

Abstract
The importance of the oncogenic transcription factor interferon regulatory factor 4 (IRF4) in hematological malignancies has been increasingly recognized. We have previously identified the B cell integration cluster (BIC), the gene encoding miR-155, as the first microRNA (miRNA)-encoding gene transcriptionally targeted by IRF4 in virus-transformed cancer cells. Activation of IRFs is prerequisite for their functions. However, how IRF4 is activated in cancer is an open question. Our phosphoproteome profiling has identified several tyrosine phosphorylation sites on IRF4 in Epstein-Barr virus (EBV)-transformed cells. Further, we show here that c-Src dramatically stimulates IRF4 phosphorylation and activity and that Y61 and Y124 are two key sites responding to c-Src-mediated activation. Consistently, c-Src is constitutively expressed and active in EBV-transformed cells. However, c-Src is unlikely to be a direct kinase for IRF4. Furthermore, we have a polyclonal antibody specific to phospho-IRF4(Y121/124) developed in rabbit. We have further shown that inhibition of c-Src activity reduces p-IRF4(Y121/124) and significantly represses transcription of the IRF4 target BIC in EBV-transformed cells. Our results therefore, for the first time, demonstrate that IRF4 is phosphorylated and activated through a c-Src-mediated pathway in virus-transformed cells. These findings will improve our understanding of IRF4 in neoplasia and will provide profound insights into the interaction of oncogenic viruses with IRF4 in the development of hematological malignancies.
INTRODUCTION
Interferon regulatory factors (IRFs) are a small but important family of transcription factors in multiple facets of host defense systems and are also involved in the regulation of tumorigenesis, cell growth, differentiation, and myeloid cell development (1). Among IRFs, IRF2 (2), -4 (3), and -7 (4) have oncogenic and transforming potentials and antiapoptotic activity (3, 5, 6). These oncogenic IRFs all intimately interact with Epstein-Barr virus (EBV) latency programs (3, 7–10), which are associated with a variety of hematological and epithelial malignancies.
IRF4, also known as multiple myeloma (MM) oncoprotein 1 (MUM1), is lymphocyte specific and is overexpressed in EBV-transformed cells (3, 7, 11, 12), MM (13, 14), and human T cell leukemia virus 1 (HTLV1)-infected cell lines and associated adult T cell lymphoma/leukemia (ATLL) (15–19). IRF4 overexpression is a hallmark of the ABC type of DLBCL and MM (20, 21) and is frequently used as a diagnostic and prognostic marker for these and other proliferative disorders (21–23). Chromosomal translocation and genetic mutation of IRF4 have been found in MM, peripheral T cell lymphomas (24), and chronic lymphocytic leukemia (CLL) (13, 25). More recently, IRF4 was shown to be expressed in all LMP1-driven tumors in mice (26). These lines of evidence underscore the importance of IRF4 in these malignancies. However, the role of IRF4 in tumorigenesis remains to be elucidated.
Importantly, we have recently identified B cell integration cluster (BIC), which encodes the oncogenic microRNA (miRNA) miR-155, as the first miRNA-encoding gene induced by IRF4 in virus-transformed cells (6). miR-155 plays important roles in innate immunity (27, 28) and is the first identified oncogenic miRNA (oncomiR) that is implicated in various types of cancers, including lymphomas (29–31), breast cancer, leukemia, pancreatic cancer, and lung cancer (32, 33). Like oncogenic IRFs, miR-155 is also associated with EBV latency (29, 34–36). Our findings therefore made a connection between these two pivotal players of cancer and immunity and have provided valuable insights into the interaction between viral oncogenesis and immune mechanisms governed by them. For example, both factors are crucial regulators of germinal center reaction (37, 38), which is implicated in lymphoma development and EBV latent infection (39). Furthermore, our microarray analysis shows that IRF4 regulates a pool of interesting genes in EBV-transformed cells (our unpublished data). Future pursuits on selected targets may disclose novel roles for IRF4 and broaden our knowledge in its interaction with viral oncogenesis and other associated cancers.
Activation of IRFs by phosphorylation is prerequisite for their functions. Serine phosphorylation of IRF4 by the kinase ROCK2 activates IRF4, leading to interleukin 17/21 (IL-17/21) production in the autoimmune response in mice (40). However, how IRF4 is activated in cancer is an open question. Many proteins involved in signal transduction are tyrosine phosphorylated. Interestingly, a few limited high-throughput profiling studies have identified several tyrosine phosphorylation sites on IRF4 in different cancer contexts, including Y191 in MM (41) and Y36, Y121, Y124, Y427, and Y439 in Hodgkin's lymphomas (42).
In this study, we have taken advantage of the high-throughput strategy phosphoproteome profiling for the first time to profile global tyrosine phosphorylation associated with EBV latency. Our results have shown that IRF4 is tyrosine phosphorylated in EBV-transformed cells and identified several phosphorylation sites. We have further shown that the tyrosine kinase c-Src promotes IRF4 phosphorylation and activation and identified Y61 and Y124 of IRF4 as two key sites responding to c-Src-mediated activation. Moreover, we show that c-Src is constitutively expressed and activated in EBV-transformed cells and that inhibition of c-Src activity represses expression of the IRF4 target BIC in these cells. These findings indicate that IRF4 is activated through a c-Src-mediated pathway in EBV-transformed cells.
MATERIALS AND METHODS
Cell lines.
Sav I, Sav III, JiJoye, P3HR1, and IB4 are EBV-transformed B cell lines. BJAB is an EBV-negative B cell line. All B cell lines are cultured in RPMI 1640 medium plus 10% fetal bovine serum (FBS) and antibiotics. 293 and 293T cells are cultured with Dulbecco's modified Eagle medium (DMEM) plus 10% FBS and antibiotics.
Phosphoproteome profiling.
