

Abstract
Human cytomegalovirus (HCMV) is a significant human pathogen that achieves lifelong persistence by establishing latent infections in undifferentiated cells of the myeloid lineage, such as CD34+ hematopoietic progenitor cells. When latency is established, viral lytic gene expression is silenced in part by a cellular intrinsic defense consisting of Daxx and histone deacetylases (HDACs) because pp71, the tegument transactivator that travels to the nucleus and inactivates this defense at the start of a lytic infection in differentiated cells, remains in the cytoplasm. Because the current in vitro and ex vivo latency models have physiological and practical limitations, we evaluated two CD34+ myeloblastic cell lines, KG-1 and Kasumi-3, for their ability to establish, maintain, and reactivate HCMV experimental latent infections. Tegument protein pp71 was cytoplasmic, and immediate-early (IE) genes were silenced as in primary CD34+ cells. However, in contrast to what occurs in primary CD34+ cells ex vivo or in NT2 and THP-1 in vitro model systems, viral IE gene expression from the laboratory-adapted AD169 genome was not induced in the presence of HDAC inhibitors in either KG-1 or Kasumi-3 cells. Furthermore, while the clinical strain FIX was able to reactivate from Kasumi-3 cells, AD169 was not, and neither strain reactivated from KG-1 cells. Thus, KG-1 and Kasumi-3 experimental latent infections differ in important parameters from those in primary CD34+ cell populations. Aspects of latency illuminated through the use of these myeloblastoid cell lines should not be considered independently but integrated with results obtained in primary cell systems when paradigms for HCMV latency are proposed.
INTRODUCTION
The prototypic betaherpesvirus, human cytomegalovirus (HCMV), is a significant worldwide pathogen infecting the majority of the population (1). Infection is subclinical in most cases but can have severe consequences in immunocompromised or immunologically naive individuals, such as AIDS patients, transplant recipients, and neonates (1, 2). Contributing to the success of this pathogen, HCMV establishes latent infections allowing for persistence in the face of robust antiviral immune responses and thus maintains a lifelong presence in its host (1, 3). HCMV establishes latency in undifferentiated cells of the myeloid lineage (4–9). Because viral DNA, but no evidence of productive replication, has been detected in peripheral blood monocytes and in the CD34+ hematopoietic progenitor cells (HPCs) from which they are derived (7, 10), it is thought that a CD34+ HPC represents at least one in vivo latent reservoir (4, 7). Therefore, primary CD34+ cell populations are currently the model of choice to study HCMV latency since known parameters of chromatin structure, viral gene repression and expression, and the differentiation dependence of reactivation are indistinguishable between natural and experimental latent infections of primary CD34+ cells. In contrast to a lytic infection in which the majority of the viral genome is transcribed in a temporally regulated gene expression cascade, transcription during natural or experimental infection of CD34+ HPCs is restricted to a limited number of loci (11). Importantly, the immediate-early (IE) genes that promote productive, lytic infection are silenced during both the establishment and maintenance of latency (1, 8, 9).
Latent virus retains the capacity to animate, or begin the expression of, lytic-phase genes (12–14), eventually leading to productive reactivation, which is a completion of the lytic replication program that allows further dissemination within and between hosts. Reactivation correlates with a change in the differentiation state of the infected cell (9) and is observed upon ex vivo terminal differentiation of either naturally (15) or experimentally (16) infected CD34+ HPCs into macrophages or dendritic cells. There is currently no efficacious vaccine for HCMV. Although antivirals that treat lytic infection exist (17), no treatment is able to target latent infections. Like primary infection, reactivation is associated with HCMV disease (1); thus, an understanding of the mechanisms underlying latency is a key step toward identifying novel therapies that attack this important aspect of the viral life cycle.
While viral genetic requirements for latency are emerging (18), molecular mechanisms that govern the establishment, maintenance, animation, or reactivation of HCMV latency remain poorly understood. One exception is the correlation between the chromatin structure of the viral major immediate-early promoter (MIEP) and the propensity for lytic-phase gene expression (19). During latency when lytic-phase genes, such as IE1, are silenced, the MIEP driving IE1 expression is associated with unacetylated histones, resembling transcriptionally silent heterochromatin (15, 16, 20). Following reactivation, when IE1 is expressed, histones associated with the MIEP are acetylated, resembling transcriptionally active euchromatin (15, 16). This mechanistically parallels the onset of lytic infection where, prior to IE gene expression, viral genomes show heterochromatic features, whereas later, when IE genes are being expressed, they are euchromatic (21–23). What initiates this switch during the transition from latency to reactivation is not known; however, the triggering step at the start of lytic infection is relatively well understood.
Silencing of the MIEP during both lytic and latent infection is accomplished in part by a cellular intrinsic immune defense mediated by proteins that localize to promyelocytic leukemia nuclear bodies (PML-NBs) such as Daxx, ATRX, PML, and Sp100 (24–33). PML-NB proteins localize with incoming viral genomes and are thought to recruit chromatin-remodeling factors including histone deacetylases (HDACs) to promote the formation of restrictive chromatin structure at the MIEP (34–37). The heterochromatin that forms at the MIEP is, for the parameters analyzed, indistinguishable from that formed at the start of a lytic infection, during natural latency in CD34+ HPCs analyzed ex vivo, or during experimental latency generated by infecting primary CD34+ HPCs in vitro.
During lytic infection, HCMV initiates the neutralization of the PML-NB intrinsic defense through delivery of the tegument protein pp71 (38). Upon entry into terminally differentiated cell types, tegument-delivered pp71 localizes to the nucleus, where it displaces the chromatin-remodeling factor ATRX and induces the proteasomal degradation of Daxx, allowing for derepression of the MIEP and initiation of IE gene expression (25, 28, 39). IE1 subsequently neutralizes PML and Sp100, further enhancing lytic gene expression (30, 31, 40). In contrast, during latent or quiescent infections, the PML-NB-mediated intrinsic defense is not overcome because tegument-delivered pp71 fails to reach the nucleus (27–29). Artificial inactivation of this defense by either depletion of Daxx or treatment with small-molecule inhibitors of HDACs relieves repression of IE gene expression when the AD169 laboratory strain is used for infection (27–29). Clinical strains FIX and TB40/E are resistant to HDAC inhibitor-mediated rescue of IE gene expression at the start of latent infections, indicating that, in addition to the cellular intrinsic defense, at least one other restriction to viral IE gene expression exists during latency (29).
Although much progress has been made toward understanding virus-host interactions during HCMV lytic infection, detailed latency studies have been complicated by the lack of tractable cellular and animal models. The extremely low frequency of latently infected cells in vivo (about 1 in 10,000 peripheral blood mononuclear cells) makes it difficult to obtain sufficient cell numbers to perform detailed latency studies using naturally infected CD34+ HPCs (41) even if loss of MRP1 in latently infected CD34+ HPCs is a means by which to sort or select cells naturally harboring latent virus, as was recently proposed (42). Furthermore, naturally latently infected cells cannot be used to answer questions regarding the initial events of latency (establishment), nor are they amenable to genetic manipulation. Thus, to date, HCMV latency has been most often studied using in vitro or ex vivo experimental infection of cell lines or primary cell models, respectively. In vitro infection of the THP-1 and NT2 cell lines has proven useful for studying early events of latency (20, 27, 39, 43–47); however, whether HCMV can truly reactivate from these cells remains an area of contention. In contrast, ex vivo experimental infection of primary CD34+ HPCs as well as CD14+ monocytes has been reported to support all stages of latency, including establishment, maintenance, animation, and reactivation, and has thus been used extensively to study HCMV latency (15, 16, 18, 29, 48–51). Although more clinically relevant than in vitro models of quiescence, primary cell models infect poorly ex vivo, and their heterogeneous nature, coupled with the difficulty associated with maintaining them in an undifferentiated state in culture, limits the utility of these models, particularly for studying long-term aspects of HCMV latency (29).
