresh L-15 medium, and centrifuged again. The cell pellet was resuspended in T cell assay medium (Dulbecco modified Eagle medium [Cellgro; Mediatech, Herndon, VA], 10% heat-inactivated fetal bovine serum [FBS; HyClone, Logan, UT], 1% penicillin-streptomycin [Cellgro; Mediatech, Herndon, VA], 1% l-glutamine, 10 mM HEPES [Cellgro; Mediatech, Herndon, VA], 0.1 mM nonessential amino acids [Invitrogen, Carlsbad, CA], sodium pyruvate, 10−6 M 2-mercaptoethanol [Cellgro; Mediatech, Herndon, VA]).
After resuspension at a concentration of 107 cells/ml, splenocytes were plated at 106/well in triplicate on 96-well round-bottom plates. T cell assay medium was supplemented with 1 μg/ml brefeldin A (GolgiPlug; BD Pharmingen, San Diego, CA) and 20 ng/ml mouse interleukin-2 (IL-2; BD Pharmingen, San Diego, CA). The β-Gal CD8 H2-Kb T cell epitope (ICPMYARV; Mimotopes, Australia) was added at 1 μg/ml to the corresponding experimental wells. Cells were stimulated for 5 h at 37°C in 10% CO2. Control cells were incubated without peptide or in the presence of phorbol myristate acetate (PMA)-ionomycin (PMA, 0.05 μg/ml; ionomycin, 1 μg/ml; Sigma, St. Louis, MO). Following the stimulation, cells were washed and stained with surface antibodies (BD Pharmingen, San Diego, CA) for 30 min at 4°C. Cells were washed with PBS–1% FBS and then permeabilized in Cytofix/Cytoperm solution at 4°C for 20 min. Cells were again thoroughly washed with 1× Perm/Wash buffer and stained intracellularly (BD Pharmingen, San Diego, CA) for 45 min at 4°C. After washing, cells were examined by flow cytometric analysis. Samples were acquired on an FC500 flow cytometer (Beckman Coulter, Miami, FL) and analyzed using FlowJo software (TreeStar, San Carlos, CA).
Histology and transgene detection.
To examine expression of nuclear β-Gal, X-Gal (5-bromo-4-chloro-3-indolyl-β-d-galactopyranoside) staining on cryosections from snap-frozen muscles was performed according to standard protocols (26). Sections were slightly counterstained with Fast Red to visualize nuclei. Muscles expressing green fluorescent protein were fixed overnight in formalin, washed in PBS for several hours, and then snap-frozen for sectioning. Cryosections were mounted in Vectashield containing DAPI (4′,6-diamidino-2-phenylindole; Vector Laboratories) to show nuclei.
RESULTS
Construction of AAV8-AAVrh32.33 hybrid capsids.
In order to map the structural determinants responsible for differential immune activation between AAV8 and AAVrh32.33, we generated a series of AAV8-AAVrh32.33 hybrid capsids by swapping individual or interacting capsid domains from one variant to the other. Figure 1A presents a sequence alignment of the two capsid variants, highlighting the nine hypervariable region (I to IX) loops, which represent the most diverse and surface-exposed regions of the capsid. Hybrids were created using PCR splicing by overlap extension (SOE), in which a first round of PCR generates the individual capsid fragments with overlapping 5′ and 3′ edges, which are then sewn together in a second PCR involving only outer primers. The diagram in Fig. 1B illustrates the composition of each of the 26 different hybrid cap genes that were constructed for this study.
Fig 1.

Comparison of the AAV8 and AAVrh32.33 capsids and the AAV8-AAVrh32.33 hybrid capsid constructs created by domain swapping. (A) Amino acid sequences of the VP1 capsid protein of AAV8 and AAVrh32.33 were aligned for comparison using ClustalX and BioEdit, version 7.0.0 (Ibis Biosciences, Carlsbad, CA). The dots in the alignment represent the amino acids that are identical to those of AAV8 VP1. The ∼ symbols indicate the amino acids that are missing at the positions in the alignment. The N termini for VP1 (ATG at M1), VP2 (ACG at T138), and VP3 (ATG at M204) are indicated. HVRs between β strands are numbered I to IX and enclosed by rectangular frames. Blue boxes, broad-swap delineations for each HVR within VP3; red boxes, narrow-swap locations for each HVR within VP3. Broad swaps contain all nonconserved amino acids within the given region. Narrow swaps contain only those amino acids that are structurally nonsuperimposable (defined as C-α atoms that are >1.0 Å apart); these are typically the most surface-exposed portions of each HVR loop. (B) Diagram of hybrid capsids constructed. Open bars, sequences from AAV8; closed bars, sequences from AAVrh32.33. The N termini of VP1, VP2, and VP3, as well as the delineations of each HVR, I to IX, are indicated at the top. Hybrid names are listed as abbreviations LM1 to LM20, with odd numbers representing hybrids composed primarily on an AAV8 backbone and even numbers representing hybrids composed on the AAVrh32.33 backbone. The swapped portion of the capsid as well as the design strategy for each set of hybrids is indicated to the right. *, hybrids that were constructed in both broad and narrow formats. (C) Superimposition of the VP3 monomers of AAV8 (green) and AAVrh32.33 (orange). Stretches of two or more C-α positions greater than 1 Å apart between the two structures were identified as variable loop regions. The major differences between AAV8 and AAVrh32.33 are located in HVRs I, IV, and V. When a second symmetry-related monomer interacts, HVR V falls in the groove between HVR VIII and HVR IV, shown in panel C to create the peaks surrounding the 3-fold axis of symmetry. The 2-fold (oval), 3-fold (triangle), and 5-fold (pentagon) axes of symmetry are indicated. In the LM14 hybrid, the inward-facing orange loop of AAVrh32.33 HVR IV is replaced with that of AAV8 (in green), opening up the gap between HVR IV and HVR VIII and exposing amino acid residues located within that groove.
The first set of hybrids created contained the swap of VP1 and VP2 unique (VP12u) regions of the capsid. Within VP3, two approaches were taken to optimize compatible swaps and promote the formation of structurally viable hybrids between two AAV capsid variants with low primary sequence identity. First, we attempted a linear design approach in which the VP3 portion of the capsid was divided into three main segments and swaps were designed at regions highly conserved between AAV8 and AAVrh32.33, hoping that swapping domains at conserved regions of the capsid would minimize structural incompatibility when the fragments were recombined. These swaps included HVR I to III, IV to VIII, and IX (Fig. 1B). It is important to note that the linear design exchanges large sections of VP3 which include both hypervariable regions and conserved portions of the capsid that flank them.