Large-scale profiling of tyrosine phosphorylation associated with EBV infection has been performed using the technique PhosphoScan by Cell Signaling Company. Whole-cell lysates from IB4 were digested, and phosphorylated peptides were enriched with IgG control or the antibody p-Tyr-100 (catalog number 8954), which recognizes the motif XyX. LTQ-Orbitrap-Velos liquid chromatography-tandem mass spectrometry (LC-MS/MS) was performed, and MS/MS spectra were evaluated against the Homo sapiens FASTA database (NCBI) using SEQUEST 3G and the SORCERER 2 platform from Sage-N Research (v4.0; Milpitas, CA), with a 5% default false-positive rate to filter the results. Duplicate runs have been performed.
Plasmids, reagents, and antibodies.
Flag-tagged IRFs were cloned in pCMV2-Flag vector. Beta interferon (IFN-β)-Luc was described in our previous work (43). Mouse c-Src cDNA constructs cloned in pCMV7.1/3×Flag were provided by Todd Miller (Stony Brook University). Anti-Flag M2 (Sigma), anti-LMP1 CS1-4 (Dako), anti-IRF4 H140 (Santa Cruz), anti-phospho-Src family (Y416 and Y527) (cell Signaling), anti-c-Src clone B12 (Santa Cruz), anti-IκBα C21 (Santa Cruz), anti-p-IκBα (S32/36) (Santa Cruz), anti-β-actin AC15 (Sigma), and anti-GAPDH (Santa Cruz) were used for Western blotting. All second antibodies were purchased from Cell Signaling. BAY11-7085 and pyrazolo-[2,3-d]pyrimidine 2 (PP2) were purchased from Sigma and EMD Millipore, respectively.
Antibody development.
Phospho-specific antibodies against IRF4 pY121 and pY124 were developed in rabbit by 21st Century Biochemicals, Inc. The designed immunogens include C-Ahx-ISDP[pY]KV[pY]RIVPE-amide, C-Ahx-ISDP[pY]KVYRIVPE-amide, and C-Ahx-ISDPYKV[pY]RIVPE-amide. The peptide fragment for immunodepletion is C-Ahx-ISDPYKVYRIVPE-amide. The antibodies recognize pY121 only, pY124 only, and both pY121 and pY124.
Promoter-reporter assay.
Cells were transfected with expression plasmids as indicated together with IFN-β-Luc and Renilla as the internal transfection control. Empty vector was used to equalize the total amounts of DNA in all transfections. Cells were collected 24 h after transfection. Luciferase activity was measured with equal amounts (10% of the total for each sample) of protein lysates with the use of a dual-luciferase assay kit (Promega).
Transfection.
293 and 293T cells were transfected with Effectene reagent (Invitrogen), and B cells were transfected with Amaxa Nucleofector kits by following the manufacturer's instructions.
RNA isolation and reverse transcriptase reactions.
Total RNA was isolated using an RNeasy minikit (Qiagen) according to the manufacturer's protocols. The eluted RNA was subjected to reverse transcriptase (RT) reactions, which were performed with the use of a GoScript RT kit (Promega) by following the manufacturer's instructions.
Real-time quantitative PCR.
Quantitative PCR (qPCR) was performed with the use of SYBR green (Applied Biosystems) on an ABI 7300 real-time PCR system with SDS version 1.3.1. All reactions were run in duplicates. Mean cycle threshold (CT) values were normalized to GAPDH (glyceraldehyde-3-phosphate dehydrogenase), yielding a normalized CT value (ΔCT). The ΔΔCT value was calculated by subtracting the respective control from the ΔCT, and the expression level was then calculated by 2 raised to the power of the respective −ΔΔCT value. Results are the average and standard error (SE) of duplicates for each sample. Primers for qPCR include the following: BIC forward, 5′-ACCAGAGACCTTACCTGTCACCTT-3′; BIC reverse, 5′-GGCATAAAGAATTTAAACCACAGATTT-3′ (44); GAPDH forward, 5′-ATGACATCAAGAAGGTGGTG-3′; GAPDH reverse, 5′-CATACCAGGAAATGAGCTTG-3′.
RESULTS
Identification of IRF4 phosphorylation sites associated with EBV infection.
Phosphoproteome profiling has identified 2,984 nonredundant (9,073 redundant) phosphorylated peptides in IB4 cells. Among these peptides, we have fortunately found five IRF4 phosphorylation sites, including Y124, -176, -191, -180, and -427 (Table 1). Consistent with our results, phosphorylation of Y124 and Y427 and of Y191 of endogenous IRF4 was also identified by phosphoproteome profiling in Hodgkin's lymphoma cells (42) and multiple myeloma cells (41), respectively. In contrast, phosphorylation of Y37, Y121, and Y439, which were identified in Hodgkin's lymphoma cells (42), was not identified in EBV-transformed cells in our assays with the same strategy.
Table 1.
Identification of phosphorylation sites of IRF4 and SFKs in EBV-transformed cells
| Protein name | Site(s) | Peptide identifieda | Avg intensity (arbitrary units) | No. of peptides identified |
|---|---|---|---|---|
| IRF4 | 124 | SQLDISDPYKVY*R | 117,487 | 1 |
| IRF4 | 176, 191 | S*WRDYVPDQPHPEIPY*QCPM#TFGPR | 93,958 | 1 |
| IRF4 | 180 | SWRDY*VPDQPHPEIPYQCPM#TFGPR | 310,990 | 4 |
| IRF4 | 180, 191 | SWRDY*VPDQPHPEIPY*QCPM#TFGPR | 105,136 | 3 |
| IRF4 | 191 | DYVPDQPHPEIPY*QCPMTFGPR | 112,356 | 1 |
| IRF4 | 427 | QLYYFAQQNSGHFLRGY*DLPEHISNPEDYHR | 278,562 | 1 |
| SFKs | 393, 416, 418, 419, 425 | LIEDNEY*TAR | 1,881,080 | 3 |
| SFKs | 436, 438, 439, 445 | WTAPEAALY*GR | 828,680 | 4 |
Asterisks (*) indicate that the left Y is phosphorylated; pound signs (#) indicate oxidized methionine.
c-Src stimulates IRF4 transcriptional activity.