With the hope of complementing the current in vitro and ex vivo systems, we along with others (52) have sought to develop immortalized human CD34+ cell lines as tractable long-term models with which to study HCMV latency and reactivation. KG-1 and Kasumi-3 are two such cell lines derived from independent acute myeloid leukemias (53, 54). KG-1 and Kasumi-3 cells offer attractive advantages over primary cell models because they can be maintained in an undifferentiated state in culture, represent a more consistent genetic background, and present the potential to construct constitutive knockdown or overexpression cell lines. Like primary HPCs, both KG-1 and Kasumi-3 cells can be induced to differentiate along the myeloid lineage (53–55). Furthermore, it has recently been reported that HCMV is able to reactivate from Kasumi-3 cells following cellular differentiation induced by the phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA) (52).
Here, we show that HCMV is able to enter both KG-1 and Kasumi-3 cells and that although the genome is maintained in these cells, viral lytic gene expression is not initiated. Consistent with what has been observed in other models of HCMV latency (29) and quiescence (27), tegument-delivered pp71 fails to localize to the nucleus of undifferentiated KG-1 and Kasumi-3 cells. In contrast, prior differentiation of these cell lines allows for nuclear localization of pp71 and the initiation of lytic gene expression. Additionally, the major PML-NB proteins ATRX, Daxx, PML, and Sp100 are present in these cells and thus available to restrict IE gene expression. However, inhibition of HDACs failed to prevent silencing of AD169 viral IE gene expression in KG-1 and Kasumi-3 cells, in contrast to its effect in primary CD34+, NT2, and THP-1 cells. Furthermore, reactivation of latent virus was not observed upon differentiation of infected KG-1 cells and was detectable only following differentiation of Kasumi-3 cells infected with a clinical, but not a laboratory-adapted, strain of HCMV. Overall, our data suggest that the KG-1 and Kasumi-3 cell lines model some, but not all, aspects of HCMV experimental latency. Thus, their utility mirrors that of NT2 and THP-1 cells, where the ease of experimentation comes at the cost of physiologic relevance.
MATERIALS AND METHODS
Cells and viruses.
KG-1 (ATCC catalog number CCL-246) and Kasumi-3 cells (ATCC catalog number CRL-2725) were maintained in Iscove's modified Dulbecco's medium (IMDM) and RPMI 1640 medium (Invitrogen), respectively, supplemented with 20% (vol/vol) fetal bovine serum (FBS). Normal human dermal fibroblasts (NHDFs; Clonetics) and 293Y cells were cultured in Dulbecco's modified Eagle medium (DMEM) (Invitrogen) supplemented with 10% (vol/vol) FBS. THP-1 monocytes (ATCC catalog number TIB-202) and 721 B cells (a gift from Bill Sugden, University of Wisconsin—Madison) were cultured in RPMI 1640 medium supplemented with 10% (vol/vol) FBS. Medium was also supplemented with 100 U/ml penicillin, 100 μg/ml streptomycin, and 0.292 mg/ml glutamine (Invitrogen). Embryonic stem cells (ESCs; WA09) were cultured as described previously (56). Primary human CD34+ hematopoietic progenitor cells derived from cord blood (Lonza) were maintained in hematopoietic progenitor growth medium (HPGM; Lonza) supplemented with 25 ng/ml recombinant human stem cell factor (SCF), 50 ng/ml recombinant human thrombopoietin (TPO), and 50 ng/ml recombinant human Fms-related tyrosine kinase 3 ligand (Flt3) (all from PeproTech Inc.), as previously described (29). To induce differentiation of KG-1 and Kasumi-3 cells, cells were treated with 10 ng/ml 12-O-tetradecanoylphorbol-13-acetate (TPA; Sigma) and 500 ng/ml ionomycin (Sigma) in X-VIVO-15 medium (Lonza) for 3 days, as previously described for KG-1 cells (55). HCMV viral strains used were AD169 and derivatives expressing IE2 or pp65 fused to green fluorescent protein (GFP) (57, 58) and GFP-expressing FIX (59). Cells were infected with HCMV in minimal volume for 60 min, followed by addition of medium to normal culture volumes. A recombinant adenovirus expressing pp71 from the EF1α promoter has been described previously (27).
Inhibitors and antibodies.
The following inhibitors were added at the time of infection with HCMV: valproic acid (VPA) (1 mM; Sigma) or sodium butyrate (8 mM; Sigma) dissolved in water or trichostatin A (TSA) (100 ng/ml; Upstate), suberoylanilide hydroxamic acid (SAHA) (500 nM; Sigma), or roscovitine (25 mM; Calbiochem) dissolved in dimethyl sulfoxide (DMSO). The following antibodies were from commercial sources: phycoerythrin (PE)-conjugated anti-CD34 (581) and allophycocyanin (APC)-conjugated anti-CD45 (HI30) or anti-CD38 (HIT2) from BD Pharmingen; anti-ATRX (H-300), anti-PML (H-238 and PG-M3), and anti-histone H3 (FL-136) from Santa Cruz Biotechnology; anti-Daxx (D7810) and anti-tubulin (DM 1A) from Sigma; anti-retinoblastoma protein (Rb; 4H1) and anti-phosphorylated Rb (Ser807/811) (9308) from Cell Signaling; anti-Sp100 (AB1380; Chemicon), anti-UL44 (CA006-100; Virusys), and anti-acetylated histone H3 lysine 4 (17-10050; Millipore). Monoclonal antibodies against pp71 (IE-233), IE1 (1B12), and pp28 (CMV157) have been described previously (28). Secondary antibodies used were Alexa Fluor 488 and 594 (Molecular Probes) for immunofluorescence and either horseradish peroxidase (HRP)-conjugated (Chemicon) or IRDye 680- and 800-conjugated (Li-Cor) secondary antibodies for Western blotting.
Flow cytometry.
Cells were collected by low-speed centrifugation, washed once in cold phosphate-buffered saline (PBS)–2% FBS, and incubated with appropriate conjugated antibodies diluted in PBS–2% FBS for 30 min on ice. Cells were then washed twice with cold PBS–2% FBS and fixed in 1% paraformaldehyde for 45 min at room temperature. Stained and unstained cell populations were analyzed using a FACSCalibur or LSRII flow cytometer (BD Biosciences) with FlowJo, version 9.4, software.
Indirect immunofluorescence.
Adherent NHDFs were grown on glass coverslips and washed in PBS prior to fixation. Nonadherent KG-1 and Kasumi-3 cells were collected by low-speed centrifugation, washed in cold PBS, and allowed to attach to water-washed coverslips for 1 h at room temperature. Alternatively, KG-1 and Kasumi-3 cells were differentiated on coverslips prior to infection. Cells were fixed in 1% paraformaldehyde in PBS and processed as previously described (28). Images were taken using either a Zeiss Axiovert 200 M or Fluoview FV1000 fluorescence microscope with a 60× objective. Images were cropped and processed using ImageJ and Adobe Photoshop CS5.1 software.
Western blotting.
Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer with protease inhibitors (see Fig. 3) or 1% SDS (see Fig. 6), and equivalent amounts of total protein were separated by SDS-PAGE and transferred to Optitran membranes (GE Healthcare). Western blots were processed as previously described (28) (see Fig. 3) or as follows (see Fig. 6): membranes were blocked with Odyssey blocking buffer (Li-Cor) and incubated with primary antibody diluted in blocking buffer plus 0.2% Tween 20. Membranes were washed with TBST (100 mM Tris, pH 8.0, 1.5 M NaCl, 0.5% Tween 20), followed by incubation with IRDye-conjugated secondary antibody diluted in blocking buffer plus 0.2% Tween 20. Following incubation, membranes were washed again in TBST and imaged using an Odyssey Fc Imager and Image Studio, version 2.1.10, software (Li-Cor).
Fig 3.