In a second approach, the rational design approach, engineering focused solely on HVRs I to IX, which are the most variable, functional, and surface-exposed regions of VP3. To create rational design hybrids, a structural analysis of the respective AAV capsids, AAV8 and AAVrh32.33, was performed to identify any interactions between hypervariable regions either within an individual VP3 monomer or between adjacent symmetry-related monomers that come together to form the intact capsid. HVRs with no notable interactions were swapped individually, including HVR I and HVR II. HVRs that appeared to interact in the folded capsid were swapped together in an attempt to maintain the integrity of those interactions within the new hybrid capsid. These swaps included combinations of HVRs IV, V, and VIII and HVRs III, VI, VII, and IX. The rational design hypothesis assumes that interactions between HVRs on the capsid surface may be critical for maintaining the appropriate secondary structure; swapping multiple HVRs together may be essential to maintain the proper folding and function of the intact capsid particle. For instance, HVRs IV and VIII from one VP3 monomer interact with HVR V on a symmetry-related monomer to form the three peaks surrounding the 3-fold axes of symmetry, a region of the capsid to which antigenicity, transduction efficiency, and receptor binding phenotypes have been mapped in structurally defined serotypes (18–21, 27) (Fig. 1C). When identifying amino acid residues that are structurally equivalent versus those that are different (defined as C-α atoms that are >1.0 Å apart), we see that HVR loops IV and V are the most structurally nonsuperimposable HVR loops between AAV8 and AAVrh32.33 (Fig. 1C). For this reason, individual domain swaps of HVR IV and HVR V were also created (Fig. 1B).
The initial swaps of HVRs were designed in a broad fashion including all nonconserved amino acids within that region (Fig. 1A). However, in many of the HVRs, the most N- and C-terminal amino acids were still structurally superimposable between AAV8 and AAVrh32.33 (defined as C-α atoms that are less than 1 Å apart). The base of each HVR loop contained amino acid differences, but these residues generally still retained the same overall main chain structure, such that only the most surface-exposed tip of the hypervariable loop was structurally nonsuperimposable between monomers. However, the different side chains engage in intra- or intersubunit interactions that are virus specific and maintain the capsid architecture. For these reasons, we predicted that swapping only the narrowest, most variable, and surface-exposed portion of each HVR loop may increase production yields and in vivo transduction, reasoning that the farther that our HVR swaps extended outwardly both 5′ and 3′ from the apex of the HVR loop, the more chances for interruption of amino acid interactions critical to maintaining the capsid architecture there would be. We also hypothesized that the broad region may be less tolerant of HVR swapping without negatively impacting proper VP folding, capsid assembly, and virus yields. Hence, for several hybrid constructs, including swaps of HVR I alone, IV alone, V alone, as well as combined swaps of HVRs IV, V, and VIII and HVRs III, VI, VII, and IX, in addition to the original broad swap design, narrow swaps were also created. The amino acids included in broad versus narrow swaps of each HVR are delineated in Fig. 1A.
All chimeric cap genes were cloned into an identical AAV packaging plasmid containing the AAV2 rep gene. The integrity of all hybrid packaging plasmids was confirmed by complete DNA sequencing (Qiagen), eliminating the concern over unexpected mutations.
Production efficiencies of AAV hybrid capsid constructs.
Due to the low sequence homology between AAV8 and AAVrh32.33, the creation of hybrid cap genes could likely impact the expression of capsid proteins, assembly of capsid particles, and/or packaging of the AAV genome. Therefore, before large-scale preparations were made, the production efficiency of each of our AAV hybrids was tested by a small-scale transfection assay. Triple transfection of HEK293 cells was performed in 6-well culture plates using our hybrid packaging construct in combination with an AAV cis-plasmid containing firefly luciferase (ffLuc) cDNA driven by the cytomegalovirus (CMV) promoter flanked by AAV2 inverted terminal repeats as well as an adenovirus helper plasmid (pAdΔF6). After transfection, both the cell pellet and the supernatant were harvested simultaneously and the production efficiency of each hybrid was assessed by quantitative real-time PCR to detect DNase-resistant particles (drp). The titers of AAV8-AAVrh32.33 hybrid vectors are shown as the number of drp per ml (Fig. 2A). All hybrids were produced in a total volume of 3 ml. Yields of some hybrid capsids varied significantly compared to those for wild-type AAV8 and AAVrh32.33 controls, which generated similar titers of approximately 2 × 1011 drp/ml.
Fig 2.

Comparison of AAV hybrid capsid genome packaging and in vitro transduction efficiencies. (A) Triple transfections of HEK293 cells were performed in 6-well plates, combining an AAV2 rep/AAV8-AAVrh32.33 hybrid cap packaging plasmid with a cis-plasmid containing CMV.ffLuc, AAV2 inverted terminal repeats, and an adenovirus helper construct (pAdΔF6). Titers of small-scale vector preparations are reported as the number of drp/ml (theoretically equivalent to the number of GCs/ml). All vectors were produced in a final volume of 3 ml. The data shown are means ± SDs from 3 wells within a 6-well plate. Results were confirmed in three separate experiments. (B and C) Small-scale vector preparations of AAV hybrid capsids encoding CMV.ffLuc were used to transduce HEK293 cells (B) or U937 human monocytes (C) in a 96-well plate at an MOI of 104. Wild-type adenovirus helper was used for cotransduction at an MOI of 102. The data shown are means ± SDs from at least 3 duplicate wells on a 96-well plate. Results were confirmed in three separate experiments both with and without adenovirus helper. No Pack, a negative control in which no packaging plasmid was added to the transfection mixture; Cells Only, a negative control of untransfected cells.
VP12u hybrids LM1 and LM2 generated titers comparable to those generated by wild-type AAV8 and AAVrh32.33, which was expected due to the higher degree of conservation in VP1 and VP2 compared to that in VP3 between these viruses. The linear design swaps LM3 to LM8 had titers below background levels, indicating that domain swaps involving large regions of VP3 encompassing both conserved regions and HVRs could not produce viable hybrids between AAV8 and AAVrh32.33. Rational design VP3 hybrids (LM9 to LM20), however, were able to generate yields significantly above the background. With the exception of LM9, swaps of single HVRs (LM9 to LM16) generated the highest titers of all VP3 hybrids, varying from 8 × 1010 − to 3 × 1011 drp/ml. The LM9 hybrid, however, showed over a log-unit decrease in titer compared to wild-type AAV8. LM9 places AAVrh32.33 HVR I onto the AAV8 backbone; the sequence of AAVrh32.33 HVR I contains a 5-amino-acid deletion relative to the AAV8 HVR I sequence (Fig. 1A). Inserting those additional 5 amino acids from AAV8 into the AAVrh32.33 backbone allowed efficient vector production with the reciprocal swap, LM10. The reduction in vector titer for the LM9 chimera may be the result of the loss of AAV8-specific side chain interactions (intra- and intersubunit), which results in loss of capsid stability (Fig. 2A).
Rational design swaps of multiple HVRs at a time (LM17 to LM20) all demonstrated 1- to 1.5-log-unit decreases in titers compared to the titer for the wild type (Fig. 2A). For these chimeras, narrowing of the regions swapped did not improve the vector titer. For instance, despite the crucial interactions of HVRs IV, V, and VIII required to assemble the 3-fold peaks on the capsid surface, hybrids containing swaps of HVRs IV, V, and VIII together showed over a log-unit decrease in titers, whereas swaps of HVR IV or HVR V alone generated titers comparable to those for the wild-type controls (Fig. 2A). This observation stems from the fact that residues within the narrow swaps are also involved in virus-specific side chain interactions with other amino acids that are altered in the chimeric viruses. Thus, in terms of production efficiency, broad versus narrow swap delineation did not lead to a significant effect on small-scale transfection titers.
In vitro transduction efficiencies of AAV hybrid vectors.