Activation of IRFs by phosphorylation is a prelude to their functions. Serine phosphorylation of IRF4 by the kinase ROCK2 activates IRF4, leading to IL-17/21 production in the autoimmune response in mice (40). To this end, PhosphoMotif Finder and Kinasephos2.0 were used to predict the tyrosine kinase(s) responsible for IRF4 phosphorylation and activation. Promoter-reporter assay results show that c-Src, but not other candidates, dramatically increases IRF4 activity. In addition to c-Src, two other kinases, Alk and TNK, also significantly increase IRF4 activity (Fig. 1A). We focus on c-Src in this study.
Fig 1.

c-Src stimulates IRF4 transcriptional activity. 293 cells in 12-well plates were transfected with 0.2 μg IRF4 and 0.2 μg kinase expression plasmids, 40 ng pGL3/IFN-β-Luc, and 10 ng Renilla. A dual-luciferase assay was performed. Results are the averages and standard errors (SE) of duplicates. Representative results from at least three independent experiments are shown. The ability of the vector control to activate the promoter construct was set to 1. (A) Identification of a potential kinase(s) for IRF4 activation. (B) c-Src kinase activity is required for stimulating IRF4 activity. (C) c-Src does not activate IRF7.
To check if the kinase activity of c-Src is required for stimulating IRF4 transcriptional activity, we have used a few c-Src mutants, including the kinase-dead mutants c-Src (K297R) and c-Src (Y418F), as well as the constitutively active form of c-Src (Y527F) to perform promoter-reporter assays. Results show that the two kinase-dead mutants failed to stimulate IRF4 activity but that c-Src (Y527F) has much greater ability to activate IRF4 than does the wild-type c-Src (Fig. 1B). These results indicate that c-Src kinase activity is required for IRF4 activation.
To define the specificity of c-Src on IRF4, we have used IRF7 as a control. Results show that c-Src failed to activate IRF7 (Fig. 1C).
Together, these data indicate that c-Src specifically activates IRF4.
Map IRF4 phosphorylation sites responsive to c-Src.
Since our phosphoproteome profiling has identified five tyrosine phosphorylation sites for IRF4 in EBV-transformed cells, we first aimed to check which of these sites are responsive to c-Src. We have also predicted other tyrosine phosphorylation sites using Netphos2.0. We have used Flag-IRF4 as the template to create a panel of tyrosine-to-phenylalanine (Y-F) or tyrosine-to-alanine (Y-A) point mutants for these sites and tested their ability to respond to c-Src. As shown in Fig. 2A in the left panel, compared to the wild-type Flag-IRF4, two single point mutants, Flag-IRF4(Y61F) and Flag-IRF4(Y124F), have significant decreases in response to c-Src stimulation, and simultaneous mutation of both sites has further decreased activity consistently in response to c-Src. All other tested point mutants have activities similar to that of the wild-type IRF4. Western blotting results show that these point mutants have similar expression levels (Fig. 2A, right). Thus, Y61 and Y124 are likely important phosphorylation sites targeted by c-Src.
Fig 2.

Identification of IRF4 tyrosine sites responding to c-Src-mediated activation. 293 cells in 12-well plates were transfected with 0.2 μg Flag-IRF4 or its point/deletion mutants, 0.2 μg c-Src, 40 ng pGL3/IFN-β-Luc, and 10 ng Renilla. A dual-luciferase assay was performed. Results are the averages and standard errors (SE) of duplicates. Representative results from at least three independent experiments are shown. The ability of the vector control to activate the promoter construct was set to 1. (A) Y61 and Y124 are crucial for IRF4 activation by c-Src. The tested Flag-IRF4 Y-F point mutants include Y61F, Y121F, Y124F, Y121/124F, Y61/124F, Y151F, Y157F, Y169F, Y180F, Y220F, and Y324F. (B) Y324 is a site critical for IRF4 function independently of phosphorylation. The tested Flag-IRF4 Y-A point mutants include Y36A, Y121A, Y124A, Y191A, Y324A, Y334A, Y427A, and Y439A. (C) c-Src targets multiple sites on IRF4. The tested Flag-IRF4 deletion mutants include 1-190, 1-255, 1-260, 1-412, 1-420, 1-445, Del(140-187), and Del(140-408).
In addition to Y124, we have found that mutation of Y324 to alanine (Y324A) completely disables IRF4 (Fig. 2B, left). However, mutation of Y324 to phenylalanine (Y324F) did not affect IRF4 activity, and all other Y-A mutants have activities similar to that of the wild-type IRF4. Western blotting results show that all the Y-A mutants have similar expression levels (Fig. 2B, right). These results indicate that Y324 is a crucial site for IRF4 active conformation and functions independently of its phosphorylation.
Further, we have generated a panel of deletion mutants for IRF4. Promoter-reporter assay results with these mutants suggest that c-Src may target multiple sites in addition to Y61 and Y124 to fully activate IRF4 (Fig. 2C, top). As indicated above, Y324 is a crucial site for IRF4 active conformation. However, some deletion mutants without this site are still functional, implying that these deletion mutants reconstitute the active conformation of the full length of IRF4. Moreover, analysis with these deletion mutants indicates that the region spanning amino acids (aa) 255 to 412 is an important domain which inhibits IRF4 activity, but the domain spanning aa 412 to 420 is crucial for its activation (Fig. 2C, top). Western blotting results show that these deletion mutants are all well expressed (Fig. 2C, bottom).
Taken together, our results indicate that c-Src targets multiple sites on IRF4 for its full activation and that Y61 and Y121 are two pivotal sites responsible for its activation by c-Src.
Development of a polyclonal antibody against p-IRF4(Y121/Y124) in rabbit.