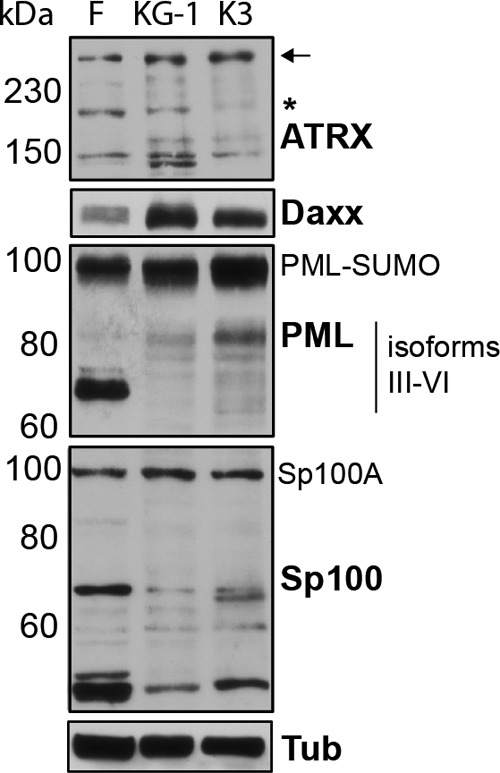
KG-1/Kasumi-3 express PML-NB proteins. Equal amounts of total protein from cell lysates of normal human dermal fibroblasts (F), KG-1, and Kasumi-3 (K3) cells were analyzed by Western blotting with antibodies specific for the indicated PML-NB components. Approximate protein sizes in kDa are indicated on the left. Tubulin (Tub) served as a loading control. Arrow and asterisk indicate the full-length ATRX and an alternative splice variant, respectively. The PML band at 100 kDa likely represents sumoylated (SUMO) PML.
Fig 6.
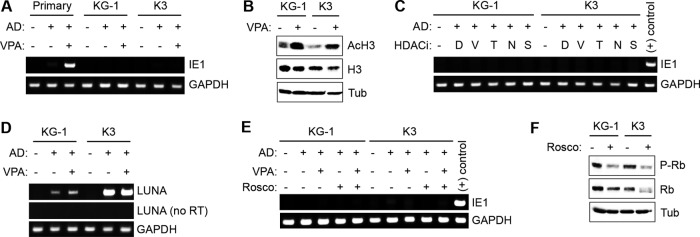
Inhibition of histone deacetylases does not enhance viral IE gene expression in KG-1 and Kasumi-3 cells. (A) Primary CD34+ (primary), KG-1, and Kasumi-3 (K3) cells were mock treated (−) or treated with VPA (+) and either mock infected (−) or infected (+) with AD169 (AD) at an MOI of 1. After 24 h, total RNA was extracted and analyzed by RT-PCR for expression of IE1. The cellular gene GAPDH served as an internal control. (B) KG-1 and K3 cells were mock treated (−) or treated with VPA (+) for 24 h. Cell lysates were analyzed by Western blotting for acetylated histone H3 (AcH3) and total histone H3 (H3). Tubulin (Tub) served as a loading control. (C) KG-1 and K3 cells were mock treated (−) or treated with DMSO (D) or HDAC inhibitors (HDACi) VPA (V), TSA (T), sodium butyrate (N), or SAHA (S) and either mock infected (−) or infected with AD169 (+) as described for panel A. Total RNA was extracted and analyzed for expression of IE1 and GAPDH as described for panel A. RNA from primary CD34+ HPCs infected with AD169 in the presence of VPA served as a positive control for the IE1 PCR. (D) KG-1 and K3 cells were infected as described for panel A, and RNA was extracted and analyzed by RT-PCR for LUNA expression with or without (no RT) reverse transcriptase. (E) KG-1 and K3 cells were mock treated (−) or treated with VPA (+) and/or roscovitine (Rosco) (+) and either mock infected (−) or infected (+) with AD169 as described for panel A. Total RNA was extracted and analyzed as described for panel A. RNA from primary CD34+ HPCs infected with AD169 in the presence of VPA served as a positive control. (F) Cell lysates from KG-1 and K3 cells mock treated (−) or treated with roscovitine (+) for 24 h were analyzed by Western blotting for phosphorylated Rb (P-Rb) and total Rb (Rb). Tubulin (Tub) served as a loading control.
Transductions and transfections.
Recombinant adenovirus transductions were performed at the indicated particle per cell ratios as previously described (27, 29). KG-1 cells were transfected with 1 μg of pSG5-pp71 or pSG5-pp71 Did2-3 DNA per 5 × 105 cells using an Amaxa Nucleofector Kit V (Lonza) according to the manufacturer's instructions.
PCR and RT-PCR.
For DNA analysis, total DNA was isolated using a Genomic DNA minikit (catalog number IB47202; IBI). DNA was amplified by PCR using a GoTaq Flexi system (M8295; Promega). For RNA analysis, total RNA was isolated using a Total RNA minikit (catalog number IB147323; IBI). RNA was quantified, and equal amounts of total RNA were treated with DNase I (M6101; Promega) and used for reverse transcription-PCR (RT-PCR; 35 to 40 cycles) using a Superscript III one-step RT-PCR system (catalog number 12574-026; Invitrogen). Primer pairs for IE1 that span the intron between exon 2 and exon 3 (29), glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (60), and LUNA (latency unique nuclear antigen) (61) have been described previously. PCR products were separated on 1.5% agarose gels, and bands were imaged and quantified using an Odyssey Fc imager with Image Studio, version 2.1.10, software (Li-Cor).
Reactivation assays.
KG-1 and Kasumi-3 cells were infected with AD169-IE2-GFP or FIX-GFP at a multiplicity of infection (MOI) of 3. After 10 days, cells were collected by low-speed centrifugation and treated with trypsin (0.5 mg/ml) for 5 min at 37°C. Cells were then returned to normal culture conditions or treated with 10 ng/ml TPA and 500 ng/ml ionomycin in X-VIVO-15 medium. After 2 days, TPA and ionomycin were washed out, and cells were cultured in DMEM supplemented with 10% FBS for an additional 3 days. Cells were then treated with trypsin and cocultured with NHDFs in DMEM. As controls, an equal number of untreated cells were lysed by sonication and plated on NHDFs. Fibroblasts were monitored for the formation of GFP-positive (GFP+) centers over the course of 2 to 3 weeks. Images were taken using a Nikon Eclipse Ti fluorescence microscope with a 10× objective. Images were cropped and processed using Adobe Photoshop CS5.1 software.
RESULTS
KG-1/Kasumi-3 cells remain CD34+ in culture and permit HCMV entry.
A significant barrier to the use of primary CD34+ HPCs for studying long-term aspects of HCMV latency is the inability to maintain them in an undifferentiated state ex vivo. For example, primary CD34+ populations rapidly differentiate in culture, with typically less than 50% of the population remaining CD34+ after 10 days (29, 49). In contrast, flow cytometry for cell surface expression (Fig. 1A) indicated that, after at least 25 days in culture, greater than 99% of KG-1 and 98% of Kasumi-3 cells remained CD34+ (Fig. 1B). Thus, KG-1 and Kasumi-3 cells overcome one impeding aspect of using primary CD34+ HPCs for HCMV latency studies. As these cells permitted the entry of both representative laboratory-adapted (AD169) and clinical (FIX) strain viruses (Fig. 1C), we analyzed these infections for other known parameters of experimental latency as defined in primary CD34+ HPCs (18, 29, 49, 62).
Fig 1.

KG-1/Kasumi-3 cells remain CD34+ in culture and permit HCMV entry. (A and B) KG-1, Kasumi-3 (K3), or THP-1 monocytes were maintained in culture for at least 25 days and then fixed, stained for CD34 and CD45 (a marker of nucleated hematopoietic cells), and analyzed by flow cytometry. (A) Representative plots depicting expression of CD34 (vertical axis) and CD45 (horizontal axis), with the percentage of cells in each quadrant indicated. (B) Quantification of the average percentage of cells staining positive for CD34. Bars show standard error. (C) KG-1 and K3 cells were infected with AD169 (AD) (MOI of 3) or FIX (MOI of 1). After 24 h, cells were treated with trypsin, fixed, and stained with an antibody against pp71. The average percentage of cells staining positive for pp71 from at least three independent experiments is shown. Bars show standard deviation.
KG-1/Kasumi-3 cells and their differentiated derivatives show differences in the subcellular localization of HCMV tegument-delivered proteins.