Our next goal was to determine the in vitro transduction ability of our small-scale hybrid vector preparations. To this end, HEK293 cells were cotransduced with AAV vector-expressing CMV.ffLuc at an MOI of 104 in combination with wild-type adenovirus at an MOI of 102. Luminescence in 96-well plates was measured and is reported as the log number of relative light units (RLUs)/s in Fig. 2B. Results show that VP12u hybrids are capable of transducing HEK293 cells to the same degree as wild-type controls, resulting in ∼7.5 × 103 to 1.1 × 104 RLU/s of luminescence. There was a direct correlation between hybrids achieving a titer of >5 × 1010 drp/ml and the ability to transduce HEK293 cells at levels significantly above the background, as hybrids LM10 to LM16 all demonstrated in vitro transduction levels at least 1 to 2 log units above the level for the negative controls (Fig. 2A and B). As with production efficiency, all linear design hybrids (LM3 to LM8), VP3 single-HVR-swap LM9, and all rational design multiple-HVR swaps (LM17 to LM20) showed background levels of transduction in HEK293 cells. Ultimately, it appears that hybrids generating titers less than 5 × 1010 drp/ml were unable to significantly transduce HEK293 cells in vitro, despite cell transduction using the same quantity of vector.
Due to the discrepancy in the ability of AAV8 and AAVrh32.33 to prime polyfunctional CD8+ T cells, we hypothesized that their transduction of antigen-presenting cells (APCs) would differ as well. Indeed, AAVrh32.33 showed significantly enhanced transduction of the JAWSII murine dendritic cell line and U937 human monocytes compared to AAV8. The ability of AAVrh32.33 to more readily transduce APCs likely augments the activation of APCs, upregulating production of proinflammatory cytokines and surface expression of MHC-II and costimulatory molecules and ultimately enhancing T cell priming (Mays and Wilson, submitted). Here, we tested our panel of AAV8-AAVrh32.33 hybrid vectors for transduction of U937 cells in vitro in order to determine whether their efficiency of APC transduction—an upstream marker of their potential ability for priming T cells—had been affected by any of the domain swaps (Fig. 2C). Initially, as seen with HEK293 cell transduction in vitro, hybrids with titers below 5 × 1010 drp/ml were unable to significantly transduce U937 cells at an MOI of 104. It is important to note that this assay would have been greatly enhanced by the ability to infect at an MOI of 106 or greater; however, the titers of small-scale hybrid preparations were limiting in this regard. Overall, results demonstrate that of the vector preparations with yields high enough to allow successful transduction of HEK293 cells, only wild-type AAVrh32.33 and those hybrids constructed on the AAVrh32.33 backbone were capable of transducing U937 cells at levels significantly above the background. AAVrh32.33 containing VP12u or VP3 HVR I, II, IV, or V from AAV8 retained the ability of wild-type AAVrh32.33 to transduce the U937 human monocyte-derived cell line in vitro. Conversely, wild-type AAV8 and AAV8 containing VP12u or VP3 HVR II, IV, or V from AAVrh32.33 were unable to significantly transduce U937 cells (Fig. 2C). This observation indicates that the domain responsible for augmenting U937 cell transduction in AAVrh32.33 does not lie fully within VP12u or VP3 HVR I, II, IV, or V.
Production efficiency of large-scale hybrid vector preparations.
In order to conclusively test the ability of our hybrid vectors to generate stable transgene expression in the muscle or to prime a transgene-specific CD8+ T cell response in vivo, we produced large amounts of the hybrid capsid vector expressing nucleus-targeted β-galactosidase (nLacZ) cDNA driven by the chicken β-actin (CB) promoter. Using muscle as a target organ, injection volumes were limited to 50 μl/mouse, requiring that the titers of our preparations be greater than or equal to 2 × 1012 GCs/ml in order to inject a dose of 1 × 1011 GC/mouse. The trend in large-scale transfection yields for AAV8-AAVrh32.33 hybrid capsid vectors was mostly consistent with those from the small-scale yields observed in Fig. 2A. The titers of hybrids LM17 to LM20 were only ∼1 to 1.5 log units below wild-type titers in small-scale preparations, while the yields from the large-scale preparations of these hybrids ranged from being ∼2.5 to 5.5 log units below wild-type titers, with the yields for some being lower than the yields obtained from small-scale preparations and barely detectable above the background. This discrepancy likely indicates the instability of these hybrid capsids and their inability to withstand the purification process. Small-scale preparations for in vitro use were not subject to purification, while their large-scale counterparts underwent two rounds of cesium chloride density gradient centrifugation.
Hybrid LM16 also demonstrated a low final titer of 1.12 × 1010 GCs/ml, approximately 2.5 log units below the wild-type titer. This low yield was not predicted by the transfection yields for the small-scale preparations, in which the titer of this hybrid was comparable to that for the wild-type controls at 1.57 × 1011 GCs/ml (Fig. 2A). Hybrid LM16 is composed of the AAVrh32.33 backbone with HVR V from AAV8. HVR V of AAV8 contains a 5-amino-acid deletion relative to the AAVrh32.33 sequence. While in the inverse swap, LM15, AAV8 tolerates the insertion of those 5 amino acids from the AAVrh32.33 sequence, deletion of the HVR V residues in AAVrh32.33 in LM16 likely causes instability because residues interacting with HVR IV, with a conformation dramatically different from HVR IV of AAV8 (Fig. 1C), are deleted. This instability could result in loss of virus during purification. This is similar to what was seen with LM9 in both small- and large-scale preparations, where insertion of HVR I from AAVrh32.33 onto AAV8 resulted in a low transfection efficiency, likely due to the 5-amino-acid deletion in the AAVrh32.33 sequence relative to the AAV8 sequence.
While the majority of hybrids with broad and narrow swaps had low to undetectable, background-level yields, LM10 was successfully produced in both the broad- and narrow-swap large-scale preparations. While the titers of the small-scale preparation (without purification) were comparable between the broad- and narrow-swap versions of LM10 (Fig. 2A), following purification the yield of the narrow-swap version of the LM10 large-scale preparation was increased over that of the broad-swap version (4.46 × 1012 GCs/ml versus 1.27 × 1012 GCs/ml, respectively). This indicates that limiting the swapped domain to only the most surface-exposed, nonsuperimposable amino acids may have had a positive effect on the stability of the hybrid capsid, allowing the narrow-swap version to withstand purification better than its broad-swap version counterpart.
In vivo performance of AAV hybrid vectors in muscle of C57BL/6 mice.
Ultimately, only the VP12u hybrids and the rational design single HVR swaps were successfully produced and carried on to in vivo testing. Hybrid vectors with yields equal to or greater than 2 × 1012 GCs/ml (LM1, LM2, the LM10 narrow-swap version, and LM13 to LM15) were then injected at a dose of 1 × 1011 GCs into the muscle of C57BL/6 mice to determine their ability to activate transgene-specific T cell responses similar to those activated by AAVrh32.33 or to allow stable β-Gal expression, as with AAV8 (Fig. 3A). The broad-swap version of LM10 as well as LM11 and LM12 generated titers which were near or below 2 × 1012 GCs/ml; these hybrids were thus injected at a dose of 5 × 1010 GCs per mouse (Fig. 3A). nLacZ-specific CD8+ T cells were quantified by MHC-I tetramer staining at days 14, 21, and 28 postinjection, while X-Gal histochemical staining was performed on muscle sections at day 28 in order to assess the degree of cellular infiltration as well as the stability of β-Gal expression in the muscle. We hypothesized that it would be possible to map the domain or domains responsible for the differential immune activation phenotypes of AAV8 and AAVrh32.33. To do so, we were looking for a set of hybrids in which the immunophenotype had been exchanged: an AAV8 hybrid capable of generating a robust nLacZ-specific T cell response and loss of β-Gal expression over time and a reciprocal AAVrh32.33 hybrid achieving minimal immune activation and allowing stable transgene expression in the muscle.