Since phosphorylation of Y124 has also been identified in Hodgkin's lymphoma cells (42), we speculate that this is an important functional phosphorylation site of IRF4 in lymphomas, and our promoter-reporter assay results support our claim (Fig. 2 and 3). Thus, we focus on this site in this study. To facilitate our research, a polyclonal antibody has been developed by 21st Century Biochemicals Inc. (Fig. 3). This antibody can specifically recognize IRF4 p-Y121 alone, p-Y124 alone, or both p-Y121 and p-Y124. However, this antibody is able to detect only phosphorylation of transiently expressed IRF4 and not endogenous phosphorylation of IRF4 (data not shown), which was detected by our phosphoproteome profiling.
Fig 3.
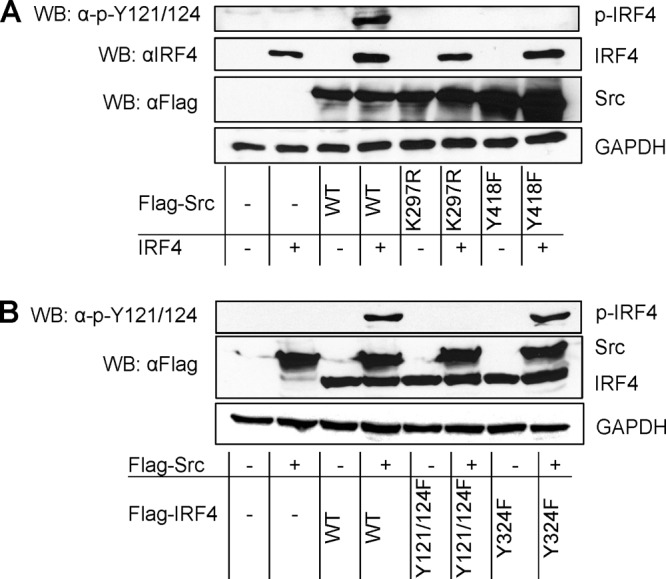
c-Src promotes tyrosine phosphorylation of IRF4. 293 cells in 60-mm dishes were transfected with 1 μg c-Src and 1 μg IRF4 expression plasmids or their mutants. Cells were collected 48 h posttransfection and analyzed for IRF4 phosphorylation with p-IRF4(Y121/124). (A) c-Src kinase activity is required for IRF4 phosphorylation. (B) c-Src targets the sites Y121/124 of IRF4. WB, Western blot.
c-Src promotes phosphorylation of IRF4 in cells.
Since c-Src specifically activates IRF4, we proposed that c-Src can promote IRF4 phosphorylation. To test this claim, we transfected 293 cells with the constructs indicated in Fig. 3A and probed IRF4 phosphorylation with the antibody p-Y121/124. Results show that the wild-type c-Src, but neither K297R nor Y418F, produces a specific band representative of IRF4 phosphorylation (Fig. 3A). These results imply that c-Src targets IRF4 Y121 and/or Y124 for phosphorylation.
To confirm that Y121 and/or Y124 is targeted by c-Src, we performed this experiment with two point mutants of IRF4, Y121/124F and Y324F, as controls. Results show that both wild-type IRF4 and IRF4(Y324F) are phosphorylated by c-Src, but IRF4(Y121/124F) did not show a specific band of phosphorylation (Fig. 3B).
Thus, our results demonstrate that c-Src promotes IRF4 phosphorylation at the sites Y121 and/or Y124, among other potential sites.
c-Src is constitutively expressed and active in EBV-transformed cells.
Since c-Src promotes IRF4 phosphorylation that is present endogenously in EBV-transformed cells, we are interested to check the level of c-Src protein and its activity in these cells. Autophosphorylation of Y418 or dephosphorylation of Y527 is required to switch c-Src from the inactive to the active form (45). Our PhosphoScan results show that Y418 of c-Src is phosphorylated in EBV-transformed cells (Table 1), indicating that c-Src is active in EBV-transformed cells. We further performed immunoblotting analysis with a specific antibody against c-Src p-Y418 for two pairs of EBV-positive cells, P3HR1 and JiJoye and Sav I and Sav III. Our results indicate that both plain c-Src and c-Src p-Y418 are present at significantly higher levels in JiJoye and Sav III cells (Fig. 4A).
Fig 4.
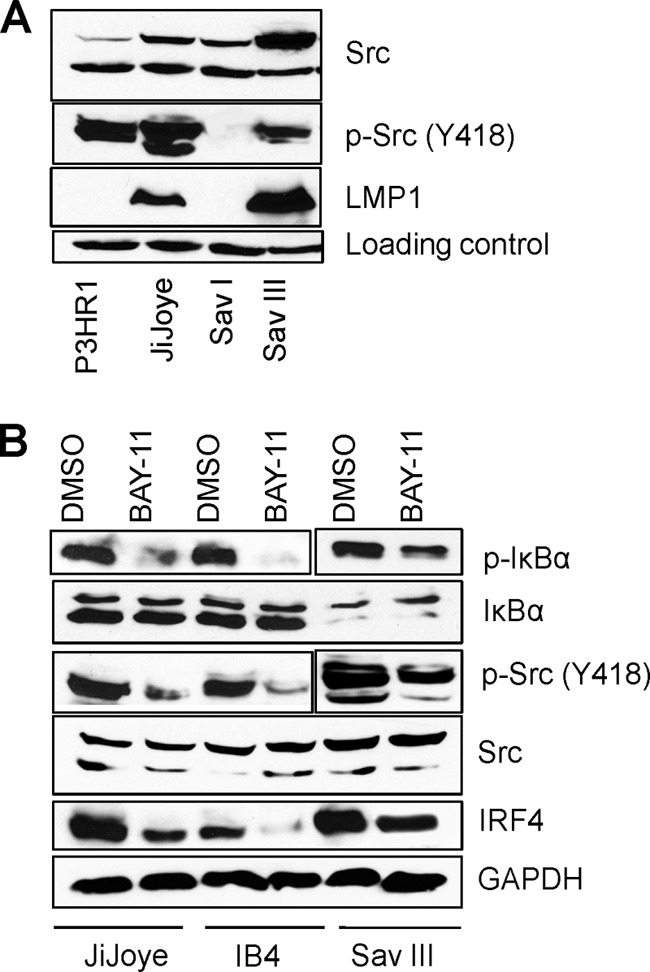
c-Src is constitutively expressed and activated in EBV-transformed cells. JiJoye, IB4, and Sav III cells were treated with 2.5 μM BAY-11, an NF-κB-specific inhibitor, for 18 h, and the levels of IRF4, c-Src, and p-Src (Y418) were evaluated by Western blotting with specific antibodies. (A) c-Src expression and activity are associated with EBV latency. (B) Inhibition of the LMP1/NF-κB pathway decreases endogenous c-Src activity in EBV-transformed cells.