Following viral entry, tegument-delivered pp71 migrates to the nucleus of differentiated dendritic cells but remains in the cytoplasm of undifferentiated primary CD34+ HPCs (29). Similarly, we found tegument-delivered pp71 in the cytoplasm of KG-1 and Kasumi-3 cell lines after infection with either AD169 (Fig. 2A) or FIX (Fig. 2B). Another tegument protein, pp65, colocalized with pp71 in the cytoplasm (Fig. 2A), raising the possibility that sequestration of tegument-delivered proteins in the cytoplasm may reflect a general defect in tegument disassembly following entry into incompletely differentiated cell types. In contrast, prior differentiation of KG-1 or Kasumi-3 cells into dendritic-like cells allowed for nuclear localization of tegument-delivered pp71 (Fig. 2C). Thus, the differences in localization of tegument-delivered proteins observed in primary CD34+ HPCs, where latency is established, compared with their differentiated dendritic cell derivatives, where productive, lytic replication initiates upon de novo infection, are also observed in the undifferentiated/differentiated pairs of KG-1 and Kasumi-3 cell lines.
Fig 2.
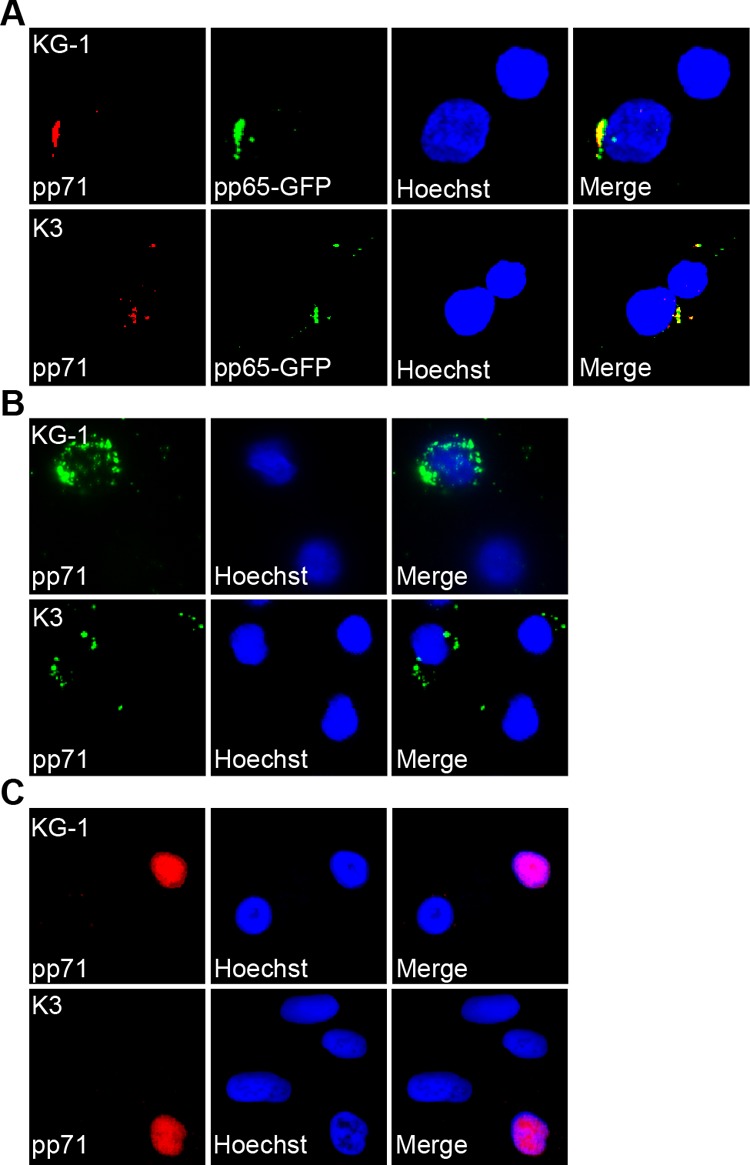
KG-1/Kasumi-3 and their differentiated derivatives show differences in the subcellular localization of HCMV tegument-delivered proteins. (A) KG-1 and Kasumi-3 (K3) cells were infected with a recombinant strain of AD169 expressing pp65 fused to GFP at an MOI of 3. After 24 h, cells were treated with trypsin, fixed, stained with an antibody against pp71, and imaged with fluorescence microscopy. (B) KG-1 and K3 cells were infected with FIX at an MOI of 1 and processed as described for panel A. (C) KG-1 and K3 cells were treated with TPA and ionomycin for 72 h to induce differentiation and then infected with AD169 at an MOI of 2 for 24 h. Cells were then fixed, stained, and imaged as above. In all panels, nuclei were counterstained with Hoechst.
KG-1/Kasumi-3 PML-NB component Daxx is targeted by pp71.
In all cell types studied, HCMV gene expression is initially silenced by a cellular intrinsic defense mediated by proteins that localize to PML-NBs, such as ATRX, Daxx, PML, and Sp100. All four of these proteins are expressed in KG-1 and Kasumi-3 cells (Fig. 3). KG-1 and Kasumi-3 cells display high Daxx levels and altered accumulation of specific ATRX, PML, and Sp100 isoforms compared to fibroblasts. Such differences were also observed in primary CD34+ cells (29). These proteins formed PML-NBs in KG-1 and Kasumi-3 cells as determined by indirect immunofluorescence microscopy (Fig. 4A to C).
Fig 4.

KG-1/Kasumi-3 cells contain PML-NBs that are modulated by de novo expressed pp71. (A to C) Actively growing normal human dermal fibroblasts (NHDFs), KG-1, and Kasumi-3 (K3) cells were fixed and stained with antibodies for PML-NB components PML and either Sp100 (A), ATRX (B), or Daxx (C) and then imaged with indirect immunofluorescence microscopy. PML is used as a marker for PML-NBs. (D) KG-1 and K3 cells were transduced with a recombinant adenovirus expressing pp71 under the control of the EF1α promoter at 1,000 particles per cell. At 48 h postransduction, cells were fixed, stained, and imaged as above. (E) KG-1 cells were transfected with a plasmid expressing wild-type pp71 or a pp71 mutant unable to bind Daxx (Did2-3). After 24 h, cells were fixed, stained, and imaged as above. In all panels, nuclei were counterstained with Hoechst.
PML and Sp100 were predominately punctate in KG-1 and Kasumi-3 cells and colocalized at PML-NBs (Fig. 4A). ATRX was also punctate and partially colocalized with PMLs although some cells also displayed diffuse nuclear staining (Fig. 4B). Interestingly, while Daxx staining in fibroblasts was punctate and predominately colocalized with PML staining, Daxx staining was diffuse in the nuclei of both KG-1 and Kasumi-3 cells (Fig. 4C). This observation agrees with what was observed in a subset of primary CD34+ cells (29) and may be the result of increased levels of Daxx oversaturating binding sites in PML-NBs. Interestingly, association with PML at PML-NBs inhibits Daxx-mediated transcriptional repression (63), suggesting that the diffuse Daxx present in KG-1 and Kasumi-3 cells may represent a more potent transcriptional repressor.
In the nucleus, pp71 initiates the inactivation of the intrinsic defense and the subsequent activation of viral productive (lytic)-phase gene expression by binding to and ultimately degrading Daxx. In contrast to the different localizations of tegument-delivered pp71 observed in undifferentiated and differentiated cells, de novo expressed pp71 localizes to the nucleus of all cell types tested to date (27, 29). Consistent with these previous observations, the de novo synthesized protein localized to the nucleus of both KG-1 and Kasumi-3 cells following transduction with recombinant adenovirus expressing pp71 (Fig. 4D). In the subset of cells successfully transduced and expressing nuclear pp71, Daxx staining was less intense. Similar observations were made after transfection of KG-1 cells with a plasmid expressing pp71 (Fig. 4E). Importantly, a pp71 mutant (Did2-3) unable to bind (64) or degrade (27) Daxx expressed from a transfected plasmid entered the nucleus but failed to diminish the intensity of Daxx staining (Fig. 4E). Taken together, our data indicate that de novo expressed pp71 is able to function properly in undifferentiated KG-1 and Kasumi-3 cells (i.e., it can degrade Daxx), but the tegument-delivered protein fails to do so during viral infection because it localizes to the cytoplasm.
Viral gene silencing during the establishment of latency is regulated differently in KG-1/Kasumi-3 cells than in primary CD34+ cells.