Fig 3.

In vivo T cell activation and stability of β-Gal expression following intramuscular injection of broad and narrow AAV8-AAVrh32.33 hybrid capsid vectors. (A) C57BL/6 mice were injected i.m. with a dose of 1 × 1011 GCs of either wild-type AAV8 or AAVrh32.33 or the panel of hybrid capsid vectors expressing CB.nLacZ. *, the broad-swap versions of LM10, LM11, and LM12 were injected at a dose of 5 × 1010 GCs/mouse to compensate for limiting titers. The percentage of nLacZ-specific CD8+ T cells present in whole blood was assayed at days 14, 21, and 28 by MHC-I tetramer staining. At day 28, X-Gal histochemistry was performed on muscle sections to monitor for cellular infiltration and the stability of β-Gal expression. Representative images were taken from 4 mice per group. The tetramer stain data represent means ± SDs from 4 mice/group. (B) C57BL/6 mice injected with 5 × 1010 GCs of AAV2/8.nLacZ, AAV2/rh32.33.nLacZ, or LM10 (AAV2/rh32.33-8_VR.I hybrids) made with either narrow or broad swaps. Representative images were taken from 4 mice per group on day 10 postinjection. Magnifications, ×10.
The functional domain(s) maps to the VP3 portion of the AAV capsid.
The majority of primary sequence diversity between AAV serotypes lies within the VP3 portion of their three overlapping capsid VPs. This overlapping region assembles the capsid surface and contains the nine most surface-exposed regions, HVRs I to IX, defined when AAV2 and AAV4 were compared (19). Therefore, we initially hypothesized that the functional region responsible for augmenting or avoiding activation of a T cell response toward the transgene product would lie within VP3. To confirm this, we compared the phenotypes of our VP12u hybrids (LM1 and LM2) in the muscle of C57BL/6 mice. The results showed that AAVrh32.33 containing VP12u from AAV8 (LM2) behaved like AAVrh32.33 in its ability to prime a robust, polyfunctional nLacZ-specific T cell response capable of clearing transduced cells, while the reciprocal hybrid containing VP12u from AAVrh32.33 and VP3 from AAV8 (LM1) showed the phenotype of AAV8, with minimal T cell activation and stable β-Gal expression in the muscle of C57BL/6 mice (Fig. 3A). Hence, in vivo results confirmed the hypothesis that the phenotype of transgene-specific T cell activation and loss of β-Gal expression in the muscle is dictated by the VP3 portion of the capsid.
VP3 HVR I from AAV8 is not sufficient to induce tolerance to AAVrh32.33 transgenes.
Having successfully mapped the functional domain to the VP3 portion of the capsid, we next aimed to further delineate the region of interest within VP3, using our panel of hybrids to assess the role of HVRs I, II, IV, and V. As discussed above, due to the 5-amino-acid deletion in HVR I of AAVrh32.33 relative to the AAV8 sequence, neither the broad- nor narrow-swap version of LM9, in which this region is switched into AAV8, could be efficiently produced. Therefore, we were able to test only LM10, which has HVR I of AAV8 on AAVrh32.33, in vivo. Due to the lower yield obtained for the broad-swap version of LM10, a maximal dose of 5 × 1010 GCs/mouse was injected intramuscularly (i.m.). This resulted in little to no muscle transduction, characterized by no β-Gal-positive muscle fibers being observed at day 10 (Fig. 3B). In the absence of the β-Gal antigen in the muscle, antigen-specific CD8+ T cells could not be primed (Fig. 3A). Therefore, the lack of β-Gal expression in the muscle at day 28 is not due to clearance of transduced cells but instead is due to a lack of muscle transduction by the broad-swap version of LM10.
To provide an equal comparison of the LM10 broad- and narrow-swap versions, the narrow-swap version of LM10 was also injected i.m. at a dose of 5 × 1010 GCs. A comparison of β-Gal expression in the muscle at day 10 revealed that the narrow-swap version of LM10 was capable of transducing muscle fibers, unlike the broad-swap version, which showed no transduction (Fig. 3B). The presence of β-Gal antigen following transduction of the narrow-swap version of LM10 corresponded to the onset of transgene-specific CD8+ T cells at day 14 (data not shown). Therefore, in addition to enhancing capsid stability during large-scale purification, the hybrid with the narrow-swap design also showed improved in vivo transduction in the muscle.
Due to significantly higher large-scale yields for the narrow-swap version of LM10, we were also able to inject this hybrid at a dose of 1 × 1011 GCs/mouse. At this dose, the narrow-swap version of LM10 generated a robust, nLacZ-specific CD8+ T cell population, resulting in the clearance of β-Gal-expressing muscle fibers by day 28 (Fig. 3A). The delayed onset and slightly lower levels of CD8+ T cell activation in the narrow-swap version of LM10 compared to those for the wild type could indicate that the level or kinetics of muscle transduction were delayed compared to that in AAVrh32.33. This was observed at the lower dose of 5 × 1010 GCs, where β-Gal expression following transfection of the narrow-swap version LM10 was less than that for wild-type AAVrh32.33 at 10 days postinjection (Fig. 3B). In conclusion, the removal of HVR I from AAVrh32.33 did not ablate T cell activation and the addition of HVR I from AAVrh32.33 into AAV8 did not confer T cell activation. Therefore, HVR I alone does not contain the domain responsible for driving differential immune activation between AAV8 and AAVrh32.33.
VP3 HVR II does not contain the functional domain.
Hybrids LM11 and LM12, which contained the reciprocal swaps of VP3 HVR II, were also compared in vivo at the lower dose of 5 × 1010 GCs, due to limiting titers. It is important to note that the portion of HVR II swapped in these hybrids is considered broad. Because we were able to successfully test both reciprocal swaps in vivo at the lower dose, there was no need to create a narrow-swap version in an attempt to improve yields. However, the trend of lower large-scale titers after purification for broad-swap-version hybrids (compared with those for the narrow-swap versions) was confirmed by hybrids LM11 and LM12.
In terms of their immunophenotype, our results indicate that AAVrh32.33 containing HVR II from AAV8 (LM12) behaved like AAVrh32.33 in its ability to prime a robust, polyfunctional nLacZ-specific T cell response capable of clearing transduced cells. The reciprocal swap, AAV8 containing HVR II from AAVrh32.33 (LM11), showed the phenotype of AAV8, with minimal T cell activation and stable β-Gal expression in the muscle of C57BL/6 mice (Fig. 3A). The delayed onset of CD8+ T cell activation to LM12 and the lower level of β-Gal-positive fibers in the muscle of mice receiving LM11 reflect the lower injection dose of 5 × 1010 GCs administered to these mice. Overall, in vivo results from the reciprocal swaps indicate that VP3 HVR II alone is not responsible for the phenotype of transgene-specific T cell activation and loss of β-Gal expression in the muscle.
VP3 HVR V of AAVrh32.33 is not sufficient to support transgene-specific CD8+ T cell priming.