Since LMP1 is absent or very low in P3HR1 cells but present at a high level in its parental JiJoye cells, these findings imply that LMP1 stimulates expression of c-Src and its activation. To verify these findings, we blocked the main LMP1 downstream pathway NF-κB using the NF-κB-specific drug BAY-11 and then checked the levels of c-Src and its phosphorylation in EBV-transformed cells. As shown in Fig. 4B, blockage of NF-κB leads to a significant decrease in c-Src (Y418) phosphorylation but did not affect the levels of plain c-Src. These data strongly suggest that the LMP1/NF-κB signaling axis activates c-Src. In fact, we have solid evidence showing that LMP1 activates IRF4 through a c-Src-mediated pathway (our unpublished data). Given that there is significant p-c-Src (Y418) in P3HR1 cells but not in Sav I cells, other EBV proteins also likely contribute to c-Src activation.
It has been reported that IRF4 is induced by the LMP1/NF-κB axis (3). Consistent with this claim, treatment of BAY-11 results in significant reduction of endogenous IRF4 levels in the tested cells (Fig. 4B).
Collectively, these results indicate that c-Src is constitutively expressed and activated in EBV-transformed cells.
Regulation of the IRF4 target BIC by c-Src in EBV-transformed cells.
Our previous findings have shown that BIC is an important transcriptional target for IRF4 in virus-transformed cells (6). Since c-Src stimulates IRF4 phosphorylation and activation, we were therefore interested to check if c-Src indirectly regulates BIC expression through activation of IRF4. To this end, we inhibited the endogenous c-Src activity in EBV-transformed cells with pyrazolo-[2,3-d]pyrimidine 2 (PP2), a c-Src-specific chemical inhibitor. Figure 5A shows that inhibition of endogenous c-Src activity significantly decreases BIC expression.
Fig 5.

c-Src regulates the IRF4 target BIC in EBV-transformed cells. (A) Inhibition of endogenous c-Src activity decreases the expression of BIC in EBV-transformed cells. JiJoye, IB4, and Sav III were treated with 20 μM PP2 or dimethyl sulfoxide (DMSO) for 48 h. RNA extraction and real-time PCR are described in details in Materials and Methods. (B and C) Inhibition of c-Src by PP2 (B) or by CSK (C) decreases IRF4 phosphorylation. 293 cells in 60-mm dishes were transfected with 1 μg of each indicated plasmid. For PP2 treatment, cells were treated with 20 μM PP2 or a DMSO control for 24 h (B). Cell lysates were prepared 48 h after transfection and subjected to Western blotting.
We then checked if inhibition of c-Src by PP2 inhibits IRF4 phosphorylation. Since the p-IRF4(Y121/124) antibody cannot detect endogenous IRF4 phosphorylation, we performed this experiment in 293 cells. To this end, we cotransfected 293 cells with combinations of expression constructs shown in Fig. 5B, and cells were then treated with PP2. IRF4 phosphorylation was checked with the antibody p-IRF4(Y121/124). As shown in Fig. 5B, PP2 treatment significantly decreases IRF4 Y121/124 phosphorylation. We also verified the results with c-Src kinase (CSK) instead of PP2 and got similar results (Fig. 5C). CSK and its homolog, CHK, can phosphorylate c-Src Y527, leading to c-Src inactivation (46).
Taken together, these results indicate that c-Src regulates IRF4 transcriptional targets through regulating IRF4 phosphorylation.
DISCUSSION
The major findings in this study are the novel discovery of IRF4 tyrosine phosphorylation in EBV-transformed cells and the unequivocal identification of a c-Src-mediated pathway for site-specific phosphorylation and activation of IRF4 (Fig. 6). IRF4 is an important oncoprotein that is overexpressed in many hematological malignancies, serves as a marker for both diagnosis and prognosis in hematological malignancies and other cancers, and is a promising therapeutic target for treatment of some of these neoplastic disorders (21, 47). Thus, study of its activation is of paramount importance.
Fig 6.
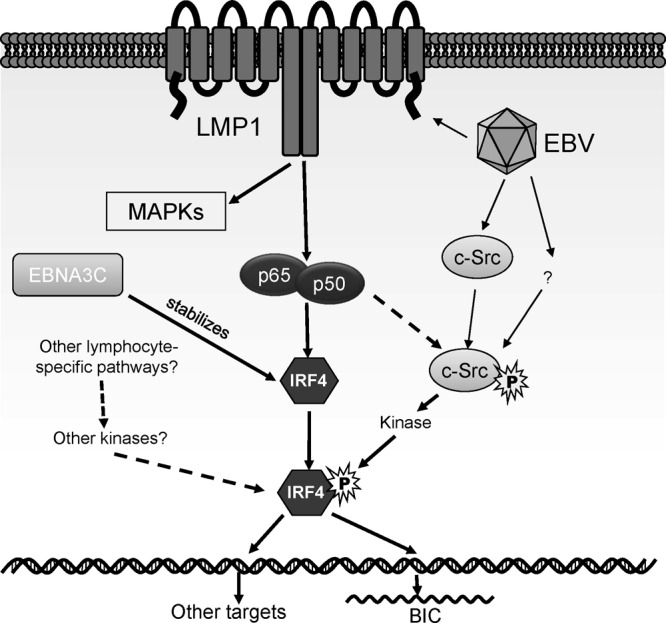
A hypothetical model for IRF4 activation in EBV latency. Our present study has disclosed that IRF4 is activated through c-Src-mediated tyrosine phosphorylation, although c-Src is unlikely to be a direct kinase for IRF4. The level of the c-Src protein is elevated in EBV type 3 latency through an unknown mechanism. c-Src is activated (phosphorylated at Y418), probably in part by the LMP1/NF-κB axis. In addition to the LMP1 pathway, other lymphocyte-specific pathways may also contribute to IRF4 activation. IRF4 expression is induced by the LMP1/NF-κB axis. In addition, a recent report shows that the IRF4 protein is stabilized by the EBV latent antigen EBNA3C (50). MAPK, mitogen-activated protein kinase.