The expression of the viral productive phase-initiating IE genes is repressed upon the establishment of experimental latency in primary CD34+ HPCs by the same intrinsic defense that initially silences it in differentiated cell types prior to being inactivated by tegument-delivered pp71 (28, 29). In KG-1 and Kasumi-3 cells, viral IE protein accumulation was not observed after infection with either AD169 (Fig. 5A) or FIX (Fig. 5B), consistent with the cytoplasmic localization of tegument-delivered pp71 and indicating that latency was established in these cells. Prior differentiation of KG-1 or Kasumi-3 cells into dendritic-like cells allowed not only for nuclear localization of tegument-delivered pp71 (Fig. 2C) but also for the accumulation of the IE2 protein as well as products of other lytic-phase genes, such as the early protein UL44 and the late protein pp28 (Fig. 5C and D).
Fig 5.

Initiation of lytic gene expression is differentiation dependent in KG-1/Kasumi-3 cells. (A and B) Undifferentiated KG-1 and Kasumi-3 (K3) cells were infected with AD169 at an MOI of 3 (A) or FIX at an MOI of 1 (B) for 24 h. Cells were then fixed, stained with an antibody against IE1, and imaged with indirect immunofluorescence microscopy. (C and D) KG-1 and K3 cells were treated with TPA and ionomycin for 72 h to induce differentiation and then infected with a recombinant strain of AD169 that expresses IE2 fused to GFP at an MOI of 2. After 72 or 96 h, cells were fixed and stained with antibodies against UL44 (C) or pp28 (D) and visualized with fluorescence microscopy. In all panels, nuclei were counterstained with Hoechst.
While the fate of viral IE gene expression (silenced) was similar in primary CD34+ HPCs and the KG-1 and Kasumi-3 cell lines, we noted an important difference regarding how this silencing was achieved. In primary CD34+ hematopoietic progenitors, NT2, or THP-1 cells infected with AD169, the silencing of IE1 transcription observed as latency is established can be counteracted by inactivating the PML-NB intrinsic defense with the histone deacetylase (HDAC) inhibitor valproic acid (VPA) (29) (Fig. 6A). However, VPA treatment failed to enhance viral IE1 transcript accumulation upon infection of either KG-1 or Kasumi-3 cells (Fig. 6A) even though it increased the level of acetylated histone H3 and thus appears to functionally inhibit HDACs in these cells (Fig. 6B). Treatment with the other HDAC inhibitors, trichostatin A (TSA), sodium butyrate, or suberoylanilide hydroxamic acid (SAHA), also failed to enhance IE1 transcription in AD169-infected KG-1 or Kasumi-3 cells (Fig. 6C). Expression of the latency-associated viral transcript LUNA (latency-unique nuclear antigen) in both KG-1 and Kasumi-3 cells in both the presence and absence of VPA (Fig. 6D) indicates that viral genomes are transcriptionally competent. The absence of IE1 transcript accumulation upon HDAC inhibition despite transcription competence of the genome indicates that viral genes are silenced in a different manner in transformed than in primary CD34+ HPCs.
In addition to the PML-NB intrinsic defense, cyclin-dependent kinase (CDK) activity inhibits IE gene expression during the onset of HCMV lytic infection, as well as during quiescent infection of NT2 cells (47), in an HDAC-independent manner (65). Though transformed cells such as KG-1 and Kasumi-3 are likely to have higher CDK activity than primary CD34+ HPCs, the CDK inhibitor roscovitine, either alone or in combination with VPA, failed to enhance IE1 transcript levels upon AD169 infection (Fig. 6E). Functional inhibition of CDK activity by roscovitine was confirmed with the observation of decreased levels of phosphorylated retinoblastoma (Rb) tumor suppressor protein in these cells (Fig. 6F). Thus, inhibition of either HDACs or CDKs or both is not sufficient to relieve repression of IE gene expression in these transformed cells, suggesting that the mechanisms by which IE gene expression is silenced in KG-1 and Kasumi-3 cells do not fully mimic what occurs during experimental latent infection of primary CD34+ HPCs cultured ex vivo. This difference compromises the utility of these cells for studying the establishment of HCMV latency.
Viral genomes are maintained in KG-1/Kasumi-3 cells, but reactivation is impaired.
Despite the significant difference in how viral IE gene expression is regulated at the start of experimental latency between KG-1 and Kasumi-3 cells versus primary CD34+ HPCs, we wanted to determine if these transformed cells modeled subsequent aspects of HCMV latency. We determined that the viral genome remained detectable in these cells for at least 10 days (Fig. 7A and C). KG-1 cells appeared to maintain the viral genome better than Kasumi-3 cells (Fig. 7B and D), and the FIX clinical strain genome appeared to be maintained better than AD169 in Kasumi-3 cells.
Fig 7.
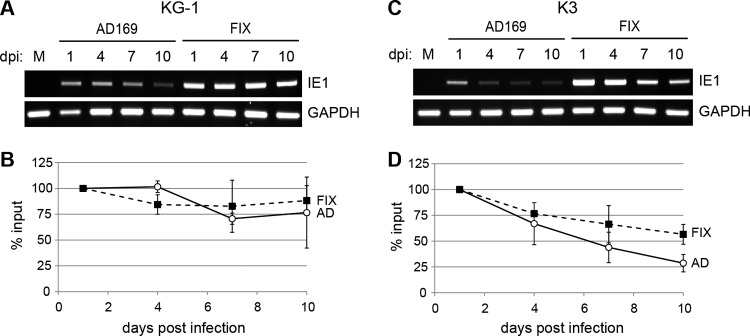
Genome maintenance in KG-1 and Kasumi-3 cells. KG-1 (A and B) and Kasumi-3 (K3) (C and D) cells were mock infected (M) or infected with AD169 or FIX at an MOI of 1. At the indicated day postinfection (dpi), cells were harvested and treated with trypsin, and total DNA was isolated and used as the template for PCR with primers specific to the viral genome (IE1) or cellular genome (GAPDH). PCR products were separated on agarose gels (A and C), and bands were quantified using Image Studio software (B and D). IE1 DNA levels were normalized to GAPDH levels and compared to the amount of viral DNA present at 1 day postinfection for each specific viral strain.
Both AD169 and FIX reactivate from latently infected primary CD34+ HPCs upon their differentiation into dendritic cells (18, 48). Surprisingly, we were unable to induce the reactivation of latent viral genomes in KG-1 cells upon their induced differentiation (Fig. 8A), and only FIX-GFP, but not AD169, was able to reactivate from Kasumi-3 cells (Fig. 8B). Lysate controls ensured that the infectious virus we observed upon differentiation of FIX-GFP-infected Kasumi-3 cells resulted mostly from true reactivation events rather than from spurious lytic replication (Fig. 9). Interestingly, primary CD34+ CD38− cells support reactivation, whereas primary CD34+ CD38+ cells do not (62). Intriguingly, both KG-1 cells (66) (Fig. 10) and Kasumi-3 cells are CD38+ (Fig. 10), indicating that reactivation of clinical strains from Kasumi-3 cells (52) (Fig. 8 and 9) deviates from the paradigm established in primary CD34+ HPCs that the CD38+ subpopulation does not support reactivation. Several negative controls demonstrated the specificity of our staining (Fig. 10). Thus, in addition to differences in viral gene silencing, there are significant differences in the reactivation phenotypes between transformed and primary CD34+ cells. In total, our data indicate that transformed KG-1 and Kasumi-3 cells mimic some, but not all, aspects of experimental HCMV latency as defined in primary CD34+ cells. Thus, aspects of latency illuminated only through the use of immortalized CD34+ cells should be considered prudently.
Fig 8.

Clinical isolates reactivate from Kasumi-3 but not KG-1 cells in response to differentiation stimuli. (A) KG-1 and (B) Kasumi-3 (K3) cells were infected with AD169-IE2GFP or FIX-GFP at an MOI of 3 and maintained in culture for 10 days. Cells were treated with trypsin and then returned to normal culture conditions or treated with TPA and ionomycin (+TPA/Iono) to induce differentiation. After 2 days, drugs were washed out, and cells were cultured an additional 3 days before coculturing with fibroblasts. For lysate controls (Lysate), an equal number of untreated cells were lysed by sonication prior to coculture. Bright-field (left) and fluorescence (right) microscopy images of cocultures were captured after 18 days.