We next tested hybrid LM15, which is AAV8 containing HVR V of AAVrh32.33. (Due to the 5-amino-acid deletion in HVR V of AAV8 relative to the sequence of AAVrh32.33, the reciprocal swap, LM16, could not withstand large-scale purification to generate usable yields.) Comparable to the findings for wild-type AAV8, injection of LM15 into the muscle of C57BL/6 mice at a dose of 1 × 1011 GCs resulted in minimal transgene-specific T cell activation, allowing stable β-Gal expression at day 28 (Fig. 3A). These results indicate that AAVrh32.33's HVR V is not sufficient to augment the priming of CD8+ T cells to the vector transgene.
The addition of VP3 HVR IV from AAV8 confers stable β-Gal expression to AAVrh32.33 in muscle.
The final hybrids generated for in vivo testing were the reciprocal swaps of HVR IV. Following i.m. injection into C57BL/6 mice at a dose of 1 × 1011 GCs, LM13 (AAV8 expressing HVR IV from AAVrh32.33) demonstrated a phenotype similar to that of wild-type AAV8, including minimal nLacZ-specific CD8+ T cell activation and stable β-Gal expression through day 28 (Fig. 3A). This result indicates that HVR IV from AAVrh32.33 alone is not sufficient to allow this hybrid to reach the threshold required for T cell activation in response to the transgene product. In the reciprocal swap, LM14 (AAVrh32.33 expressing HVR IV of AAV8), wild-type levels of nLacZ-specific CD8+ T cells were detectable by MHC-I tetramer staining at 14 days postinjection. Interestingly, however, instead of increasing over time like wild-type AAVrh32.33, the percentage of nLacZ-specific CD8+ T cells progressively declined from day 14 through day 28 (Fig. 3A). This decrease in T cell frequency over time correlated with stable expression of β-Gal in the muscle (Fig. 4A). To allow a closer look at both expression and cellular infiltration, Fig. 4A and B show representative tissue sections from wild-type AAV8, AAVrh32.33, and LM14 at both ×4 and ×20 magnifications, respectively. Due to the unequal distribution of β-Gal-positive fibers throughout the muscle sections, the ×4 objective allows a better overall picture of the extent of positivity between groups, illustrating the notable difference in the stability of transgene expression between wild-type AAVrh32.33 and LM14 at days 28 and 48. With the ×20 magnification, it is clear that muscles into which AAV8 was injected contain little to no cellular infiltrates, in contrast to those into which AAVrh32.33 was injected, which show a robust degree of cellular infiltration present in the muscle at day 28. The LM14 hybrid (AAVrh32.33 containing HVR IV from AAV8) showed an increased degree of cellular infiltration over that observed with AAV8; however, it was markedly less than that observed with wild-type AAVrh32.33 and appeared to have no effect on the stability of β-Gal-positive fibers (Fig. 3). A detailed comparison of the phenotypes for AAV8, AAVrh32.33, and the LM14 hybrid is documented in Table 1. Slight preparation-to-preparation variations in the kinetics of T cell responses and β-Gal clearance were observed following the use of different AAVrh32.33 vector lots, where AAVrh32.33 showed a slightly less immunogenic phenotype in the muscle following production by PEI-MAX transfection and total lysate harvest (Table 1). However, the stability of transgene expression observed with AAV8 and the LM14 hybrid (AAVrh32.33-8_VR.IV) could not be achieved with any AAVrh32.33 vector preparations (Fig. 3 and Table 1). It is important to note that the phenotypic variation observed for AAVrh32.33 under various production and purification methods was not observed for other vectors, including AAV8. In both cases, AAV8 resulted in minimal T cell activation or cellular infiltration and stable β-Gal expression in muscle.
Fig 4.
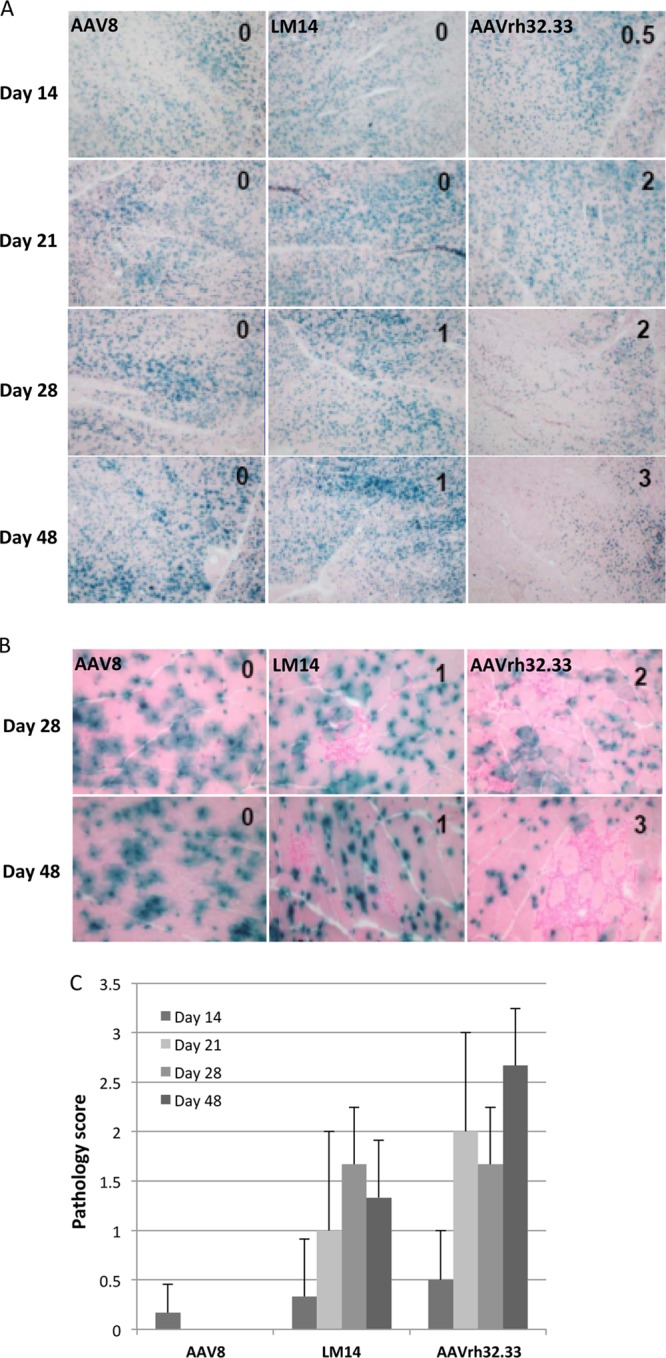
Time course of expression from AAV2/8, AAV2/rh32.33, and LM14 (AAV2/rh32.33-8_VR.IV). (A to C) C57BL/6 mice were i.m. injected with 1011 GCs of AAV2/8, AAV2/rh32.33, or LM14 (AAV2/rh32.33-8_VR.IV) expressing nLacZ. All vectors were produced using the identical production and purification method of PEI transfection on cell stacks, followed by iodixanol gradient purification. Muscles were harvested for X-Gal histochemistry on days 14, 21, 28, and 48. Infiltration in muscle sections was scored as normal (score, 0), mild (score, 1), moderate (score, 2), or severe (score, 3), and scores are given at the upper left of each image. (A) Images were taken to assess the levels and stability of β-Gal expression. Magnification, ×4. (B) Images were taken at days 28 and 48 to show cellular infiltration. Magnification, ×20. (C) Average pathology scores are plotted to compare the mean ± SD for each group.