c-Src is the first described proto-oncogene which plays important roles in tumor cell proliferation and metastasis (45) and is overexpressed as well as highly activated in many human cancers and cell lines derived from these cancers (48). Inhibition of c-Src kinase activity results in the suppression of primary tumor growth and metastasis. Thus, c-Src is an important pharmacological target for cancer treatment (46, 48). Some Src-specific inhibitors have now been used in clinical treatment of cancers, including breast carcinoma and prostate and pancreatic cancers (46). Importantly, we present evidence that c-Src is also overexpressed, activated, and functional in EBV-transformed cells, and our data from BAY-11 inhibition suggest that the activity of c-Src partially results from the LMP1/NF-κB signaling axis (Fig. 4 and 5). However, BAY-11 may not inhibit NF-κB very specifically. Moreover, the LMP1/NF-κB axis does not contribute to c-Src overexpression. Thus, following this study, more investigation is needed to verify these observations and to disclose the mechanism underlying c-Src induction and activation in this setting.
c-Src-mediated activation of IRF4 may not be limited to the context of EBV infection; in fact, we have preliminary data showing that IRF4 is also likely activated by c-Src in the context of HTLV1 infection (data not shown). Moreover, in addition to c-Src, our results also show that Alk and TNK significantly enhance IRF4 transcriptional activity (Fig. 1), suggesting that these two kinases may contribute to full activation of IRF4 or may have unique roles in activation of IRF4 in distinct cancer contexts.
Although IRF4 was shown to interact with c-Src in a yeast two-hybrid high-throughput screening (49), we failed to verify their interaction by coimmunoprecipitation assays despite many attempts. Therefore, c-Src is unlikely to be a direct kinase for IRF4. Further study is necessary to confirm this claim and to identify the c-Src-targeted direct kinase(s) for IRF4.
Our mutation analysis has shown that c-Src targets multiple sites on IRF4 and that Y61 and Y124 are two key sites responsible for IRF4 activation (Fig. 2). Although phosphorylation of Y61 has not been identified by phosphoproteome profiling, phosphorylation of Y124 has also been identified in Hodgkin's lymphomas (42). Thus, Y124 is a convincing phosphorylation site at least important for IRF4-associated lymphomas.
Our results have identified that the region spanning aa 255 to 412 is an important domain which inhibits IRF4 activity but that the domain spanning aa 412 to 420 is crucial for its activation (Fig. 2C). These results may provide additional details for previously identified functional domains for IRF4 (37). Further analysis with more deletion mutants is needed to define definite IRF4 functional domains.
Virus-associated cancers are endemic to the underserved minority community in South Florida and are increasing and expected to become significantly more prevalent in the United States. In future long-term pursuits, we will delineate the interaction of IRF4 with viral malignancies from aspects of oncogenesis as well as immunity, in both of which IRF4 is a crucial player (21, 47). These will include the identification of the potential lymphocyte-specific signaling pathways leading to IRF4 activation and a systematic and in-depth analysis of the functional role of IRF4 in viral oncogenesis, such as its potential activity in dampening IFN and other innate immune signaling pathways. These studies will highlight the importance of IRF4 in viral oncogenesis and will provide profound insights into the interaction of virus with this cellular factor in the development of cancer. Eventually, we expect to discover novel signaling pathways and novel molecules, such as the direct kinase(s) for IRF4, which may open up unique opportunities for therapeutic treatments of virus-associated diseases and cancers.
ACKNOWLEDGMENTS
This work is supported by the State of Florida Biomedical Research Programs (1BN-07) and the American Society of Hematology Scholar Award to S.N.