Fig 9.
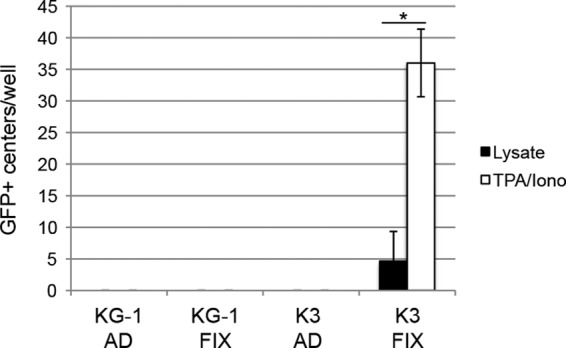
Quantitation of HCMV reactivation from latency in KG-1 and Kasumi-3 cells. The numbers of GFP+ centers from reactivation assays (Fig. 8) of KG-1 or Kasumi-3 (K3) cells infected with AD169-IE2GFP (AD) or FIX-GFP (FIX) were counted after 17 to 20 days of coculture with fibroblasts. GFP+ centers were defined as clusters of three or more GFP+ fibroblasts. The average number of GFP+ centers per well from three independent biological replicates is shown. Bars show standard error. The asterisk indicates a statistically significant difference using a paired Student t test (P < 0.001).
Fig 10.

KG-1 and Kasumi-3 cells are CD38+. KG-1, Kasumi-3 (K3), embryonic stem cells (ESC), 293Y, and the B cell lymphoma cell line 721 (721) were stained for CD38 and CD34 and analyzed by flow cytometry. (A) Representative dot plots displaying the percentage of cells in each quadrant. (B to D) The average percentage of cells staining positive for CD38 (B), CD34 (C), or both CD38 and CD34 (D), as determined by flow cytometry, is displayed with standard error.
DISCUSSION
Viral latency prevents immunological clearance of HCMV infections. As no current antivirals prevent or clear latent infections, pharmacologic clearance of HCMV is also unachievable. Understanding the molecular mechanisms that underlie establishment, maintenance, animation, and reactivation of HCMV latency is a crucial first step in developing prophylactic or therapeutic regimens targeting latency. Studies using naturally latently infected cells and those employing current ex vivo and in vitro models have laid a solid foundation for our understanding of HCMV latency. However, technical limitations associated with these models hamper their ability to elucidate detailed molecular mechanisms.
In an effort to complement existing ex vivo and in vitro models, we have evaluated the potential of two CD34+ tumor cell lines, KG-1 and Kasumi-3, to model parameters of HCMV experimental latency observed in primary CD34+ HPCs. A previous report proposed Kasumi-3 cells as a viable and tractable model for HCMV latency (52). Our work presented here detects significant differences between immortal and primary CD34+ cells, suggesting that the viability of Kasumi-3 and KG-1 cells as models for individual aspects of true latency should be considered independently and prompting us to urge caution when developing latency models based solely on data acquired in immortalized cells.
The many differences between the previous study (52) and the present one include the substantial lytic-phase gene expression (IE1 transcripts and UL99 [pp28] late protein) and viral DNA replication after infection of these undifferentiated cells observed in the earlier study but not here. The prior study employed infections at a very high multiplicity, used centrifugal enhancement, and selected for virus-encoded reporter-expressing cells. We observed arguably comparable infection rates using 10-fold less virus without centrifugal enhancement or reporter selection and perhaps thus avoided spurious lytic infection.
We found certain parameters of experimental latency, including the cytoplasmic sequestration of tegument-delivered pp71, and the ability to maintain the viral genome over time to be in common between these transformed and primary CD34+ HPCs. In fact, we utilized these tumor cells to demonstrate, for the first time, colocalization of two cytoplasmically sequestered tegument-delivered proteins (Fig. 2A). This observation may indicate that tegument disassembly may be different in undifferentiated cells than in their differentiated counterparts and perhaps the root cause for the different infectious programs initiated upon infection of these two different classes of cells. Thus, for exploring molecular mechanisms controlling these specific latent parameters (and perhaps others), KG-1 and Kasumi-3 cells appear to represent tractable alternatives to primary cells, with clear advantages in terms of their cost effectiveness and potential utility for exploring cellular contributions to latency using genetic approaches.
However, experimental latency in KG-1 and Kasumi-3 cells differs in at least two significant ways from the paradigms established in primary CD34+ HPCs. First, silencing of lytic-phase gene expression is not reversed in KG-1 or Kasumi-3 cells by HDAC inhibition prior to infection with AD169, in contrast to the effect in primary CD34+ HPCs, NT2, and THP-1 cells. Second, KG-1 cells fail to support reactivation while Kasumi-3 cells do. Curiously, Kasumi-3 cells reactivate despite their cell surface expression of CD38, which was shown in primary cells to correlate with the inability to reactivate (62). Thus, two defining components of latency, the silencing of productive-phase gene expression and reactivation, are regulated differently in these transformed cell lines than in primary CD34+ HPCs.
Our data suggest that KG-1 and Kasumi-3 cells have an HDAC-independent mechanism to silence IE gene expression from AD169, which we suspect is in addition to the PML-NB intrinsic defense although we currently cannot test this hypothesis. Interestingly, we have also detected an HDAC-independent suppression of IE gene expression at the start of experimental latent infections of primary CD34+ HPCs when the clinical strain viruses FIX and TB40/E are used for infections (29). It is possible that these represent the same restrictive mechanism and that it simply is, or becomes, activated in transformed CD34+ cells after infection with laboratory-adapted or clinical strain viruses but that in primary cells it is active only with clinical strains. Were this to be true, then KG-1 and Kasumi-3 cells could represent viable models for the initial silencing of IE gene expression when latency is established, albeit less useful ones than primary cells because of the inability to contrast laboratory-adapted and clinical strain infections. Alternatively, HDAC-independent silencing in KG-1 and Kasumi-3 cells may be a result of transformation, which would render them less useful for studies of the initial silencing of viral IE gene expression when latency is established and perhaps for studies other latency parameters as well.
The inability of AD169 to reactivate from either cell line is puzzling as this viral strain is hyperreactive in primary CD34+ HPCs (18). AD169 infects these cells with low efficiency (Fig. 1C), and viral genomes are lost over time (Fig. 7). However, no infectious progeny were ever detected in differentiated cohorts or in lysate controls (Fig. 9), leading us to speculate that the inability to reactivate results from more than just inefficient infection and/or substantial genome loss. The clinical strain FIX reactivated efficiently from Kasumi-3 cells, as did the clinical strain TB40/E in a previous report (52). Curiously, reactivation failed to correlate with CD38 status as both cells showed cell surface expression of this marker.
The exact site or sites of natural HCMV latency remain undefined. Cells expressing CD34 on their surfaces represent a heterologous mixture of multiple types, one or more of which could be a true latent reservoir, and CD34-negative cells could also harbor latent virus. Thus, the differences described here between transformed CD34+ HPCs and their primary counterparts do not necessarily mean that the transformed cells display nonnatural molecular details for HCMV latency. For example, KG-1 and Kasumi-3 cells may represent a long-term latency reservoir where viral IE gene expression is more stringently suppressed and where reactivation requires more than just differentiation. However, these tumor cells undoubtedly show numerous genetic alterations compared to the cells from which they are derived and which HCMV would naturally infect. Thus, results obtained in these cell lines should be interpreted with caution.
Kasumi-3 and KG-1 cells appear commensurate with the other immortal cell line models used to study aspects of HCMV latency or quiescence, NT2 and THP-1 cells. Each has its own idiosyncrasies. NT2 cells are adherent and larger and thus infect more efficiently and are more amenable to imaging approaches, yet they spontaneously differentiate and fail to reactivate. THP-1 cells remain undifferentiated but fail to efficiently reactivate. Importantly for the study of latency establishment, both NT2 and THP-1 faithfully mimic the silencing yet HDAC inhibitor responsiveness of AD169 IE gene expression observed in primary CD34+ cells. As detailed above, Kasumi-3 and KG-1 cells do not mimic this responsiveness, diminishing their relevance.