Table 1.
Detailed comparison of AAV2/8, AAV2/rh32.33, and LM14a
| Construct | T cell onset | T cell kinetics | Infiltration | Expression |
|---|---|---|---|---|
| AAV8 | Little to none | None | Little to none | Stable |
| AVrh32.33 | ||||
| Old method | High at day 14 | Dramatic increase from days 14–21 | Severe | Cleared by day 64 |
| New method | High at day 14 | Minor increase from days 14–21 or remain the same | Moderate to severe | Decreased from days 21–48; low levels remain at day 105 |
| LM14 | High at day 14 | Decrease after day 14 | Mild to moderate | Slight decrease from days 48–64; moderate levels remain at day 105 |
Mice were i.m. injected with 1011 GCs of AAV2/8, AAV2/rh32.33, or LM14 (AAV2/rh32.33-8_VR.IV) expressing nLacZ. Vectors produced by the old method were produced via 40-plate transfection using CaCl2 and HBS, followed by vector harvest from the cell pellet and cesium gradient purification. In the new method, vectors were produced by PEI-MAX transfection on cell stacks, followed by total lysate harvest and iodixanol gradient purification. AAV8 and LM14 were produced by the new method. Muscles were harvested for histochemistry on days 14, 21, 28, 48, and 105. Infiltration in muscle sections was scored as normal (score, 0), mild (score, 1), moderate (score, 2), or severe (score, 3). T cell assays were performed on days 14, 21, and 28 postinjection.
To assess the quality of the T cell response to LM14 versus that to AAVrh32.33, intracellular cytokine staining was performed to determine the percentage of gamma interferon (IFN-γ) production by CD8+ T cells. We observed that the LM14 mutant did generate a reduced percentage of IFN-γ-positive (IFN-γ+) CD8+ T cells compared to that for AAVrh32.33, most notably on days 14 and 21 (Fig. 5). The kinetics of this response were also delayed in comparison to those for the control group. Taken together, these findings suggest that decreased T cell functionality may account for the phenotype observed. While nLacZ-specific CD8+ T cells are primed following LM14 delivery to muscle, a lower percentage of these CD8+ T cells is able to secrete IFN-γ. A lack of T cell functionality may prevent clearance of transduced cells, leading to T cell exhaustion in the presence of persistent antigen expression, and further studies will be necessary to test this hypothesis.
Fig 5.

Percentage of IFN-γ+ CD8+ T cells generated by AAV2/rh32.33 versus LM14 (AAV2/rh32.33-8_VR.IV). C57BL/6 mice were i.m. injected with 1011 GCs of AAV2/rh32.33 or LM14 (AAV2/rh32.33-8_VR.IV) expressing nLacZ. All vectors were produced using the identical production and purification method of PEI transfection on cell stacks, followed by iodixanol gradient purification. (A) Intracellular cytokine staining was performed to assess the percentage of IFN-γ+ CD8+ T cells at days 14, 21, 28, and 48. (B) Representative flow plots with the average ± SD for each group.
DISCUSSION
In addition to their functional diversity, AAV8 and AAVrh32.33 are two of the most phylogenetically and structurally diverse capsid variants within the Dependovirus genus of the Parvoviridae family (18–21). While the primary amino acid sequences of AAV2 and AAV8 differ by only 16%, the amino acid sequences of both AAV2 and AAV8 differ from the AAVrh32.33 amino acid sequence by over 32%. As a close homologue of AAV4, AAVrh32.33 is an engineered hybrid between natural rhesus isolates AAVrh32 and AAVrh33. It was found to be much less seroprevalent than AAV2, AAV7, and AAV8 (13), decreasing the problem of preexisting neutralizing antibodies to the capsid and making it a potential candidate for gene transfer. However, unlike AAV8, AAVrh32.33 vectors induce strong T cell responses to transgene products, which, while a beneficial feature for use in vaccine development (28), are not desired in gene therapy applications. Ultimately, by mapping the domain responsible for augmenting cellular immunity to AAVrh32.33 transgenes or inducing tolerance to AAV8-encoded proteins, those domains could be either removed from or inserted into the AAVrh32.33 capsid, respectively, resulting in an optimized vector with the combined properties of ablated cellular immunogenicity and low seroprevalence.
The production efficiencies of AAV hybrid capsids confirmed that domain swaps involving large regions of VP3 encompassing both conserved regions and HVRs could not produce viable hybrids between the structurally diverse AAV8 and AAVrh32.33. This finding was expected, given the low degree of VP3 sequence homology between AAV8 and AAVrh32.33 and our hypothesis that swapping large regions, without consideration of neighboring interactions, could disrupt the tertiary structure and prevent proper capsid assembly. The incorporation of rational design, swapping single or interacting HVRs, was more successful in the generation of viable hybrid vectors. As expected, swaps of single HVRs generated the highest titers, a result which was anticipated, as they exchanged the smallest amount of capsid VP, thus minimizing negative effects on folding and architecture. Hybrids with multiple-HVR swaps showed 1- to 1.5-log-unit decreases in titers compared to the wild type, indicating that the swapping of multiple HVRs simultaneously is not able to properly maintain capsid architecture and is therefore unable to preserve critical HVR interactions. The small- and large-scale production efficiencies of LM9 and LM16 demonstrated that swapping a domain that contains deletions relative to the backbone sequence has a negative impact on the ability of the capsid to properly assemble and successfully package the genome, resulting in either poor transfection efficiency or capsid instability during purification.
Recent studies have demonstrated that a virally encoded assembly-activating protein (AAP) is required to assist in AAV capsid assembly (29, 30). Mutation of the AAP-encoding sequence itself or regions of the capsid with which it interacts, for instance, in the C terminus of VP3, was able to disrupt capsid assembly (29). The AAPs from AAV1 and AAV2 were able to complement each other in the assembly of AAV1 and AAV2 capsids. However, assembly of the more divergent AAV5 had more specific requirements (30). This was supported by sequence alignments of predicted AAP proteins, where AAP1 and AAP2 shared a high degree of homology but AAP5 did not. The predicted AAP8 also shared a high degree of homology with AAP1 and AAP2, while AAP4 was divergent. As capsid variant AAVrh32.33 was the most closely related to AAV4, it is possible that the AAPs of AAVrh32.33 and AAV8 are not interchangeable, as seen with AAV5 and AAV1 or AAV2. If this is the case, swapping regions of the capsid that include the AAP sequence itself or regions interacting with the AAP may inhibit proper AAP function and capsid assembly. This may be another explanation for poor capsid assembly or stability in our hybrids. Further studies will be necessary to evaluate this point.