Footnotes
Published ahead of print 26 June 2013
REFERENCES
- 1.Takaoka A, Tamura T, Taniguchi T. 2008. Interferon regulatory factor family of transcription factors and regulation of oncogenesis. Cancer Sci. 99:467–478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harada H, Kitagawa M, Tanaka N, Yamamoto H, Harada K, Ishihara M, Taniguchi T. 1993. Anti-oncogenic and oncogenic potentials of interferon regulatory factor-1 and factor-2. Science 259:971–974 [DOI] [PubMed] [Google Scholar]
- 3.Xu D, Zhao L, Del Valle L, Miklossy J, Zhang L. 2008. Interferon regulatory factor 4 is involved in Epstein-Barr virus-mediated transformation of human B lymphocytes. J. Virol. 82:6251–6258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang L, Zhang J, Lambert Q, Der CJ, Del Valle L, Miklossy J, Khalili K, Zhou Y, Pagano JS. 2004. Interferon regulatory factor 7 is associated with Epstein-Barr virus-transformed central nervous system lymphoma and has oncogenic properties. J. Virol. 78:12987–12995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lohoff M, Mittrucker HW, Brustle A, Sommer F, Casper B, Huber M, Ferrick DA, Duncan GS, Mak TW. 2004. Enhanced TCR-induced apoptosis in interferon regulatory factor 4-deficient CD4(+) Th cells. J. Exp. Med. 200:247–253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang L, Toomey NL, Diaz LA, Walker G, Ramos JC, Barber GN, Ning S. 2011. Oncogenic IRFs provide a survival advantage for EBV- or HTLV1-transformed cells through induction of BIC expression. J. Virol. 85:8328–8337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martin HJ, Lee JM, Walls D, Hayward SD. 2007. Manipulation of the Toll-like receptor 7 signaling pathway by Epstein-Barr virus. J. Virol. 81:9748–9758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schaefer BC, Paulson E, Strominger JL, Speck SH. 1997. Constitutive activation of Epstein-Barr virus (EBV) nuclear antigen 1 gene transcription by IRF1 and IRF2 during restricted EBV latency. Mol. Cell. Biol. 17:873–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang L, Pagano JS. 1997. IRF7, a new interferon regulatory factor associated with Epstein-Barr virus latency. Mol. Cell. Biol. 17:5748–5757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang L, Pagano JS. 1999. Interferon regulatory factor 2 represses the Epstein-Barr virus BamHI Q latency promoter in type III latency. Mol. Cell. Biol. 19:3216–3223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cahir-McFarland ED, Carter K, Rosenwald A, Giltnane JM, Henrickson SE, Staudt LM, Kieff E. 2004. Role of NF-κB in cell survival and transcription of latent membrane protein 1-expressing or Epstein-Barr virus latency III-infected cells. J. Virol. 78:4108–4119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Spender LC, Lucchesi W, Bodelon G, Bilancio A, Karstegl CE, Asano T, Dittrich-Breiholz O, Kracht M, Vanhaesebroeck B, Farrell PJ. 2006. Cell target genes of Epstein-Barr virus transcription factor EBNA-2: induction of the p55a regulatory subunit of PI3-kinase and its role in survival of EREB2.5 cells. J. Gen. Virol. 87:2859–2867 [DOI] [PubMed] [Google Scholar]
- 13.Iida S, Rao PH, Butler M, Corradini P, Boccadoro M, Klein B, Chaganti RSK, la-Favera R. 1997. Deregulation of MUM1/IRF4 by chromosomal translocation in multiple myeloma. Nat. Genet. 17:226–230 [DOI] [PubMed] [Google Scholar]
- 14.Shaffer AL, Emre NCT, Lamy L, Ngo VN, Wright G, Xiao W, Powell J, Dave S, Yu X, Zhao H, Zeng Y, Chen B, Epstein J, Staudt LM. 2008. IRF4 addiction in multiple myeloma. Nature 454:226–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mamane Y, Grandvaux N, Hernandez E, Sharma S, Innocente SA, Lee JM, Azimi N, Lin R, Hiscott J. 2002. Repression of IRF-4 target genes in human T cell leukemia virus-1 infection. Oncogene 21:6751–6765 [DOI] [PubMed] [Google Scholar]
- 16.Mamane Y, Sharma S, Grandvaux N, Hernandez E, Hiscott J. 2002. IRF-4 activities in HTLV-I-induced T cell leukemogenesis. J. Interferon Cytokine Res. 22:135–143 [DOI] [PubMed] [Google Scholar]
- 17.Ramos JC, Ruiz P, Jr, Ratner L, Reis IM, Brites C, Pedroso C, Byrne GEJ, Toomey NL, Andela V, Harhaj EW, Lossos IS, Harrington WJ., Jr 2007. IRF4 and c-Rel expression in antiviral-resistant adult T-cell leukemia/lymphoma. Blood 109:3060–3068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sharma S, Grandvaux N, Mamane Y, Genin P, Azimi N, Waldmann T, Hiscott J. 2002. Regulation of IFN regulatory factor 4 expression in human T cell leukemia virus-I-transformed T cells. J. Immunol. 169:3120–3130 [DOI] [PubMed] [Google Scholar]
- 19.Sharma S, Mamane Y, Grandvaux N, Bartlett J, Petropoulos L, Lin R, Hiscott J. 2000. Activation and regulation of interferon regulatory factor 4 in HTLV type 1-infected T lymphocytes. AIDS Res. Hum. Retroviruses 16:1613–1622 [DOI] [PubMed] [Google Scholar]
- 20.Rui L, Schmitz R, Ceribelli M, Staudt LM. 2011. Malignant pirates of the immune system. Nat. Immunol. 12:933–940 [DOI] [PubMed] [Google Scholar]
- 21.Shaffer AL, Emre NC, Romesser PB, Staudt LM. 2009. IRF4: immunity. Malignancy! Therapy? Clin. Cancer Res. 15:2954–2961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chang CC, Lorek J, Sabath DE, Li Y, Chitambar CR, Logan B, Kampalath B, Cleveland RP. 2002. Expression of MUM1/IRF4 correlates with clinical outcome in patients with B-cell chronic lymphocytic leukemia. Blood 100:4671–4675 [DOI] [PubMed] [Google Scholar]
- 23.Sundram U, Harvell JD, Rouse RV, Natkunam Y. 2003. Expression of the B-cell proliferation marker MUM1 by melanocytic lesions and comparison with S100, gp100 (HMB45), and MelanA. Mod. Pathol. 16:802–810 [DOI] [PubMed] [Google Scholar]