Finally, we recently characterized HCMV experimental latency in vitro in cultures of human embryonic stem cells (ESCs) (56). Unlike the transformed CD34+ cells tested here, our results in ESCs were, for every parameter tested, indistinguishable from those of experimental latent infections in primary CD34+ HPCs. ESC cells are adherent, easily maintained in an undifferentiated state, genetically malleable, show HDAC inhibitor responsiveness for AD169 IE gene expression, and reactivate virus upon differentiation. Though their propagation is somewhat laborious, they appear to represent the best compromise between convenience and accuracy. As multiple latency models are now in use (49, 50, 52, 56, 67, 68), our challenge will be to utilize each to its advantage while integrating results obtained in independent systems into an accurate model for natural HCMV latency.
ACKNOWLEDGMENTS
We thank Phil Balandyk for expert technical assistance, Bill Sugden for 721 cells, Rhiannon Penkert for ESCs and helpful discussions and suggestions, and Eain Murphy for helpful discussions.
This work was supported by National Institutes of Health grant AI074984 (to R.F.K.). R.F.K. is a Vilas Fellow and a Burroughs Wellcome Fund Investigator in the Pathogenesis of Infectious Disease. E.R.A. is an NSF Graduate Research Fellow.
Footnotes
Published ahead of print 3 July 2013
REFERENCES
- 1.Mocarski ES, Shenk T, Pass RF. 2007. Cytomegaloviruses, p 2701–2772 In Knipe DM, Howley PM, Griffin DE, Lamb RA, Martin MA, Roizman B, Straus SE. (ed), Fields virology, 5th ed. Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 2.Boeckh M, Geballe AP. 2011. Cytomegalovirus: pathogen, paradigm, and puzzle. J. Clin. Invest. 121:1673–1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goodrum F, Caviness K, Zagallo P. 2012. Human cytomegalovirus persistence. Cell Microbiol. 14:644–655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hahn G, Jores R, Mocarski ES. 1998. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. U. S. A. 95:3937–3942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Khaiboullina SF, Maciejewski JP, Crapnell K, Spallone PA, Dean Stock A, Pari GS, Zanjani ED, Jeor SS. 2004. Human cytomegalovirus persists in myeloid progenitors and is passed to the myeloid progeny in a latent form. Br. J. Haematol. 126:410–417 [DOI] [PubMed] [Google Scholar]
- 6.Kondo K, Kaneshima H, Mocarski ES. 1994. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc. Natl. Acad. Sci. U. S. A. 91:11879–11883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mendelson M, Monard S, Sissons P, Sinclair J. 1996. Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J. Gen. Virol. 77:3099–3102 [DOI] [PubMed] [Google Scholar]
- 8.Sinclair J. 2008. Human cytomegalovirus: Latency and reactivation in the myeloid lineage. J. Clin. Virol. 41:180–185 [DOI] [PubMed] [Google Scholar]
- 9.Sinclair J, Sissons P. 2006. Latency and reactivation of human cytomegalovirus. J. Gen. Virol. 87:1763–1779 [DOI] [PubMed] [Google Scholar]
- 10.Taylor-Wiedeman J, Sissons JG, Borysiewicz LK, Sinclair JH. 1991. Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J. Gen. Virol. 72:2059–2064 [DOI] [PubMed] [Google Scholar]
- 11.Slobedman B, Cao JZ, Avdic S, Webster B, McAllery S, Cheung AK, Tan JC, Abendroth A. 2010. Human cytomegalovirus latent infection and associated viral gene expression. Future Microbiol. 5:883–900 [DOI] [PubMed] [Google Scholar]
- 12.Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC. 2012. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of latent HSV-1 in neurons. PLoS Pathog. 8:e1002540. 10.1371/journal.ppat.1002540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Penkert RR, Kalejta RF. 2011. Tegument protein control of latent herpesvirus establishment and animation. Herpesviridae 2:3. 10.1186/2042-4280-2-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wilson AC, Mohr I. 2012. A cultured affair: HSV latency and reactivation in neurons. Trends Microbiol. 20:604–611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Reeves MB, MacAry PA, Lehner PJ, Sissons JG, Sinclair JH. 2005. Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc. Natl. Acad. Sci. U. S. A. 102:4140–4145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reeves MB, Lehner PJ, Sissons JG, Sinclair JH. 2005. An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J. Gen. Virol. 86:2949–2954 [DOI] [PubMed] [Google Scholar]
- 17.Biron KK. 2006. Antiviral drugs for cytomegalovirus diseases. Antiviral Res. 71:154–163 [DOI] [PubMed] [Google Scholar]
- 18.Goodrum F, Reeves M, Sinclair J, High K, Shenk T. 2007. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110:937–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sinclair J. 2010. Chromatin structure regulates human cytomegalovirus gene expression during latency, reactivation and lytic infection. Biochim. Biophys. Acta 1799:286–295 [DOI] [PubMed] [Google Scholar]
- 20.Murphy JC, Fischle W, Verdin E, Sinclair JH. 2002. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J. 21:1112–1120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cuevas-Bennett C, Shenk T. 2008. Dynamic histone H3 acetylation and methylation at human cytomegalovirus promoters during replication in fibroblasts. J. Virol. 82:9525–9536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Groves IJ, Reeves MB, Sinclair JH. 2009. Lytic infection of permissive cells with human cytomegalovirus is regulated by an intrinsic ‘pre-immediate-early' repression of viral gene expression mediated by histone post-translational modification. J. Gen. Virol. 90:2364–2374 [DOI] [PubMed] [Google Scholar]
- 23.Nitzsche A, Steinhausser C, Mucke K, Paulus C, Nevels M. 2012. Histone H3 lysine 4 methylation marks postreplicative human cytomegalovirus chromatin. J. Virol. 86:9817–9827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adler M, Tavalai N, Muller R, Stamminger T. 2011. Human cytomegalovirus immediate-early gene expression is restricted by the nuclear domain 10 component Sp100. J. Gen. Virol. 92:1532–1538 [DOI] [PubMed] [Google Scholar]
- 25.Lukashchuk V, McFarlane S, Everett RD, Preston CM. 2008. Human cytomegalovirus protein pp71 displaces the chromatin-associated factor ATRX from nuclear domain 10 at early stages of infection. J. Virol. 82:12543–12554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McFarlane S, Preston CM. 2011. Human cytomegalovirus immediate early gene expression in the osteosarcoma line U2OS is repressed by the cell protein ATRX. Virus Res. 157:47–53 [DOI] [PubMed] [Google Scholar]
- 27.Saffert RT, Kalejta RF. 2007. Human cytomegalovirus gene expression is silenced by Daxx-mediated intrinsic immune defense in model latent infections established in vitro. J. Virol. 81:9109–9120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Saffert RT, Kalejta RF. 2006. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 80:3863–3871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Saffert RT, Penkert RR, Kalejta RF. 2010. Cellular and viral control over the initial events of human cytomegalovirus experimental latency in CD34+ cells. J. Virol. 84:5594–5604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tavalai N, Adler M, Scherer M, Riedl Y, Stamminger T. 2011. Evidence for a dual antiviral role of the major nuclear domain 10 component Sp100 during the immediate-early and late phases of the human cytomegalovirus replication cycle. J. Virol. 85:9447–9458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tavalai N, Papior P, Rechter S, Leis M, Stamminger T. 2006. Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J. Virol. 80:8006–8018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tavalai N, Stamminger T. 2009. Interplay between herpesvirus infection and host defense by PML nuclear bodies. Viruses 1:1240–1264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woodhall DL, Groves IJ, Reeves MB, Wilkinson G, Sinclair JH. 2006. Human Daxx-mediated repression of human cytomegalovirus gene expression correlates with a repressive chromatin structure around the major immediate early promoter. J. Biol. Chem. 281:37652–37660 [DOI] [PubMed] [Google Scholar]