In terms of their transduction ability, hybrids generating titers of less than 5 × 1010 drp/ml were unable to significantly transduce HEK293 or U937 cells in vitro, despite cell transduction using the same quantity of vector. Of those with high enough yields, the ability to transduce U937 monocytes was retained only by wild-type AAVrh32.33 and hybrids constructed on the AAVrh32.33 background. It is important to note that in the fully assembled capsid particle, many HVRs interact with each other to form structures that may be functionally important. Therefore, it is possible that functional domains do not map entirely to one linear coordinate but that multiple regions throughout VP3 interact in the folded capsid to form a domain that is conformationally important. We attempted to address this possibility through the construction of rationally designed, multiple-HVR hybrids, which simultaneously swapped all HVRs localized to specific capsid regions, for example, HVRs IV, V, and VIII forming the 3-fold peaks. However, the yields of these hybrids in the small-scale experiments were not sufficient to allow further study. The observation that grafting single domains of AAVrh32.33 onto an AAV8 capsid did not increase U397 transduction is likely because transduction efficiency may require more complex structural interactions. What we have shown is that a single HVR from AAVrh32.33 is not sufficient for transduction, although it may still be necessary. Furthermore, APC transduction is not the only upstream event contributing to the priming of a functional T cell response, such as activation of innate immunity.
In vivo studies of VP12u hybrids (LM1 and LM2) confirmed that the functional domain mapped to the VP3 portion of the AAV capsid. Within VP3, swaps of HVR I, HVR II, and HVR V were not sufficient to transfer the phenotype between AAV8 and AAVrh32.33. From our studies comparing broad and narrow swaps of HVR I in hybrid LM10, we were able to conclude that making a structural determination of the most surface-exposed, nonsuperimposable residues within a given HVR loop is critical in optimizing hybrid design. Extending too far into the broad region of the HVR loop may begin to have a negative effect on both capsid stability and in vivo transduction. It is important to note, however, that additional evidence is needed to confirm whether the capsid is truly destabilized. As an alternative to stability, this phenotype may also be influenced by uncoating, or it could mean that an interaction involving HVR I was not possible because the correct disposition of amino acids for AAVrh32.33, for instance, amino acids affecting receptor attachment, was not present.
An interesting observation was made with hybrid LM14, where the addition of AAV8's HVR IV created an AAVrh32.33 vector capable of achieving stable β-Gal expression in the muscle. Because the hybrid with the reciprocal swap (LM13) did not gain the ability to generate T cells, we can conclude that either HVR IV of AAVrh32.33 is not involved in the generation of cellular immunity or this domain by itself is simply not sufficient to reach the threshold required to activate an immune response. There is likely redundancy in the structural domains contributing to this phenotype or a need for interaction between multiple HVRs, which leads us to several hypotheses to explain the behavior of hybrid LM14. First, insertion of the HVR IV loop of AAV8 could be inducing a mechanism of tolerance, such as regulatory T cell activation, which limits the effectiveness of the CD8+ effector T cells which are initially primed by domains within the AAVrh32.33 backbone. Removal of this domain from the reciprocal swap, LM13, may not completely break tolerance if the domain responsible for this phenotype involves more than one capsid surface loop. Second, this region of AAVrh32.33 may be responsible, in part, for some degree of innate immune activation. Removal of HVR IV from AAVrh32.33 could result in a decrease in innate activation such that the hybrid falls below a threshold required to generate a functional CD8+ T cell response capable of clearing transgene-expressing cells. In the absence of sufficient innate immune activation, T cell priming may not incorporate proper costimulation, resulting in a population of anergic T cells, which are deleted over time. Conversely, the reciprocal swap would then gain some degree of innate immune activation, but that may not be sufficient to reach the threshold required to prime CD8+ T cells.
Alternatively, a third hypothesis relies more on structural conjecture. When looking at the overlay of VP3 monomers for AAV8 and AAVrh32.33, we see that HVR IV of AAVrh32.33 falls inward, covering the gap between HVRs IV and VIII (Fig. 1C). This gap is where HVR V from an adjacent monomer will be positioned when two 3-fold symmetry-related monomers come together to form the peaks surrounding the 3-fold axis of symmetry. Therefore, our third hypothesis is that the removal of HVR IV from AAVrh32.33 opens up the gap between HVRs IV and VIII, allowing some of the less surface-exposed residues of AAVrh32.33, which were normally blocked, to become exposed. Exposure of these additional residues in AAVrh32.33 may account for an enhanced degree of transduction, resulting in a higher antigen load. Chronic, high-level antigen exposure is known to result in CD8+ T cell exhaustion, which is marked by an initial onset of CD8+ T cell priming but a progressive loss in the functionality of those T cells over time (31). Therefore, in this case we hypothesize that CD8+ T cells are being primed by LM14, but the greater degree of transduction and antigen expression is resulting in T cell exhaustion and those primed T cells successively lose their ability to secrete IFN-γ, tumor necrosis factor alpha, and IL-2 and eventually undergo deletion over time. Our findings that the percentage of IFN-γ+ CD8+ T cells is reduced in response to LM14 in comparison to that to AAVrh32.33 may support this hypothesis. Studies are under way to address each of these three hypotheses in more detail.
In conclusion, we attempted to map the structural domain responsible for driving differential immune activation toward the transgene product through the study of genetic chimeras. In this study, we confirmed that it is possible to create viable hybrids between two of the most structurally diverse AAV capsid variants within the family. Our findings have confirmed that this functional T cell-activating domain lies within the VP3 portion of the capsid, excluding specific HVRs I, II, and V. Interestingly, the addition of HVR IV, which is a candidate domain for conferring T cell responses, from AAV8 onto AAVrh32.33 results in an AAVrh32.33 vector capable of generating stable, high-level β-Gal expression in the muscle of C57BL/6 mice. Studies are under way to further characterize the dysfunctional T cell response elicited by this vector. Ultimately, understanding the contribution of capsid structure to the transgene immune response is crucial in determining which serotypes are the best candidates for safe and efficacious gene transfer. In this study, the generation of an AAVrh32.33-based vector with the combined properties of low seroprevalence and robust, stable transgene expression is promising for the field of muscle-directed gene therapy.
ACKNOWLEDGMENTS
We thank the Animal Models Core and Cell Morphology Core (University of Pennsylvania Gene Therapy Program) for help with the animal studies, processing of tissue samples, and histological staining. We also thank Penn Vector at the University of Pennsylvania for providing the vectors used in this research.
This work was funded by an SRA from ReGenX Biosciences (J.M.W.), NIH R01GM082946 (M.A.-M.), NIH T32AR053461 (L.E.M.), and T32AI007324 (L.E.M.).
J.M.W. is a consultant to ReGenX Holdings and is a founder of, holds equity in, and receives a grant from affiliates of ReGenX Holdings; in addition, he is an inventor on patents licensed to various biopharmaceutical companies, including affiliates of ReGenX Holdings.