- 24.Feldman AL, Law M, Remstein ED, Macon WR, Erickson LA, Grogg KL, Kurtin PJ, Dogan A. 2009. Recurrent translocations involving the IRF4 oncogene locus in peripheral T-cell lymphomas. Leukemia 23:574–580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Havelange V, Pekarsky Y, Nakamura T, Palamarchuk A, Alder H, Rassenti L, Kipps T, Croce CM. 2011. IRF4 mutations in chronic lymphocytic leukemia. Blood 118:2827–2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang B, Kracker S, Yasuda T, Casola S, Vanneman M, Homig-Holzel C, Wang Z, Derudder E, Li S, Chakraborty T, Cotter SE, Koyama S, Currie T, Freeman GJ, Kutok JL, Rodig SJ, Dranoff G, Rajewsky K. 2012. Immune surveillance and therapy of lymphomas driven by Epstein-Barr virus protein LMP1 in a mouse model. Cell 148:739–751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pedersen I, David M. 2008. MicroRNAs in the immune response. Cytokine 43:391–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Xiao C, Rajewsky K. 2009. MicroRNA control in the immune system: basic principles. Cell 136:26–36 [DOI] [PubMed] [Google Scholar]
- 29.Kluiver J, Haralambieva E, de Jong D, Blokzijl T, Jacobs S, Kroesen BJ, Poppema S, van den Berg A. 2006. Lack of BIC and microRNA miR-155 expression in primary cases of Burkitt lymphoma. Genes Chromosomes Cancer 45:147–153 [DOI] [PubMed] [Google Scholar]
- 30.Lin Z, Flemington EK. 2011. miRNAs in the pathogenesis of oncogenic human viruses. Cancer Lett. 305:186–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.van den Berg A, Kroesen BJ, Kooistra K, de Jong D, Briggs J, Blokzijl T, Jacobs S, Kluiver J, Diepstra A, Maggio E, Poppema S. 2003. High expression of B-cell receptor inducible gene BIC in all subtypes of Hodgkin lymphoma. Genes Chromosomes Cancer 37:20–28 [DOI] [PubMed] [Google Scholar]
- 32.Cho WC. 2007. OncomiRs: the discovery and progress of microRNAs in cancers. Mol. Cancer 6:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garzon R, Croce CM. 2008. MicroRNAs in normal and malignant hematopoiesis. Curr. Opin. Hematol. 15:352–358 [DOI] [PubMed] [Google Scholar]
- 34.Cameron JE, Fewell C, Yin Q, McBride J, Wang X, Lin Z, Flemington EK. 2008. Epstein-Barr virus growth/latency III program alters cellular microRNA expression. Virology 382:257–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jiang J, Lee EJ, Schmittgen TD. 2006. Increased expression of microRNA-155 in Epstein-Barr virus transformed lymphoblastoid cell lines. Genes Chromosomes Cancer 45:103–106 [DOI] [PubMed] [Google Scholar]
- 36.Yin Q, McBride J, Fewell C, Lacey M, Wang X, Lin Z, Cameron J, Flemington EK. 2008. MicroRNA-155 is an Epstein-Barr virus-induced gene that modulates Epstein-Barr virus-regulated gene expression pathways. J. Virol. 82:5295–5306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.De Silva NS, Simonetti G, Heise N, Klein U. 2012. The diverse roles of IRF4 in late germinal center B-cell differentiation. Immunol. Rev. 247:73–92 [DOI] [PubMed] [Google Scholar]
- 38.Thai T-H, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL, Schmidt-Supprian M, Rajewsky N, Yancopoulos G, Rao A, Rajewsky K. 2007. Regulation of the germinal center response by microRNA-155. Science 316:604–608 [DOI] [PubMed] [Google Scholar]
- 39.Roughan JE, Thorley-Lawson DA. 2009. The intersection of Epstein-Barr virus with the germinal center. J. Virol. 83:3968–3976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Biswas PS, Gupta S, Chang E, Song L, Stirzaker RA, Liao JK, Bhagat G, Pernis AB. 2010. Phosphorylation of IRF4 by ROCK2 regulates IL-17 and IL-21 production and the development of autoimmunity in mice. J. Clin. Invest. 120:3280–3295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.St-Germain JR, Taylor P, Tong J, Jin LL, Nikolic A, Stewart II, Ewing RM, Dharsee M, Li Z, Trudel S, Moran MF. 2009. Multiple myeloma phosphotyrosine proteomic profile associated with FGFR3 expression, ligand activation, and drug inhibition. Proc. Natl. Acad. Sci. U. S. A. 106:20127–20132 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gu TL, Cherry J, Tucker M, Wu J, Reeves C, Polakiewicz RD. 2010. Identification of activated Tnk1 kinase in Hodgkin's lymphoma. Leukemia 24:861–865 [DOI] [PubMed] [Google Scholar]
- 43.Ning S, Huye LE, Pagano JS. 2005. Regulation of the transcriptional activity of the IRF7 promoter by a pathway independent of interferon signaling. J. Biol. Chem. 280:12262–12270 [DOI] [PubMed] [Google Scholar]
- 44.O'Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. 2007. MicroRNA-155 is induced during the macrophage inflammatory response. Proc. Natl. Acad. Sci. U. S. A. 104:1604–1609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Alvarez RH, Kantarjian HM, Cortes JE. 2006. The role of Src in solid and hematologic malignancies. Cancer 107:1918–1929 [DOI] [PubMed] [Google Scholar]
- 46.Rucci N, Susa M, Teti A. 2008. Inhibition of protein kinase c-Src as a therapeutic approach for cancer and bone metastases. Anticancer Agents Med. Chem. 8:342–349 [DOI] [PubMed] [Google Scholar]
- 47.Gualco G, Weiss LM, Bacchi CE. 2010. MUM1/IRF4: a review. Appl. Immunohistochem. Mol. Morphol. 18:301–310 [DOI] [PubMed] [Google Scholar]
- 48.Irby RB, Yeatman TJ. 2000. Role of Src expression and activation in human cancer. Oncogene 19:5636–5642 [DOI] [PubMed] [Google Scholar]
- 49.Mamane Y, Hiscott J. 2001. Ph.D. thesis. McGill University, Montreal, Canada [Google Scholar]
- 50.Banerjee S, Lu J, Cai Q, Saha A, Jha HC, Dzeng RK, Robertson ES. 2013. The EBV latent antigen 3C inhibits apoptosis through targeted regulation of interferon regulatory factors 4 and 8. PLoS Pathog. 9:e1003314. 10.1371/journal.ppat.1003314 [DOI] [PMC free article] [PubMed] [Google Scholar]