- 34.Ishov AM, Stenberg RM, Maul GG. 1997. Human cytomegalovirus immediate early interaction with host nuclear structures: definition of an immediate transcript environment. J. Cell Biol. 138:5–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Maul GG. 2008. Initiation of cytomegalovirus infection at ND10. Curr. Top. Microbiol. Immunol. 325:117–132 [DOI] [PubMed] [Google Scholar]
- 36.Saffert RT, Kalejta RF. 2008. Promyelocytic leukemia-nuclear body proteins: herpesvirus enemies, accomplices, or both? Future Virol. 3:265–277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tavalai N, Stamminger T. 2011. Intrinsic cellular defense mechanisms targeting human cytomegalovirus. Virus Res. 157:128–133 [DOI] [PubMed] [Google Scholar]
- 38.Penkert RR, Kalejta RF. 2012. Tale of a tegument transactivator: the past, present and future of human CMV pp71. Future Virol. 7:855–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Penkert RR, Kalejta RF. 2010. Nuclear localization of tegument-delivered pp71 in human cytomegalovirus-infected cells is facilitated by one or more factors present in terminally differentiated fibroblasts. J. Virol. 84:9853–9863 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tavalai N, Papior P, Rechter S, Stamminger T. 2008. Nuclear domain 10 components promyelocytic leukemia protein and hDaxx independently contribute to an intrinsic antiviral defense against human cytomegalovirus infection. J. Virol. 82:126–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Slobedman B, Mocarski ES. 1999. Quantitative analysis of latent human cytomegalovirus. J. Virol. 73:4806–4812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weekes MP, Tan SY, Poole E, Talbot S, Antrobus R, Smith DL, Montag C, Gygi SP, Sinclair JH, Lehner PJ. 2013. Latency-associated degradation of the MRP1 drug transporter during latent human cytomegalovirus infection. Science 340:199–202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ioudinkova E, Arcangeletti MC, Rynditch A, De Conto F, Motta F, Covan S, Pinardi F, Razin SV, Chezzi C. 2006. Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene 384:120–128 [DOI] [PubMed] [Google Scholar]
- 44.Lee CH, Lee GC, Chan YJ, Chiou CJ, Ahn JH, Hayward GS. 1999. Factors affecting human cytomegalovirus gene expression in human monocyte cell lines. Mol. Cells 9:37–44 [PubMed] [Google Scholar]
- 45.Meier JL. 2001. Reactivation of the human cytomegalovirus major immediate-early regulatory region and viral replication in embryonal NTera2 cells: role of trichostatin A, retinoic acid, and deletion of the 21-base-pair repeats and modulator. J. Virol. 75:1581–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Meier JL, Stinski MF. 1996. Regulation of human cytomegalovirus immediate-early gene expression. Intervirology 39:331–342 [DOI] [PubMed] [Google Scholar]
- 47.Zydek M, Hagemeier C, Wiebusch L. 2010. Cyclin-dependent kinase activity controls the onset of the HCMV lytic cycle. PLoS Pathog. 6:e1001096. 10.1371/journal.ppat.1001096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cheung AK, Gottlieb DJ, Plachter B, Pepperl-Klindworth S, Avdic S, Cunningham AL, Abendroth A, Slobedman B. 2009. The role of the human cytomegalovirus UL111A gene in down-regulating CD4+ T-cell recognition of latently infected cells: implications for virus elimination during latency. Blood 114:4128–4137 [DOI] [PubMed] [Google Scholar]
- 49.Goodrum FD, Jordan CT, High K, Shenk T. 2002. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc. Natl. Acad. Sci. U. S. A. 99:16255–16260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hargett D, Shenk TE. 2010. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc. Natl. Acad. Sci. U. S. A. 107:20039–20044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Reeves MB, Sinclair JH. 2010. Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J. Gen. Virol. 91:599–604 [DOI] [PubMed] [Google Scholar]
- 52.O'Connor CM, Murphy EA. 2012. A myeloid progenitor cell line capable of supporting human cytomegalovirus latency and reactivation, resulting in infectious progeny. J. Virol. 86:9854–9865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Asou H, Suzukawa K, Kita K, Nakase K, Ueda H, Morishita K, Kamada N. 1996. Establishment of an undifferentiated leukemia cell line (Kasumi-3) with t(3;7)(q27;q22) and activation of the EVI1 gene. Jpn. J. Cancer Res. 87:269–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Koeffler HP, Golde DW. 1978. Acute myelogenous leukemia: a human cell line responsive to colony-stimulating activity. Science 200:1153–1154 [DOI] [PubMed] [Google Scholar]
- 55.Teobald I, Dunnion DJ, Whitbread M, Curnow SJ, Browning MJ. 2008. Phenotypic and functional differentiation of KG-1 into dendritic-like cells. Immunobiology 213:75–86 [DOI] [PubMed] [Google Scholar]
- 56.Penkert RR, Kalejta RF. 2013. Human embryonic stem cell lines model experimental human cytomegalovirus latency. mBio 4(3):e00298–13. 10.1128/mBio.00298-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Ibig-Rehm Y, Gotte M, Gabriel D, Woodhall D, Shea A, Brown NE, Compton T, Feire AL. 2011. High-content screening to distinguish between attachment and post-attachment steps of human cytomegalovirus entry into fibroblasts and epithelial cells. Antiviral Res. 89:246–256 [DOI] [PubMed] [Google Scholar]
- 58.Sanchez V, Clark CL, Yen JY, Dwarakanath R, Spector DH. 2002. Viable human cytomegalovirus recombinant virus with an internal deletion of the IE2 86 gene affects late stages of viral replication. J. Virol. 76:2973–2989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Murphy E, Yu D, Grimwood J, Schmutz J, Dickson M, Jarvis MA, Hahn G, Nelson JA, Myers RM, Shenk TE. 2003. Coding potential of laboratory and clinical strains of human cytomegalovirus. Proc. Natl. Acad. Sci. U. S. A. 100:14976–14981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Juckem LK, Boehme KW, Feire AL, Compton T. 2008. Differential initiation of innate immune responses induced by human cytomegalovirus entry into fibroblast cells. J. Immunol. 180:4965–4977 [DOI] [PubMed] [Google Scholar]
- 61.Bego M, Maciejewski J, Khaiboullina S, Pari G, St Jeor S. 2005. Characterization of an antisense transcript spanning the UL81-82 locus of human cytomegalovirus. J. Virol. 79:11022–11034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Goodrum F, Jordan CT, Terhune SS, High K, Shenk T. 2004. Differential outcomes of human cytomegalovirus infection in primitive hematopoietic cell subpopulations. Blood 104:687–695 [DOI] [PubMed] [Google Scholar]
- 63.Li H, Leo C, Zhu J, Wu X, O'Neil J, Park EJ, Chen JD. 2000. Sequestration and inhibition of Daxx-mediated transcriptional repression by PML. Mol. Cell Biol. 20:1784–1796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Hofmann H, Sindre H, Stamminger T. 2002. Functional interaction between the pp71 protein of human cytomegalovirus and the PML-interacting protein human Daxx. J. Virol. 76:5769–5783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zydek M, Uecker R, Tavalai N, Stamminger T, Hagemeier C, Wiebusch L. 2011. General blockade of human cytomegalovirus immediate-early mRNA expression in the S/G2 phase by a nuclear, Daxx- and PML-independent mechanism. J. Gen. Virol. 92:2757–2769 [DOI] [PubMed] [Google Scholar]
- 66.Hu X, Moscinski LC, Zuckerman KS. 1999. Transforming growth factor beta inhibits growth of more differentiated myeloid leukemia cells and retinoblastoma protein phosphorylation at serine 795. Exp. Hematol. 27:605–614 [DOI] [PubMed] [Google Scholar]
- 67.Huang MM, Kew VG, Jestice K, Wills MR, Reeves MB. 2012. Efficient human cytomegalovirus reactivation is maturation dependent in the Langerhans dendritic cell lineage and can be studied using a CD14+ experimental latency model. J. Virol. 86:8507–8515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Keyes LR, Hargett D, Soland M, Bego MG, Rossetto CC, Almeida-Porada G, St Jeor S. 2012. HCMV protein LUNA is required for viral reactivation from latently infected primary CD14+ cells. PLoS One 7:e52827. 10.1371/journal.pone.0052827 [DOI] [PMC free article] [PubMed] [Google Scholar]