Footnotes
Published ahead of print 29 June 2013
REFERENCES
- 1.Kotin RM. 1994. Prospects for the use of adeno-associated virus as a vector for human gene therapy. Hum. Gene Ther. 5:793–801 [DOI] [PubMed] [Google Scholar]
- 2.Atchison RW, Casto BC, Hammon WM. 1965. Adenovirus-associated defective virus particles. Science 149:754–756 [DOI] [PubMed] [Google Scholar]
- 3.Chiorini JA, Kim F, Yang L, Kotin RM. 1999. Cloning and characterization of adeno-associated virus type 5. J. Virol. 73:1309–1319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chiorini JA, Yang L, Liu Y, Safer B, Kotin RM. 1997. Cloning of adeno-associated virus type 4 (AAV4) and generation of recombinant AAV4 particles. J. Virol. 71:6823–6833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gao G, Alvira MR, Somanathan S, Lu Y, Vandenberghe LH, Rux JJ, Calcedo R, Sanmiguel J, Abbas Z, Wilson JM. 2003. Adeno-associated viruses undergo substantial evolution in primates during natural infections. Proc. Natl. Acad. Sci. U. S. A. 100:6081–6086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gao G, Vandenberghe LH, Alvira MR, Lu Y, Calcedo R, Zhou X, Wilson JM. 2004. Clades of adeno-associated viruses are widely disseminated in human tissues. J. Virol. 78:6381–6388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gao GP, Alvira MR, Wang L, Calcedo R, Johnston J, Wilson JM. 2002. Novel adeno-associated viruses from rhesus monkeys as vectors for human gene therapy. Proc. Natl. Acad. Sci. U. S. A. 99:11854–11859 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Melnick JL, Mayor HD, Smith KO, Rapp F. 1965. Association of 20-millimicron particles with adenoviruses. J. Bacteriol. 90:271–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mori S, Wang L, Takeuchi T, Kanda T. 2004. Two novel adeno-associated viruses from cynomolgus monkey: pseudotyping characterization of capsid protein. Virology 330:375–383 [DOI] [PubMed] [Google Scholar]
- 10.Muramatsu S, Mizukami H, Young NS, Brown KE. 1996. Nucleotide sequencing and generation of an infectious clone of adeno-associated virus 3. Virology 221:208–217 [DOI] [PubMed] [Google Scholar]
- 11.Rutledge EA, Halbert CL, Russell DW. 1998. Infectious clones and vectors derived from adeno-associated virus (AAV) serotypes other than AAV type 2. J. Virol. 72:309–319 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao G, Wang Q, Calcedo R, Mays L, Bell P, Wang L, Vandenberghe LH, Grant R, Sanmiguel J, Furth EE, Wilson JM. 2009. Adeno-associated virus-mediated gene transfer to nonhuman primate liver can elicit destructive transgene-specific T cell responses. Hum. Gene Ther. 20:930–942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Calcedo R, Vandenberghe LH, Gao G, Lin J, Wilson JM. 2009. Worldwide epidemiology of neutralizing antibodies to adeno-associated viruses. J. Infect. Dis. 199:381–390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mays LE, Vandenberghe LH, Xiao R, Bell P, Nam HJ, Agbandje-McKenna M, Wilson JM. 2009. Adeno-associated virus capsid structure drives CD4-dependent CD8+ T cell response to vector encoded proteins. J. Immunol. 182:6051–6060 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sanlioglu S, Monick MM, Luleci G, Hunninghake GW, Engelhardt JF. 2001. Rate limiting steps of AAV transduction and implications for human gene therapy. Curr. Gene Ther. 1:137–147 [DOI] [PubMed] [Google Scholar]
- 16.Hauck B, Xiao W. 2003. Characterization of tissue tropism determinants of adeno-associated virus type 1. J. Virol. 77:2768–2774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Xie Q, Bu W, Bhatia S, Hare J, Somasundaram T, Azzi A, Chapman MS. 2002. The atomic structure of adeno-associated virus (AAV-2), a vector for human gene therapy. Proc. Natl. Acad. Sci. U. S. A. 99:10405–10410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nam HJ, Lane MD, Padron E, Gurda B, McKenna R, Kohlbrenner E, Aslanidi G, Byrne B, Muzyczka N, Zolotukhin S, Agbandje-McKenna M. 2007. Structure of adeno-associated virus serotype 8, a gene therapy vector. J. Virol. 81:12260–12271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Govindasamy L, Padron E, McKenna R, Muzyczka N, Kaludov N, Chiorini JA, Agbandje-McKenna M. 2006. Structurally mapping the diverse phenotype of adeno-associated virus serotype 4. J. Virol. 80:11556–11570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lochrie MA, Tatsuno GP, Christie B, McDonnell JW, Zhou S, Surosky R, Pierce GF, Colosi P. 2006. Mutations on the external surfaces of adeno-associated virus type 2 capsids that affect transduction and neutralization. J. Virol. 80:821–834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wu P, Xiao W, Conlon T, Hughes J, Agbandje-McKenna M, Ferkol T, Flotte T, Muzyczka N. 2000. Mutational analysis of the adeno-associated virus type 2 (AAV2) capsid gene and construction of AAV2 vectors with altered tropism. J. Virol. 74:8635–8647 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horton RM, Cai ZL, Ho SN, Pease LR. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 8:528–535 [PubMed] [Google Scholar]
- 23.Wang L, Calcedo R, Nichols TC, Bellinger DA, Dillow A, Verma IM, Wilson JM. 2005. Sustained correction of disease in naive and AAV2-pretreated hemophilia B dogs: AAV2/8-mediated, liver-directed gene therapy. Blood 105:3079–3086 [DOI] [PubMed] [Google Scholar]
- 24.Krissinel E, Henrick K. 2004. Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensions. Acta Crystallogr. D Biol. Crystallogr. 60:2256–2268 [DOI] [PubMed] [Google Scholar]
- 25.Emsley P, Cowtan K. 2004. Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60:2126–2132 [DOI] [PubMed] [Google Scholar]
- 26.Bell P, Limberis M, Gao G, Wu D, Bove MS, Sanmiguel JC, Wilson JM. 2005. An optimized protocol for detection of E. coli beta-galactosidase in lung tissue following gene transfer. Histochem. Cell Biol. 124:77–85 [DOI] [PubMed] [Google Scholar]
- 27.Pulicherla N, Shen S, Yadav S, Debbink K, Govindasamy L, Agbandje-McKenna M, Asokan A. 2011. Engineering liver-detargeted AAV9 vectors for cardiac and musculoskeletal gene transfer. Mol. Ther. 19:1070–1078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li X, Cao H, Wang Q, Di B, Wang M, Lu J, Pan L, Yang L, Mei M, Pan X, Li G, Wang L. 2012. Novel AAV-based genetic vaccines encoding truncated dengue virus envelope proteins elicit humoral immune responses in mice. Microbes Infect. 14:1000–1007 [DOI] [PubMed] [Google Scholar]
- 29.Naumer M, Sonntag F, Schmidt K, Nieto K, Panke C, Davey NE, Popa-Wagner R, Kleinschmidt JA. 2012. Properties of the adeno-associated virus assembly-activating protein. J. Virol. 86:13038–13048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sonntag F, Kother K, Schmidt K, Weghofer M, Raupp C, Nieto K, Kuck A, Gerlach B, Bottcher B, Muller OJ, Lux K, Horer M, Kleinschmidt JA. 2011. The assembly-activating protein promotes capsid assembly of different adeno-associated virus serotypes. J. Virol. 85:12686–12697 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wherry EJ, Blattman JN, Murali-Krishna K, van der Most R, Ahmed R. 2003. Viral persistence alters CD8 T-cell immunodominance and tissue distribution and results in distinct stages of functional impairment. J. Virol. 77:4911–4927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mays LE, Wang L, Lin J, Bell P, Crawford A, Wherry EJ, Wilson JM. 19 June 2013. AAV8 induces tolerance in murine muscle as a result of poor APC transduction, T cell exhaustion and minimal MHCI upregulation on target cells. Mol. Ther. 10.1038/mt.2013.134 [DOI] [PMC free article] [PubMed] [Google Scholar]